

Abstract
Members of the chicken ovalbumin upstream promoter-transcription factor (COUP-TF) subfamily of orphan nuclear receptors, which minimally includes COUP-TFI and ARP1, are highly expressed in brain and are generally considered to be constitutive repressors of transcription. We have used a yeast two-hybrid system to isolate proteins expressed in brain that interact with ARP1. One of the proteins isolated in this screen was Ear2, another orphan receptor that has been suggested to be a member of the COUP-TF subfamily. Here we demonstrate that ARP1 and Ear2 form heterodimers in solution and on directly repeated response elements with high efficiency and a specificity differing from that of homodimeric complexes composed of either receptor. ARP1 and Ear2 were observed to interact in mammalian cells, and the tissue distribution of Ear2 transcripts was found to overlap precisely with the expression pattern of ARP1 in several mouse tissues and embryonal carcinoma cell lines. Heterodimeric interactions between ARP1 and Ear2 may define a distinct pathway of orphan receptor signaling.
COUP-TF1 orphan nuclear receptors have been reported to interfere with the signaling pathways of several nuclear receptors including retinoic acid (1–5), thyroid hormone (3), estrogen (6–9), and vitamin D3 (4) receptors, as well as peroxisome proliferator-activated receptor α (10, 11). In addition, COUP-TFs negatively modulate hepatic nuclear factor 4- (12, 13), RZR-/ROR- (14), and Nur77 (15)-mediated transcriptional activation. Four mechanisms have been postulated to underlie modulation of multiple signaling pathways by COUP-TF subfamily members: 1) competition for DNA response element binding, 2) inactive heterocomplex formation including titration of retinoid X receptor (RXR), 3) active silencing of basal transcription, and 4) transcriptional transrepression (16). Of these potential mechanisms, competition for binding to response elements is well documented and is based upon the ability of COUP-TF proteins to bind a large variety of response elements (16). COUP-TFI and ARP1 are known to form homodimers in solution and to bind promiscuously as such to direct repeats (DR) of the canonical half-site AGGTCA exhibiting the highest affinity for a DR spaced by 1 base pair (DR1; Refs. 1 and 3).
ARP1 and COUP-TFI share extensive identity, particularly in the ligand and DNA binding domains (Refs. 12, 17, and 18; see also Fig. 1). Ear2 is less well conserved with either protein in these domains (Fig. 1), which has led some to suggest that Ear2 may not be a member of COUP-TF subfamily of nuclear receptors (16). However, based on compelling evolutionary arguments, Escriva and co-workers (19) have placed Ear2 within the COUP-TF subfamily. Moreover, other groups consider Ear2 to be a member of COUP-TF subfamily based on the capacity of this protein to form homodimeric, DNA binding complexes and to repress ligand-dependent activation of target genes mediated by other nuclear receptors, such as retinoic acid (1, 20) and estrogen (7–9) receptors, in a manner similar to that of COUP-TFI and ARP1.
Fig. 1. Relationship between COUP-TF family members.
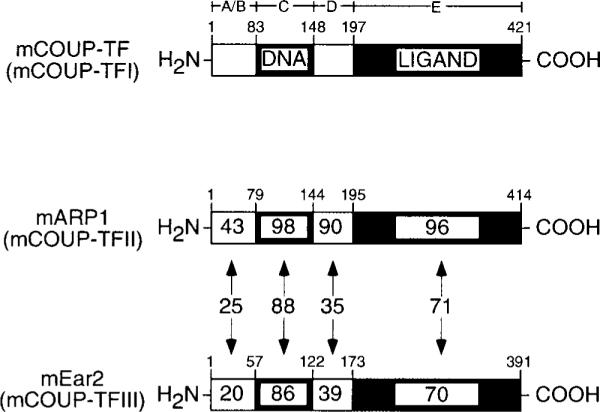
The amino acid identities between domains of COUP-TFI and both ARP1 and Ear2 are indicated as percentages within each schematic domain. The corresponding identities between ARP1 and Ear2 are indicated within double-headed arrows. Alignments were carried out using Clustal X (version 1.63b) and the following GenBank™ accession numbers: COUP-TFI, gi466468; ARP1, gi482927; Ear2, gi482930.
COUP-TFI and ARP1 play important roles in development, particularly in patterning of the nervous system in Xenopus (21), Drosophila (22, 23), and mammals (24, 25). COUP-TFI null animals exhibit perinatal lethality, possibly arising from malformations of the glossopharyngeal ganglion and associated IXth cranial nerve resulting in an inability to feed properly (25). Given the overall similarity between COUP-TFI and ARP1, both in terms of structural conservation and overlapping patterns of expression in the developing central nervous system (24), it seems remarkable that ARP1 does not compensate for the lack of COUP-TFI expression in these animals (25). Knock-out of the ARP1 gene in the mouse is apparently embryonic lethal (16).
In this report, we demonstrate that Ear2 and ARP1 form heterodimers in yeast and in vitro, both in solution and on directly repeated response elements. Moreover, these orphan receptors were shown to interact in mammalian cells and to be coexpressed in the embryo and in two embryonal carcinoma cell lines as well as in several adult tissues suggesting that ARP1•Ear2 heterodimeric complexes may play a role(s) in embryonic development and in the adult organism.
MATERIALS AND METHODS
Plasmids and Constructs
The bait vectors (pBTM116 and pBL1) for the yeast two-hybrid screen and the yeast reporter strains L40 (26) and PL3 (27) were all generous gifts from Drs. R. Losson and P. Chambon (IGBMC, Illkirch, France). Human ARP1 (ARP1, Ref. 28) was a kind gift from Dr. H. Nakshatri, and NR1 was a kind gift from Dr. S. Nakanishi. A fragment corresponding to the hinge and putative ligand binding domain of ARP1 (regions D and E, amino acids 144–414) was amplified by PCR and cloned into both pBTM116 and pBL1 (26), encoding LexA DBD-ARP1 DE and ER DBD-ARP1 DE fusion proteins, respectively. Receptor bait constructs for mouse RXRα (amino acids 132–467) and human RARγ (amino acids 90–454) have been described previously (29). An unrelated bait corresponding to the carboxyl tail (amino acids 835–938) of the NR1 subunit of the N-methyl-D-aspartate receptor (30) was similarly prepared for these studies. All fragments amplified by PCR were verified by sequence analysis.
A mammalian expression vector encoding an ER DBD/ARP1 DE fusion protein was constructed by inserting the EcoRI fragment of pBL1-ARP1 DE into the EcoRI site of pTL1 (31). A mammalian expression vector encoding the DE region of mouse Ear2 (amino acids 122–390) fused to the transcriptional activation domain of GAL4 (GAL4 AD; amino acids 768–881) was prepared by insertion of the corresponding Ear2 fragment into pACT2 (CLONTECH, Palo Alto, CA), yielding pACT2-Ear2 DE. This plasmid was then digested with HindIII, excising the GAL4 AD-Ear2 DE fragment, which was subsequently cloned into the eukaryotic expression vector, pTL1. An amino-terminal truncation mutant of ARP1, mycARP1ΔAB, with a myc epitope tag at the amino terminus was prepared by PCR amplification of an appropriate fragment and insertion into pTL1. Full-length Ear2, fused to an HA epitope tag and the GAL 4 AD, was excised from the library plasmid (pACT2) by digestion with HindIII, and the resulting fragment was ligated into pTL1 creating HA-Ear2. The latter two constructs were prepared for use in DNA binding experiments designed to distinguish between heterodimeric complexes of intermediate electrophoretic mobility and homodimeric complexes composed of either receptor. The ER-responsive CAT reporter, 17-mer-ERE-globin-CAT, has been described previously (32).
GST fusion proteins were prepared for ARP1 DE (GST-ARP1 DE) and Ear2 DE (GST-Ear2 DE) by PCR amplification of the corresponding fragments and subcloning into pGEX-2T (Amersham Pharmacia Biotech, Uppsala, Sweden) that had been previously digested with BamHI and EcoRI. The integrity of all constructs was verified by sequence analysis.
Yeast Two-hybrid Screen
Saccharomyces cerevisiae L40 reporter strain expressing ARP1 (D and E regions) was transformed with a mouse brain cDNA library (CLONTECH) fused to the GAL4 AD on a leucine-selectable vector, pACT2. The screen for ARP1-interacting clones was conducted in synthetic medium lacking tryptophan, leucine, and histidine and supplemented with 20 mM 3-aminotriazole. The plasmids from positive clones containing the brain cDNAs were rescued, shuttled in Escherichia coli, and sequenced using the dideoxynucleotide dye terminator method on an Applied Biosystems, Inc., model 373 or 377 sequencer (Applied Biosystems, Inc., Foster City, CA) at the Central Services Laboratory of the Oregon State University Center for Gene Research and Biotechnology.
β-Galactosidase Assays
Filter assays were utilized to confirm the positive clones. For quantitative purposes the positive clones were grown in synthetic medium to an A600 of approximately 2 units, and β-galactosidase activity was assayed as described by Kippert (33), and modified by Avram and Bakalinsky (34). Ligand-dependent assays were carried out by inclusion of 1 μM 9-cis-RA or identical amounts of vehicle in the culture medium.
Protein-Protein Interaction Assays
GST pull-down experiments were conducted as described previously (29, 35).
Electrophoretic Mobility Shift Assays
EMSAs were conducted essentially as described previously (31). ARP1ΔAB and HA-Ear2 were expressed by in vitro transcription/translation in rabbit reticulocyte lysates (Promega). In each case the receptors used for gel retardation assays were translated in parallel in the presence of [35S]methionine for the purpose of quantification. The DR1(G) response element upper strand had the following sequence (canonical half-sites are bold and underlined): 5′-TCGAGGGTAGAGGTCAGAGGTCACTCG-3′; DR2(G) through DR5(G) were synthesized with identical flanking and repeated hexanucleotide sequence but with the following inter-repeat spacers (5′ to 3′): GA (DR2), CGA (DR3), CGAA (DR4), and CCGAA (DR5). The DR(T) series of oligonucleotides were identical to the DR(G) series except that the repeated hexanucleotide had the sequence AGTTCA.
Cell Culture and Transfections
Human embryonic kidney (HEK) 293 cells (ATCC CRL 1573) were cultured in minimum essential medium with Earle's salts (Sigma) supplemented with 10% fetal bovine serum (Summit, Boulder, CO) and 4 mM glutamine (Life Technologies, Inc.). Cells were grown to 50–60% confluence and transiently transfected using the calcium phosphate method. Each 100-mm plate was cotransfected with 1 μg of expression vector encoding the ER DBD or ER DBD/ARP1 DE, 1 μg of GAL4 AD, or GAL4 AD/Ear2 DE, 2 μg of the 17-mer-ERE-Globin-CAT reporter construct, and 10 ng of pCMV-SPORT-βgal (Life Technologies, Inc.) encoding β-galactosidase which was used to normalize for transfection efficiency. Cells were harvested 48 h after transfection, and extracts were prepared using standard techniques. β-Galactosidase activity in cellular extracts was quantified using a colorimetric assay, and CAT activity was determined by thin layer chromatography.
Reverse Transcription-Polymerase Chain Reaction
First strand cDNA was synthesized using 1 μg of RNA (Ambion, Austin, TX) and 100 ng of random hexamer primers (Promega) in a buffer containing 50 mM Tris-HCl, pH 8.3, 75 mM KCl, 3 mM MgCl2, 10 mM dithiothreitol, 100 μM concentrations of each dNTP, and 200 units of Moloney murine leukemia virus reverse transcriptase (Life Technologies, Inc.) in a total volume of 25 μl, and the reactions were carried out for 30 min at 37 °C. One μl of each reverse transcription reaction was subjected to PCR amplification using ARP1-, Ear2-, and 36B4 (36)-specific primers. ARP1 and Ear2 amplifications were carried out for 25 cycles, whereas amplification of 36B4 was conducted for 19 cycles. Amplification of 36B4 was used as a control for the quantity of cDNA present in each sample as well in normalization of gel loading and Southern blotting. Note that, in general, one cycle consisted of the following steps: 94 °C × 30 s, 50 °C for 45 s, 72 °C for 60 s; however, the annealing temperature was varied for each set of primers to optimize amplification. Amplification products were separated on a 2% agarose gel and then transferred to ZetaProbe membranes (Bio-Rad). The blots were probed with specific, end-labeled oligonucleotides corresponding to internal sequence in the PCR products. RT-PCR analyses of ARP1 and Ear2 expression in F9 and P19 embryonal carcinoma cells treated in monolayers with either vehicle or trans-retinoic acid for 24 h were carried out as described above.
RESULTS
Ear2 Interacts with ARP1 in a Yeast Two-hybrid System
A fragment encoding the ARP1 hinge region and putative ligand binding domain was used as a bait in a yeast two-hybrid screen to identify proteins expressed in brain that interact with ARP1, which is highly expressed in brain (Refs. 18, 24, 37, and this report). A protein found to interact with ARP1 was Ear2, another member of steroid hormone receptor superfamily, which is closely related to COUP-TF subfamily, that includes COUP-TFI and ARP1 (Fig. 1).
The Ear2 clone isolated in our screen contains the entire open reading frame identified by Jonk and co-workers (18). In addition, the present clone contains 183 base pairs upstream of the putative initiator methionine previously described (18). However, as there is no in frame stop codon(s) in this upstream sequence, it is possible that the actual Ear2 protein is extended in the amino terminus beyond the previously identified initiator methionine (18).
To evaluate the specificity of ARP1-Ear2 interaction, we examined interactions of Ear2 with RAR, RXR, and an unrelated bait, the carboxyl terminus of the NR1 subunit of the N-methyl-D-aspartate receptor (30). The interaction of Ear2 with ARP1 was specific inasmuch as Ear2 did not interact with RXRα or RARα in the presence or absence of 9-cis-retinoic acid (Fig. 2). Similarly, Ear2 did not interact with the carboxyl terminus of NR1 (Fig. 2).
Fig. 2. Interaction between Ear2 and ARP1, RXRα, RARγ, and NR1 in yeast.

LexA DBD/receptor fusion baits (ARP1, RXRα, RARγ, and NR1) were coexpressed together with Gal4 AD/Ear2 (pACT2:Ear2) or GAL4 AD (pACT2) and examined for the ability to activate LexAop-lacZ reporter in the yeast strain L40. Ligand-dependent interactions were examined in the presence of 1 μM 9-cis-retinoic acid (9cRA). The results shown represent the mean ± S.E. of three independent experiments in which individual transformants were assayed.
Ear2 and ARP1 Interact in Vitro
A qualitative, in vitro protein-protein interaction study was conducted to confirm the ARP1-Ear2 interaction observed in yeast and to investigate the ability of each protein to self-associate. The hinge and putative LDB regions of each protein were fused to glutathione S-transferase (GST), creating GST-ARP1 DE and GST-Ear2 DE, and both proteins were expressed in bacteria for use in standard GST pull-down experiments (29, 35). [35S]Met-labeled Ear2 interacted with GST-ARP1 DE (Fig. 3A, lane 3) confirming the interaction observed in yeast. [35S]Met-labeled Ear2 was also found to interact with itself (GST-Ear2 DE, lane 4) with efficiency nearly identical to that with which it interacted with GST-ARP1 DE (lane 3) suggesting that Ear2 may form heterodimeric and homodimeric complexes with equal facility. Similarly, [35S]Met-labeled ARP1ΔAB interacted with both GST-Ear2 DE and -ARP1 DE fusion proteins (Fig. 3B, lanes 3 and 4, respectively) but not with glutathione charged with GST alone (lane 2). These in vitro findings corroborate results obtained in yeast (Fig. 2) and also suggest that ARP1 and Ear2 participate in heterodimeric interactions in solution with an apparent efficiency comparable to that of the homodimerization involving either receptor.
Fig. 3. In vitro interaction between ARP1 and Ear2.

GST/ARP1 DE and GST/EAR2 DE were bound to glutathione-Sepharose and used as affinity matrices to examine the interaction with the in vitro translated [35S]methionine-labeled Ear2 (A) and ARP1 ΔAB (B). Approximately 10% of input [35S]methionine-labeled protein was retained on GST fusion proteins in all cases. Shown are representative experiments that were replicated 4–6 times.
ARP1 and Ear2 Form Heterodimers on Directly Repeated Response Elements
COUP-TFI, ARP1, and Ear2 form homodimers in solution and bind promiscuously to direct repeats (DR) of the canonical half-site, AGGTCA, with varied inter-repeat spacing. However, all of these receptors exhibit a degree of selectivity for DRs spaced by a single base pair (DR1) (1, 3). In addition, COUP-TFI and ARP1 bind as a heterodimeric complex to DR elements composed of varied spacing (3). Based on our observation that ARP1 and Ear2 form heterodimers in yeast (Fig. 2) and in solution (Fig. 3), we conducted electrophoretic mobility shift assays (EMSAs) to determine if ARP1 and Ear2 bind to directly repeated response elements as a heterodimeric complex. mycARP1ΔAB homodimeric complexes bound in a protein concentration-dependent manner to the DR1 probe (Fig. 4, lanes 1–3; complex C5). HA-Ear2 formed two complexes, C1 and C2, that exhibited differential electrophoretic mobility on this probe (Fig. 4, lanes 4 and 5). The two species of HA-Ear2, which bound to this probe in a protein concentration-dependent manner, most likely arose from correct and internal initiation of HA-Ear2 translation in rabbit reticulocyte lysates (data not shown). At comparable amounts of protein, HA-Ear2 (C1 and C2) clearly bound to the DR1 probe less efficiently than mycARP1ΔAB (C5; compare lanes 1–3 to 4–6 of Fig. 4). However, incubation of a fixed amount of mycARP1ΔAB with increasing amounts of HA-Ear2 resulted in the complete titration of the mycARP1ΔAB homodimeric complex (C5) into mycARP1ΔAB·HA-Ear2 heterodimeric complexes, C3 and C4 (Fig. 4, lanes 7–9). When excessive amounts of HA-Ear2 were used, the appearance of HA-Ear2 homodimeric complexes (C1 and C2) were also evident (Fig. 4, lanes 8–9). Similarly, titration of a fixed amount of HA-Ear2 with increasing amounts of mycARP1ΔAB quantitatively shifted HA-Ear2 homodimeric complexes (C1 and C2) into mycARP1ΔAB·HA-Ear2 heterodimeric complexes (C3 and C4; Fig. 4, lanes 10–12). The homodimeric mycARP1ΔAB complex (C5) was apparent when this protein was used in excess (Fig. 4, lanes 11 and 12). Complexes C1/C2 and C5 were supershifted with anti-HA and anti-myc antibodies, respectively, whereas both antibodies supershifted complexes C3 and C4, confirming the identity of each complex (data not shown).
Fig. 4. Formation of ARP1 and Ear2 homodimers and heterodimers on the DR1(G) probe.
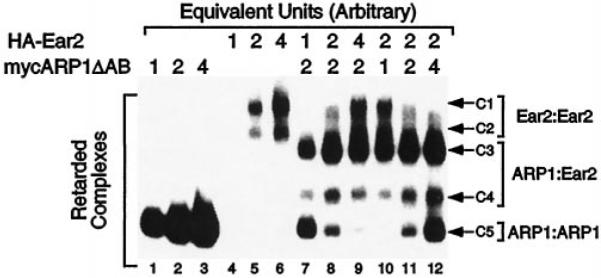
Lanes 1–3 and 4–6 contain increasing amounts of mycARP1ΔAB and HA-Ear2 as indicated. Lanes 7–9 represent titration of a fixed amount of mycARP1ΔAB with increasing amounts of HA-Ear2, and lanes 10–12 correspond to the converse titration. The position of homodimeric HA-Ear2 (C1 and C2) and mycARP1ΔAB (C5) complexes as well as heterodimeric mycARP1ΔAB·HA-Ear2 complexes (C3 and C4) are indicated to the right of this EMSA gel. Electrophoresis was carried out for about 1 h after the free probe ran out of the gel to maximize resolution of the multiple complexes. A representative experiment is shown which was replicated five times. Note that the amount of reticulocyte lysate in each lane was held constant by addition of unprogrammed lysate.
ARP1·Ear2 Heterodimeric Complexes Bind Directly Repeated Response Elements with Specificity Differing from Homodimers of Either Receptor
We next sought to determine if the capacity of ARP1 and Ear2 to bind directly repeated response elements as homodimeric and/or heterodimeric complexes was a function of the length of the inter-repeat spacer. mycARP1ΔAB homodimers bound to probes with spacing ranging from 1 to 5 base pairs (complex C5; Fig. 5A, lanes 1, 4, 7, 10, and 13) with somewhat varied efficiency (DR2 ≥ DR1 > DR5 > DR3 > DR4). Conversely, HA-Ear2 did not appear to bind any of these probes with the exception of DR1, to which HA-Ear2 homodimers bound weakly at the relatively low amount of HA-Ear2 protein used in these experiments (Fig. 5B, lanes 2, 5, 8, 11, and 14). mycARP1ΔAB·HA-Ear2 heterodimeric complexes (C3 and C4) bound to probes of all spacings with a distinct preference for DR1 and DR2 (Fig. 5A, lanes 3, 6, 9, 12, and 15). Thus, mycARP1ΔAB×HA-Ear2 heterodimeric complexes exhibited a DNA binding specificity (DR1 ≥ DR2 > DR4 > DR5 > DR3) that was clearly distinct from that of HA-Ear2 homodimers (which bound weakly to DR1 only) and subtly distinct from mycARP1ΔAB homodimeric complexes (see above).
Fig. 5. ARP1 and Ear2 homodimer and heterodimer formation on DR(G) (A) and DR(T) (B) series of probes of varied spacing.
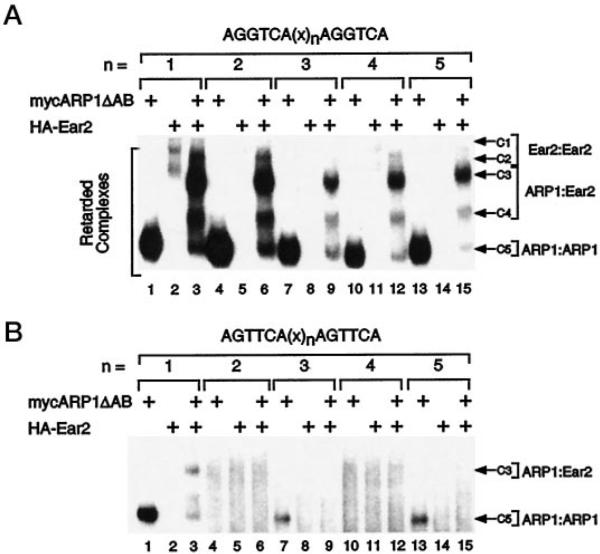
The amounts of mycARP1ΔAB and HA-Ear2 used in these EMSA experiments correspond to 1 and 2 arbitrary units, respectively (see Fig. 4). Identification of complexes is as described in the legend to Fig. 4. The length of the inter-repeat spacer is given by n, and the sequence of this spacer is given under “Materials and Methods.” Note that the autoradiograph corresponding to B was exposed to film longer than that of A to allow visualization of weak complexes. Shown is a representative experiment that was replicated five (A) or three (B) times.
Methylation interference studies have implicated a role for the guanines present in each half-site of a DR1-type response element in the specific interaction of COUP-TFI with DNA (38). To address the potential role of the second guanine (G2) in each AGGTCA half-site in DNA recognition by ARP1·ARP1, ARP1·Ear2, and Ear2·Ear2 complexes, EMSAs were carried out using a series of probes in which G2 in each half-site was mutated to thymidine (see Fig. 5B). This G2 → T mutation had a dramatic effect on the efficiency of both mycARP1ΔAB homodimer and mycARP1ΔAB·HA-Ear2 heterodimer binding to probes of all spacing (Fig. 5B). However, mycARP1ΔAB homodimers did bind to the “T” series probe with a single base pair spacer, DR1(T) (Fig. 5B, lane 1), and with lower efficiency to DR5(T) (lane 13) and DR3(T) (lane 7). In contrast, mycARP1ΔAB·HA-Ear2 heterodimer binding was not evident on any of the “T” series probes with the possible exception of DR1(T) (Fig. 5B, lane 3). However, HA-Ear2 appeared to inhibit mycARP1ΔAB homodimer binding to DR1(T), DR3(T), and DR5(T) (Fig. 5B, lanes 3, 9, and 15, respectively), the latter of which is identical to the RARE in the RARβ promoter (39). These findings suggest that mycARP1ΔAB and HA-Ear2 may form a heteromeric complex in solution that is not competent for DNA binding to the T series of response elements. Consistent with this hypothesis, mycARP1ΔAB and HA-Ear2 interacted in DNA-binding independent, GST pull-down experiments (Fig. 3, A and B).
ARP1 and Ear2 Interact in Mammalian Cells
Although ARP1 and Ear2 interact in yeast and in vitro, both in solution and on various directly repeated response elements, a physiologically relevant interaction between these two proteins would only occur in cells in which both proteins were expressed. Thus, cotransfection experiments were carried out to determine if the two proteins are capable of interacting in cells using a mammalian two-hybrid system. HEK 293 cells were cotransfected with a plasmid (ER DBD-ARP1 DE) encoding ARP1 DE fused to ER DBD together with a second plasmid (Ear2 DE-GAL4 AD) encoding Ear2 DE fused to GAL4 AD. The previously described 17-mer-ERE-globin-CAT (32) was used as a reporter construct in these studies. ER DBD alone (Fig. 6, lane 4) or when cotransfected with the GAL4 AD (lane 5) failed to activate the reporter construct. Similarly, Ear2 DE-GAL4 AD did not activate the reporter plasmid when cotransfected with the ER DBD (Fig. 6, lane 6). However, cotransfection of ER DBD-ARP1 DE together with Ear2 DE-GAL4 AD resulted in strong induction of the reporter suggesting that Ear2 and ARP1 are capable of interaction in mammalian cells.
Fig. 6. ARP1 and Ear2 interact in HEK 293 cells.

Cells were cotransfected with the indicated mammalian expression vectors and the 17-mer-ERE-globin-CAT reporter. Cells were harvested 24 h after transfection, and extracts were prepared using standard techniques. Transfection efficiency was normalized across treatments using a cotransfected β-galactosidase expression vector, and CAT activity was determined using [14C]chloramphenicol and acetyl-CoA. Acetylated and unacetylated [14C]chloramphenicol were separated by thin layer chromatography and visualized by autoradiography. A representative experiment is shown that was replicated three times.
Ear2 and ARP1 Expression Patterns Overlap in Several Mouse Tissues
Based on the observation that ARP1 and Ear2 interact in yeast, in solution, on DNA, and in mammalian cells, we next determined if the corresponding genes, Ear2 and Arp1, may exhibit overlapping patterns of expression in several mouse tissues that would facilitate a biologically relevant interaction between the two proteins.
Jonk and co-workers (18) have previously observed, by in situ hybridization, high level Arp1 expression in embryonal brain, lung, and kidney, whereas transcripts derived from the Ear2 gene were found to be present at lower levels and in several tissues in the developing embryo (8.5–14.5 d.p.c.). We have examined Arp1 and Ear2 expression in several tissues of the adult mouse by RT-PCR. The expression patterns of both genes were found to overlap precisely with the highest level of expression in brain, lung, kidney, and in the embryo (10–12 d.p.c.; Fig. 7A). Neither gene appeared to be expressed in thymus, spleen, and testis (Fig. 7A). Although these studies demonstrate that Arp1 and Ear2 genes are expressed in similar tissues, one cannot conclude from these data that the two orphan receptors are expressed in the same cell type(s) that would obviously be required for a physiologically relevant interaction to occur. To address the possibility that Arp1 and Ear2 may be coexpressed in a single cell type, RT-PCR analyses were conducted using RNA derived from two embryonal carcinoma cell lines, F9 and P19. These analyses revealed that Ear2 was constitutively expressed in both P19 and F9 cells and was unaffected by treatment of either cell line with all-trans-retinoic acid (RA; Fig. 7B), consistent with the finding of Jonk and co-workers (18). Arp1 was also found to be constitutively expressed in both cells lines and did not appear to be induced by treatment with RA (Fig. 7C), in contrast to previous studies (18). However, the treatment protocol employed in the current studies involved exposure to RA for only 24 h, whereas Jonk and co-workers (18) observed maximal induction of Arp1 in P19 cells by Northern analysis after a 72-h treatment with RA.
Fig. 7. Tissue distribution of ARP1 and Ear2 transcripts.
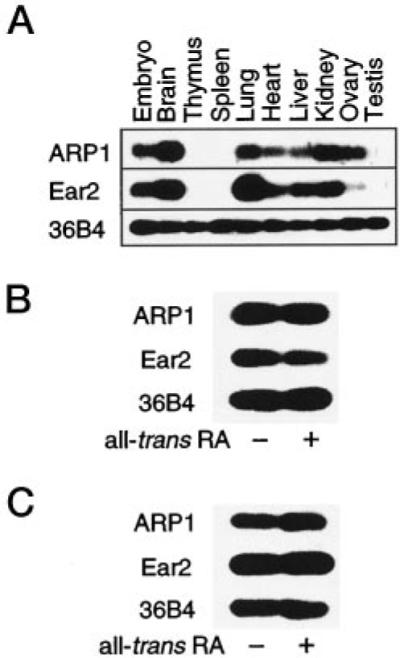
A, ARP1 and Ear2 transcripts were detected in the embryo (10–12 d.p.c.) and the indicated adult mouse tissues by RT-PCR and Southern blotting as described under “Materials and Methods.” An amplified fragment of the ubiquitously expressed 36B4 gene was used for normalization purposes (36). B and C, RT-PCR/Southern analyses of Arp1, Ear2, and 36B4 expression in F9 and P19 embryonal carcinoma cells, respectively. Each cell type was treated with vehicle (0.1% v/v ethanol) or 1 μM all-trans-RA as indicated for 24 h prior to preparation of RNA.
DISCUSSION
ARP1 and Ear2 were shown to interact in yeast, in vitro (both in solution and on various directly repeated response elements), and in mammalian cells. The two orphan receptors exhibit overlapping patterns of tissue expression and are coexpressed in two pluripotent, embryonal carcinoma cell lines. ARP1 and Ear2 clearly are capable of interaction and, at least in the cell lines and possibly the tissues examined, these two proteins have the opportunity to do so.
In addition to the ARP1·Ear2 complex described in this report, we have also observed the formation of COUP-TFI·ARP1 as well as COUP-TFI·Ear2 complexes on a DR1-type response element (data not shown). Considered together, these finding suggest that the dimerization interfaces of all COUP-TF family members are competent for both homodimerization and heterodimerization within the subfamily as well as with other nuclear receptors (1, 3, 4). Heterodimerization within the COUP-TF subfamily has been previously demonstrated between COUP-TFI and ARP1 (3). However, COUP-TFI·ARP1 heterodimeric complexes apparently did not exhibit differences with regard to either DNA binding specificity or efficiency of formation on various response elements when compared with homodimers of either receptor (3). In contrast, the present studies demonstrated that ARP1·Ear2 heterodimeric complexes exhibited a response element specificity that was distinct from that of homodimers composed of either receptor. This was most clearly evident in the case of Ear2 homodimers that bound weakly to a DR1(G) probe and did not interact detectably with any other probe tested. ARP1·Ear2 heterodimeric complexes bound to DR1(G) and DR2(G) strongly but also bound to the DR4(G) and DR5(G) probes and, to a lesser extent, the heterodimer bound to DR3(G). ARP1 homodimers bound very efficiently to all direct repeats of the “G” series with a modest preference for DR2(G) and DR1(G), a DNA binding specificity that was quite similar to that of ARP1·Ear2 heterodimeric complexes. However, ARP1 homodimers and ARP1·Ear2 heterodimers exhibited differential capacities to bind to direct repeats of the T series. For example, ARP1 homodimers bound efficiently to a DR1(T) probe and more weakly to DR5(T) and DR3(T), whereas ARP1·Ear2 heterodimeric complexes bound weakly to the DR1(T) probe only. Ear2 strongly inhibited ARP1 homodimer binding to the T series of probes suggesting that ARP1·Ear2 heterodimerization resulted in a complex that was incapable of binding to T series probes. This “inactive” heterodimeric complex may be speculated to have important implications in the signaling pathways of both receptors. For example, one may envision that ARP1 homodimeric complexes are capable of regulating the expression of promoters containing either DR(G)-type response elements or DR(T)-type response elements with inter-repeat spacing of 1, 3, or 5, the latter of which corresponds to the classical retinoic acid response element (39). In contrast, both ARP1·Ear2 heterodimers and Ear2 homodimeric complexes may only be capable of regulating DR(G)-type response elements albeit with differential capacities. However, relative overexpression of Ear2 in a given cell type at a given time may disrupt ARP1 homodimer-mediated regulation of the aforementioned DR(T)-containing promoters by the formation of ARP1·Ear2 heterodimeric complexes that are inactive with respect to DR(T)-type response elements. In addition, such an event could conceivably represent a gain of function with respect to Ear2 as ARP1·Ear2 heterodimeric complexes bind efficiently to DR(G)-type response elements with inter-repeat spacing ranging from at least 1 to 5, whereas Ear2 homodimers appear to bind efficiently only to DR1(G)-type elements. Thus, external and/or autocrine factors that act to perturb the stoichiometric balance of COUP-TF orphan receptors within a single cell may alter this regulatory dynamic and provide a mechanism by which these proteins may regulate the expression of a broader network of genes in a combinatorial fashion. Two such stimuli have been reported and may be relevant to this example as follows: expression of both ARP1 and COUPTFI has been demonstrated to be positively regulated by treatment with retinoic acid (18), whereas Sonic Hedgehog has been shown to induce expression of APR1 (40). Either of these treatments could alter the balance of COUP-TF family members expressed in a particular cell type, and it will be of interest to determine if such stoichiometric alterations ultimately dictate the scope of target genes subject to transcriptional regulation by these proteins.
Finally, members of the COUP-TF subfamily of nuclear receptors are generally considered to be repressors of transcription, possibly through interactions with silencing mediator of retinoid and thyroid hormone receptors (SMRT, see Ref. 41), nuclear receptor corepressor (NCoR, see Ref. 41), and/or splice variants of nuclear receptor corepressor (42). However, it is important to note that COUP-TF proteins may also activate transcription in some promoter and/or cellular contexts. Indeed, COUP-TFI, purified from HeLa cells, was originally characterized as a positive factor in the regulation of the ovalbumin promoter by O'Malley and colleagues (43–46), who have also demonstrated that COUP-TFI, presumably in a phosphorylated form, was a transcriptional activator when fused to a heterologous DNA binding domain (47). Other groups have subsequently demonstrated that COUP-TF can also function as a transcriptional activator in transient transfection experiments (48, 49) and in vitro (50). These findings, when considered together with the possibility that an activating ligand(s) for COUP-TFs may exist, raise the possibility that homodimeric or heterodimeric complexes composed of COUP-TF family members may positively regulate transcription of target genes in a cell-specific manner. Although the potential role of APR1·Ear2 complexes in either transcriptional activation or repression remains to be established, the results presented herein demonstrate that such heterodimeric complexes may play a role in the COUP-TF signaling pathways.
Acknowledgments
We gratefully thank H. Nakshatri, P. Chambon, R. Losson, P. Kastner, A. Krust, T. Lufkin, and S. Nakanishi for plasmid constructs and reagents and A. Fields, A. Son, and J. Webster for expert technical assistance.
This work was supported in part by the American Heart Association Grant 9640219N (to M. L.), the Oregon State University College of Pharmacy, and the Laboratory of Molecular Pharmacology.
Footnotes
The abbreviations used are: COUP-TF, chicken ovalbumin upstream promoter-transcription factor; AD, activation domain; RXR, retinoid X receptor; DR, direct repeat; RAR, retinoic acid receptor; 9-cis-RA, 9-cis-retinoic acid; RT, reverse transcription; PCR, polymerase chain reaction; NR1, NR1 subunit of N-methyl-D-aspartate receptor; HEK, human embryonic kidney; CAT, chloramphenicol acetyltransferase; ER, human estrogen receptor; ERE, estrogen response element; DBD, DNA binding domain; d.p.c., days postcoitum; GST, glutathione S-transferase; HA, hemagglutinin; EMSA, electrophoretic mobility shift assays.
REFERENCES
- 1.Kliewer SA, Umesono K, Heyman RA, Mangelsdorf DJ, Dyck JA, Evans RM. Proc. Natl. Acad. Sci. U. S. A. 1992;89:1448–1452. doi: 10.1073/pnas.89.4.1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tran P, Zhang X-K, Salbert G, Hermann T, Lehmann JM, Pfahl M. Mol. Cell. Biol. 1992;12:4666–4676. doi: 10.1128/mcb.12.10.4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cooney AJ, Tsai SY, O'Malley BW, Tsai M-J. Mol. Cell. Biol. 1992;12:4153–4163. doi: 10.1128/mcb.12.9.4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cooney AJ, Leng X, Tsai SY, O'Malley BW, Tsai M-J. J. Biol. Chem. 1993;268:4152–4160. [PubMed] [Google Scholar]
- 5.Leng X, Cooney AJ, Tsai SY, Tsai M-J. Mol. Cell. Biol. 1996;16:2332–2340. doi: 10.1128/mcb.16.5.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shigeta H, Newbold RR, McLachlan JA, Teng C. Mol. Reprod. Dev. 1996;45:21–30. doi: 10.1002/(SICI)1098-2795(199609)45:1<21::AID-MRD3>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 7.Klinge C, Silver BF, Driscoll MD, Sathyan G, Bambara RA, Hilf R. J. Biol. Chem. 1997;272:31465–31474. doi: 10.1074/jbc.272.50.31465. [DOI] [PubMed] [Google Scholar]
- 8.Chu K, Zingg HH. J. Mol. Endocrinol. 1997;19:163–172. doi: 10.1677/jme.0.0190163. [DOI] [PubMed] [Google Scholar]
- 9.Chu K, Boutin J-M, Breton C, Zingg HH. Mol. Cell. Endocrinol. 1998;137:145–154. doi: 10.1016/s0303-7207(97)00241-4. [DOI] [PubMed] [Google Scholar]
- 10.Miyajima N, Kadowasi Y, Fukushige S, Semba K, Yamanashi Y, Matsubara K, Toyoshima K, Yamamoto T. Nucleic Acids Res. 1988;16:11057–11074. doi: 10.1093/nar/16.23.11057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Palmer CNA, Hsu M-H, Griffin KJ, Johnson EF. J. Biol. Chem. 1995;270:16114–16121. doi: 10.1074/jbc.270.27.16114. [DOI] [PubMed] [Google Scholar]
- 12.Ladias JAA, Hadzopoulou-Cladaras M, Kardassis D, Cardot P, Cheng J, Zannis V, Caldaras C. J. Biol. Chem. 1992;267:15849–15860. [PubMed] [Google Scholar]
- 13.Mietus-Snyder M, Sladek F, Ginsburg G, Kuo F, Ladias JAA, Darnell JE, Karathanasis SK. Mol. Cell. Biol. 1992;12:1798–1718. doi: 10.1128/mcb.12.4.1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schrader M, Danielsson C, Wiesenberg I, Carlberg C. J. Biol. Chem. 1996;271:19732–19736. doi: 10.1074/jbc.271.33.19732. [DOI] [PubMed] [Google Scholar]
- 15.Wu Q, Li Y, Agadir A, Lee M-O, Liu Y, Zhang X-K. EMBO J. 1997;16:1656–1669. doi: 10.1093/emboj/16.7.1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsai SY, Tsai M-J. Endocr. Rev. 1997;18:229–240. doi: 10.1210/edrv.18.2.0294. [DOI] [PubMed] [Google Scholar]
- 17.Wang LH, Ing NH, Tsai SY, O'Malley BW, Tsai M-J. Gene Expr. 1991;1:207–216. [PMC free article] [PubMed] [Google Scholar]
- 18.Jonk LJC, de Jonge EJ, Pals CEGM, Wissink S, Vervaart JMA, Schoorlemmer J, Kruijer W. Mech. Dev. 1994;47:81–97. doi: 10.1016/0925-4773(94)90098-1. [DOI] [PubMed] [Google Scholar]
- 19.Escriva H, Safi R, Hanni C, Langlois M-C, Saumitou-Laprade P, Stehelini D, Capron A, Pierce R, Laudet V. Proc. Natl. Acad. Sci. U. S. A. 1997;94:6803–6808. doi: 10.1073/pnas.94.13.6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Islam TC, Toftgard R. Biochem. Biophys. Res. Commun. 1994;203:545–552. doi: 10.1006/bbrc.1994.2217. [DOI] [PubMed] [Google Scholar]
- 21.van der Wees J, Matharu PJ, de Roos K, Destree OH, Godsave SF, Durston AJ, Sweeney GE. Mech. Dev. 1996;54:173–184. doi: 10.1016/0925-4773(95)00471-8. [DOI] [PubMed] [Google Scholar]
- 22.Mlodzik M, Hiromi Y, Weber U, Goodman CS, Rubin GM. Cell. 1990;60:211–224. doi: 10.1016/0092-8674(90)90737-y. [DOI] [PubMed] [Google Scholar]
- 23.Fjose A, Nornes S, Weber U, Mlodzik M. EMBO J. 1993;12:1403–1414. doi: 10.1002/j.1460-2075.1993.tb05784.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qiu Y, Cooney A, Kuratani S, DeMayo FJ, Tsai SY, Tsai M-J. Proc. Natl. Acad. Sci. U. S. A. 1994;91:4451–4455. doi: 10.1073/pnas.91.10.4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qiu Y, Pereira FA, DeMayo FJ, Lydon JP, Tsai SY, Tsai MJ. Genes Dev. 1997;11:1925–1937. doi: 10.1101/gad.11.15.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Le Douarin B, Pierrat B, vom Baur E, Chambon P, Losson R. Nucleic Acids Res. 1995;23:876–878. doi: 10.1093/nar/23.5.876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pierrat B, Heery D, Lemoine Y, Losson R. Gene (Amst.) 1992;119:237–245. doi: 10.1016/0378-1119(92)90277-v. [DOI] [PubMed] [Google Scholar]
- 28.Nakshatri H, Chambon P. J. Biol. Chem. 1994;269:890–902. [PubMed] [Google Scholar]
- 29.Dowell P, Ishmael J, Avram D, Peterson V, Nevrivy D, Leid M. J. Biol. Chem. 1997;272:33435–33443. doi: 10.1074/jbc.272.52.33435. [DOI] [PubMed] [Google Scholar]
- 30.Moriyoshi K, Masu M, Ishii T, Shigemoto R, Mizuno N, Nakanishi S. Nature. 1991;354:31–37. doi: 10.1038/354031a0. [DOI] [PubMed] [Google Scholar]
- 31.Leid M, Kastner P, Lyons R, Nakshatri H, Saunders M, Zacharewski T, Chen J-Y, Staub A, Garnier J-M, Mader S, Chambon P. Cell. 1992;68:377–395. doi: 10.1016/0092-8674(92)90478-u. [DOI] [PubMed] [Google Scholar]
- 32.Nagpal S, Saunders M, Kastner P, Durand B, Nakshatri H, Chambon P. Cell. 1992;70:1007–1019. doi: 10.1016/0092-8674(92)90250-g. [DOI] [PubMed] [Google Scholar]
- 33.Kippert F. FEMS Microbiol. Lett. 1995;128:201–206. doi: 10.1111/j.1574-6968.1995.tb07523.x. [DOI] [PubMed] [Google Scholar]
- 34.Avram D, Bakalinsky A. Genetics. 1996;144:511–521. doi: 10.1093/genetics/144.2.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dowell P, Peterson VJ, Zabriskie TM, Leid M. J. Biol. Chem. 1997;272:2013–2020. doi: 10.1074/jbc.272.3.2013. [DOI] [PubMed] [Google Scholar]
- 36.Kronczynska AM, Coutts M, Makrides S, Brawerman G. Nucleic Acids Res. 1989;17:6408. doi: 10.1093/nar/17.15.6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lopes da Silva S, Cox JJ, Jonk LJ, Kruijer W, Burbach JP. Brain Res. Mol. Brain Res. 1995;30:131–136. doi: 10.1016/0169-328x(94)00289-q. [DOI] [PubMed] [Google Scholar]
- 38.Hwung Y-P, Wang L-H, Tsai SY, Tsai M-J. J. Biol. Chem. 1988;263:13470–13474. [PubMed] [Google Scholar]
- 39.de The H, Vivanco-Ruiz MM, Tiollais P, Stunnenberg H, Dejean A. Nature. 1990;343:177–180. doi: 10.1038/343177a0. [DOI] [PubMed] [Google Scholar]
- 40.Krishnan V, Pereira FA, Qiu Y, Chen C-H, Beachy PA, Tsai SY, Tsai M-J. Science. 1997;278:1947–1950. doi: 10.1126/science.278.5345.1947. [DOI] [PubMed] [Google Scholar]
- 41.Shibata H, Nawaz Z, Tsai SY, O'Malley B, Tsai M-J. Mol. Endocrinol. 1997;11:714–724. doi: 10.1210/mend.11.6.0002. [DOI] [PubMed] [Google Scholar]
- 42.Bailey PJ, Dowhan DH, Franke K, Burke LJ, Downes M, Muscat GEO. J. Steroid Biochem. Mol. Biol. 1997;63:165–174. doi: 10.1016/s0960-0760(97)00079-4. [DOI] [PubMed] [Google Scholar]
- 43.Pastorcic M, Wang H, Elbrecht A, Tsai SY, Tsai MJ, O'Malley BW. Mol. Cell. Biol. 1986;6:2784–2791. doi: 10.1128/mcb.6.8.2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pastorcic M, Bagchi MK, Tsai SY, Tsai MJ, O'Malley BW. Nucleic Acids Res. 1989;17:6693–6711. doi: 10.1093/nar/17.16.6693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang LH, Tsai SY, Sagami I, Tsai MJ, O'Malley BW. J. Biol. Chem. 1987;262:16080–16086. [PubMed] [Google Scholar]
- 46.Sagami I, Tsai SY, Wang H, Tsai M-J, O'Malley BW. Mol. Cell. Biol. 1986;6:4259–4267. doi: 10.1128/mcb.6.12.4259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Power RF, Lydon JP, Conneely OM, O'Malley BW. Science. 1991;252:1546–1548. doi: 10.1126/science.2047861. [DOI] [PubMed] [Google Scholar]
- 48.Kimura A, Nishiyori A, Murakami T, Tsukamoto T, Hata S, Osumi T, Okamura R, Mori M, Takiguchi M. J. Biol. Chem. 1993;268:11125–11133. [PubMed] [Google Scholar]
- 49.Lu XP, Salbert G, Pfahl M. Mol. Endocrinol. 1994;8:1774–1788. doi: 10.1210/mend.8.12.7708064. [DOI] [PubMed] [Google Scholar]
- 50.Malik S, Karathanasis S. Nucleic Acids Res. 1995;23:1536–1543. doi: 10.1093/nar/23.9.1536. [DOI] [PMC free article] [PubMed] [Google Scholar]