

Abstract
Duchenne muscular dystrophy (DMD) is characterized by muscle degeneration and structural defects in the neuromuscular synapse that are caused by mutations in dystrophin. Whether aberrant neuromuscular synapse structure is an indirect consequence of muscle degeneration or a direct result of loss of dystrophin function is not known. Rational design of truncated dystrophins has enabled the design of expression cassettes highly effective at preventing muscle degeneration in mouse models of DMD using gene therapy. Here we examined the functional capacity of a minidystrophin (minidysGFP) and a microdystrophin (microdystrophinΔR4–R23) transgene on the maturation and maintenance of neuromuscular junctions (NMJ) in mdx mice. We found that minidysGFP prevents fragmentation and the loss of postsynaptic folds at the NMJ. In contrast, microdystrophinΔR4–R23 was unable to prevent synapse fragmentation in the limb muscles despite preventing muscle degeneration, although fragmentation was observed to temporally correlate with the formation of ringed fibers. Surprisingly, microdystrophinΔR4–R23 increased the length of synaptic folds in the diaphragm muscles of mdx mice independent of muscle degeneration or the formation of ringed fibers. We also demonstrate that the number and depth of synaptic folds influences the density of voltage-gated sodium channels at the neuromuscular synapse in mdx, microdystrophinΔR4–R23/mdx and mdx:utrophin double knockout mice. Together, these data suggest that maintenance of the neuromuscular synapse is governed through its lateral association with the muscle cytoskeleton, and that dystrophin has a direct role in promoting the maturation of synaptic folds to allow more sodium channels into the junction.
Introduction
Duchenne muscular dystrophy (DMD) is a lethal X-linked disease characterized by progressive wasting of skeletal muscles, which become highly susceptible to contraction-induced injuries, the development of cardiomyopathy and in many cases impairment in cognition that are caused by mutations in the dystrophin gene (Anderson et al., 2002; Emery and Muntoni, 2003; Koenig et al., 1987). Dystrophin is concentrated in the postsynaptic surface of the neuromuscular junctions (NMJ) and gamma-amino butyric acid (GABAergic) synapses (Knuesel et al., 1999; Sealock et al., 1991). Dystrophin is not required for synapse formation, but the maintenance of these synapses is impaired in the mdx mouse model of DMD (Knuesel et al., 1999; Lyons and Slater, 1991). NMJs are fragmented in mdx mice, and have fewer folds in the postsynaptic apparatus (Lyons and Slater, 1991). However, these structural defects of the NMJ are temporally coincident with muscle degeneration in mdx mice. Whether fragmentation is a direct consequence of the absence of dystrophin (and its associated proteins) or is secondary to muscle degeneration is unclear (Grady et al., 1997; Lyons and Slater, 1991; Marques et al., 2007a; Minatel et al., 2003; Torres and Duchen, 1987).
The distinctive structural features of the mammalian neuromuscular synapse ensure that nerve-evoked stimulation of the muscle exceeds that required to generate an action potential in the sarcolemma (Slater, 2003). The neuromuscular synapse is characterized by the direct apposition between vesicle release sites (called active zones) in the nerve terminal with nicotinic acetylcholine receptors (AChRs) that cluster on the crests of the junctional folds in the postsynaptic membrane (Sanes and Lichtman, 1999). The high safety factor of the NMJ relies on the number of quanta released by the nerve terminal and the number of functional AChRs on the postsynaptic membrane, which are usually in excess (Chang et al., 1975; Harris and Ribchester, 1979). In addition, the troughs of the postsynaptic folds contain a high concentration of voltage gated sodium channels that reduce the threshold required to evoke endplate currents (Sealock et al., 1991; Wood and Slater, 1997). The number of quanta decreases with repeat stimulation during normal muscle activity in vivo (Hennig and Lomo, 1985), but the folds help ensure that the safety of transmission is maintained (Wood and Slater, 1997). Neuromuscular synapses in humans are smaller than murine synapses and the folds are more extensive (Slater, 2003). The direct alignment between active zones and junctional folds is lost in many neuromuscular synapses in DMD, because the folds are not mature or maintained (Lyons and Slater, 1991). Synaptic transmission becomes more variable with age in the mdx mouse model of DMD (Carlson and Roshek, 2001), and is likely to be more deleterious in DMD patients because of the greater reliability of synapse function on fold maturation in humans (Slater, 2003).
Dystrophin is a large protein (427 kD) that consists of an amino terminal actin-binding domain, a central rod domain, a cysteine rich region and a C-terminal domain (Fig. 1A) (Abmayr et al., 2006; Hoffman et al., 1987; Koenig and Kunkel, 1990; Koenig et al., 1988). The central rod domain contains a second actin-binding domain, 4 hinge regions and 24 spectrin-like repeats that form α-helical coiled-coils predicted to provide flexibility and elasticity to dystrophin (Bennett and Gilligan, 1993; Brown and Lucy, 1993; Koenig and Kunkel, 1990; Koenig et al., 1988; Winder, 1997). Structure-function analyses of dystrophins expressed in the more mild form of DMD called Becker muscular dystrophy demonstrate that large in-frame deletions in the central rod domain can minimally affect the functional capacity of dystrophin (England et al., 1990). We have previously developed a number of truncated dystrophins that are highly effective at preventing muscle necrosis in mdx mice (Chamberlain, 2002; Crawford et al., 2000; Harper et al., 2002; Phelps et al., 1995). Two promising candidates for gene therapy of DMD that we developed are ΔH2-R19/ΔCT minidystrophin and microdystrophinΔR4–R23 (Fig. 1A). The minidystrophin studied here has 8 spectrin-like repeats in the central rod domain and an in-frame eGFP in place of the C-terminal domain (minidysGFP) for purposes of tracking the protein in cell transplantation experiments (Li et al., 2006). This minidystrophin is similar to, but more functional than, an exon 17–48 deletion that prevents muscle degeneration and the fragmentation of neuromuscular synapses in mdx mice (Harper et al., 2002; Phelps et al., 1995; Rafael et al., 2000). MicrodystrophinΔR4–R23 has only 4 spectrin-like repeats in the central rod domain and can be delivered to large regions of muscle via intravascular injection using recombinant adeno-associated viral vectors (rAAV)(Gregorevic et al., 2004; Harper et al., 2002).
Figure 1.

Structure of truncated dystrophins and their effect on neuromuscular synapse stabilization in mdx mice. A) ABD, actin binding domain. CR, cysteine-rich domain. CT, C-terminal domain. R, spectrin-like repeats. The numbers in the shaded boxes represent the hinge regions. GFP, enhanced green fluorescent protein. B) Topographic view of the acetylcholine receptor (AChR) clusters in the postsynaptic membrane of the neuromuscular junction stained with α-bungarotoxin (α-BTX; in red) on teased gastrocnemius muscles at 3 months of age. Note that synapses were fragmented in mdx fibers that have central nuclei (DAPI in blue), whereas synapses in microdystrophinΔR4–R23/mdx mice were fragmented on myofibers with peripheral nuclei. Dashed lines outline the muscle fiber. Scale bar = 15 μm.
In the present study we examined whether truncated dystrophin transgenes could maintain the NMJ in mdx mice. We found that minidysGFP was highly effective at preventing synapse fragmentation and loss of synaptic folds in mdx mice. However, postsynaptic structure was abnormal in microdystrophinΔR4–R23/mdx despite the absence of muscle necrosis. Taken together, these results suggest that synapse fragmentation and muscle fiber degeneration/regeneration are separable events.
Results
Fragmented neuromuscular synapses in microdystrophinΔR4–R23/mdx mice
Nicotinic acetylcholine receptor (AChR) clusters in the adult NMJ form a complex tertiary structure (Marques et al., 2000). To determine the effect of modified dystrophin forms that hold promise for gene therapy, we compared NMJs of wild type, mdx, minidysGFP/mdx, and microdystrophinΔR4–R23/mdx mice. At three months of age, endplates of wild-type mice usually (95%) had three or fewer continuous regions of AChR clusters, viewed when stained with α–bungarotoxin (BTX) on teased wholemount muscle fibers and frozen sections (Fig. 1, Table 1). Therefore, we classified neuromuscular synapses as “continuous” if they presented with 3 or less AChR cluster regions or “discontinuous” if they presented with more than 3 regions of AChR clusters. Muscles of wild-type, and minidysGFP/mdx mice had continuous synapses almost exclusively on skeletal muscle fibers with peripheral nuclei (Fig. 1B). Fragmented synapses were found almost exclusively on mdx skeletal muscle fibers with nuclei that lie close together within the center of the fiber, indicative of muscle degeneration/regeneration (Fig. 1B)(Lyons and Slater, 1991). The percentage of continuous synapses in mdx mice was significantly reduced compared to wild-type mice in all muscles examined (P < 0.001) (Table. 1). MinidysGFP prevented the fragmentation of synapses in mdx muscles (P < 0.001 compared to mdx mice; Table 1). Surprisingly, most fragmented NMJs in microdystrophinΔR4–R23/mdx gastrocnemius muscles were present on muscle fibers that had only peripheral nuclei (Fig. 1B). The percentage of continuous junctions in microdystrophinΔR4–R23/mdx gastrocnemius and quadriceps muscles was significantly reduced compared to wild-type mice, despite having few muscle fibers that had undergone degeneration or regeneration (P < 0.001 compared to mdx; Table 1). Approximately 70% of gastrocnemius muscle fibers with peripheral nuclei had fragmented synapses (Table 1). MicrodystrophinΔR4–R23 was able to prevent the fragmentation of synapses in the mdx diaphragm (Table 1). Thus, synapses were fragmented in some microdystrophinΔR4–R23/mdx limb muscles in the absence of muscle degeneration and regeneration.
Table 1.
Percentage of Continuous Synapses and Central Myonuclei
| WT | mdx | ΔR4–R23 | MiniGFP | ||
|---|---|---|---|---|---|
| % Continuous Synapses: | Diaphragm | 95 | 48a | 93b | 94e |
| Gastrocnemius | 98 | 11c | 22c | 95e | |
| Quadriceps | 93 | 6c | 18b,c | 96e | |
| % Central Nuclei: | Diaphragm | <1 | 39c | 3e | <1e |
| Gastrocnemius | <1 | 79c | 8e | 1e | |
| Quadriceps | <1 | 89c | 8a,e | 1e | |
| % Fragmented synapses* on muscles without central nuclei: | Diaphragm | 4 | 13 | 6 | 5 |
| Gastrocnemius | 1 | 10 | 70b,c | 4 | |
| Quadriceps | 6 | 5 | 74b,c | 3 |
Fragmented synapses are synapses with 3 or more separate domains.
WT, wild-type. ΔR4–R23, microdystrophinΔR4–R23 mice. MiniGFP, minidysGFP mice.
Significantly different from wild-type (P < 0.01).
Significantly different from mdx (P < 0.01).
Significantly different from wild-type (P < 0.001).
Significantly different from wild-type (P < 0.05).
Significantly different from mdx (P < 0.001).
Synapse fragmentation in microdystrophinΔR4–R23/mdx mice is secondary to ringed fibers
To determine if fragmentation of neuromuscular synapses occurred secondarily to ultrastructural defects in the skeletal muscles of microdystrophinΔR4–R23/mdx mice, we examined the muscles using electron microscopy. We found ringed fibers in the gastrocnemius and quadriceps muscles as previously described (Banks et al., 2008), where most synapses were fragmented (Fig. 2). Ringed fibers are where the peripheral myofibrils form rings around the central myofibrils (Fig. 2D). Ringed fibers are common in human muscles and become more prevalent with age, injury and disease (Engel and Franzini-Armstrong 2004). Ringed fibers can be experimentally induced by cutting the point between insertion and origin of the muscle, such as a tenotomy (Pena et al., 2001). We had previously found that myotendinous strain injury led to ringed fibers in the limb muscles of microdystrophinΔR4–R23/mdx transgenic mice (Banks et al., 2008). Longitudinal sections of microdystrophinΔR4–R23/mdx gastrocnemius muscles revealed regions of sarcolemma that were ruffled in appearance in approximately 43% of fibers at 3 months of age (Fig. 2E). We did not find ringed fibers in the diaphragm muscle fibers where synapses were continuous (Fig. 2F), which is consistent with our previous study showing no myotendinous strain injury in the diaphragm and no ringed fibers (Banks et al., 2008). We did not find ringed fibers in wild-type, mdx or minidysGFP mice (Fig. 2).
Figure 2.

Synapses fragmentation was coincident with ringed fiber formation in microdystrophinΔR4–R23/mdx mice. Shown are electron microscopy images of two adjacent muscle fibers from A) wild-type, B) mdx, C) MinidysGFP/mdx D) microdystrophinΔR4–R23/mdx mice. White arrows show the ringed fibers in microdystrophinΔR4–R23/mdx mice. E) Longitudinal section of a muscle fiber from microdystrophinΔR4–R23/mdx mice showing the instability of the sarcolemma (black arrows). F) Ringed fibers were not found in the diaphragm in microdystrophinΔR4–R23/mdx mice. Scale bars = 2 μm.
Synapses in mdx mice begin to fragment at approximately 3–4 weeks of age coincident with muscle degeneration. At birth we found no indication of synapse fragmentation or the formation of ringed fibers in wild-type, mdx or microdystrophinΔR4–R23/mdx gastrocnemius muscles (results not shown). At 2 weeks of age, neuromuscular synapses were continuous in wild-type and mdx mice, but were fragmented in some of the microdystrophinΔR4–R23/mdx gastrocnemius muscles. (Fig. 3). Approximately 4% of the synapses in microdystrophinΔR4–R23/mdx gastrocnemius muscles were fragmented at 2 weeks of age. At the same time, ringed fibers appeared in approximately 4% of the microdystrophinΔR4–R23/mdx gastrocnemius muscle fibers at 2 weeks of age (Fig. 3). Thus, fragmentation of the neuromuscular junctions in microdystrophinΔR4–R23/mdx mice was independent from muscle degeneration, but coincident with the formation of ringed fibers.
Figure 3.

Synapses begin to fragment in microdystrophinΔR4–R23/mdx mice before mdx mice. A) Topographic view of AChR clusters stained with α-BTX at 2 weeks of age. Scale bar = 15 μm. B) A ringed fiber in microdystrophinΔR4–R23/mdx gastrocnemius muscle at 2 weeks of age. Arrowhead points to the sarcolemma. Arrow points to the interface between ringed fibers and normally oriented longitudinal fibers. Scale bar = 1 μm.
Ultrastructural analyses of neuromuscular synapses in microdystrophinΔR4–R23/mdx mice
To test whether truncated dystrophins could have a direct affect on the maturation of NMJs, we examined synapses in the diaphragm muscles that do not have ringed fibers. Neuromuscular synapses in wild-type mice have folds in the postsynaptic membrane adjacent to active zones in the presynaptic nerve terminal (Fig. 4). MicrodystrophinΔR4–R23 and minidysGFP were concentrated in the postsynaptic folds of mdx mice similar to full-length dystrophin in wild-type muscles (Supplementary Figure 1). The number and length of junctional folds at neuromuscular synapses was modestly reduced in mdx diaphragm compared to wild-type muscles (P = 0.0133 and P = 0.0274 respectively from 46 junctions from n = 4 mice; Fig. 4). Both microdystrophinΔR4–R23 and minidysGFP were able to maintain the normal number of synaptic folds in mdx diaphragm (Fig. 4). However, the folds extended significantly further into the postsynaptic membrane in microdystrophinΔR4–R23/mdx mice (P < 0.001; Fig. 4). MinidysGFP maintained the normal length of the folds in mdx mice (Fig. 4). Thus, the length of synaptic folds was significantly increased in microdystrophinΔR4–R23/mdx mice independent from degenerating fibers or ringed fibers.
Figure 4.

A) Ultrastructure of neuromuscular synapses in wild-type, mdx, microdystrophinΔR4–R23/mdx mice and minidysGFP/mdx mice. Scale bar = 2μm. B) The graph shows the mean +/− SD number of folds per μm of postsynaptic membrane juxtaposed to the presynaptic cleft. The number of folds/μm was significantly reduced in mdx mice compared to wild-type mice (P < 0.05). C) The mean +/− SD depth of the folds was significantly reduced in mdx diaphragm (*P < 0.05), and significantly increased in microdystrophinΔR4–R23/mdx diaphragms (***P < 0.001).
The dystrophin-glycoprotein complex, nNOS and utrophin at the neuromuscular synapse of microdystrophinΔR4–R23/mdx mice
Most components of the dystrophin glycoprotein complex (DGC) are required for the maturation and maintenance of the neuromuscular junction (Banks et al., 2003; Shiao et al., 2004), and many of these proteins are reduced or absent in mdx mice (Deconinck et al., 1997; Grady et al., 1997). We next examined whether components of the DGC could affect the maturation and maintenance of neuromuscular synapses in microdystrophinΔR4–R23/mdx mice. We found that β-dystroglycan, α-dystrobrevin, and α-syntrophin were concentrated at the neuromuscular synapse in minidysGFP and microdystrophinΔR4–R23/mdx gastrocnemius muscles (Supplementary Fig. 2). Neuronal nitric oxide synthase (nNOS) was reduced from neuromuscular synapses in mdx mice, as previously described (Shiao et al., 2004). nNOS expression was similarly reduced from the sarcolemma and NMJ in both minidysGFP and microdystrophinΔR4–R23/mdx mice (Fig. 5). Considering neuromuscular synapses are continuous in minidysGFP mice, it is unlikely that the reduction of nNOS alone leads to fragmentation of neuromuscular synapses. Utrophin was expressed extrasynaptically in microdystrophinΔR4–R23/mdx gastrocnemius muscles, but was restricted to the NMJs in minidysGFP muscles (Fig. 5). However, utrophin is also expressed extrasynaptically in mdx mice expressing dystrophin that lacks the C-terminal domain, which have continuous synapses (Crawford et al., 2000). Thus, we found no evidence that the DGC, nNOS or utrophin contributed to abnormal synapse maturation and maintenance in microdystrophinΔR4–R23/mdx mice.
Figure 5.

Localization of nNOS and utrophin in transverse sections of the gastrocnemius muscles. Antibody staining is shown in green and αBTX in red. Scale bar = 30 μm.
Concentration of sodium channels in synaptic folds
Junctional folds reduce the threshold required for a nerve-evoked endplate potential by concentrating voltage gated sodium channels to the synapse (Bailey et al., 2003; Wood and Slater, 1997). Dystrophin colocalizes with voltage gated sodium channels at the troughs of synaptic folds (Sealock et al., 1991)(Fig. 6). To test whether dystrophin expression affects the localization of voltage gated sodium channels we examined their distribution in transverse frozen sections. We found that voltage gated sodium channels localized beneath the synaptic gutter containing AChR clusters in wild-type, mdx, microdystrophinΔR4–R23/mdx and minidysGFP/mdx mice (Fig 6A). (Fig 6A). Thus, voltage gated sodium channels appear to localize to the NMJ independent from dystrophin.
Figure 6.
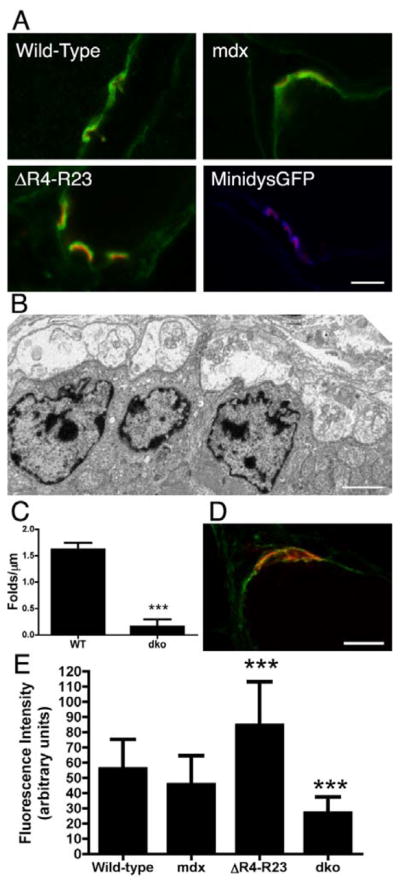
Synaptic folds influence the density of voltage gated sodium channels at the NMJ. A) Voltage gated sodium channels concentrate to the postsynaptic folds of wild-type, mdx, microdystrophinΔR4–R23/mdx mice and minidysGFP/mdx mice. Voltage gated sodium channels are shown in green or blue and AChR clusters are shown in red. B) Electron micrograph of a NMJ from mdx/utrophin double knockout mice (mdx:utrn−/−) showing few folds in the postsynaptic membrane. Scale bar = 2μm. C) Mean +/− S.D. number of folds per μm of postsynaptic membrane in mdx:utrn−/− mice. dko is mdx:utrn−/− mice. ***P < 0.001. D) Synapses in mdx:utrn−/− mice have a low density of voltage gated sodium channels. Sodium channels are in green and AChR are in red. Scale bar = 10 μm. E) Bars represent the mean +/− S.D. immunofluorescence intensity of sodium channels at the NMJs from wild-type, mdx, microdystrophinΔR4–R23/mdx and mdx:utrn−/− gastrocnemius muscles. ***P < 0.001 compared to wild-type.
Synaptic folds are required to concentrate voltage-gated sodium channels to developing mammalian synapses (Bailey et al., 2003). This suggests that changes in synaptic folding in dystrophic muscles or in the microdystrophinΔR4–R23 transgenic mice would lead to changes in the density of voltage-gated sodium channels at the NMJ. To test this hypothesis, we quantitated the sodium channel immunofluorescence staining in the postsynaptic region in transverse frozen sections from wild-type, mdx, microdystrophinΔR4–R23/mdx and mdx/utrophin double knockout (mdx:utrn−/−) gastrocnemius muscles. All sections and images were processed identically to allow a direct comparison between the mice (see methods). Synaptic folding was significantly reduced in mdx:utrn−/− mice (P < 0.001; Fig. 6B,C), as previously reported (Grady et al., 1997). The reduction in synaptic folding in mdx:utrn−/− mice correlated with a significant reduction in density of voltage-gated sodium channels (P < 0.001; Fig. 6D,E). We found that the density of voltage-gated sodium channels in mdx mice was moderately reduced compared to wild-type mice, although this was not significant (P = 0.0618; Fig. 6E). The density of sodium channels in microdystrophinΔR4–R23/mdx synapses was significantly increased compared to wild-type (P < 0.001; Fig. 6E). Thus, the number and depth of synaptic folds influenced the density of voltage-gated sodium channels at the neuromuscular junction.
Discussion
The direct apposition between vesicle release sites in the presynaptic nerve terminal with AChR clusters on the crests of junctional folds in the postsynaptic muscle membrane help provide the specializations necessary for efficient synaptic transmission (Slater, 2003). A high concentration of voltage gated sodium channels in the postsynaptic folds of the NMJ also ensures that the post-synaptic potential evokes an action potential, thus leading to muscle contraction (Wood and Slater, 1997). The maturation and maintenance of the NMJ is impaired in mdx mice, where the synapses are fragmented and many of the postsynaptic folds are lost (Lyons and Slater, 1991). These synaptic abnormalities are generally assumed to result from muscle degeneration (Grady et al., 1997; Lyons and Slater, 1991; Marques et al., 2007a; Minatel et al., 2003; Torres and Duchen, 1987), but the functional role of dystrophin at the NMJ is not known. Here, we showed that microdystrophinΔR4–R23 was unable to prevent the fragmentation of synapses in the gastrocnemius muscles of mdx mice and surprisingly increased the depth of the folds in the postsynaptic membrane, even though muscle degeneration was prevented.
Synapse fragmentation in mdx mice
In the present study, we found that synapses in microdystrophinΔR4–R23/mdx mice were fragmented independent from muscle degeneration, but fragmentation was coincident with the formation of ringed fibers, which we have previously demonstrated were brought about by chronic myotendinous strain injury (Banks et al., 2008). The sarcolemma appeared ruffled during the formation of ringed fibers in microdystrophinΔR4–R23/mdx mice. From these results, we suggest that the stability of the neuromuscular synapse depends on its lateral association with the muscle cytoskeleton. A demonstration of this lateral association has been found in denervation experiments that show changes in the orientation of costameres that extend through the NMJ (Bezakova and Lomo, 2001). Consistent with this proposal, the lateral edges of the postsynaptic clefts in mice lacking either α-dystrobrevin or α-syntrophin are frayed (Adams et al., 2000; Grady et al., 2000). Thus, it is likely that destabilization of the sarcolemma and cytoskeleton during muscle degeneration in mdx mice leads to synapse fragmentation.
While synapses were fragmented in most limb muscles in microdystrophinΔR4–R23/mdx mice, the endplates were continuous in the diaphragm muscles. We previously found no myotendinous strain injury or ringed fibers in the diaphragm muscles further supporting the notion that ringed fibers lead to synapse fragmentation (Banks et al., 2008). It is possible that expression levels of microdystrophinΔR4–R23 could have contributed to the muscle to muscle differences in the transgenic mice. Harper et al., 2002 had previously shown that more microdystrophinΔR4–R23 is expressed in the limbs than the diaphragms in the same mice. However, we suggest this possibility is unlikely because dystrophin expression in wild-type mice is also more in the limbs compared to the diaphragm (Harper et al., 2002). Furthermore, the increase in microdystrophinΔR4–R23 expression compared to wild-type dystrophin is greater in the diaphragm than the limbs (Harper et al., 2002). And finally, the expression of minidysGFP is similar to microdystrophinΔR4–R23 and we have currently found no abnormalities in minidysGFP transgenic mice (Harper et al., 2002; Li et al., 2002).
Most components of the DGC are required for NMJ maturation and stabilization (Banks et al., 2003). Each truncated dystrophin in this study was able to restore several members of the DGC to the NMJ, except for nNOS. The absence of nNOS at the sarcolemma of mdx mice has been proposed to contribute to synapse fragmentation (Shiao et al., 2004). Expression of an nNOS transgene mitigates muscle degeneration and prevents the fragmentation of NMJs in mdx mice (Shiao et al., 2004; Wehling et al., 2001). We found here that the reduction of nNOS from the sarcolemma in minidysGFP mice did not correlate with the fragmentation of neuromuscular synapses. Thus, normal levels of nNOS are not required to prevent synapse fragmentation in mdx mice.
Dystrophin is present in a subset of GABAergic synapses in the CNS allowing the possibility that altered motoneuron function or motor neuropathy could contribute to the fragmentation of the neuromuscular synapses in mdx mice (Graciotti et al., 2008; Knuesel et al., 1999; Kueh et al., 2008; Marques et al., 2007b). Here we show using a muscle-specific human α-skeletal actin promoter that minidysGFP expression selectively in skeletal muscle prevents the fragmentation of the neuromuscular synapse in mdx mice. Therefore, muscle-specific expression of dystrophin is sufficient to maintain the structural specializations of the neuromuscular synapse in mdx mice.
Effect of dystrophin on synaptic fold maturation
The number of folds in the postsynaptic membrane is significantly reduced in mdx mice (Lyons and Slater, 1991), a change proposed to result from muscle degeneration. In contrast, however, muscle degeneration in γ-sarcoglycan knockout mice does not lead to synaptic abnormalities (Hack et al., 1998). It is interesting to note that dystrophin localization is unaffected in γ-sarcoglycan knockout mice (Hack et al., 1998). In the present study, we found that the microdystrophinΔR4–R23 transgene significantly increased the length of folds independent from muscle degeneration or ringed fibers. Thus, dystrophin can promote synaptic folding, which suggests the reduced folding in mdx mice may result from the local loss of dystrophin and the DGC rather than muscle degeneration.
We found that the number and depth of synaptic folds influenced the density of voltage-gated sodium channels in the postsynaptic membrane. This is consistent with previous findings that show sodium channels are excluded from developing mammalian NMJs and chick NMJs with no synaptic folds (Bailey et al., 2003). Thus, reduced synaptic folding in dystrophic muscles could contribute to the functional deficits in synaptic transmission in mdx mice (Carlson and Roshek, 2001; Wood and Slater, 1997). Deficits in synaptic transmission are likely to be exacerbated in DMD because of the greater reliance of synaptic transmission on fold maturation in humans (Slater, 2003). The increased depth of synaptic folds in microdystrophinΔR4–R23/mdx mice could increase the safety factor of synaptic transmission by allowing more voltage gated sodium channels into the NMJ.
Our results show that minidysGFP, but not microdystrophinΔR4–R23 maintains the structure of synapses in the limb muscles. It is not clear whether the ability of truncated dystrophins to maintain synapse structure depends on the size of the rod domain or the composition of the rod domain. Future experiments will decipher whether the number of spectrin repeats could influence neuromuscular synapse structure. It is also possible that the hinge composition (minidysGFP has hinge 3 whereas microdystrophinΔR4–R23 has hinge 2) could influence neuromuscular synapse structure.
In conclusion, we have found that minidysGFP can prevent the structural abnormalities of the neuromuscular synapse in mdx mice. We suggest that synapse fragmentation in mdx muscles is dependent on a lateral association with the subsarcolemmal cytoskeleton, and that dystrophin can promote the maturation of synaptic folds to allow more sodium channels into the junction.
Materials and Methods
Mice
We utilized C57Bl/10 wildtype mice, mdx mice and mdx mice expressing various truncated dystrophin transgenes. The truncated dystrophin transgenes are presented in figure 1A and include a dystrophin construct that lacks spectrin-like repeats 4–23 in the central rod domain (microdystrophinΔR4–R23; (Harper et al., 2002)), a dystrophin construct that lacks spectrin-like repeats 9 to 16 and has an eGFP fusion protein in place of the C-terminal domain (minidysGFP; (Li et al., 2006)). All truncated dystrophin transgenes use a human α-skeletal actin promoter (Crawford et al., 2000). Mice were genotyped as previously described (Harper et al., 2002). All mice were compared at 3 months of age unless otherwise shown. All experiments are in accordance with the institute of animal care and use committee (IACUC) of the University of Washington.
Quantification of neuromuscular synapses
Neuromuscular synapses were analyzed in both wholemount immunofluorescence stained muscles and in frozen OCT tissue sections. For wholemount preparations, skeletal muscle fibers were fixed for 2 hours in 2% paraformaldehyde at 4°C. Immediately following fixation, individual muscle fibers were teased apart using a fine needle. The fibers were then incubated 0.1M glycine for 30 minutes, blocked in blocking solution (1% BSA, 0.05% TritonX-100 in 1x PBS) for 1 hour and stained with TRITC conjugated α-bungarotoxin (αBTX; 1:500; Molecular Probes) for 1 hour. The fibers were washed 3 times in blocking solution after staining and mounted on slides with antifade mounting media containing DAPI (Vector Labs). For tissue sections, diaphragm, gastrocnemius and quadriceps skeletal muscle was immediately frozen in OCT placed in 2-methylebutane in liquid nitrogen. For analyses of neuromuscular synapses, 40 μm thick longitudinal sections were blocked in blocking solution for 30 min at 4°C, stained with TRITC conjugated αBTX (αBTX; 1:800) for 30 minutes at room temperature and washed 3 times in 1x PBS. All wholemount preparations and sections were analyzed using a Delta Vision deconvolution microscope. Synapses were classified as continuous if they presented with 3 or less continuous regions of AChR clustering and discontinuous if they presented with more than 3 regions of AChR clustering. More than 50 synapses were analyzed on each diaphragm, gastrocnemius and quadriceps skeletal muscle on n = 4 mice. We compared the proportion of continuous synapses using a Students t-test.
Gross muscle morphology and morphometry
For analyses dystrophin-associated proteins, 10 μm thick transverse frozen sections of gastrocnemius muscle were taken. For analyses of dystrophin and dystrophin glycoproteins the sections were incubated in blocking solution for 30 min and stained with primary antibodies. The antibodies included a rabbit polyclonal to dystrophin (N-terminus of dystrophin; 1:800), rabbit polycolonal anti-α-dystrobrevin (Transduction laboratories; 1:200), rabbit polyclonal anti-Syn17 (α-syntrophin; 1:200; (Peters et al., 1997)), rabbit polyclonal anti-Utrophin A (1:200), rabbit polyclonal anti-nNOS (Alexis; 1:200), mouse monoclonal anti-sodium channel (PAN; clone K58/35; SIGMA; 1:100). The sections were then washed 5 times in 1xPBS and incubated for 30 minutes in a combination of TRITC conjugated αBTX (Molecular Probes; 1:500) and Alexa 488 rabbit polyclonal or mouse monoclonal secondary antibody, or Alexa 633 rabbit polyclonal antibody (Molecular Probes; 1:800). The sections were washed 5 times in 1xPBS, mounted in anti-fade mounting media containing DAPI (Vector Labs). All fluorescent sections were analyzed using a DeltaVision fluorescence microscope.
Quantitation of sodium channel density in the postsynaptic folds
Frozen sections of wild-type, mdx, microdystrophinΔR4–R23/mdx, and mdx:utrn−/−gastrocnemius muscles were immunostained in parallel for voltage-gated sodium channels and AChR clusters as described above. We examined approximately 40 junctions from n = 4 mice. All images were taken with the Leica SL confocal microscope (Exton, PA) using identical parameters. The images were analyzed with Image J (NIH), by encompassing the NMJs and comparing the integrated density of the immunostaining divided by the area of the image being examined to give the “mean grey value”, which is the optical density (or fluorescence intensity) of the selected region (NIH). We subtracted the background mean grey value to obtain the mean grey value of sodium channel immunostaining at the NMJ, which is graphed in Figure 6.
Electron Microscopy
Small (approximately 1mm3) regions of diaphragm or quadriceps muscles from 3 month old mice were fixed in half strength Karnovskys fixative for at least 24 hours at 4°C. Fixed muscles were washed in 1x phosphate buffer (pH7.1), postfixed in 1% (w/v) aqueous osmium tetroxide containing 1.5% (w/v) potassium ferrocyanide (Kopriwa, 1984), stained en bloc with uranyl acetate, dehydrated through acetone into Epon resin, embedded, and polymerized at 60°C for 24 h. Ultrathin sections were cut at ~ 80nm, stained with Reynolds lead citrate (Reynolds, 1963), viewed and photographed with a Hitachi (Tokyo, Japan) H600 transmission electron microscope. The junctional fold number and lengths were quantitated using Image J (NIH) and compared using a Students t-test using the Prism statistics program. We quantitated the number of folds by dividing the number of folds in the synaptic cleft by the distance where the nerve terminal contacts the muscle. We quantitated the length of folds by measuring the distance from the synaptic cleft to the furthest region each fold extended into the muscle using the line measure tool in the Image J program. We examined at least 42 junctions from n = 4 mice.
Supplementary Material
Acknowledgments
We are grateful to Leonard Meuse for animal husbandry. We are also grateful for Froehner and Chamberlain lab members for critically reviewing the manuscript. We would also like to thank Franque Remington, Judith Bousman and Bobbie Schneider for electron microscopy at the Fred Hutchinson Cancer Research Institute. In addition, we thank Greg Martin at the Keck Imaging Center University of Washington for help with confocal microscopy. This work was supported by grants from the National Institutes of Health (PO1 NS 046788; R37 AR040864). GBB was supported by a CJ Martin PostDoctoral Fellowship from the National Health and Medical Research Council of Australia (372212).
References
- Abmayr S, Chamberlain JS. The structure and function of dystrophin. In: Winder S, editor. The Molecular Mechanisms of Muscular Dystrophy. Landes Bioscience; Georgetown: 2006. [Google Scholar]
- Adams ME, Kramarcy N, Krall SP, Rossi SG, Rotundo RL, Sealock R, Froehner SC. Absence of alpha-syntrophin leads to structurally aberrant neuromuscular synapses deficient in utrophin. J Cell Biol. 2000;150:1385–1398. doi: 10.1083/jcb.150.6.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JL, Head SI, Rae C, Morley JW. Brain function in Duchenne muscular dystrophy. Brain. 2002;125:4–13. doi: 10.1093/brain/awf012. [DOI] [PubMed] [Google Scholar]
- Bailey SJ, Stocksley MA, Buckel A, Young C, Slater CR. Voltage-gated sodium channels and ankyrinG occupy a different postsynaptic domain from acetylcholine receptors from an early stage of neuromuscular junction maturation in rats. J Neurosci. 2003;23:2102–2111. doi: 10.1523/JNEUROSCI.23-06-02102.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks GB, Combs AC, Chamberlain JR, Chamberlain JS. Hum Mol Genet. 2008. Molecular and cellular adaptations to chronic myotendinous strain injury in mdx mice expressing a truncated dystrophin. Accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks GB, Fuhrer C, Adams ME, Froehner SC. The postsynaptic submembrane machinery at the neuromuscular junction: requirement for rapsyn and the utrophin/dystrophin-associated complex. J Neurocytol. 2003;32:709–726. doi: 10.1023/B:NEUR.0000020619.24681.2b. [DOI] [PubMed] [Google Scholar]
- Bennett V, Gilligan DM. The spectrin-based membrane skeleton and micron-scale organization of the plasma membrane. Annu Rev Cell Biol. 1993;9:27–66. doi: 10.1146/annurev.cb.09.110193.000331. [DOI] [PubMed] [Google Scholar]
- Bezakova G, Lomo T. Muscle activity and muscle agrin regulate the organization of cytoskeletal proteins and attached acetylcholine receptor (AchR) aggregates in skeletal muscle fibers. J Cell Biol. 2001;153:1453–1463. doi: 10.1083/jcb.153.7.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SC, Lucy JA. Dystrophin as a mechanochemical transducer in skeletal muscle. Bioessays. 1993;15:413–419. doi: 10.1002/bies.950150608. [DOI] [PubMed] [Google Scholar]
- Carlson CG, Roshek DM. Adult dystrophic (mdx) endplates exhibit reduced quantal size and enhanced quantal variation. Pflugers Arch. 2001;442:369–75. doi: 10.1007/s004240100561. [DOI] [PubMed] [Google Scholar]
- Chamberlain JS. Gene therapy of muscular dystrophy. Hum Mol Genet. 2002;11:2355–2362. doi: 10.1093/hmg/11.20.2355. [DOI] [PubMed] [Google Scholar]
- Chang CC, Chuang ST, Huang MC. Effects of chronic treatment with various neuromuscular blocking agents on the number and distribution of acetylcholine receptors in the rat diaphragm. J Physiol. 1975;250:161–173. doi: 10.1113/jphysiol.1975.sp011047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford GE, Faulkner JA, Crosbie RH, Campbell KP, Froehner SC, Chamberlain JS. Assembly of the dystrophin-associated protein complex does not require the dystrophin COOH-terminal domain. J Cell Biol. 2000;150:1399–1410. doi: 10.1083/jcb.150.6.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deconinck AE, Potter AC, Tinsley JM, Wood SJ, Vater R, Young C, Metzinger L, Vincent A, Slater CR, Davies KE. Postsynaptic abnormalities at the neuromuscular junctions of utrophin-deficient mice. J Cell Biol. 1997;136:883–894. doi: 10.1083/jcb.136.4.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery AE, Muntoni . Duchenne Muscular Dystrophy. 3. Oxford University Press; Oxford: 2003. [Google Scholar]
- Engel AG, Franzini-Armstrong C. Basic and Clinical. McGraw-Hill; New York: 2004. Myology. [Google Scholar]
- England SB, Nicholson LV, Johnson MA, Forrest SM, Love DR, Zubrzycka-Gaarn EE, Bulman DE, Harris JB, Davies KE. Very mild muscular dystrophy associated with the deletion of 46% of dystrophin. Nature. 1990;343:180–182. doi: 10.1038/343180a0. [DOI] [PubMed] [Google Scholar]
- Graciotti L, Minelli A, Minciacchi D, Procopio A, Fulgenzi G. GABAergic miniature spontaneous activity is increased in the CA1 hippocampal region of dystrophic mdx mice. Neuromuscul Disord. 2008 doi: 10.1016/j.nmd.2007.11.009. [DOI] [PubMed] [Google Scholar]
- Grady RM, Teng H, Nichol MC, Cunningham JC, Wilkinson RS, Sanes JR. Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: a model for Duchenne muscular dystrophy. Cell. 1997;90:729–738. doi: 10.1016/s0092-8674(00)80533-4. [DOI] [PubMed] [Google Scholar]
- Grady RM, Zhou H, Cunningham JM, Henry MD, Campbell KP, Sanes JR. Maturation and maintenance of the neuromuscular synapse: genetic evidence for roles of the dystrophin--glycoprotein complex. Neuron. 2000;25:279–293. doi: 10.1016/s0896-6273(00)80894-6. [DOI] [PubMed] [Google Scholar]
- Gregorevic P, Blankinship MJ, Allen JM, Crawford RW, Meuse L, Miller DG, Russell DW, Chamberlain JS. Systemic delivery of genes to striated muscles using adeno-associated viral vectors. Nat Med. 2004;10:828–834. doi: 10.1038/nm1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hack AA, Ly CT, Jiang F, Clendenin CJ, Sigrist KS, Wollmann RL, McNally EM. Gamma-sarcoglycan deficiency leads to muscle membrane defects and apoptosis independent of dystrophin. J Cell Biol. 1998;142:1279–1287. doi: 10.1083/jcb.142.5.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper SQ, Hauser MA, DelloRusso C, Duan D, Crawford RW, Phelps SF, Harper HA, Robinson AS, Engelhardt JF, Brooks SV, Chamberlain JS. Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nat Med. 2002;8:253–261. doi: 10.1038/nm0302-253. [DOI] [PubMed] [Google Scholar]
- Harris JB, Ribchester RR. The relationship between end-plate size and transmitter release in normal and dystrophic muscles of the mouse. J Physiol. 1979;296:245–265. doi: 10.1113/jphysiol.1979.sp013003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennig R, Lomo T. Firing patterns of motor units in normal rats. Nature. 1985;314:164–166. doi: 10.1038/314164a0. [DOI] [PubMed] [Google Scholar]
- Hoffman EP, Brown RH, Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- Knuesel I, Mastrocola M, Zuellig RA, Bornhauser B, Schaub MC, Fritschy JM. Short communication: altered synaptic clustering of GABAA receptors in mice lacking dystrophin (mdx mice) Eur J Neurosci. 1999;11:4457–4462. doi: 10.1046/j.1460-9568.1999.00887.x. [DOI] [PubMed] [Google Scholar]
- Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C, Kunkel LM. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50:509–517. doi: 10.1016/0092-8674(87)90504-6. [DOI] [PubMed] [Google Scholar]
- Koenig M, Kunkel LM. Detailed analysis of the repeat domain of dystrophin reveals four potential hinge segments that may confer flexibility. J Biol Chem. 1990;265:4560–4566. [PubMed] [Google Scholar]
- Koenig M, Monaco AP, Kunkel LM. The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell. 1988;53:219–226. doi: 10.1016/0092-8674(88)90383-2. [DOI] [PubMed] [Google Scholar]
- Kopriwa BM. Block-staining tissues with potassium ferrocyanide-reduced osmium tetroxide and lead aspartate for electron microscopic radioautography. J Histochem Cytochem. 1984;32:552–554. doi: 10.1177/32.5.6201530. [DOI] [PubMed] [Google Scholar]
- Kueh SL, Head SI, Morley JW. GABA(A) receptor expression and inhibitory post-synaptic currents in cerebellar Purkinje cells in dystrophin-deficient mdx mice. Clin Exp Pharmacol Physiol. 2008;35:207–210. doi: 10.1111/j.1440-1681.2007.04816.x. [DOI] [PubMed] [Google Scholar]
- Li S, Kimura E, Ng R, Fall BM, Meuse L, Reyes M, Faulkner JA, Chamberlain JS. A highly functional mini-dystrophin/GFP fusion gene for cell and gene therapy studies of Duchenne muscular dystrophy. Hum Mol Genet. 2006;15(10):1610–1622. doi: 10.1093/hmg/ddl082. [DOI] [PubMed] [Google Scholar]
- Lyons PR, Slater CR. Structure and function of the neuromuscular junction in young adult mdx mice. J Neurocytol. 1991;20:969–981. doi: 10.1007/BF01187915. [DOI] [PubMed] [Google Scholar]
- Marques MJ, Conchello JA, Lichtman JW. From plaque to pretzel: fold formation and acetylcholine receptor loss at the developing neuromuscular junction. J Neurosci. 2000;20:3663–3675. doi: 10.1523/JNEUROSCI.20-10-03663.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques MJ, Pertille A, Carvalho CL, Santo Neto H. Acetylcholine receptor organization at the dystrophic extraocular muscle neuromuscular junction. Anat Rec (Hoboken) 2007a;290:846–854. doi: 10.1002/ar.20525. [DOI] [PubMed] [Google Scholar]
- Marques MJ, Taniguti AP, Minatel E, Neto HS. Nerve terminal contributes to acetylcholine receptor organization at the dystrophic neuromuscular junction of mdx mice. Anat Rec (Hoboken) 2007b;290:181–187. doi: 10.1002/ar.20421. [DOI] [PubMed] [Google Scholar]
- Minatel E, Neto HS, Marques MJ. Acetylcholine receptor distribution and synapse elimination at the developing neuromuscular junction of mdx mice. Muscle Nerve. 2003;28:561–569. doi: 10.1002/mus.10416. [DOI] [PubMed] [Google Scholar]
- Pena J, Luque E, Noguera F, Jimena I, Vaamonde R. Experimental induction of ring fibers in regenerating skeletal muscle. Pathol Res Pract. 2001;197:21–27. doi: 10.1078/0344-0338-00004. [DOI] [PubMed] [Google Scholar]
- Peters MF, Adams ME, Froehner SC. Differential association of syntrophin pairs with the dystrophin complex. J Cell Biol. 1997;138:81–93. doi: 10.1083/jcb.138.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelps SF, Hauser MA, Cole NM, Rafael JA, Hinkle RT, Faulkner JA, Chamberlain JS. Expression of full-length and truncated dystrophin mini-genes in transgenic mdx mice. Hum Mol Genet. 1995;4:1251–1258. doi: 10.1093/hmg/4.8.1251. [DOI] [PubMed] [Google Scholar]
- Rafael JA, Townsend ER, Squire SE, Potter AC, Chamberlain JS, Davies KE. Dystrophin and utrophin influence fiber type composition and post-synaptic membrane structure. Hum Mol Genet. 2000;9:1357–1367. doi: 10.1093/hmg/9.9.1357. [DOI] [PubMed] [Google Scholar]
- Reynolds ES. The use of lead citrate at high pH as an electron-opaque stain in electron microscopy. J Cell Biol. 1963;17:208–212. doi: 10.1083/jcb.17.1.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW. Development of the vertebrate neuromuscular junction. Annu Rev Neurosci. 1999;22:389–442. doi: 10.1146/annurev.neuro.22.1.389. [DOI] [PubMed] [Google Scholar]
- Sealock R, Butler MH, Kramarcy NR, Gao KX, Murnane AA, Douville K, Froehner SC. Localization of dystrophin relative to acetylcholine receptor domains in electric tissue and adult and cultured skeletal muscle. J Cell Biol. 1991;113:1133–1144. doi: 10.1083/jcb.113.5.1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiao T, Fond A, Deng B, Wehling-Henricks M, Adams ME, Froehner SC, Tidball JG. Defects in neuromuscular junction structure in dystrophic muscle are corrected by expression of a NOS transgene in dystrophin-deficient muscles, but not in muscles lacking alpha- and beta1-syntrophins. Hum Mol Genet. 2004;13:1873–1884. doi: 10.1093/hmg/ddh204. [DOI] [PubMed] [Google Scholar]
- Slater CR. Structural determinants of the reliability of synaptic transmission at the vertebrate neuromuscular junction. J Neurocytol. 2003;32:505–522. doi: 10.1023/B:NEUR.0000020607.17881.9b. [DOI] [PubMed] [Google Scholar]
- Torres LF, Duchen LW. The mutant mdx: inherited myopathy in the mouse. Morphological studies of nerves, muscles and end-plates. Brain. 1987;110 (Pt 2):269–299. doi: 10.1093/brain/110.2.269. [DOI] [PubMed] [Google Scholar]
- Wehling M, Spencer MJ, Tidball JG. A nitric oxide synthase transgene ameliorates muscular dystrophy in mdx mice. J Cell Biol. 2001;155:123–131. doi: 10.1083/jcb.200105110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winder SJ. The membrane-cytoskeleton interface: the role of dystrophin and utrophin. J Muscle Res Cell Motil. 1997;18:617–629. doi: 10.1023/a:1018627705273. [DOI] [PubMed] [Google Scholar]
- Wood SJ, Slater CR. The contribution of postsynaptic folds to the safety factor for neuromuscular transmission in rat fast- and slow-twitch muscles. J Physiol. 1997;500 (Pt 1):165–176. doi: 10.1113/jphysiol.1997.sp022007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.