

Summary
Long-lived humoral immune responses are a hallmark of thymus-dependent immunity. The cellular basis for enduring antibody-mediated immunity is long-lived memory B cells and plasma cells (PCs). Both of these cell populations acquire longevity as a result of antigen-specific, CD40–dependent, cognate interactions with helper T cells within germinal centers (GCs). At the molecular level, defined functional domains of CD40 control the post-GC fate of B cells. PC precursors that emerge from these GC reactions are highly proliferative and terminally differentiate to end-stage cells within the bone marrow (BM). The striking phenotypic similarities between the PC precursors and the putative malignant cell in multiple myeloma (MM) suggests that MM may result from the transformation of PC precursors. Within the domain of autoimmune disease, recent studies have shown that dysregulated migration of PCs to the BM may impact immune homeostasis and the development of lupus. Understanding the processes of normal PC differentiation will provide strategic insights into identifying therapeutic targets for the treatment of differentiated B-cell disorders.
Introduction
During the mid-twentieth century, F. MacFarlane Burnet proposed an elegant theory that has been the underpinning of modern cellular immunology. In contrast to an instructional methodology, Burnet's Clonal Selection theory envisions a vast repertoire of B-cell receptor specificities, amongst which only those B cells best suited to recognize a pathogen are selected to grow and differentiate. The selection of clones by an invading pathogen is just the beginning of a long process, culminating in the transformation of the clonally selected B cells to terminally differentiated plasma cells (PCs), whose only function is to produce neutralizing immunoglobulin (Ig). A tenet of the Clonal Selection theory predicts the massive generation of clonal diversity during B-cell formation, without engendering a loss of self-tolerance or loss of cell-cycle control. As Burnet predicted, we now know that the resolution of immune regulation occurs largely at the level of single cell-fate decisions. These decision steps transpire throughout the lifespan of B cells. The defining cellular and molecular events that regulate the final steps in terminal B-cell differentiation are just beginning to be resolved. An understanding of the events that control terminal B-cell differentiation will provide opportunities to design strategies for therapeutic intervention in cancer and autoimmunity.
Generation of short-lived and long-lived plasma cells
PCs represent the terminal stage of differentiation for all antigen activated B cells. Following encounter with antigen, B cells undergo a transformation process, acquiring the ability to produce copious amounts of antibodies capable of neutralizing pathogenic antigens (1). As such, the characteristics of PCs include a larger cytoplasm-to-nucleus ratio than resting naïve B cells and increased amounts of rough endoplasmic reticulum. Both the signals that PCs receive during postgerminal center (post-GC) differentiation and the microenvironmental niche, which they occupy, influence whether they persist for only a few days or perhaps for many years. As terminally differentiated cells, PCs remain quiescent, unable to proliferate and generate daughter cells.
The quality of B-cell activation results in the formation of either short-lived or long-lived PCs. B cells activated in the absence of T-cell help [T-independent (TI) response] predominantly become short-lived PCs that reside within the extra-follicular regions of secondary lymphoid organs, such as the spleen (2, 3). Immunization via the TI antigen, NP-Ficoll, results in a peak proliferation of B cells at day 5, followed by a rapid loss of the majority of these cells (4). Antibody production was measured by serum enzyme-linked immunosorbent assay, and PC numbers also peak around day 5 and then quickly wane thereafter. As a characteristic, TI responses generate no memory B cells and very few, if any, long-lived PCs (4, 5).
B cells that encounter antigen in the presence of T cells providing help through the ligation of CD40 [T-dependent (TD) responses] generate prolonged responses characterized by the presence of GCs (6–9). T cells provide help to B cells via ligation of CD40 on the surface of B cells (Fig. 1).
Fig. 1. Short- and long-lived plasma cell (PC) formation.
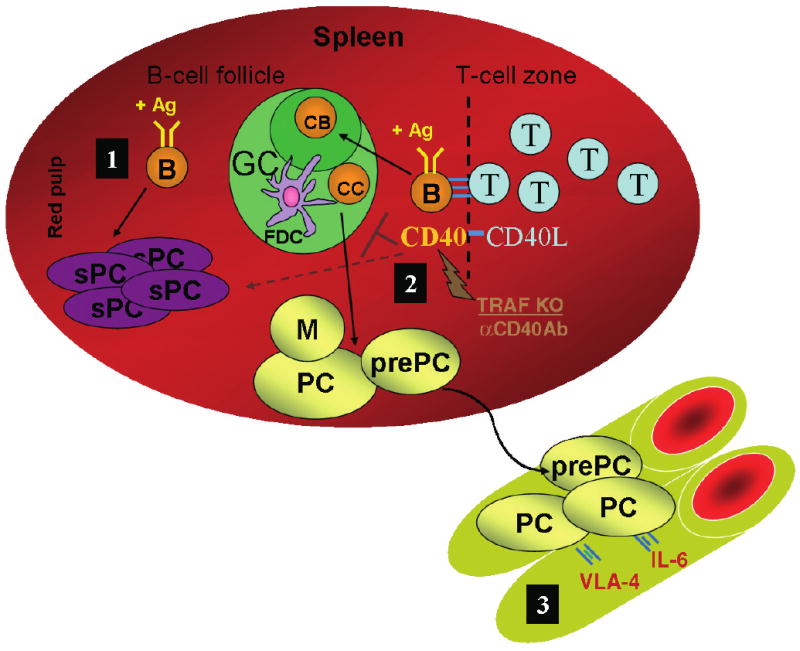
B cells activated by antigen (Ag) within secondary lymphoid organs, such as the spleen, may differentiate into either short-lived or long-lived PCs (1). B cells that encounter Ag in the absence of T-cell help (T-independent response) primarily differentiate to short-lived PCs (sPC) (2). B cells that encounter Ag along with CD40 ligation provided by helper T cells (T-dependent response) may be incorporated within a germinal center (GC). Within the dark zone of the GC, B cells called centroblasts (CB) rapidly proliferate, giving rise to daughter cells called centrocytes (CC). Centrocytes compete for binding to Ag presented via follicular dendritic cells (FDC), leading to affinity maturation of the antibody response. Centrocytes that successfully compete further mature as either memory B cells (M), long-lived PCs or PC precursors (prePC). Inhibition of CD40 signaling via the disruption of tumor necrosis factor receptor-associated factor (TRAF) binding aborts the GC, resulting in a primarily short-lived antibody response. Thus, both memory and long-lived PC formation are regulated by CD40. Interestingly, the GC response is also aborted by supraoptimal CD40 stimulation provided via either an agonistic anti-CD40 antibody or in vivo T-cell priming (αCD40Ab) (3). Optimal CD40 signaling results in the trafficking of long-lived PCs and PC precursors to the bone marrow (BM). BM PCs persist for extended periods, perhaps existing for a lifetime. PC precursors give rise to long-lived and short-lived PCs within the BM. Interactions between the BM environment and the PC regulate longevity and function. Some of these interactions are cell-contact mediated, such as binding of PC very late activation antigen-4 (VLA-4) to either vascular cellular adhesion molecule-1 (VCAM-1) on BM stromal cells or fibronectin on the extracellular matrix. Other interactions are mediated by soluble factors, such as interleukin-6 (IL-6).
Blocking interactions between CD40, expressed on B cells, and its ligand CD154, expressed on the surface of activated T cells, inhibits TD immune responses and terminates GCs (9–11). GCs initiate within the follicles of secondary lymphoid organs around day 7, when extrafollicular TI responses begin to wane. Within the GC, the activated B cells called centroblasts proliferate rapidly and endure processes of somatic hypermutation and isotype switching. Somatic hypermutation (SHM) alters the specificity of the B-cell receptor (BCR) by introducing point mutations within the hypervariable regions of the Ig genes. The daughter cells of centroblasts, centrocytes, inherit varying specificities to the activating antigen. Centrocytes must then endure the process of affinity maturation involving competition for survival signals based on the affinity of the BCR for antigen. Those that compete successfully and receive survival signals exit the GC and continue to mature as either long-lived PCs or memory B cells (7, 11). Interestingly, the addition of an agonistic CD40 signal to mimic T-cell help during a TI response (i.e. NP-Ficoll stimulation) increases the magnitude of the antibody response, yet fails to induce the onset of GCs (4, 12). Hence, signals beyond the engagement of CD40 are essential for B cells to terminally differentiate to long-lived PCs.
Blocking CD40 signaling not only inhibits GC formation but also inhibits subsequent affinity maturation, isotype switching, and formation of memory cells and long-lived PCs. Recent studies have suggested that each of these differentiating events may be controlled by distinct functional domains of the CD40 cytoplasmic tail. Ligation of CD40 by CD154 results in a recruitment of tumor necrosis factor receptor-associated factors (TRAFs) 1, 2, 3, 5, and 6 to the cytoplasmic domain of CD40 (13–15). Ablation of the TRAF 2/3- and 6-binding sites results in the loss of GC formation (16). However, GCs do form if either the TRAF 2/3- or 6-binding site is available (16). Moreover, signals via CD40 control the maturation and selection of centroblasts and centrocytes. The ablation of TRAF 6-, but not TRAF 2/3-, binding sites results in a loss of affinity maturation and long-lived PC formation (16). Using a similar approach, Yasui and coworkers (17) demonstrated that both TRAF 6- and TRAF 2/3/5-binding sites were required for GC formation. These studies utilized different forms of mutant human CD40 molecules, which probably account for the differential requirement of TRAF 2/3 binding for GC responses. Recent studies also demonstrate that an excess CD40 signal results in an aborted GC response. Immunization with NP(4-Hydroxy-3-nitrophenylacetyl)-KLH (keyhole limpet hemocyanin) induces signal 1 via BCR ligation, signal 2 via helper T-cell CD40 stimulation and eventual GC formation. Administration of an agonistic anti-CD40 antibody (FGK115) in addition to NP-KLH stimulation provides supraoptimal CD40 signaling and aborts the normal follicular GC response, resulting in an extrafollicular response (18). The duration of the extrafollicular response is reduced compared with non-FGK-treated responses. Interestingly, like αCD40, the hyperimmunization of the helper T-cell compartment can also accelerate GC responses and cause an enhanced differentiation of B cells to PCs. These studies demonstrate that the intensity of the CD40 signal can exert a dramatic impact on the quality of the humoral immune response.
B cells that do receive an optimal CD40 signal and survive affinity maturation within the GC may become long-lived PCs (1). Long-lived PCs are characterized by the downregulation of several cell-surface markers. This loss includes surface CD45, B220, major histocompatibility complex (MHC) class II, CD19, and most notably the BCR (1, 19, 20). Leukocyte markers that PCs continue to express and that are useful in their phenotype characterization include the chemokine receptor CXCR4, the integrins, very late activation antigen-4 (VLA-4) and leukocyte function-associated antigen-1 (LFA-1), the hyaluronic acid receptor CD44, the glycoprotein Ly6C, and an adhesion/growth factor receptor CD138, also known as syndecan-1 (1). Of course, the most striking characteristic of long-lived PCs is their ability to produce antigen-specific antibody for extended periods of time. While short-lived PCs may survive for only a few days, long-lived PCs persist for at least 1 year (20). Following the induction of an immune response to ovalbumin (OVA), B220− PCs isolated from murine bone marrow (BM) were transferred to naïve recipients. Absent from antigen stimulation, these BM PCs continued to produce OVA-specific antibody for greater than 3 months. This response did not reflect contamination with memory B cells, as they were separated via the expression of B220. In yet another murine study regarding the longevity of antibody production, long-lived PCs were also observed (19). Following in vivo depletion by irradiation of naïve and memory B cells 60 days post challenge with lymphocytic choriomeningitis virus (LCMV), PCs persisted for periods greater than 1 year. Again, these results were found to be exempt from the activities of naïve B cells and memory B cells. Within humans, antibody titers to viral pathogens have been shown to persist for decades (21). As recently suggested, the persistent antibody titers may be derived via polyclonal stimulation of memory B cells (22). Long-lived antibody titers may also be due in part to long-lived PCs that reside within human BM for many decades, secreting protective antibody.
The course of B-cell differentiation can be tracked by the sequential expression and silencing of transcription factors that control B-cell fate. A number of studies underscore the central role that B lymphocyte-induced maturation protein 1 (BLIMP-1) plays in the PC-transformation process (23, 24). BLIMP-1 expression results in a repression of Bcl-6 and B-cell lineage-specific activator (BSAP, encoded by PAX5), both of which are required for earlier B-cell maturation stages. BLIMP-1 also represses a wide variety of other genes including activation-induced cytidine deaminase (AID), which is required for SHM and isotype class switching (25), and MHC Class II transactivator (CIITA), which is required for MHC class II expression (26). Inhibition of BLIMP-1 expression leads to a loss of PC formation. Other transcription factors necessary for PC development include X-box binding protein 1 and interferon regulatory factor 4 (27, 28). The regulatory interactions of these factors for PC formation are complex and have already been reviewed in great detail elsewhere (1). Thus, we will not review their function here. However, it should be emphasized the importance of these transcription factors in PC development.
Long-lived PCs may persist within the spleen but primarily reside within the BM. Early studies by Benner and colleagues (29, 30) reported that over 80% of serum Ig arises from long-lived PCs residing in the BM. Whether antigen is a pathogen component or a neoantigen in adjuvant affects the location of long-lived PCs and their longevity. Immunization with viral pathogens, such as LCMV, results in long-lived PC formation within both the spleen and BM (19). Following LCMV challenge of BALB/c mice, thousands of PCs persist within both the spleen and BM. Approximately 3 × 104 PCs persist within the spleen, while more than 2 × 104 PCs reside within the BM for the life of the animal (19). Non-replicating antigens, such as OVA, result in an initial burst of short-lived PCs within the spleen followed by an accumulation of PCs within the BM that persist for periods greater than 3 months (20). The variability between the numbers of long-lived PCs within the spleen and BM may reflect environmental differences at these sites as well as intrinsic constraints on splenic PCs vs. those that traffick to the BM (21). Sze et al. (5) indicate that the peak level of PCs within the spleen measures between 100 and 5000 cell/mm2. However, the spleen only retains between 20 and 100 long-lived PCs/mm2 beyond 10 days. This loss of PCs may reflect the competition for a limited number of survival niches within splenic architecture. The majority of long-lived PCs generated within the spleen eventually traffick to the BM, where they must also compete for survival niches. Similar to the spleen, the levels of PCs found within the BM remain stable over time, suggesting a limited carrying capacity. Yet, in contrast to the spleen, the total capacity for PCs within the BM may be 400 000 cells or approximately 0.5% of all mononucleated BM cells (21). It remains unclear whether the greater number of PCs found within the BM reflects intrinsic longevity of cells that traffick to the BM or a greater number of survival niches as compared with the spleen.
PCs may also be derived from long-lived memory B cells derived from GC centrocytes. Memory B cells contain high-affinity BCRs, enabling them to respond in vivo to minute amounts of antigen. Yet, memory B cells can persist for long periods of time in the absence of continual antigen stimulation. The longevity of memory B cells was demonstrated most recently in a study with murine B cells capable of switching the specificity of their BCR via an interferon-induced Cre recombinase from an affinity for the antigen NP to phycoerythrin (PE) (31). NP-binding memory B cells were generated in vivo followed by the induction of BCR switching via interferon injection. The persistence of the resulting PE-switched memory B cells was then observed in the absence of immunization with the PE antigen. The PE-switched memory cells persisted for periods greater than 100 days (31). Moreover, upon PE antigen immunization, the switched memory cells quickly differentiated to PCs secreting PE-specific antibody (31). Other studies have similarly demonstrated that memory B cells differentiate into PCs upon secondary encounter with antigen. In vitro cultures of naïve vs. memory B cells with interleukin-4 (IL-4), IL-10 and either CD40 or BCR ligation resulted in the presence of PCs in each culture (32, 33). However, the memory cell cultures resulted in five- to eight-fold more PCs than the naïve B-cell cultures (32). Moreover, while the naïve cell cultures produced predominantly IgM B-cell blasts, the memory cell cultures produced PCs secreting IgG and IgM. Thus, memory B cells show a bias towards producing new PCs capable of producing large amounts of antibody.
The inherent longevity of the PCs resulting from memory cell stimulation remains unknown. Limited evidence may suggest that these memory cell-derived PCs may only survive for short periods of time. Barrington et al. (34) recently studied the production of NP-specific antibody and PCs following adoptive transfer of memory B cells with antigen and T-cell help. The level of serum antibody derived from transferred memory B cells decreased with time within the spleen and BM. Memory cells transferred to wildtype (WT) recipients results in NP-specific antibody titers of 22.7 × 103 at week 2 and only 6.4 × 103 at week 16 (34). The decrease in antibody titers was more pronounced in secondary recipients lacking complement receptor 2 (CR2KO) on their stromal surfaces, falling from 16.7 × 103 at week 2 to 2.9 × 103 at week 16 (34). The level of antibody titer within the CR2KO mice at week 16 was very similar to background levels within non-immunized mice. The antibody deficit observed within the CR2KO mice correlated with a concomitant loss of memory B cells within the spleen and BM, compared with WT controls (34). The loss of antibody titers within the WT and CR2KO mice may reflect the short life of PCs derived from memory B-cell activation. Moreover, the increased deficit of antibody titer observed within the CR2KO mice may result from an inhibited memory cell response, with a lower rate of short-lived PC production. Previous studies have demonstrated that optimal memory cell responses require CR2 expression (35). Perhaps memory B cells produce long-lived PCs, following antigen activation. However, it seems clear, given the significant decrease in antibody titers, following adoptive transfer, that memory cells predominantly produce short-lived PCs. Neutralization of a pathogen following memory cell activation via short-lived PC progeny would allow for the termination of excess antibody production. Upon re-infections, successive rounds of primary B-cell responses leading to memory B-cell formation and activation would replace the short-lived PC compartment. Thus, the neutralization of a pathogen would only require memory B-cell activation, leading to short-lived PC formation. However, without direct examination of proliferation rates of memory B cells and survival of PC progeny within secondary recipients, the longevity of PC progeny cannot be determined.
In summary, PCs may be generated as either short-lived or long-lived cells. PCs may be derived from antigen-induced B-cell activation, with or without cognate T-cell help. PCs may be derived from activated B cells, memory B cells, or GC centrocytes. The majority of long-lived PCs reside within the BM and probably represent second or third generation descendents of GC centrocytes. Thus, the ultimate quality and quantity of a PC population depends largely on the type of B-cell activation and anatomical site of residence. The formation of a PC population will largely depend on interactions between earlier stages of development and environmental regulation.
A novel intermediate: plasma cell precursors
To date, discrete steps in lineage commitment between a GC B cell and a memory or PC have not been defined. Recently, we and various other groups exploiting both human and murine experimental systems have begun to identify post-GC B cells that are direct precursors to PCs (36–39). In contrast to fully differentiated BM PCs, murine PC precursors continue to express many cell-surface markers (39). PC precursors retain the expression of the BCR, MHC class II, and B220 receptors, albeit at lower levels than observed on splenic B cells. Precursors are antigen-experienced B cells exhibited by an activated profile including the expression of CD80 and CD86. The expression of CD138, a marker of PCs, distinguishes precursors from splenic B cells. PC precursors express receptors capable of interacting with BM stroma. These surface markers include VLA-4, CD44, LFA-1, and IL-6 receptor (39). An ability to interact with stromal elements probably fosters the longevity of PC precursor residence within the BM.
PC precursors generated at secondary lymphoid sites initially appear within the BM, 2 weeks following induction of an immune response. Once within the BM environment, precursors rapidly proliferate, giving rise to new PCs within 7 days (39). The PC progeny of precursors produce antigen-specific antibody within the BM compartment and survive for greater than 6 months. PC precursors are also long-lived, persisting within the BM for a period of at least 6 months (39). Inhibition of precursor proliferation via irradiation or mitomycin C treatment ablates the formation of new PCs (19, 39). Hence, PC precursors rapidly proliferate within the BM, and this proliferation is critical for the differentiation to PCs. Identification of mechanisms regulating PC precursor proliferation and differentiation is the subject of ongoing studies. Isolation of PC precursors confirms the existence of a post-GC B-cell lineage that promotes short- and long-lived PC formation. Separation of the precursor population, based on CD138 and CD44 expression, revealed subpopulations with varying capabilities. CD138+CD44− and CD138−CD44+ PC precursors give rise to long-lived PCs, while CD138+CD44+ precursors generate only short-lived PCs (39). Interestingly, CD138−CD44− PC precursors produce very few, if any, PCs, and their functional identity is still unresolved (39). The phenotype of the precursors exhibits intermediate characteristics between GC B cells and PCs, while also inferring an ability to interact with the BM environment. This ability to communicate with BM stroma may be central to the regulation of PC precursors, and thus the formation of long-lived PCs. Considering this supposition, continued expression of the BCR by precursors within the BM may indicate a susceptibility to BCR-mediated control mechanisms. Perhaps even post-GC PC precursors are subject to positive or negative selection within the BM, providing a means through which to regulate affinity or autoreactivity of individual antigen-specific PC populations.
Homing properties of plasma cells
The specific microenvironment occupied by PCs has been implicated as a determinant in cellular lifespan and survival, and hence the capacity to produce Ig for long periods of time (21, 40, 41). Migration to specific microenvironmental niches is controlled by chemokines and their receptors. Recent studies have demonstrated that members of the chemokine family play an integral role in mediating T and B lymphocyte migration within primary and secondary lymphoid compartments (42–44). Chemokines make up a large superfamily of approximately 50 members, and they function primarily as chemoattractants towards leukocyte and lymphocyte populations. Similar to their cousins of the cytokine superfamily, chemokines are small proteins (8–10 kDa) that share a conserved structure that includes a core β barrel and three antiparallel β-strands. The structure of this core is maintained by disulfide bonds between four cysteine residues. The position of these four cysteines can vary within each molecule, leading to the establishment of two major subfamilies of chemokines known as ‘CXC’ and ‘CC’ chemokines. The initial two cysteine residues are separated by another residue in CXC chemokines, hence distinguishing them from the CC chemokines. The action of specific chemokines is regulated by the selective expression of chemokine receptors, 7-transmembrane G-protein-coupled molecules, and is defined by virtue of its ligand binding. Thus, receptors that preferentially bind CXC chemokines are identified as CXCRs, while those that preferentially bind CC chemokines are recognized as CCRs. The coordinated expression of chemokines and their receptors orchestrate the movement of naïve B cells to follicles, antigen-reactive B cells to the T-cell zone within spleen, and PC localization to BM.
Burkitt lymphoma receptor 1 or CXCR5 is a chemokine receptor that is upregulated on B cells during their transition from an immature (IgMhiIgD−) to mature (IgM+IgD+) stage (45). CXCL13 [also known as B lymphocyte chemoattractant (BLC)/B-cell-attracting chemokine-1 (BCA-1)] has been identified as a ligand for CXCR5, and it is abundantly expressed in splenic follicles (46, 47). Mice that are genetically deficient in CXCR5 do not develop inguinal lymph nodes and Peyer's patches, and the splenic architecture is significantly compromised (45). In the absence of CXCR5, mature B cells lose their capacity to migrate into discrete follicles. Even so, CXCR5-deficient animals have the capacity to generate ectopic GCs, leading to the selection of affinity-matured B cells (48). Interestingly, when CXCR5-deficient B cells were adoptively transferred into WT recipients, homing to follicles in lymph nodes appeared normal. In contrast, studies using CXCL13-deficient mice demonstrated a requirement for this chemokine and its receptor in B-cell homing to both splenic and lymph node follicles (49). These results suggest that while CXCR5 is necessary for lymph node organogenesis, other chemokine receptors that bind CXCL13 can assist in the recruitment and organization of B cells into follicles. Importantly, antigen-reactive B cells that terminally differentiate to PCs downregulate CXCR5 expression, causing a loss in their responsiveness towards CXCL13, and thus release of these cells from being retained within follicles (50–53).
Previous work has also shown that while mature follicular B cells do not express CCR7, activation through CD40 can induce CCR7 (54). Differentiation to a PC fate results in the downregulation of CCR7 expression, and as a consequence, show decreased responsiveness to the chemokines, CCL19 and CCL21 (51). Similar to CXCR5, these findings have led to the conclusion that the pattern of chemokine receptor expression on differentiating B cells function to retain cells within the B-cell follicle (CXCR5, CCR7) or to initiate migration to other sites, such as the BM (CXCR4).
While many chemokine receptors are able to bind multiple chemokines, CXCR4 binds only CXCL12 (42, 55). CXCR4 and CXCL12 have well-established roles in directing peripheral cell types to the BM. CXCL12 is highly expressed by BM stromal cells (50, 56, 57), and it was shown more recently to be expressed by sinusoidal endothelial cells (58, 59). The biologic function of CXCR4–CXCL12 interactions in guiding cellular movement to the BM was revealed by studies demonstrating that transgenic overexpression of CXCR4 on CD4+ T cells induced homing to the BM (60). Only recently has the role of CXCR4 and CXCL12 in B-cell differentiation been studied. While naïve B cells constitutively express CXCR4, these levels are upregulated as differentiating B cells commit to a PC fate (50). Combined, the decrease in CXCR5 and increase in CXCR4 expression on PCs allow emigration from splenic red pulp and homing to BM. Evidence for the critical role of CXCR4 in navigating PCs to the BM comes from CXCR4−/− mice. While CXCR4-deficient mice die perinatally, studies using CXCR4−/− fetal liver chimeras have demonstrated that the frequency of PCs in the BM is <30% compared with WT controls (50, 61, 62). However, the few CXCR4−/− PCs that did migrate to the BM suggest that other chemokine interactions participate in PC localization. PCs generated extra-follicularly express CXCR4, yet fail to migrate (51), and this finding implies that chemokines have distinct functions depending on the nature of the PC (i.e. short-lived or long-lived).
The role of CXCR3 and its multiple ligands, CXCL9, CXCL10, and CXCL11, has been implicated in the migration of lymphocytes, primarily T cells, to inflammatory sites (63–65). Contrary to CXCR5 expression, follicular B cells do not express CXCR3 (66). However, ex vivo chemotaxis assays of OVA-specific PCs have shown that early splenic and BM PCs can migrate in response to these inflammatory chemokines (67). These findings suggest that CXCR3 may also act as a mediator in guiding PCs into the BM, or in the context of autoimmunity, CXCR3 may promote the migration of autoreactive PCs to target organs known to be involved in disease pathogenesis. To date, there are no in vivo experiments on the expression and role of CXCR3 in mediating PCs to inflamed tissues.
Interaction of plasma cells with the bone marrow environment
A population of long-lived PCs may be observed within the BM, approximately 2 weeks following immunization. Both Ahmed et al. (19) and Radbruch et al. (20) demonstrate the longevity of antibody production from long-lived PCs in the absence of continued antigen stimulation. The contribution of BM PCs over the life of an organism can thus be quite significant. As the site of BM PC residence, the BM environment contributes to the regulation of humoral immune memory. This contribution comes in the form of competition between long-lived BM PC, PC precursors, and other resident BM cells for access to survival factors within environmental niches. A number of studies illustrate the ability of BM stroma to support PC formation (68–71). In one study, early PCs with a phenotype of CD38++CD19+VLA5− were isolated from human peripheral blood and were cultured in vitro (69). Within 2 days, virtually all of the early PCs died. However, the addition of the BM stromal cell line, KM-102, rescued a great number of these early PCs and promoted the continued maturation towards a fully differentiated BM PC, illustrated via increased CD38 surface expression (69). These studies also demonstrated that IL-6 derived from the BM stroma promoted the formation of mature BM PCs (69). Recent studies have also elucidated a number of environmental factors promoting PC longevity within the BM. E-selectin/P-selectin-deficient mice (E/P−/−) contain an easily isolated population of PCs from lymph nodes (72). In the presence of primary BM stromal cells, E/P−/− PCs and WT murine BM PCs continued to produce antibody for at least 3 weeks (70). In the absence of BM stroma, antibody titers could only be detected for approximately 7 days. The recovery of cultured PCs as a percentage of input at day 3 was 35.8% with stromal cells but only 2.1% without stromal cells. Soluble factors derived from the BM stroma seem to affect PC longevity. Use of stromal cells unable to produce IL-6 resulted in a loss of antibody production by approximately day 7 (70). Moreover, stromal cells cocultured with PCs upregulate the expression of IL-6 mRNA compared with stromal cells cultured in the absence of PCs (70). Contact between PCs and BM stroma also seems to affect longevity. PC cocultured with stromal cells in the presence of a blocking antibody to the adhesion molecule VLA-4 resulted in a 70% reduction of antibody titers by day 7 (70). These studies clearly indicate that both soluble factors and cell–cell contact contribute to the duration and differentiation of PCs. They also demonstrate the ability of BM PC and perhaps PC precursors to modify the BM environment. The interactions between BM PC or PC precursors and BM stroma promote the formation and persistence of long-lived humoral immunity.
Resident populations other than PCs are also managed by BM stroma. Memory cells within the BM can be phenotypically and functionally separated from PCs via B220 expression (20). Mice defective in the expression of complement receptor 2 (CR2−/−) exhibit abnormal numbers of memory B cells within the BM (34). The numbers of memory B cells and their PC progeny greatly decreases in the absence of CR2 (34). However, the direct influence of the CR2 on PC longevity has yet to be separated from the effect on the memory B-cell population within the BM. Exactly how these two B-cell populations interact within the BM has yet to be examined.
Long-lived plasma cells and maturation of humoral memory
The persistence of immune complexes influences the magnitude and quality of memory B-cell responses. Numerous studies suggest that the presentation of antigen via antigen–antibody (Ag–Ab) complexes, bound by Fc receptors (FcR) and CRs, facilitate the duration and affinity of the memory B-cell responses (34, 35, 73). However, the form of antigen immunization may determine the requirement for FcR and/or CR Ag–Ab complexes. Prolonged antigen exposure in the form of multiple antigen immunizations, or in the context of an adjuvant, alleviates the requirement for Ag–Ab complex deposition (35, 73). For example, secondary immune responses to NP were compared between WT and CR2KO mice. When primary immunization consisted of NP-bovine serum albumin (BSA) in phosphate-buffered saline, antibody titers in CR2KO mice following secondary challenge were almost absent, while WT mice exhibited a significant increase in titer (35). In contrast, primary immunization with NP-BSA in alum completely restored NP-specific antibody titer levels in CR2KO mice to WT levels, following secondary antigen challenge (35). Thus, while Ag–Ab complexes bound to FcR and CR generate optimal secondary responses, in the absence of these interactions, prolonged exposure to antigen can compensate for this deficit.
In addition to affinity selection of B cells in the GC, affinity maturation of the immune response may occur within the immune BM. Kelsoe and colleagues (74) have shown that dissolution of GCs late in the immune response does not impede further affinity selection. We have demonstrated that both short-lived and long-lived PCs traffick to the BM, following a primary response (39), and they may be targets for affinity maturation. Perhaps PC precursors are subject to selection within the BM based on the antigen affinity of their BCR. Competitive selection between PC precursors would engender BM PCs encoding high-affinity Ig genes and account for the maturation of circulating antibody. This reasoning may explain the observations of Kelsoe et al. (74)
Contribution of plasma cells to disease
Autoimmunity and some hematological malignancies may be because of dysregulated differentiation of B cells to PCs. The spleens of autoimmune mice contain unusually large numbers of PCs (75). Moreover, nonlymphoid sites within autoimmune mice, such as the kidneys, may also abnormally harbor populations of PCs (76). Mature B-cell malignancies, especially multiple myeloma (MM), are characterized by the accumulation of clonotypic malignant antibody secreting cells within lymphoid organs (77). Often, malignancies are derived from genetic translocations that result in a loss of cell-cycle regulation. Recent studies have illustrated circumstances sufficient to generate lymphomas in vivo. Mice deficient in the p53 tumor suppressor and the NHEJ DNA repair protein Ku spontaneously develop lymphomas characterized by translocations between the IgH and c-myc loci (78). Cycles of replication lead to the amplification of the IgH/c-myc translocation and protection from degradation through telomere capture (78). This type of translocation often accompanies the diagnosis of BM PC malignancies (79). Whether the translocations associated with PC malignancies occur along a similar pathway as observed with the p53/Ku-deficient mice remains to be determined. However, given that PCs are by definition sessile, replication events leading to the amplification of genetic translocations would be unlikely. Rather, the target of genetic transformation may more likely be the proliferating progenitor of BM PCs, the PC precursor. Thus, BM mechanisms regulating PC precursor function may play an important role in preventing disease.
Multiple myeloma
MM is an incurable B-cell malignancy that accounts for approximately 1% of all cancer-related deaths in Western countries (80). It is the second most common malignancy of the blood in the United States, affecting 40 000 people at any one time; approximately 13 000 new diagnoses are made each year (81). The disorder is characterized by malignant PC accumulations in the BM and the aberrant production of Ig, usually monoclonal IgG or IgA. Common clinical features of MM include susceptibility to bacterial infections, anemia, osteolytic lesions, and renal insufficiency. A condition known as monoclonal gammopathy of undetermined significance (MGUS) is fairly common in the US, with an estimated 1 million patients affected by the disorder. Significantly, 25% of MGUS patients progress to MM, although malignant conversion is difficult to predict and may not follow MGUS presentation for several decades (81). Recent advances in therapy for MM have been extending the survival period of MM patients, but the average life expectancy from diagnosis remains about 30–48 months. Chemotherapeutic approaches predominate, as the malignant cell progenitors have proven highly radio-resistant. The current therapies include induction chemotherapy (VAD: vincristin, adriamycin, and dexamethasone) proceeding to high-dose therapy and some form of stem cell rescue, with autologous BM transplantation being predominant (82). Additional alternative therapies include the application of thalidomide in advanced cases of myeloma. Thalidomide appears to influence the disease through anti-angiogenic effects, downregulation of tumor necrosis factor-α, and alteration of adhesion molecules in the BM microenvironment (83). Remarkably, thalidomide has induced strong and lasting responses in patients suffering relapses post high-dose chemotherapy, extending survival periods beyond 5 years in some cases (84). New therapies, currently enrolled in Food & Drug Administration clinical trials, will provide novel avenues of treatment. Velcade (Millennium Pharmaceuticals Inc., Cambridge, MA, USA) is currently being tested in a multicenter phase III trial with MM patients. Velcade inhibits proteosome activity, which disrupts the cell cycle and leads to malignant cell apoptosis (85). Despite these aggressive approaches, only small percentages of patients diagnosed with MM have survived beyond 5 years post diagnosis. One of the critical elements preventing improvements in therapy is the lack of a defined clinical target. The precise cellular origin of malignancy and the differentiation stage of the responsible B lymphocytes is ill defined (86–88). Although malignant cells may be almost eradicated from the BM by chemotherapy, drug-resistant myeloma precursor cells persist (89). In spite of the disease prevalence and lethality, the molecular pathogenesis and the steps involved in transformation from MGUS to MM remain unclear (81).
The first step of the proposed multistep transformation process is immortalization. The malignant PCs observed in MM are localized to the BM in the earlier stages of the disease, and most closely resemble long-lived PCs. These cells have undergone antigen selection outside the BM, as evidenced by their isotype-switched and somatically hypermutated Ig genes. Despite their similarities to long-lived PCs, myeloma cells have significantly lower rates of Ig secretion compared with normal PCs. Therefore, it appears that the critical neoplastic transformation events take place after or do not interfere with most of the normal B-cell differentiation process, leading to long-lived PCs (86). The most common oncogenic events in MM involve translocations into the IgH locus, commonly in switch regions (90). Clearly, the diverse nature of tumor cells found in MM lends credence to a complex multiple step transformation process, and the development of malignant clones arises from a differentiation process closely tied to the normal B-cell differentiation pathway.
Terminal B-cell development and multiple myeloma malignancy
Once immortalized, myeloma cells have to become established in the BM, as early tumor growth is entirely dependent on the BM microenvironment, especially the paracrine support of IL-6 provided by BM stromal cells. Normal BM PCs derive from plasmablastic cells generated in peripheral lymphoid tissues, following antigen stimulation. From periphery to BM, centrocytic B cells must differentiate into low-rate Ig-secreting plasmablastic cells, migrate to the BM, and proliferate as immature plasmablasts (91). Differentiation of BM plasmablastic cells into high-rate Ig-secreting PCs is the final step in long-lived BM PC production. Cytokines IL-2, IL-4, and IL-10 demonstrate B-cell growth factor activity, with IL-10 capable of inducing complete differentiation of B cells into Ig-secreting PCs along with CD40 activation. IL-6 is a critical cytokine supporting the proliferation of normal plasmablasts in vitro, and plays an important role in the development of plasmablasts into mature PCs. While identified as the primary differentiation factor for plasmablasts in normal BM, IL-6 fails to stimulate Ig production in myeloma cells (normally low Ig-secreting cells) (80). IL-6, however, is critical for the proliferation of early myeloma cells. The adhesion of normal B cells or myeloma cells to BM stromal cells, mediated by VLA-4 on the B cells/myeloma cells binding to vascular cellular adhesion molecule-1 (VCAM-1) on BM stromal cells is critical. This cell-to-cell interaction upregulates IL-6 secretion from the BM stromal cells, mediating the differentiation of normal B cells, but inducing the proliferation of myeloma cells (82). The fact that IL-6 fails to induce profuse Ig production in myeloma cells while still serving as a critical survival/proliferation factor for myeloma cells suggests that the myeloma precursor is an incompletely differentiated PC, still retaining plasmablast characteristics (87). Numerous studies have demonstrated the presence of circulating clonotypic B lymphocytes in peripheral blood samples from patients with active myeloma (92–95). These cells represent an upstream population of late-stage B cells that may serve as a drug-resistant reservoir of myeloma precursors, capable of expanding and differentiating into a malignant PC tumor. Thus, clonotypic myeloma B lymphocytes share many characteristics with normal PC precursors, including an ability to interact with stromal cells via VLA-4 and IL-6 receptor expression and to generate new BM PCs (38, 39, 82).
B-cell transcription factors associated with multiple myeloma
In a recent report from Nagy et al. (96), B-cell-specific transcription factor expression was analyzed in malignant cells from MM patients and PC leukemia (PCL) patients. Myeloma cells were found to express BLIMP-1, oct-2, PU.1, and Spi-B, and were shown to not express BSAP (96). BLIMP-1 is required for PC differentiation, and represses c-myc (leading to the cessation of the cell cycle in normal PCs), CIITA (leading to the downregulation of MHC class II on PCs), and Pax-5 (leading to the downregulation of BSAP) (1). Activity of oct-2 in myeloma cells correlates with its role in normal PC development, as oct-2 binds octamer motifs in Ig enhancer and promoter regions and stimulates Ig production. PU.1 and Spi-B are transcription factors belonging to the ets family, and their expression in myeloma cells in this study contrasted with their notable absence in PCL samples. Established PC lines studied in this research also lacked expression of PU.1 and Spi-B. PU.1 has been shown to regulate the expression of various integrins involved in homing and engraftment of stem cells to BM stroma (97). The expression of PU.1 in myeloma cells helps to explain the homing of the tumor to the BM and may be indicative of their stage of development. Spi-B has high homology with PU.1, but in knockout experiments in mice, it has been shown to play a role in affinity maturation and the GC reaction (98). Retention of both of these ets family member transcription factors may be further evidence of the origin of the myeloma precursor as a plasmablastic cell exiting the GC.
Myeloma clonotypic origin
Studies analyzing the nature of circulating clonal cells in B-cell proliferative disorders showed that MGUS patients in their study, in contrast with MM patients, commonly lacked circulating clonal PCs, as determined by immunofluorescence microscopy and flow cytometry. Clonal cells were found in the peripheral blood of MGUS patients by flow cytometry, but these cells lacked typical PC morphology, resembling instead plasmablastic PC precursors, presumably in transition from distal sites to the BM for continued differentiation into mature PCs (99). Further evidence that the malignant lesion in myeloma occurs at the post-GC plasmablast stage comes from studies on the role of isotype switching in myeloma development. A variety of isotype-switched myelomas have been described, including secretory IgG, IgD, IgE, and IgM variants. Although the mechanisms involved in isotype switching are still poorly understood, the fundamental contribution of switching to the development of malignancy is clearly suspected. As isotype switching appears to introduce double-strand breaks and involves enzymes associated with RNA splicing, it is possible that foreign DNA from another chromosome can become involved, resulting in an aberrant translocation event (100). Indeed, up to 74% of myeloma patient samples show rearrangements of the Ig heavy chain locus, involving partner loci that include cyclin D3, c-myc, Bcl-2, and FGFR3. Isotype switching normally takes place near the end of the GC reaction. Translocations involving switch regions of the IgH locus are recognized as the most common karyotypic abnormality in MM (100).
Myeloma precursor: targets of transformation
The abnormally high IL-6 production seen in all cases of MM and the dependence of myeloma cells on IL-6 as a growth and antiapoptotic factor are critical components of disease pathogenesis (80, 86, 87). Exploring the B-cell differentiation pathway has produced some major advances in the field of MM research. However, the myeloma precursor has yet to be identified. Recent advancements indicate a similarity of clonotypic B cells with PC precursors, a normal counterpart. Both cell types express similar phenotypes including CD45, CD44, and class-switched surface Ig expression (38, 39, 86, 101–103). Both myeloma B cells and PC precursors represent a post-GC stage of B-cell development. CD45+ PC precursors and myeloma B cells can also produce PCs, following adoptive transfer (38, 39). However, unlike PC precursors, the lack of a characteristic MM precursor phenotype prevents targeted studies of the environmental factors promoting disease progression. Identification and isolation of MM progenitors will provide the next leap forward to understanding the molecular pathogenesis of disease and provide novel avenues for the development of therapeutics in the treatment of MM (Table 1).
Table 1. Characteristics of normal and malignant plasma cells and precursors.
| PC precursor | Putative myeloma precursor | PC | Myeloma tumor | |
|---|---|---|---|---|
| Phenotype | ||||
| Surface immunoglobulin | ++ | ++ | − | − |
| IL-6R | ++ | ++(?)† | ++ | ++ |
| VLA-4 | ++ | ++(?)† | ++ | ++ |
| Bone marrow homing/localization | Yes | Yes | Yes | Yes |
| Proliferation potential | + | + | − | +/− |
| Antigen selection | ||||
| Isotype switched | Yes | Yes | Yes | Yes |
| Somatic mutation | Yes | Yes | Yes | Yes |
| B-cell-specific transcription factor expression | ||||
| XBP-1 expression | +/−(?)* | +/−(?)† | ++ | ++ |
| Pax-5 (BSAP) expression | +/−(?)* | +/−(?)† | − | − |
| BLIMP-1 expression | +/−(?)* | +/−(?)† | ++ | ++(?)‡ |
IL-6R, interleukin-6R; PC, plasma cell; VLA-4, very late activation antigen-4; XBP-1, X-box binding protein 1; BSAP, B-cell lineage-specific activator; BLIMP-1, B lymphocyte-induced maturation protein 1.
The expression profile of transcription factors for PC precursors has not yet been reported. Given their intermediate stage of development, precursors may express both B cell and PC transcription factors.
A definitive phenotype of myeloma precursors has not yet been elucidated. Given the importance of BM stroma for myeloma tumor progression, it is likely that MM precursors express IL-6 receptor and VLA-4.
Identification of transcription factors expressed by myeloma tumors is a continuing process, with varying results depending on the assay and sample.
Systemic lupus erythematosus
Long-lived, affinity-matured humoral immune responses are essential for enduring protection of the host from pathogens. However, the persistent production of pathogenic autoantibodies by the host mediates the chronic, destructive clinical manifestations of antibody-mediated autoimmune diseases, like systemic lupus erythematosus (SLE) (104–107). SLE is considered to be the prototypical systemic autoimmune disease. It involves multiple organ systems and is characterized by the uncontrolled differentiation of PCs that secrete IgG autoantibodies (108, 109). These pathogenic autoantibody-producing PCs are primarily specific for nuclear constituents such as DNA, histones, and small nuclear ribonucleoproteins (75). As a result, long-lived autoreactive BM PCs are generated that play a crucial role in the continuous production of somatically mutated, isotype-switched autoantibodies. Normally, these autoantibody-producing PCs that emerge during B-cell differentiation are deleted, anergized, or undergo receptor editing in a last ditch effort to create a nonself-reacting Ig receptor. In both, patients with SLE and lupus-prone mice, a predisposition to B-cell hyperactivity appears to override these tolerance mechanisms. Despite the presence of pathogenic autoantibodies and tissue pathology, the defining molecular and cellular events leading to SLE remain unclear. Loss of self-tolerance and disease pathogenesis in SLE are thought to emerge from mutations or polymorphisms in genes that influence lymphocyte signaling, the clearance of immune complexes, and apoptosis (110, 111). The genetic basis for susceptibility to SLE is extremely complex, involving contributions from both MHC class II and non-MHC genes. Study of MRL mice that carry mutations in the lpr (Fas, CD95) and gld (Fas ligand, CD95L) genes has revealed that full expression of lupus-like disease can be achieved via a single genetic lesion (112, 113). In contrast, linkage studies from the New Zealand hybrid model have identified multiple non-MHC genes that, when combined, mediate susceptibility to SLE (75, 110, 114). These studies highlight that SLE develops from multiple abnormalities in immunoregulation.
Defective plasma cell migration in lupus
The NZM2410 model has been used to extensively map the genes responsible for SLE. NZM2410 mice were originally generated by breeding NZW and NZB/W mice (115), and thus they represent an inbred strain that has acquired specific disease susceptibility loci from the NZB and NZW genomes. Both female and male NZM mice develop severe glomerulonephritis with 85% penetrance and a 50% mortality rate at approximately 6 months of age (115, 116). Three recessive loci, each comprised of approximately 100 genes, strongly associated with SLE susceptibility have been identified in the NZM2410 strain (Sle1, Sle2, and Sle3 located on chromosomes 1, 4, and 7, respectively) (116). The generation of B6 congenic mice carrying these susceptibility loci individually and combined has provided important insights into the biological pathways leading to disease pathogenesis (117). It is hypothesized that Sle1 initiates the loss of self-tolerance, mediating the development of antinuclear antibodies. Sle2 and Sle3 appear to contribute to disease progression by lowering the activation threshold of B cells, resulting in hyperproliferative and hyper-Ig secretory responses. B6 congenic mice carrying each of these loci separately have only partial, nonpathogenic phenotypes, while congenic mice expressing all three loci reconstitutes the autoimmune pathology of NZM2410 mice (118). How the combination of these inherited traits influences the normal regulation of PC differentiation is unclear.
Our laboratory has initiated studies in lupus-prone mice to examine potential defects in B-cell differentiation that may result in the loss of censoring the development of autoreactive B lymphocytes. It has been shown that autoreactive B cells clonally expand and differentiate to produce isotype-switched, high-affinity autoantibodies – a process that mimics normal T-cell-dependent B-cell responses (119). As many autoantibodies produced by lupus-prone mice are somatically mutated and isotype-switched, it has been assumed that high levels of persisting serum autoantibodies are generated via the development of long-lived PCs residing in the BM. To evaluate this hypothesis, we determined the tissue distribution of antinuclear PCs from a cohort of autoimmune mice. Contrary to the behavior of normal long-lived PCs, and PCs from autoimmune MRLlpr/lpr and BXSB mice, we have found that PCs from NZM2410 mice accumulate in the spleen but are virtually absent in the BM (Erickson et al. submitted for publication). Unexpectedly, although these pathogenic PCs are restricted to the splenic environment, the vast majority of them are long-lived. Based on ex vivo migration studies, a loss in responsiveness of NZM PCs to CXCL12 is at least one mechanism responsible for the lack of BM homing. Furthermore, the loss of CXCL12-induced migration is not a trait expressed by resting lymphocytes, but it is an acquired deficiency manifested by post-GC B cells in the NZM strain. Using a panel of B6 congenic strains, we further established that this deficiency is not conferred by an individual SLE susceptibility locus but is owing to the epistatic interactions amongst Sle1, Sle2, and Sle3 that mediate this deficiency.
To date, these findings are the first to implicate B-cell chemokine receptor dysregulation in antibody-mediated autoimmunity. However, the role of chemokines in the pathogenesis of other autoimmune diseases should not be overlooked. Numerous studies have demonstrated aberrant expression of chemokines and their receptors in the chronic inflammation of multiple sclerosis, rheumatoid arthritis, Type 1 diabetes, Graves' disease, and Sjögren's syndrome (120). Emerging from these studies is the hypothesis that chemokines and their receptors may serve as a mechanism in the initiation or progression of autoimmunity by altering the natural homing properties of lymphocytes. In the case of NZM mice, dysregulation in CXCL12 responsiveness may contribute to disease pathogenesis in several ways. First, the accumulation of PCs within the spleen of NZM animals demonstrates that longevity of PCs can still be maintained, even in the absence of BM PCs. It is noteworthy that these findings indicate that CXCL12–CXCR4 interactions are not required for the persistence of PCs, as they are unresponsive to CXCL12. One possibility is that an Sle locus alters the microenvironment of the spleen to support the maintenance of long-lived PCs. Interestingly, Cassese and coworkers (76) have recently shown that the kidney of NZB/W mice is a major reservoir of PCs, regardless of antigen specificity, indicating that peripheral tissues other than secondary lymphoid organs can maintain PC homeostasis. Second, one reason PCs accumulate in the spleens of NZM mice may be owing to follicular B-cell differentiation being hyperaccelerated. Thus, the transitional period from a GC B cell to a PC fate may occur hastily, compromising positive selection and allowing the escape of self-reactive clones committed to a PC fate. We propose that this loss of self-tolerance transpires at the PC precursor level where selection of these precursors, through continued expression of BCR, has gone awry by the inability to exit the spleen. Thus, competition for higher affinity long-lived PCs, while neglecting autoantibody-secreting cells, believed to take place in the BM is prevented.
Currently, the most effective regimen for treating SLE is the administration of the alkylating agent, cyclophosphamide (CTX). Studies in both human and murine lupus models have demonstrated that treatment with CTX significantly reduces the progression of glomerulonephritis (121–124). While most therapeutic strategies for treating SLE are immunosuppressive in nature, these approaches generally affect only proliferating cells and have little impact on pre-existing long-lived PCs. Thus, identifying molecular targets specific to long-lived PCs will greatly assist in devising more effective therapies for antibody-mediated autoimmune diseases.
Conclusion
Long-lived PCs are critical in sustaining humoral immune responses to invading pathogens. Numerous steps are involved in the differentiation of an antigen-experienced B cell to a PC. Early regulation of PC commitment involves signaling through the BCR and costimulatory molecules, such as CD40. These signals lead to changes in chemokine receptor expression that direct PC movement. Concomitant with these events is the sequential expression and silencing of transcription factors that control B-cell fate. The identification of early PC progenitors should provide the opportunity to discover additional factors that govern long-lived humoral immunity, and the PC progenitors are attractive candidates for the normal counterpart of transformed MM progenitors. The microenvironment is also a determinant in the growth and survival of PCs. As long-lived PCs predominantly reside in the BM, defects in homing to this organ may contribute to disease pathogenesis in autoimmunity. Common to both MM and SLE is the establishment of long-lived PCs that have circumvented the normal differentiation pathway, leading to malignancy and the loss of self-tolerance (Fig. 2).
Fig. 2. Defects in B-cell differentiation contribute to disease pathogenesis.

(1). Upon encountering antigen (Ag), B cells downregulate CXCR5 expression, causing a loss in CXCL13 responsiveness. The expression of CCR7 guides antigen-experienced B cells to the interface of the periarteriolar lymphatic sheath, where they receive T-cell help. As B cells differentiate to plasma cells (PCs), an increase in CXCR4 expression navigates their movement to the bone marrow (BM) (2). Defective migration of PCs towards CXCL12, such as in NZM mice, results in the accumulation of autoreactive PCs in the spleen and a concordant loss in BM PCs. The inability of PCs to home to the BM may prevent the natural selection of higher affinity long-lived PCs in the BM, thereby contributing to the loss of self-tolerance (3). Early progenitors of long-lived PCs (prePC) exhibit striking phenotypic similarities to the myeloma precursor, suggesting that the genetic transformation of PC precursors during differentiation may lead to malignancy (proMM).
Acknowledgments
R.J. Noelle is supported by grants from the National Institutes of Health (AI026296, AI048667, AI049580, AI052211, and CA082542). L.D. Erickson is supported by a grant from the American Cancer Society (PF LIB-0202101).
References
- 1.Calame KL. Plasma cells: finding new light at the end of B cell development. Nat Immunol. 2001;2:1103–1108. doi: 10.1038/ni1201-1103. [DOI] [PubMed] [Google Scholar]
- 2.Vos Q, Lees A, Wu ZQ, Snapper CM, Mond JJ. B-cell activation by T-cell-independent type 2 antigens as an integral part of the humoral immune response to pathogenic microorganisms. Immunol Rev. 2000;176:154–170. doi: 10.1034/j.1600-065x.2000.00607.x. [DOI] [PubMed] [Google Scholar]
- 3.Stein KE. Thymus-independent and thymus-dependent responses to polysaccharide antigens. J Infect Dis. 1992;165:S49–S52. doi: 10.1093/infdis/165-supplement_1-s49. [DOI] [PubMed] [Google Scholar]
- 4.Erickson LD, et al. B cell immunopoiesis: visualizing the impact of CD40 engagement on the course of T cell-independent immune responses in an Ig transgenic system. Eur J Immunol. 2000;30:3121–3131. doi: 10.1002/1521-4141(200011)30:11<3121::AID-IMMU3121>3.0.CO;2-M. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sze DM, Toellner KM, Garcia de Vinuesa C, Taylor DR, MacLennan IC. Intrinsic constraint on plasmablast growth and extrinsic limits of plasma cell survival. J Exp Med. 2000;192:813–821. doi: 10.1084/jem.192.6.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Foy T, Aruffo A, Bajorath J, Buhlmann JE, Noelle RJ. Immune regulation by CD40 and its ligand gp39. Annu Rev Immunol. 1996;14:591–617. doi: 10.1146/annurev.immunol.14.1.591. [DOI] [PubMed] [Google Scholar]
- 7.MacLennan IC. Germinal centers. Annu Rev Immunol. 1994;12:117–139. doi: 10.1146/annurev.iy.12.040194.001001. [DOI] [PubMed] [Google Scholar]
- 8.Kelsoe G. In situ studies of the germinal center reaction. Adv Immunol. 1995;60:267–288. doi: 10.1016/s0065-2776(08)60587-8. [DOI] [PubMed] [Google Scholar]
- 9.Banchereau J, et al. The CD40 antigen and its ligand. Annu Rev Immunol. 1994;12:881–922. doi: 10.1146/annurev.iy.12.040194.004313. Review. [DOI] [PubMed] [Google Scholar]
- 10.Noelle RJ. CD40 and its ligand in host defense. Immunity. 1996;4:415–419. doi: 10.1016/s1074-7613(00)80408-2. [DOI] [PubMed] [Google Scholar]
- 11.Gray D, et al. B–T lymphocyte interactions in the generation and survival of memory cells. Immunol Rev. 1996;150:45–61. doi: 10.1111/j.1600-065x.1996.tb00695.x. [DOI] [PubMed] [Google Scholar]
- 12.Garcia de Vinuesa C, MacLennan IC, Holman M, Klaus GG. Anti-CD40 antibody enhances responses to polysaccharide without mimicking T cell help. Eur J Immunol. 1999;29:3216–3224. doi: 10.1002/(SICI)1521-4141(199910)29:10<3216::AID-IMMU3216>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 13.Kuhne MR, et al. Assembly and regulation of the CD40 receptor complex in human B cells. J Exp Med. 1997;186:337–342. doi: 10.1084/jem.186.2.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pullen SS, Miller HG, Everdeen DS, Dang TTA, Crute JJ, Kehry MR. CD40-tumor necrosis factor receptor-associated factor (TRAF) interactions: regulation of CD40 signaling through multiple TRAF binding sites and TRAF hetero-oligomerization. Biochemistry. 1998;37:11836–11845. doi: 10.1021/bi981067q. [DOI] [PubMed] [Google Scholar]
- 15.Pullen SS, Dang TT, Crute JJ, Kehry MR. CD40 signaling through tumor necrosis factor receptor-associated factors (TRAFs). Binding site specificity and activation of downstream pathways by distinct TRAFs. J Biol Chem. 1999;274:14246–14254. doi: 10.1074/jbc.274.20.14246. [DOI] [PubMed] [Google Scholar]
- 16.Ahonen CL, et al. The CD40-TRAF6 axis controls affinity maturation and the generation of long-lived plasma cells. Nat Immunol. 2002;3:451–456. doi: 10.1038/ni792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yasui T, et al. Dissection of B cell differentiation during primary immune responses in mice with altered CD40 signals. Int Immunol. 2002;14:319–329. doi: 10.1093/intimm/14.3.319. [DOI] [PubMed] [Google Scholar]
- 18.Erickson LD, et al. Short-circuiting long-lived humoral immunity by the heightened engagement of CD40. J Clin Invest. 2002;109:613–620. doi: 10.1172/JCI14110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Slifka MK, Antia R, Whitmire JK, Ahmed R. Humoral immunity due to long-lived plasma cells. Immunity. 1998;8:363–372. doi: 10.1016/s1074-7613(00)80541-5. [DOI] [PubMed] [Google Scholar]
- 20.Manz RA, Lohning M, Cassese G, Thiel A, Radbruch A. Survival of long-lived plasma cells is independent of antigen. Int Immunol. 1998;10:1703–1711. doi: 10.1093/intimm/10.11.1703. [DOI] [PubMed] [Google Scholar]
- 21.Manz RA, Arce S, Cassese G, Hauser AE, Hiepe F, Radbruch A. Humoral immunity and long-lived plasma cells. Curr Opin Immunol. 2002;14:517–521. doi: 10.1016/s0952-7915(02)00356-4. [DOI] [PubMed] [Google Scholar]
- 22.Bernasconi NL, Traggiai E, Lanzavecchia A. Maintenance of serological memory by polyclonal activation of human memory B cells. Science. 2002;298:2199–2202. doi: 10.1126/science.1076071. [DOI] [PubMed] [Google Scholar]
- 23.Angelin-Duclos C, Cattoretti G, Lin KI, Calame K. Commitment of B lymphocytes to a plasma cell fate is associated with Blimp-1 expression in vivo. J Immunol. 2000;165:5462–5471. doi: 10.4049/jimmunol.165.10.5462. [DOI] [PubMed] [Google Scholar]
- 24.Turner CA, Jr, Mack DH, Davis MM. Blimp-1, a novel zinc finger-containing protein that can drive the maturation of B lymphocytes into immunoglobulin-secreting cells. Cell. 1994;77:297–306. doi: 10.1016/0092-8674(94)90321-2. [DOI] [PubMed] [Google Scholar]
- 25.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 26.Piskurich JF, Lin KI, Lin Y, Wang Y, Ting JP, Calame K. BLIMP-I mediates extinction of major histocompatibility class II transactivator expression in plasma cells. Nat Immunol. 2000;1:526–532. doi: 10.1038/82788. [DOI] [PubMed] [Google Scholar]
- 27.Reimold AM, et al. Plasma cell differentiation requires the transcription factor XBP-1. Nature. 2001;412:300–307. doi: 10.1038/35085509. [DOI] [PubMed] [Google Scholar]
- 28.Falini B, et al. A monoclonal antibody (MUM1p) detects expression of the MUM1/IRF4 protein in a subset of germinal center B cells, plasma cells, and activated T cells. Blood. 2000;95:2084–2092. [PubMed] [Google Scholar]
- 29.Benner R, Hijmans W, Haaijman JJ. The bone marrow: the major source of serum immunoglobulins, but still a neglected site of antibody formation. Clin Exp Immunol. 1981;46:1–8. [PMC free article] [PubMed] [Google Scholar]
- 30.Koch G, Osmond DG, Julius MH, Benner R. The mechanism of thymus-dependent antibody formation in bone marrow. J Immunol. 1981;126:1447–1451. [PubMed] [Google Scholar]
- 31.Maruyama M, Lam KP, Rajewsky K. Memory B-cell persistence is independent of persisting immunizing antigen. Nature. 2000;407:636–642. doi: 10.1038/35036600. [DOI] [PubMed] [Google Scholar]
- 32.Arpin C, Banchereau J, Liu YJ. Memory B cells are biased towards terminal differentiation: a strategy that may prevent repertoire freezing. J Exp Med. 1997;186:931–940. doi: 10.1084/jem.186.6.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tangye SG, Avery DT, Hodgkin PD. A division-linked mechanism for the rapid generation of Ig-secreting cells from human memory B cells. J Immunol. 2003;170:261–269. doi: 10.4049/jimmunol.170.1.261. [DOI] [PubMed] [Google Scholar]
- 34.Barrington RA, Pozdnyakova O, Zafari MR, Benjamin CD, Carroll MC. B lymphocyte memory: role of stromal cell complement and FcgammaRIIB receptors. J Exp Med. 2002;196:1189–1199. doi: 10.1084/jem.20021110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu X, et al. Impaired affinity maturation in Cr2−/− mice is rescued by adjuvants without improvement in germinal center development. J Immunol. 2000;165:3119–3127. doi: 10.4049/jimmunol.165.6.3119. [DOI] [PubMed] [Google Scholar]
- 36.Billadeau D, Ahmann G, Greipp P, Van Ness B. The bone marrow of multiple myeloma patients contains B cell populations at different stages of differentiation that are clonally related to the malignant plasma cell. J Exp Med. 1993;178:1023–1031. doi: 10.1084/jem.178.3.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yaccoby S, Epstein J. The proliferative potential of myeloma plasma cells manifest in the SCID-hu host. Blood. 1999;94:3576–3582. [PubMed] [Google Scholar]
- 38.Pilarski LM, Belch AR. Clonotypic myeloma cells able to xenograft myeloma to nonobese diabetic severe combined immunodeficient mice copurify with CD34(+) hematopoietic progenitors. Clin Cancer Res. 2002;8:3198–3204. [PubMed] [Google Scholar]
- 39.O'Connor BP, Cascalho M, Noelle RJ. Short-lived and long-lived bone marrow plasma cells are derived from a novel precursor population. J Exp Med. 2002;195:737–745. doi: 10.1084/jem.20011626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Freitas AA, Rocha B. Population biology of lymphocytes: the flight for survival. Annu Rev Immunol. 2000;18:83–111. doi: 10.1146/annurev.immunol.18.1.83. [DOI] [PubMed] [Google Scholar]
- 41.Antia R, Pilyugin SS, Ahmed R. Models of immune memory on the role of cross-reactive stimulation, competition, and homeostasis in maintaining immune memory. Proc Natl Acad Sci USA. 1998;95:14926–14931. doi: 10.1073/pnas.95.25.14926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baggiolini M, Dewald B, Moser B. Human chemokines: an update. Annu Rev Immunol. 1997;15:675–705. doi: 10.1146/annurev.immunol.15.1.675. [DOI] [PubMed] [Google Scholar]
- 43.Zlotnik A, Morales J, Hedrick JA. Recent advances in chemokines and chemokine receptors. Crit Rev Immunol. 1999;19:1–47. [PubMed] [Google Scholar]
- 44.Mackay CR. Chemokines: immunology's high impact factors. Nat Immunol. 2001;2:95–101. doi: 10.1038/84298. [DOI] [PubMed] [Google Scholar]
- 45.Forster R, Mattis AE, Kremmer E, Wolf E, Brem G, Lipp M. A putative chemokine receptor, BLR1, directs B cell migration to defined lymphoid organs and specific anatomic compartments of the spleen. Cell. 1996;87:1037–1047. doi: 10.1016/s0092-8674(00)81798-5. [DOI] [PubMed] [Google Scholar]
- 46.Gunn MD, Ngo VN, Ansel KM, Ekland EH, Cyster JG, Williams LT. A B-cell-homing chemokine made in lymphoid follicles activates Burkitt's lymphoma receptor-1. Nature. 1998;391:799–803. doi: 10.1038/35876. [DOI] [PubMed] [Google Scholar]
- 47.Legler DF, Loetscher M, Roos RS, Clark-Lewis I, Baggiolini M, Moser B. B cell-attracting chemokine 1, a human CXC chemokine expressed in lymphoid tissues, selectively attracts B lymphocytes via BLR1/CXCR5. J Exp Med. 1998;187:655–660. doi: 10.1084/jem.187.4.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Voigt I, Camacho SA, de Boer BA, Lipp M, Forster R, Berek C. CXCR5-deficient mice develop functional germinal centers in the splenic T cell zone. Eur J Immunol. 2000;30:560–567. doi: 10.1002/1521-4141(200002)30:2<560::AID-IMMU560>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 49.Ansel KM, et al. A chemokine-driven positive feedback loop organizes lymphoid follicles. Nature. 2000;406:309–314. doi: 10.1038/35018581. [DOI] [PubMed] [Google Scholar]
- 50.Hargreaves DC, et al. A coordinated change in chemokine responsiveness guides plasma cell movements. J Exp Med. 2001;194:45–56. doi: 10.1084/jem.194.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wehrli N, et al. Changing responsiveness to chemokines allows medullary plasmablasts to leave lymph nodes. Eur J Immunol. 2001;31:609–616. doi: 10.1002/1521-4141(200102)31:2<609::aid-immu609>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 52.Kaiser E, Forster R, Wolf I, Ebensperger C, Kuehl WM, Lipp M. The G protein-coupled receptor BLR1 is involved in murine B cell differentiation and is also expressed in neuronal tissues. Eur J Immunol. 1993;23:2532–2539. doi: 10.1002/eji.1830231023. [DOI] [PubMed] [Google Scholar]
- 53.Forster R, Emrich T, Kremmer E, Lipp M. Expression of the G-protein-coupled receptor BLR1 defines mature, recirculating B cells and a subset of T-helper memory cells. Blood. 1994;84:830–840. [PubMed] [Google Scholar]
- 54.Roy MP, Kim CH, Butcher EC. Cytokine control of memory B cell homing machinery. J Immunol. 2002;169:1676–1682. doi: 10.4049/jimmunol.169.4.1676. [DOI] [PubMed] [Google Scholar]
- 55.Rollins BJ. Chemokines. Blood. 1997;90:909–928. [PubMed] [Google Scholar]
- 56.Bleul CC, Fuhlbrigge RC, Casasnovas JM, Aiuti A, Springer TA. A highly efficacious lymphocyte chemoattractant, stromal cell-derived factor 1 (SDF-1) J Exp Med. 1996;184:1101–1109. doi: 10.1084/jem.184.3.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.D'Apuzzo M, et al. The chemokine SDF-1, stromal cell-derived factor 1, attracts early stage B cell precursors via the chemokine receptor CXCR4. Eur J Immunol. 1997;27:1788–1793. doi: 10.1002/eji.1830270729. [DOI] [PubMed] [Google Scholar]
- 58.Imai K, et al. Selective secretion of chemoattractants for haemopoietic progenitor cells by bone marrow endothelial cells: a possible role in homing of haemopoietic progenitor cells to bone marrow. Br J Haematol. 1999;106:905–911. doi: 10.1046/j.1365-2141.1999.01644.x. [DOI] [PubMed] [Google Scholar]
- 59.Peled A, et al. The chemokine SDF-1 stimulates integrin-mediated arrest of CD34(+) cells on vascular endothelium under shear flow. J Clin Invest. 1999;104:1199–1211. doi: 10.1172/JCI7615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sawada S, et al. Disturbed CD4+ T cell homeostasis and in vitro HIV-1 susceptibility in transgenic mice expressing T cell line-tropic HIV-1 receptors. J Exp Med. 1998;187:1439–1449. doi: 10.1084/jem.187.9.1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kawabata K, et al. A cell-autonomous requirement for CXCR4 in long-term lymphoid and myeloid reconstitution. Proc Natl Acad Sci USA. 1999;96:5663–5667. doi: 10.1073/pnas.96.10.5663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ma Q, Jones D, Springer TA. The chemokine receptor CXCR4 is required for the retention of B lineage and granulocytic precursors within the bone marrow microenvironment. Immunity. 1999;10:463–471. doi: 10.1016/s1074-7613(00)80046-1. [DOI] [PubMed] [Google Scholar]
- 63.Qin S, et al. The chemokine receptors CXCR3 and CCR5 mark subsets of T cells associated with certain inflammatory reactions. J Clin Invest. 1998;101:746–754. doi: 10.1172/JCI1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zlotnik A, Yoshie O. Chemokines: a new classification system and their role in immunity. Immunity. 2000;12:121–127. doi: 10.1016/s1074-7613(00)80165-x. [DOI] [PubMed] [Google Scholar]
- 65.Baggiolini M. Chemokines and leukocyte traffic. Nature. 1998;392:565–568. doi: 10.1038/33340. [DOI] [PubMed] [Google Scholar]
- 66.Bowman EP, et al. Developmental switches in chemokine response profiles during B cell differentiation and maturation. J Exp Med. 2000;191:1303–1318. doi: 10.1084/jem.191.8.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hauser AE, et al. Chemotactic responsiveness toward ligands for CXCR3 and CXCR4 is regulated on plasma blasts during the time course of a memory immune response. J Immunol. 2002;169:1277–1282. doi: 10.4049/jimmunol.169.3.1277. [DOI] [PubMed] [Google Scholar]
- 68.Vincent T, Jourdan M, Sy MS, Klein B, Mechti N. Hyaluronic acid induces survival and proliferation of human myeloma cells through an interleukin-6-mediated pathway involving the phosphorylation of retinoblastoma protein. J Biol Chem. 2001;276:14728–14736. doi: 10.1074/jbc.M003965200. [DOI] [PubMed] [Google Scholar]
- 69.Kawano MM, Mihara K, Huang N, Tsujimoto T, Kuramoto A. Differentiation of early plasma cells on bone marrow stromal cells requires interleukin-6 for escaping from apoptosis. Blood. 1995;85:487–494. [PubMed] [Google Scholar]
- 70.Minges Wols HA, Underhill GH, Kansas GS, Witte PL. The role of bone marrow-derived stromal cells in the maintenance of plasma cell longevity. J Immunol. 2002;169:4213–4221. doi: 10.4049/jimmunol.169.8.4213. [DOI] [PubMed] [Google Scholar]
- 71.Jego G, Bataille R, Pellat-Deceunynck C. Interleukin-6 is a growth factor for nonmalignant human plasmablasts. Blood. 2001;97:1817–1822. doi: 10.1182/blood.v97.6.1817. [DOI] [PubMed] [Google Scholar]
- 72.Underhill GH, Minges Wols HA, Fornek JL, Witte PL, Kansas GS, Minges-Wols HA. IgG plasma cells display a unique spectrum of leukocyte adhesion and homing molecules. Blood. 2002;99:2905–2912. doi: 10.1182/blood.v99.8.2905. [DOI] [PubMed] [Google Scholar]
- 73.Qin D, et al. Fc gamma receptor IIB on follicular dendritic cells regulates the B cell recall response. J Immunol. 2000;164:6268–6275. doi: 10.4049/jimmunol.164.12.6268. [DOI] [PubMed] [Google Scholar]
- 74.Takahashi Y, Dutta PR, Cerasoli DM, Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. V. Affinity maturation develops in two stages of clonal selection. J Exp Med. 1998;187:885–895. doi: 10.1084/jem.187.6.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vyse TJ, Kotzin BL. Genetic susceptibility to systemic lupus erythematosus. Annu Rev Immunol. 1998;16:261–292. doi: 10.1146/annurev.immunol.16.1.261. [DOI] [PubMed] [Google Scholar]
- 76.Cassese G, et al. Inflamed kidneys of NZB/W mice are a major site for the homeostasis of plasma cells. Eur J Immunol. 2001;31:2726–2732. doi: 10.1002/1521-4141(200109)31:9<2726::aid-immu2726>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 77.Shaffer AL, Yu X, He Y, Boldrick J, Chan EP, Staudt LM. BCL-6 represses genes that function in lymphocyte differentiation, inflammation, and cell cycle control. Immunity. 2000;13:199–212. doi: 10.1016/s1074-7613(00)00020-0. [DOI] [PubMed] [Google Scholar]
- 78.Difilippantonio MJ, et al. Evidence for replicative repair of DNA double-strand breaks leading to oncogenic translocation and gene amplification. J Exp Med. 2002;196:469–480. doi: 10.1084/jem.20020851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shaffer AL, Rosenwald A, Staudt LM. Lymphoid malignancies: the dark side of B-cell differentiation. Nat Rev Immunol. 2002;2:920–932. doi: 10.1038/nri953. [DOI] [PubMed] [Google Scholar]
- 80.Bataille R, Harousseau JL. Multiple myeloma. N Engl J Med. 1997;336:1657–1664. doi: 10.1056/NEJM199706053362307. [DOI] [PubMed] [Google Scholar]
- 81.Rettig MB, et al. Kaposi's sarcoma-associated herpesvirus infection of bone marrow dendritic cells from multiple myeloma patients. Science. 1997;276:1851–1854. doi: 10.1126/science.276.5320.1851. [DOI] [PubMed] [Google Scholar]
- 82.Gado K, Domjan G, Hegyesi H, Falus A. Role of INTERLEUKIN-6 in the pathogenesis of multiple myeloma. Cell Biol Int. 2000;24:195–209. doi: 10.1006/cbir.2000.0497. [DOI] [PubMed] [Google Scholar]
- 83.Richardson P, Hideshima T, Anderson K. Thalidomide: emerging role in cancer medicine. Annu Rev Med. 2002;53:629–657. doi: 10.1146/annurev.med.53.082901.104043. [DOI] [PubMed] [Google Scholar]
- 84.Podczaski E, Cain J. Multiple myeloma. Clin Obstet Gynecol. 2002;45:928–938. doi: 10.1097/00003081-200209000-00038. [DOI] [PubMed] [Google Scholar]
- 85.Anderson KC. Moving disease biology from the lab to the clinic. Cancer. 2003;97:796–801. doi: 10.1002/cncr.11137. [DOI] [PubMed] [Google Scholar]
- 86.Hallek M, Bergsagel PL, Anderson KC. Multiple myeloma: increasing evidence for a multistep transformation process. Blood. 1998;91:3–21. [PMC free article] [PubMed] [Google Scholar]
- 87.Jego G, et al. Reactive plasmacytoses are expansions of plasmablasts retaining the capacity to differentiate into plasma cells. Blood. 1999;94:701–712. [PubMed] [Google Scholar]
- 88.Fujii R, Ishikawa H, Mahmoud MS, Asaoku H, Kawano MM. MPC-1-CD49e-immature myeloma cells include CD45+ subpopulations that can proliferate in response to IL-6 in human myelomas. Br J Haematol. 1999;105:131–140. [PubMed] [Google Scholar]
- 89.Arpin C, et al. The normal counterpart of IgD myeloma cells in germinal center displays extensively mutated IgVH gene, Cmu-Cdelta switch, and lambda light chain expression. J Exp Med. 1998;187:1169–1178. doi: 10.1084/jem.187.8.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bergsagel PL, Chesi M, Nardini E, Brents LA, Kirby SL, Kuehl WM. Promiscuous translocations into immunoglobulin heavy chain switch regions in multiple myeloma. Proc Natl Acad Sci USA. 1996;93:13931–13936. doi: 10.1073/pnas.93.24.13931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hardy RR, Hayakawa K. B cell development pathways. Annu Rev Immunol. 2001;19:595–621. doi: 10.1146/annurev.immunol.19.1.595. [DOI] [PubMed] [Google Scholar]
- 92.Szczepek AJ, Bergsagel PL, Axelsson L, Brown CB, Belch AR, Pilarski LM. CD34+ cells in the blood of patients with multiple myeloma express CD19 and IgH mRNA and have patient-specific IgH VDJ gene rearrangements. Blood. 1997;89:1824–1833. [PubMed] [Google Scholar]
- 93.Bergsagel PL, Smith AM, Szczepek A, Mant MJ, Belch AR, Pilarski LM. In multiple myeloma, clonotypic B lymphocytes are detectable among CD19+ peripheral blood cells expressing CD38, CD56, and monotypic Ig light chain. Blood. 1995;85:436–447. [PubMed] [Google Scholar]
- 94.Bergsagel PL, Masellis Smith A, Belch AR, Pilarski LM. The blood B-cells and bone marrow plasma cells in patients with multiple myeloma share identical IgH rearrangements. Curr Top Microbiol Immunol. 1995;194:17–24. doi: 10.1007/978-3-642-79275-5_3. [DOI] [PubMed] [Google Scholar]
- 95.Rottenburger C, et al. Clonotypic CD20+ and CD19+ B cells in peripheral blood of patients with multiple myeloma post high-dose therapy and peripheral blood stem cell transplantation. Br J Haematol. 1999;106:545–552. doi: 10.1046/j.1365-2141.1999.01548.x. [DOI] [PubMed] [Google Scholar]
- 96.Nagy M, Chapuis B, Matthes T. Expression of transcription factors Pu.1, Spi-B, Blimp-1, BSAP and oct-2 in normal human plasma cells and in multiple myeloma cells. Br J Haematol. 2002;116:429–435. doi: 10.1046/j.1365-2141.2002.03271.x. [DOI] [PubMed] [Google Scholar]
- 97.Fisher RC, Lovelock JD, Scott EW. A critical role for PU.1 in homing and long-term engraftment by hematopoietic stem cells in the bone marrow. Blood. 1999;94:1283–1290. [PubMed] [Google Scholar]
- 98.Kim N, Martin TE, Simon MC, Storb U. The transcription factor Spi-B is not required for somatic hypermutation. Mol Immunol. 2003;39:577–583. doi: 10.1016/s0161-5890(02)00201-8. [DOI] [PubMed] [Google Scholar]
- 99.Billadeau D, et al. Clonal circulating cells are common in plasma cell proliferative disorders: a comparison of monoclonal gammopathy of undetermined significance, smoldering multiple myeloma, and active myeloma. Blood. 1996;88:289–296. [PubMed] [Google Scholar]
- 100.Fenton JA, Pratt G, Rawstron AC, Morgan GJ. Isotype class switching and the pathogenesis of multiple myeloma. Hematol Oncol. 2002;20:75–85. doi: 10.1002/hon.688. [DOI] [PubMed] [Google Scholar]
- 101.Pilarski LM, Masellis-Smith A, Szczepek A, Mant MJ, Belch AR. Circulating clonotypic B cells in the biology of multiple myeloma: speculations on the origin of myeloma. Leuk Lymphoma. 1996;22:375–383. doi: 10.3109/10428199609054775. [DOI] [PubMed] [Google Scholar]
- 102.Reiman T, et al. Persistent preswitch clonotypic myeloma cells correlate with decreased survival: evidence for isotype switching within the myeloma clone. Blood. 2001;98:2791–2799. doi: 10.1182/blood.v98.9.2791. [DOI] [PubMed] [Google Scholar]
- 103.Taylor BJ, et al. Intraclonal homogeneity of clonotypic immunoglobulin m and diversity of nonclinical post-switch isotypes in multiple myeloma: insights into the evolution of the myeloma clone. Clin Cancer Res. 2002;8:502–513. [PubMed] [Google Scholar]
- 104.Odendahl M, et al. Disturbed peripheral B lymphocyte homeostasis in systemic lupus erythematosus. J Immunol. 2000;165:5970–5979. doi: 10.4049/jimmunol.165.10.5970. [DOI] [PubMed] [Google Scholar]
- 105.Putterman C, Deocharan B, Diamond B. Molecular analysis of the autoantibody response in peptide-induced autoimmunity. J Immunol. 2000;164:2542–2549. doi: 10.4049/jimmunol.164.5.2542. [DOI] [PubMed] [Google Scholar]
- 106.Jacobi AM, Hansen A, Burmester GR, Dorner T, Lipsky PE. Enhanced mutational activity and disturbed selection of mutations in V(H) gene rearrangements in a patient with systemic lupus erythematosus. Autoimmunity. 2000;33:61–76. doi: 10.3109/08916930108994110. [DOI] [PubMed] [Google Scholar]
- 107.Manheimer-Lory AJ, Zandman-Goddard G, Davidson A, Aranow C, Diamond B. Lupus-specific antibodies reveal an altered pattern of somatic mutation. J Clin Invest. 1997;100:2538–2546. doi: 10.1172/JCI119796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lipsky PE. Systemic lupus erythematosus: an autoimmune disease of B cell hyperactivity. Nat Immunol. 2001;2:764–766. doi: 10.1038/ni0901-764. [DOI] [PubMed] [Google Scholar]
- 109.Davidson A, Diamond B. Autoimmune diseases. N Engl J Med. 2001;345:340–350. doi: 10.1056/NEJM200108023450506. [DOI] [PubMed] [Google Scholar]
- 110.Wakeland EK, Wandstrat AE, Liu K, Morel L. Genetic dissection of systemic lupus erythematosus. Curr Opin Immunol. 1999;11:701–707. doi: 10.1016/s0952-7915(99)00039-4. [DOI] [PubMed] [Google Scholar]
- 111.Drake CG, Rozzo SJ, Vyse TJ, Palmer E, Kotzin BL. Genetic contributions to lupus-like disease in (NZB × NZW)F1 mice. Immunol Rev. 1995;144:51–74. doi: 10.1111/j.1600-065x.1995.tb00065.x. [DOI] [PubMed] [Google Scholar]
- 112.Cohen PL, Eisenberg RA. Lpr and gld: single gene models of systemic autoimmunity and lymphoproliferative disease. Annu Rev Immunol. 1991;9:243–269. doi: 10.1146/annurev.iy.09.040191.001331. [DOI] [PubMed] [Google Scholar]
- 113.Kono DH, Theofilopoulos AN. Genetics of systemic autoimmunity in mouse models of lupus. Int Rev Immunol. 2000;19:367–387. doi: 10.3109/08830180009055504. [DOI] [PubMed] [Google Scholar]
- 114.Mohan C. Murine lupus genetics: lessons learned. Curr Opin Rheumatol. 2001;13:352–360. doi: 10.1097/00002281-200109000-00003. [DOI] [PubMed] [Google Scholar]
- 115.Rudofsky UH, Evans BD, Balaban SL, Mottironi VD, Gabrielsen AE. Differences in expression of lupus nephritis in New Zealand mixed H-2z homozygous inbred strains of mice derived from New Zealand black and New Zealand white mice. Origins and initial characterization. Lab Invest. 1993;68:419–426. [PubMed] [Google Scholar]
- 116.Morel L, Rudofsky UH, Longmate JA, Schiffenbauer J, Wakeland EK. Polygenic control of susceptibility to murine systemic lupus erythematosus. Immunity. 1994;1:219–229. doi: 10.1016/1074-7613(94)90100-7. [DOI] [PubMed] [Google Scholar]
- 117.Morel L, Yu Y, Blenman KR, Caldwell RA, Wakeland EK. Production of congenic mouse strains carrying genomic intervals containing SLE-susceptibility genes derived from the SLE-prone NZM2410 strain. Mamm Genome. 1996;7:335–339. doi: 10.1007/s003359900098. [DOI] [PubMed] [Google Scholar]
- 118.Morel L, et al. Genetic reconstitution of systemic lupus erythematosus immunopathology with polycongenic murine strains. Proc Natl Acad Sci USA. 2000;97:6670–6675. doi: 10.1073/pnas.97.12.6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Radic MZ, Mackle J, Erikson J, Mol C, Anderson WF, Weigert M. Residues that mediate DNA binding of autoimmune antibodies. J Immunol. 1993;150:4966–4977. [PubMed] [Google Scholar]
- 120.Godessart N, Kunkel SL. Chemokines in autoimmune disease. Curr Opin Immunol. 2001;13:670–675. doi: 10.1016/s0952-7915(01)00277-1. [DOI] [PubMed] [Google Scholar]
- 121.Russell PJ, Hicks JD, Burnet FM. Cyclophosphamide treatment of kidney disease in (NZB × NZW)F1 mice. Lancet. 1966;1:1280–1284. [PubMed] [Google Scholar]
- 122.Russell PJ, Hicks JD. Cyclophosphamide treatment of renal disease in (NZB × NZW)F1 hybrid mice. Lancet. 1968;1:440–441. doi: 10.1016/s0140-6736(68)92778-5. [DOI] [PubMed] [Google Scholar]
- 123.Boumpas DT, et al. Controlled trial of pulse methylprednisolone versus two regimens of pulse cyclophosphamide in severe lupus nephritis. Lancet. 1992;340:741–745. doi: 10.1016/0140-6736(92)92292-n. [DOI] [PubMed] [Google Scholar]
- 124.Austin HA, III, et al. Therapy of lupus nephritis. Controlled trial of prednisone and cytotoxic drugs. N Engl J Med. 1986;314:614–619. doi: 10.1056/NEJM198603063141004. [DOI] [PubMed] [Google Scholar]