

Abstract
Glutathione (GSH) is an essential antioxidant responsible for the maintenance of intracellular redox homeostasis. As tumors outgrow their blood supply and become hypoxic, their redox homeostasis is challenged by the production of nitric oxide and reactive oxygen species (ROS). In gliomas, the sustained import of l-cystine via the l-cystine/l-glutamate exchanger, system xc−, is rate-limiting for the synthesis of GSH. We show that hypoxia causes a significant increase in NO and ROS but without affecting glioma cell growth. This is explained by a concomitant increase in the utilization of GSH, which is accompanied by an increase in the cell-surface expression of xCT, the catalytic subunit of system xc−, and l-cystine uptake. Growth was inhibited when GSH synthesis was blocked by buthionine sulfoximine (BSO), an inhibitor of the enzyme required for GSH synthesis, or when cells were deprived of l-cystine. These findings suggest that glioma cells show an increased requirement for GSH to maintain growth under hypoxic conditions. Therefore, approaches that limit GSH synthesis such as blocking system xc− may be considered as an adjuvant to radiation or chemotherapy.
Keywords: Amino Acid Transport, Antioxidant, Cell-surface Protein, Glutathione, Neurochemistry, Nitric Oxide, Oxygen Radicals, Cystine/Glutamate Exchanger, System xc−
Introduction
Cellular antioxidants are important for the protection of cells against reactive nitrogen species and reactive oxygen species (RNS2/ROS) generated endogenously or through exogenously imparted stress. GSH (l-γ-glutamyl-l-cysteinylglycine) is one of the most abundant antioxidants in the central nervous system with concentrations in the low millimolar range (1–3). GSH is synthesized from l-cysteine, l-glutamate, and glycine, with l-cysteine being rate-limiting. l-Cysteine is provided through the import of l-cystine (the reduced form of l-cysteine) via the Na+-independent l-cystine/l-glutamate exchanger, system xc− (4–6). System xc− is a member of the family of heteromeric amino acid transporters composed of a regulatory heavy subunit, 4F2hc/CD98, and a catalytic light subunit, xCT, which confers the specificity of this transport system. System xc− mediates the electroneutral uptake of l-cystine in exchange for the release of l-glutamate at a 1:1 stoichiometry. Extracellular l-cystine is rapidly reduced intracellularly to l-cysteine and incorporated into GSH, which is necessary to neutralize increased RNS/ROS as a result of changes in oxygen tension (7, 8). During glioma expansion, oxygen becomes limiting due to poor tumor vasculature (9, 10). In fact, tumor oxygen tension has been reported to be as low as 0.1%, resulting in tumor regions that are under chronic hypoxic conditions (11–14). System xc− has been shown to be up-regulated following oxidative stress (14, 15). Therefore, the synthesis of GSH may becomes critical for the protection of gliomas against oxidative damage (14–18).
In this study, we examined the role of system xc−-mediated l-cystine uptake in providing glioma cells with sufficient l-cysteine for the synthesis of GSH. Additionally, we compared the biological importance of GSH in glioma cell growth under hypoxic (2% O2) and normoxic (21% O2) conditions. We show that in hypoxia, glioma cells increase NO and ROS production, which leads to a concomitant increase in l-cystine uptake via system xc−, as well as an enhanced cell-surface expression of the xCT subunit. Sustained GSH synthesis becomes more critical for the support of glioma cell growth under hypoxic conditions compared with normoxic conditions. This is demonstrated by a ∼3-fold increase in the utilization of GSH and an enhanced sensitivity of glioma cell growth to the inhibition of GSH synthesis by BSO.
EXPERIMENTAL PROCEDURES
Cell Culture
D54-MG cells (World Health Organization grade IV) were a gift from Dr. D. D. Bigner (Duke University, Durham, NC). Routine mycoplasma tests were performed to ensure the absence of contamination. Cells were grown in DMEM/F12 (Media Tech, University of Alabama at Birmingham Media Preparation Facility) and supplemented with 2 mm glutamine (Media Tech) and 7% FBS (HyClone, Logan, UT) at 37 °C with 10% CO2 and balanced with ambient air. For some experiments, cells were grown in 1× DMEM (Invitrogen catalog no. 231013-024) supplemented with 0.5 mm sodium pyruvate, 2 mm glutamine, and 7% FBS (Media Tech, University of Alabama at Birmingham Media Preparation Facility). For hypoxic conditions, cells were grown at 37 °C in a trigas incubator in which 2% O2 was maintained by purging the chamber with 100% N2 and supplementing with 10% CO2. The pH of the media was tested regularly with pH strips, and a pH indicator was included in the media. The pH was maintained at 7.4 under hypoxic and normoxic conditions.
Drugs
All drugs were purchased from Sigma unless specified otherwise. (S)-4-Carboxyphenylglycine was purchased from Tocris Bioscience (Ellisville, MO).
Cell Proliferation
Proliferation was assessed by seeding 10,000 cells into each well of a 12-well plate (Fisher). Cells were harvested using 0.05% trypsin and resuspended in 10 ml of standard bath solution (125 mm NaCl, 5 mm KCl, 1.2 mm MgSO4, 1 mm CaCl2, 1.6 mm Na2HPO4, 0.4 mm NaH2PO4, 10.5 mm glucose, and 32.5 mm HEPES). The pH was adjusted to 7.4 using NaOH, and osmolarity was measured at ∼300 mosm. Three readings were made on specified days using a Coulter Counter cell sizer (Beckman Coulter, Inc., Miami, FL). Cell number was recorded per 500 μl, and the mean cell number was normalized to Day 0.
Western Blotting
Confluent plates of D54-MG cells were lysed using radioimmune precipitation assay buffer supplemented with protease and phosphatase inhibitors (1:100). Protein analysis was performed using the Bio-Rad DC protein assay kit. 25 μg of protein was mixed with 6× sample buffer (60% glycerol, 300 mm Tris (pH 6.8), 12 mm EDTA, 12% SDS, 864 mm 2-mercaptoethanol, and 0.05% bromphenol blue) and boiled for 3 min. Samples were loaded into a 10% pre-cast SDS-polyacrylamide gel (Bio-Rad). Gels were run at 100 V for 90 min and transferred at 200 mA for 120 min at room temperature onto polyvinylidene fluoride paper (Millipore, Bedford, MA). Membranes were blocked in blocking buffer (5% nonfat dried milk in TBST (TBS plus 0.1% Tween 20)). Blots were probed with goat anti-xCT primary antibody (0.06 μg/ml; Abcam, Cambridge, MA) overnight at 4 °C. Blots were also probed with mouse anti-GAPDH antibody (0.05 μg/ml; Abcam). Following primary antibody incubation, blots were washed four times for 5 min each with TBST. Next, membranes were incubated with HRP-conjugated secondary antibodies (2 μg/0.5 ml, Santa Cruz Biotechnology Inc., Santa Cruz, CA) for 1 h at room temperature, followed by another wash period (four times for 5 min each with TBST), and developed using enhanced chemiluminescence (ECL, Amersham Biosciences). Membranes were exposed using Kodak Image Station 4000MM.
Cytoplasmic and Nuclear Protein Extraction
D54-MG cells were harvested and washed with 1× PBS. NE-PER nuclear and cytoplasmic extraction reagents (NER and CER I, respectively; Pierce) were used to isolate protein fractions. The protocol recommended by the manufacturer was followed with some modification. CER I and NER were supplemented with protease and phosphatase inhibitors (1:50). Proteins were examined by Western blotting and probed with mouse anti-hypoxia-inducible factor 1α (HIF-1α) antibody (1.3 μg/ml; Abcam) and mouse anti-histone 1 antibody (1 μg/ml; Millipore) for 1 h at room temperature. To confirm proper separation between cytoplasmic and nuclear protein, blots were also probed with rabbit anti-α-tubulin antibody (0.2 μg/ml; Abcam).
Biotinylation
To prevent endocytosis of surface proteins, this assay was performed at 4 °C. Cells were washed with standard bath solution supplemented with 1 mm CaCl2. After washing, 1.5 mg/ml Sulfo-NHS-Biotin (Pierce) was added and allowed to incubate for 30 min with occasional gentle rocking. Biotinylation was quenched with standard bath solution supplemented with 100 mm glycine and 1 mm CaCl2 (pH 8.0). Cells were washed once with standard bath solution and lysed in radioimmune precipitation assay buffer supplemented with protease and phosphatase inhibitors (1:100). Protein analysis was performed using the Bio-Rad DC protein assay kit. A 2.5-mg/ml protein stock was prepared, and 0.4 ml of protein was incubated with 200 μl of streptavidin-agarose beads (Pierce) overnight at 4 °C. The bound fraction was gently washed five times with radioimmune precipitation assay buffer, resuspended in 50 μl of 6× sample buffer, and boiled for 10 min to separate surface protein from beads. Samples were processed by Western blotting. Blots were probed with mouse anti-Na+/K+-ATPase primary antibody (1 μg/ml; Millipore) for 1 h at room temperature and with goat anti-xCT antibody overnight at 4 °C.
l-Cystine Uptake
l-Cystine uptake was performed using l-[14C]cystine as described previously with modifications (4). Uptake was performed using 2 μCi of l-[14C]cystine (PerkinElmer Life Sciences) with 100 μm l-cystine and was measured over 3 min. Uptake was normalized to protein, which was measured using the Better Bradford protein assay (Thermo Fisher Scientific).
Glutathione Assay
Reduced glutathione was measured using the QuantiChromTM glutathione assay kit (DIGT-250, BioAssay Systems, Hayward, CA). The protocol as directed by manufacturer was followed. The QuantiChromTM glutathione assay kit measures reduced GSH. D54-MG cells grown under hypoxic or normoxic conditions were harvested and sonicated in a solution containing 50 mm NaH2PO4 and 1 mm EDTA. Lysates were centrifuged at 10,000 × g for 15 min at 4 °C, and the supernatant was collected for assay. First, samples were mixed with an equal volume of Reagent A (H3PO4, H2SO4, Na2WO4·2H2O, CH3CH2OH, and 5,5′-dithiobis(2-nitrobenzoic acid)), vortexed, and centrifuged for 5 min at 14,000 rpm. Next, 200 μl of sample/Reagent A mixture was aliquoted into wells of a 96-well plate, and 100 μl of Reagent B KH2PO4 was added to each sample/Reagent A-containing well. The plate was incubated for 25 min at room temperature and read for absorption at 450 nm (A). GSH concentration was calculated using the following formula: ((Asample − Ablank)/(Acalibrator − Ablank)) × 100 × n = GSH (μm). The calibrator was equal to 100 μm glutathione, and the blank was water alone. GSH was normalized to the protein concentration, which was measured with the Bio-Rad DC protein assay kit.
NO/ROS Detection
D54-MG cells were plated onto coverslips and grown under hypoxic conditions for 0, 24, 48, and 96 h. Cells were first washed twice with Hanks' balanced salt solution containing Ca2+/Mg2+ (wash buffer). They were loaded with 1 μm CM-H2DCFDA, a ROS dye (Invitrogen C6827), or 2.5 μm DAF-FM, a NO indicator dye (Invitrogen D23844), and 1 μm Hoechst 33342 (Invitrogen H3570) for 15 min at 37 °C. CM-H2DCFDA detects hydrogen peroxide, superoxide anion, and the hydroxyl radical. The loading buffer used was the same as wash buffer. Next, cells were washed three times with wash buffer and allowed to recover for 10 min at 37 °C. This was followed by fixation with 4% paraformaldehyde for 20 min, and 20× images were later acquired using a Zeiss Axiovert 200M microscope.
Data Analysis
Results were graphed using Origin Version 7.5 (MicroCal Software, North Hampton, MA) and analyzed using InStat 3.00 (GraphPad Software, San Diego, CA). Significance was determined using two-way analysis of variance (ANOVA), followed by Tukey's post hoc test. For all data sets comparing the mean of only two groups, an unpaired t test was employed. Details of statistical analysis used in each figure can be found in the figure legends.
RESULTS
Glioma Cells Experience Hypoxia at 2% O2
To maintain redox homeostasis, adequate synthesis of antioxidants (specifically GSH) is critical for tumor cell survival (19). Previous studies show that changes in the oxygen tension within and around the tumor microenvironment lead to tumor hypoxia and modification of the redox status by paradoxically challenging tumor cells oxidatively and/or nitrosatively (15, 17, 18). To mimic hypoxic conditions, cells were grown at 37 °C in a trigas incubator with 2% O2, 10% CO2, and 88% N2. For comparison, normoxic conditions were achieved in an incubator in which the temperature was set to 37 °C with 10% CO2 and balanced with ambient air. To show that D54-MG cells were indeed responding to hypoxic conditions, we examined a classical cellular response to hypoxia, namely an increase in HIF-1α. HIF-1α is the regulated subunit of the HIF-1 transcription factor. Activated HIF-1α translocates to the nucleus, where it binds to its response element, and induces transcription of a number of genes involved in the cellular response to hypoxia (20). D54-MG cells were cultured under hypoxic conditions at defined time points, and nuclear and cytoplasmic proteins were isolated and examined by Western blotting and probed for HIF-1α expression (Fig. 1A). To ensure efficient separation of nuclear and cytoplasmic proteins, membranes were also probed for α-tubulin. Following densitometric analysis, HIF-1α bands were normalized to histone 1. After 5 h of hypoxia, HIF-1α expression increased significantly (p < 0.001); it remained elevated for 24 h and returned to basal levels by 48 h (Fig. 1B). These data demonstrate that 2% O2 is sufficient to induce a hypoxic response in D54-MG cells.
FIGURE 1.

Increased nuclear HIF-1α expression in response to 2% O2. A, representative blot of nuclear expression of HIF-1α after treatment with 2% O2. B, densitometric analysis of four independent experiments. Two-way ANOVA, followed by Tukey's post hoc analysis, was used to determine significance. **, p < 0.01; ***, p < 0.001 (n = 4).
Increased Utilization of GSH under Hypoxic Conditions
Thiol-reduced GSH acts as an electron donor to reduce oxidized proteins, with the product being disulfide-oxidized GSSG (21, 22). As l-cysteine is rate-limiting for the synthesis of GSH, we first examined the dependence of GSH synthesis on the availability of extracellular l-cystine under hypoxic and normoxic conditions (7). D54-MG glioma cells were cultured under 2 or 21% O2 for 96 h, and at 72 h were depleted of intracellular GSH by removing extracellular l-cystine 24 h before measuring GSH. After GSH depletion, glioma cells were treated with increasing concentrations of l-cystine for 6 h, followed by GSH measurement, which was normalized to protein concentration. Results show a concentration-dependent increase in intracellular GSH with increasing concentrations of l-cystine. The half-maximal concentrations of l-cystine required under hypoxic and normoxic conditions were 12 and 9 μm, respectively (Fig. 2A). Next, we examined the GSH concentration under hypoxic and normoxic conditions over time. D54-MG cells were grown under hypoxic or normoxic conditions, and after 72 h, the culture medium was changed to medium without l-cystine. After 24 h, 100 μm l-cystine was added at defined time points, and GSH was measured and normalized to protein concentrations. The results show no significant difference in GSH concentration (Fig. 2B). This may be explained by an elevated rate of GSH consumption. To assess how quickly GSH is consumed, D54-MG cells were grown under hypoxic and normoxic conditions for 96 h. The culture medium was changed to medium containing 0 μm l-cystine at 1, 3, 6, 12, and 24 h prior to determining the remaining GSH concentration. This was done to inhibit cellular resynthesis of GSH (Fig. 2C). These data were well fit by exponential decay function. These fits yielded a decay time of 2.87 h for hypoxic conditions compared with 8.09 h for normoxic conditions, a significant difference (p < 0.05) (Fig. 2C). These data suggest that under hypoxic conditions, GSH is consumed approximately three times faster, possibly due to an increased requirement for the reduction of oxidized proteins and/or entry of GSH into the γ-glutamyl cycle to release amino.
FIGURE 2.

Hypoxia increases the utilization of GSH. A, l-cystine-dependent GSH synthesis under normoxic (21% O2) or hypoxic (2% O2) conditions. B, GSH concentration as a function of time after the addition of 100 μm l-cystine. C, rate of GSH utilization as a function of time after the exclusion of l-cystine. A two-sample t test was used to analyze the difference between IC50 values for GSH and the decay constant (n = 4).
Increased Sensitivity to the Inhibition of GSH Synthesis under Hypoxic Conditions
The ability to maintain homeostatic balance between free radical production and detoxification by antioxidants is critical for the survival of most cell types, and the loss of that imbalance can lead to cell death (23). Therefore, we examined the importance of GSH in glioma cell growth under hypoxic and normoxic conditions. To do so, the cell number was measured over 4 days in the presence of 100 μm l-cystine, 0 μm l-cystine, or 0 μm l-cystine plus GSH ethyl ester (GSHee), which is a cell-permeant form of reduced GSH that has been shown to increase intracellular GSH (24). Growth was determined using a Coulter Counter cell sizer, which enabled us to count the cell number at defined time points. In addition, a trypan blue exclusion assay demonstrated that cell viability was unaffected under hypoxic and normoxic conditions (data not shown). If GSH production from l-cystine is required for cell growth, supplementing the medium with GSHee alone would be sufficient to maintain growth under either condition. After 4 days under hypoxic conditions, there was a 5-fold increase in cell number in the absence of l-cystine compared with a 18-fold increase under control conditions, a significant difference (p < 0.001). The addition of 5 mm GSHee completely restored growth to control conditions at days 3 and 4 (Fig. 3A). Likewise, under normoxic conditions, glioma cell numbers by days 2, 3, and 4 were significantly reduced in the absence of l-cystine but were restored to control levels by GSHee (Fig. 3B). Indeed, glioma cell growth was unaffected by hypoxic conditions, supporting the notion that for cells to maintain normal growth rates under hypoxic and normoxic conditions, sufficient concentrations of GSH must be maintained.
FIGURE 3.
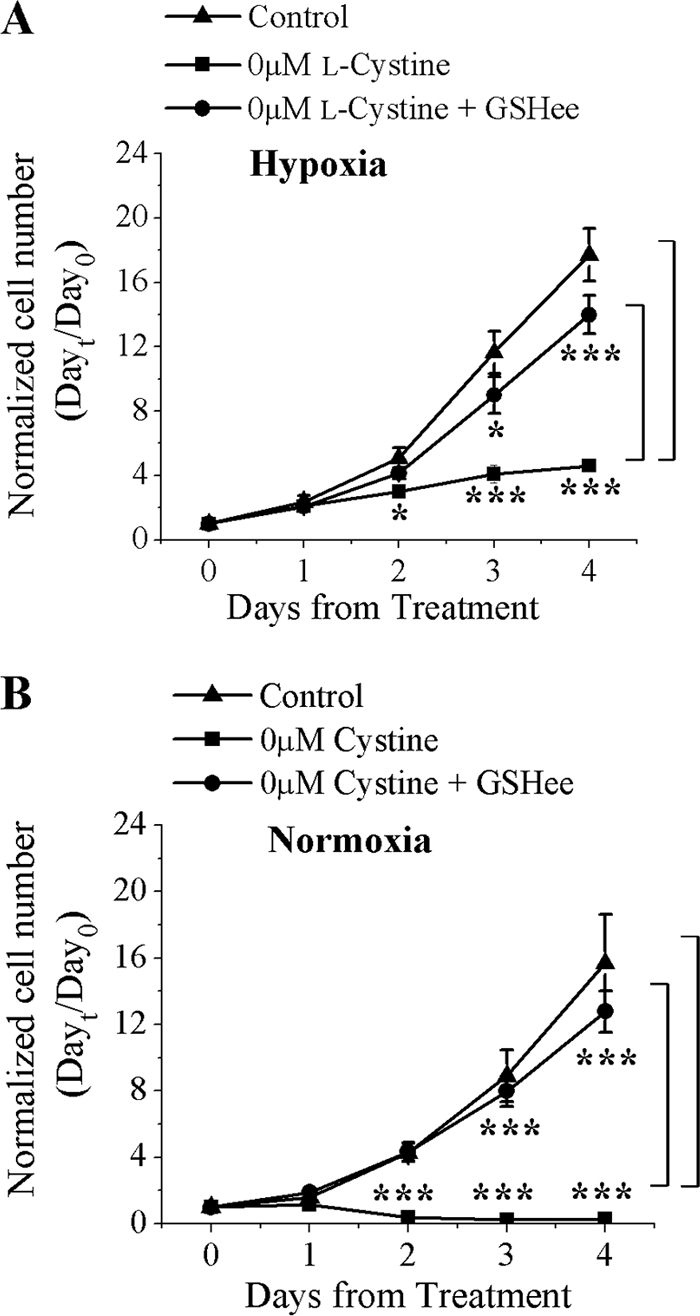
Glioma cell growth is dependent on GSH. A and B, glioma cell growth under normoxic (21% O2) or hypoxic (2% O2) conditions is dependent on GSH in the absence of l-cystine. Two-way ANOVA, followed by Tukey's post hoc analysis, was used to determine significance. *, p < 0.05; ***, p < 0.001 (n = 4).
To further examine the requirement of GSH for glioma cell growth, particularly under hypoxic conditions, we examined the effects of blocking GSH synthesis with BSO, an inhibitor of γ-glutamylcysteine synthetase, which is the rate-limiting enzyme in GSH synthesis. We confirmed that BSO does effectively inhibit GSH synthesis (data not shown). Under hypoxic and normoxic conditions and in the presence of 100 μm l-cystine, BSO inhibited glioma cell growth with IC50 values of 258 and 119 μm, respectively (Fig. 4A). However, in the presence of 10 μm l-cystine, there was an overall increased sensitivity to BSO under both hypoxic and normoxic conditions compared with cells grown in 100 μm l-cystine. Statistical analysis revealed that BSO in the presence of 10 μm l-cystine had a significantly lower IC50 (1 μm) under hypoxic conditions compared with the IC50 (5 μm) under normoxic conditions (p < 0.05) (Fig. 4B). To show that the effect of BSO is due to decreased intracellular GSH rather than other nonspecific actions, we treated glioma cells with 30 μm BSO with and without 3 mm GSHee. Under normoxic and hypoxic conditions and in the presence of 10 μm l-cystine, BSO decreased cell numbers by 95 and 96%, respectively. Furthermore, exogenous application of 3 mm GSHee completely restored growth to control levels (Fig. 4C). These results further indicate that GSH plays a critical role in glioma cell growth under hypoxic conditions and particularly at physiological concentrations of l-cystine.
FIGURE 4.

Increased sensitivity of glioma cell growth to the inhibition of GSH. A and B, inhibition of GSH synthesis by BSO in 100 μm l-cystine under normoxic (21% O2) or hypoxic (2% O2) conditions (n = 5). B, increased sensitivity to the inhibition of GSH synthesis by BSO in 10 μm l-cystine under normoxic or hypoxic conditions (n = 6). C, treatment of D54-MG cells with 3 mm GSHee restores growth inhibition by BSO under normoxic or hypoxic conditions (n = 3). A two-sample t test was used to analyze the difference between IC50 values for BSO. ***, p < 0.001.
Hypoxia-induced NO and ROS
Hypoxia has been shown to lead to increases in free radical production, notably ROS (17, 18). We examined changes in free radical production in response to hypoxic conditions as well as the effectiveness of its neutralization by GSH. To assay for ROS production, D54-MG cells were cultured under hypoxic conditions for 48 h. Glioma cells were loaded with a ROS indicator dye, CM-H2DCFDA, and Hoechst 33342 to stain nuclei. CM-H2DCFDA is initially non-fluorescent, and once it permeates live cells, it is cleaved by nonspecific intracellular esterases. In the presence of ROS, the reduced fluorescein compound is oxidized and has excitation/emission maxima of ∼495/529 nm. Representative images are shown in Fig. 5A, and the analyses of cells that emitted a green fluorescence are shown in Fig. 5A.1. After 48 h of hypoxia, ROS detection significantly increased from 7.7 to 41.6% (Fig. 5A.1). The specificity of CM-H2DCFDA was assessed by treating glioma cells under hypoxic conditions with GSH, a well characterized scavenger of ROS (25, 26). To determine whether GSH is able to neutralize hypoxia-induced ROS, cells were cultured with 3 mm GSHee under hypoxic conditions. GSHee significantly reduced hypoxia-induced ROS by 70% (Fig. 5A.1). Similar results were observed at 96 h (data not shown).
FIGURE 5.

Hypoxia-induced ROS and NO. A and B, representative images of ROS- and NO-positive D54-MG cells as detected by indicator dyes CM-H2DCFDA and DAF-FM, respectively. Glioma cells were grown under normoxic (21% O2) or hypoxic (2% O2) conditions for 0 and 48 h with and without 3 mm GSHee. A.1 and B.1, analysis of ROS- and NO-positive D54-MG cells, respectively. Two-way ANOVA, followed by Tukey's post hoc analysis, was used to determine significance. ***, p < 0.001; **, p < 0.01 (n = 3). Scale bars = 50 μm.
Secondary classes of free radicals are generated under hypoxic conditions (15). NO is capable of reacting with oxygen radicals such as O2− to form peroxynitrite (ONOO−) and nitrogen dioxide (NO2) (27). NO intermediates (specifically NO2) can further react with NO to form dinitrogen trioxide (N2O3), a potent RNS that imparts nitrosative stress (28). NO production was assessed by loading cells with a NO indicator dye, DAF-FM diacetate, which is a cell-permeant dye that, once inside the cell, it is deacetylated by esterases to form DAF-FM. In the presence of NO, DAF-FM forms a fluorescent benzotriazole derivative with excitation/emission maxima of ∼495/515 nm. Glioma cells grown under hypoxic conditions also demonstrated a significant increase in NO production (Fig. 5B). After 48 h of hypoxia, NO detection significantly increased from 7.3 to 35.8% (Fig. 5B.1). Incubation with 3 mm GSHee also significantly reduced NO production after 48 h of hypoxia by 65% (Fig. 5B.1). Similar findings were observed at 96 h (data not shown). These data suggest that GSH is capable of fully neutralizing RNS/ROS generated by hypoxic conditions.
Inhibition of System xc− Decreases Glioma Growth
Inhibition of system xc− under normoxic conditions decreases glioma cell growth and intracellular GSH (16). To further determine the significance of GSH in glioma cell growth under hypoxic conditions, we inhibited system xc− using two inhibitors, (S)-4-carboxyphenylglycine (S4CPG) and sulfasalazine (SAS). S4CPG and SAS have been shown to effectively inhibit l-cystine uptake and to decrease tumor growth (4, 16, 29–31). In addition, the effect of SAS on tumor growth has been shown to be independent of NF-κB and specifically due to l-cystine starvation (30, 32). First, dose responses for both inhibitors were established in the presence of either 100 or 10 μm l-cystine. In 100 μm l-cystine, S4CPG decreased growth under hypoxic and normoxic conditions with IC50 values of 145 and 126 μm, respectively (supplemental Fig. 1A). In the presence of 10 μm l-cystine, there was an increase in the sensitivity of glioma cells to S4CPG with IC50 values of 0.80 μm under hypoxic conditions and 2 μm under normoxic conditions (supplemental Fig. 1B). In the presence of 100 μm l-cystine, the IC50 values for SAS under hypoxic and normoxic conditions were 440 and 315 μm, respectively (supplemental Fig. 1C). In addition, lowering the extracellular l-cystine to 10 μm increased the overall sensitivity of glioma cells to SAS under both hypoxic and normoxic conditions with IC50 values of 32 and 40 μm, respectively (supplemental Fig. 1D). These dose responses establish the efficacy of S4CPG and SAS for the inhibition of glioma cell growth at both high and low concentrations of l-cystine.
To examine whether GSHee could rescue growth inhibition by S4CPG and SAS under hypoxic and normoxic conditions, we used 1 mm GSHee and drug concentrations of S4CPG and SAS that resulted in >80% growth inhibition, a growth inhibition similar to that seen in the absence of l-cystine. Under hypoxic conditions and in the presence of 100 μm l-cystine, S4CPG and SAS decreased the cell number by 99 and 84%, respectively (Fig. 6, A and B). Although 1 mm GSHee significantly increased the cell number in the presence of both S4CPG and SAS, only S4CPG restored growth completely to control levels (Fig. 6, A and B). Under normoxic conditions and in the presence of 100 μm l-cystine, 500 μm S4CPG and SAS decreased the cell number by 98 and 95%, respectively (Fig. 6, A and B). Furthermore, exogenous application of 1 mm GSHee restored growth to control levels in the presence of either drug.
FIGURE 6.

GSH restores growth inhibition by SAS and S4CPG. A and B, inhibition of system xc− with 500 μm S4CPG or SAS under normoxic (21% O2) or hypoxic (2% O2) conditions. 1 mm GSHee restored growth inhibition of S4CPG and SAS to control levels. Two-way ANOVA, followed by Tukey's post hoc analysis, was used to determine significance, ***, p < 0.001; **, p < 0.01; *, p < 0.05 (n = 3).
Hypoxia Increases l-[14C]Cystine Uptake and Cell-surface Expression of the xCT Subunit of System xc−
Previous reports demonstrate that NO donors such as 3-nitroso-N-acetylpenicillamine and S-nitrosoglutathione and ROS donors such as xanthine/xanthine oxidase and H2O2 increase system xc− activity (40). This led us to examine how l-[14C]cystine uptake through system xc− is affected by hypoxia. After 72 h under hypoxic or normoxic conditions, glioma cells were washed with Na+-independent uptake solution to eliminate the contribution of Na+-dependent uptake systems. This was followed by the addition of Na+-independent uptake solution containing 2 μCi of l-[14C]cystine and 100 μm l-cystine, and uptake was measured over 3 min. D54-MG cells grown under hypoxic conditions took up ∼30% more l-cystine than cells grown under normoxic conditions (Fig. 7). Concentrations as low as 250 μm S4CPG and SAS equally decreased l-cystine uptake in D54-MG cells (16). To investigate a possible up-regulation of any competing l-cystine transporters, we measured l-cystine uptake in the presence of a high dose of SAS to maximize inhibition of l-cystine uptake. D54-MG cells were grown under hypoxic conditions for 72 h, and uptake was measured in the presence of 750 μm SAS or vehicle. The results show that in the presence of SAS, l-cystine uptake was decreased by ∼67% under hypoxic conditions and by 63% under normoxic conditions (Fig. 7). This suggests that under hypoxic conditions, the majority of l-cystine transport is mediated through system xc−, and the enhanced uptake may be due to an enhanced expression of system xc−.
FIGURE 7.

Hypoxia enhances l-[14C]cystine uptake via system xc−. A, uptake of l-[14C]cystine in D54-MG cells cultured under normoxic (21% O2) or hypoxic (2% O2) conditions for 72 h with and without 750 μm SAS. Two-way ANOVA, followed by Tukey's post hoc analysis, was used to determine significance, ***, p < 0.001; *, p < 0.05 (n = 8).
To investigate the effects of hypoxia on xCT expression, we examined total protein after glioma cells were grown under hypoxic conditions for defined periods. Cell lysates were collected, subjected to Western blotting, and probed for xCT and GAPDH (Fig. 8A). Through densitometric analysis, we determined that hypoxia had no effect on total protein (Fig. 8B). Next, we examined cell-surface xCT expression using a biotinylation assay. D54-MG cells were grown in 2% O2 for defined periods. At the end of the last time point, cells were brought to 4 °C to stop endocytosis, and surface proteins were biotin-labeled and streptavidin-coupled. Cells were lysed and collected for Western blotting. Blots were probed with xCT and normalized to Na+/K+-ATPase (Fig. 8C). Our results show a 3–4-fold increase in cell-surface expression of the xCT subunit at 48 and 96 h, respectively (Fig. 8D). These findings suggest that following hypoxia, increased surface expression of xCT leads to enhanced l-cystine uptake.
FIGURE 8.

Hypoxia increases surface expression of xCT in D54-MG cells. A and C, representative blots of D54-MG cells grown under hypoxic (2% O2) conditions and probed for xCT. A, total protein; C, surface protein. B and D, densitometric analysis of four independent experiments. B, total; D, surface Two-way ANOVA, followed by Tukey's post hoc analysis, was used to determine significance. *, p < 0.05 (n = 4).
DISCUSSION
Our results demonstrate that under hypoxic conditions, glioma cells exhibit an increased dependence on GSH for cell growth, particularly under conditions of limited l-cystine availability. GSH maintains the thiol redox potential in cells, neutralizes free radicals, and serves as a reservoir for intracellular l-cysteine (33–36). We have shown that in the presence of 10 μm l-cystine, BSO inhibits glioma cell growth and that GSHee completely returns growth to control levels. This suggests that the primary role for GSH in glioma cells is redox regulation rather than protein synthesis. It is possible that under hypoxic conditions, there is an increase in oxidized proteins, possibly ribonucleotide reductase. Ribonucleotide reductase is an enzyme that catalyzes the formation of deoxyribonucleotides from ribonucleotides and is required for DNA synthesis and cell cycle progression (21, 22, 37). These findings are supported by data showing that under hypoxic conditions, there are indeed increases in both NO and ROS. Despite these increases, in glioma cell growth was unabated provided cells were maintained under conditions that supported the de novo synthesis of GSH. Interestingly, some cell types are capable of synthesizing GSH from l-methionine in the absence of l-cystine by going through the trans-sulfuration pathway (38). In fact, some cancers are dependent on the availability of l-methionine for the synthesis of GSH exclusively, and in its absence, growth is stunted (39). However, D54-MG cells are unable to substitute l-methionine for l-cystine (data not shown), making l-cystine critical for GSH synthesis and glioma cell survival.
We hypothesize that glioma cells adjust to increased levels of NO and ROS by increasing the uptake of l-cystine to provide sufficient substrate for GSH synthesis. This is consistent with previous findings that demonstrate that increased nitrosative and oxidative stress increases system xc− activity and xCT expression in retinal ganglion cells (40). Furthermore, IL-1β potentiates hypoxic neuronal cell death via a functional increase in system xc− activity (41). Likewise, we also found that glioma cells grown under hypoxic conditions for 72 h showed enhanced l-[14C]cystine uptake and that SAS decreased uptake by >50%. The inhibitory effects of SAS on system xc−-mediated l-cystine uptake are in agreement with previous reports showing that SAS reduces system xc− activity in gliomas and not in astrocytes and neurons (4). These cell types depend mainly on l-cystine/l-cysteine transport via the Na+-dependent excitatory amino acid transporter systems (4, 42, 43).
The increased system xc− activity under hypoxic conditions reported in this study is contrary to findings in human fibroblasts and mouse peritoneal macrophages, where hypoxia reduces l-cystine uptake (44, 45). It is possible that fibroblasts and macrophages, unlike glioma cells, show a differential dependence on system xc−-mediated l-cystine uptake in response to low oxygen. However, the increased l-cystine uptake in gliomas is readily explained by our finding that hypoxia increases cell-surface expression of xCT by 3-fold. Indeed, the increase at the protein level is larger than the increase in l-cystine transport recorded here, suggesting that not all xCT subunits participate in l-cystine transport. It is possible, for example, that not all surface xCT associates with CD98, which is required to compose a functional transporter. From a mechanistic point of view, we suggest that glioma cells maintain a cytoplasmic reservoir of xCT, which is recruited to the plasma membrane on demand to meet its redox needs, i.e. GSH production. This likely represents an adaptation to the cell's biological microenvironment, where oxygen tension has been shown to vary considerably from 2–21% in normal tissue to as low as 0.1% in tumors (11–14).
Although hypoxia increased l-cystine uptake, there was no increase in GSH concentrations over time. This can be readily explained by the increased utilization of GSH observed under hypoxic conditions. GSH metabolism in the γ-glutamyl cycle and increased γ-glutamyl transpeptidase activity are possible mechanisms of GSH consumption (33, 46). Interestingly, γ-glutamyl transpeptidase expression positively correlates with high-grade glioma, and increased oxidative stress may increase γ-glutamyl transpeptidase expression and/or activity (47, 48). Consistent with an enhanced need for GSH under hypoxic conditions, glioma cells in the presence of low l-cystine and under hypoxic conditions were also more sensitive to BSO, an inhibitor of GSH synthesis. These findings suggest that system xc−-mediated l-cystine uptake gains even greater importance in the growth control of gliomas as they outgrow their blood supply and the tumor experiences hypoxia. Although hypoxia was the only exogenously imparted stress in this study, one can extrapolate from our findings that under hypoxic conditions, the ability of glioma cells to maintain homeostatic GSH levels may render them more resistant to radiation and chemotherapeutic approaches. Of note, radiation damage is due mainly to the generation of hydroxyl radicals, which are effectively neutralized by GSH, and many gliomas are indeed highly resistant to radiation therapy (49, 50). Similarly, resistance to chemotherapeutic drugs is common in gliomas and is thought to be due to the activity of the multidrug-resistant gene that encodes a transporter that requires conjugation of the compound to GSH to be transported (51). Hence, GSH production under hypoxic conditions is likely to enhance both radiation and chemoresistance of gliomas. This raises the question of whether an increase in free radical production in response to radiation therapy and chemotherapeutic drugs signals glioma cells to increase system xc− to combat its new redox status. Previous findings suggest that SAS may be an excellent drug candidate to target system xc− in gliomas (16). Our findings suggest that the target is indeed up-regulated under hypoxic conditions. A strong argument can be made that the inhibition of system xc− via SAS or similar drugs should be considered as adjuvant treatment for patients undergoing radiation and/or chemotherapy to enhance treatment effectiveness.
This work was supported, in whole or in part, by National Institutes of Health Grants RO1 NS052634 and T32MH18882.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. 1.
- RNS
- reactive nitrogen species
- ROS
- reactive oxygen species
- BSO
- buthionine sulfoximine
- HIF-1α
- hypoxia-inducible factor 1α
- ANOVA
- analysis of variance
- GSHee
- GSH ethyl ester
- S4CPG
- (S)-4-carboxyphenylglycine
- SAS
- sulfasalazine
- DAF-FM
- (4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate)
- CM-H2DCFDA
- 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate acetyl ester.
REFERENCES
- 1.Anderson M. E., Underwood M., Bridges R. J., Meister A. (1989) FASEB J. 3, 2527–2531 [DOI] [PubMed] [Google Scholar]
- 2.Choi C., Zhao C., Dimitrov I., Douglas D., Coupland N. J., Kalra S., Hawesa H., Davis J. (2009) J. Magn. Reson. 198, 160–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Terpstra M., Henry P. G., Gruetter R. (2003) Magn. Reson. Med. 50, 19–23 [DOI] [PubMed] [Google Scholar]
- 4.Ye Z. C., Rothstein J. D., Sontheimer H. (1999) J. Neurosci. 19, 10767–10777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cho Y., Bannai S. (1990) J. Neurochem. 55, 2091–2097 [DOI] [PubMed] [Google Scholar]
- 6.Kato S., Negishi K., Mawatari K., Kuo C. H. (1992) Neuroscience 48, 903–914 [DOI] [PubMed] [Google Scholar]
- 7.Lu S. C. (2009) Mol. Aspects Med. 30, 42–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lakshmi V. M., Hsu F. F., Zenser T. V. (2003) Chem. Res. Toxicol. 16, 367–374 [DOI] [PubMed] [Google Scholar]
- 9.Hockel M., Vaupel P. (2001) Semin. Oncol. 28, 36–41 [PubMed] [Google Scholar]
- 10.Yuan F., Salehi H. A., Boucher Y., Vasthare U. S., Tuma R. F., Jain R. K. (1994) Cancer Res. 54, 4564–4568 [PubMed] [Google Scholar]
- 11.Rampling R., Cruickshank G., Lewis A. D., Fitzsimmons S. A., Workman P. (1994) Int. J. Radiat. Oncol. Biol. Phys. 29, 427–431 [DOI] [PubMed] [Google Scholar]
- 12.Nordsmark M., Bentzen S. M., Overgaard J. (1994) Acta Oncol. 33, 383–389 [DOI] [PubMed] [Google Scholar]
- 13.Erecińska M., Silver I. A. (2001) Respir. Physiol. 128, 263–276 [DOI] [PubMed] [Google Scholar]
- 14.Papandreou I., Powell A., Lim A. L., Denko N. (2005) Mutat. Res. 569, 87–100 [DOI] [PubMed] [Google Scholar]
- 15.Yamamoto Y., König P., Henrich M., Dedio J., Kummer W. (2006) Cell Tissue Res. 325, 3–11 [DOI] [PubMed] [Google Scholar]
- 16.Chung W. J., Lyons S. A., Nelson G. M., Hamza H., Gladson C. L., Gillespie G. Y., Sontheimer H. (2005) J. Neurosci. 25, 7101–7110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chandel N. S., McClintock D. S., Feliciano C. E., Wood T. M., Melendez J. A., Rodriguez A. M., Schumacker P. T. (2000) J. Biol. Chem. 275, 25130–25138 [DOI] [PubMed] [Google Scholar]
- 18.Guzy R. D., Hoyos B., Robin E., Chen H., Liu L., Mansfield K. D., Simon M. C., Hammerling U., Schumacker P. T. (2005) Cell Metab. 1, 401–408 [DOI] [PubMed] [Google Scholar]
- 19.Shih A. Y., Erb H., Sun X., Toda S., Kalivas P. W., Murphy T. H. (2006) J. Neurosci. 26, 10514–10523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang G. L., Semenza G. L. (1993) J. Biol. Chem. 268, 21513–21518 [PubMed] [Google Scholar]
- 21.Luthman M., Eriksson S., Holmgren A., Thelander L. (1979) Proc. Natl. Acad. Sci. U.S.A. 76, 2158–2162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holmgren A. (1976) Proc. Natl. Acad. Sci. U.S.A. 73, 2275–2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang J., Wu L. J., Tashiro S., Onodera S., Ikejima T. (2008) Free Radic. Res. 42, 1–11 [DOI] [PubMed] [Google Scholar]
- 24.Anderson M. F., Nilsson M., Sims N. R. (2004) Neurochem. Int. 44, 153–159 [DOI] [PubMed] [Google Scholar]
- 25.Dringen R. (2000) Prog. Neurobiol. 62, 649–671 [DOI] [PubMed] [Google Scholar]
- 26.Na N., Chandel N. S., Litvan J., Ridge K. M. (2010) FASEB J. 24, 799–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Radi R., Beckman J. S., Bush K. M., Freeman B. A. (1991) J. Biol. Chem. 266, 4244–4250 [PubMed] [Google Scholar]
- 28.Ridnour L. A., Thomas D. D., Mancardi D., Espey M. G., Miranda K. M., Paolocci N., Feelisch M., Fukuto J., Wink D. A. (2004) Biol. Chem. 385, 1–10 [DOI] [PubMed] [Google Scholar]
- 29.Guan J., Lo M., Dockery P., Mahon S., Karp C. M., Buckley A. R., Lam S., Gout P. W., Wang Y. Z. (2009) Cancer Chemother. Pharmacol. 64, 463–472 [DOI] [PubMed] [Google Scholar]
- 30.Doxsee D. W., Gout P. W., Kurita T., Lo M., Buckley A. R., Wang Y., Xue H., Karp C. M., Cutz J. C., Cunha G. R., Wang Y. Z. (2007) Prostate 67, 162–171 [DOI] [PubMed] [Google Scholar]
- 31.Narang V. S., Pauletti G. M., Gout P. W., Buckley D. J., Buckley A. R. (2007) Chemotherapy 53, 210–217 [DOI] [PubMed] [Google Scholar]
- 32.Chung W. J., Sontheimer H. (2009) J. Neurochem. 110, 182–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meister A., Tate S. S. (1976) Annu. Rev. Biochem. 45, 559–604 [DOI] [PubMed] [Google Scholar]
- 34.Burk R. F., Patel K., Lane J. M. (1983) Biochem. J. 215, 441–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tateishi N., Higashi T., Naruse A., Nakashima K., Shiozaki H. (1977) J. Nutr. 107, 51–60 [DOI] [PubMed] [Google Scholar]
- 36.Cho E. S., Johnson N., Snider B. C. (1984) J. Nutr. 114, 1853–1862 [DOI] [PubMed] [Google Scholar]
- 37.Herrick J., Sclavi B. (2007) Mol. Microbiol. 63, 22–34 [DOI] [PubMed] [Google Scholar]
- 38.Schmidt C. L., Allen F. W., Tarver H. (1940) Science 91, 18–19 [DOI] [PubMed] [Google Scholar]
- 39.Zhang W., Braun A., Bauman Z., Olteanu H., Madzelan P., Banerjee R. (2005) Cancer Res. 65, 1554–1560 [DOI] [PubMed] [Google Scholar]
- 40.Dun Y., Mysona B., Van Ells T., Amarnath L., Ola M. S., Ganapathy V., Smith S. B. (2006) Cell Tissue Res. 324, 189–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fogal B., Li J., Lobner D., McCullough L. D., Hewett S. J. (2007) J. Neurosci. 27, 10094–10105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Flynn J., McBean G. J. (2000) Neurochem. Int. 36, 513–521 [DOI] [PubMed] [Google Scholar]
- 43.McBean G. J., Flynn J. (2001) Biochem. Soc. Trans. 29, 6–22 [DOI] [PubMed] [Google Scholar]
- 44.Bannai S., Sato H., Ishii T., Sugita Y. (1989) J. Biol. Chem. 264, 18480–18484 [PubMed] [Google Scholar]
- 45.Sato H., Kuriyama-Matsumura K., Hashimoto T., Sasaki H., Wang H., Ishii T., Mann G. E., Bannai S. (2001) J. Biol. Chem. 276, 10407–10412 [DOI] [PubMed] [Google Scholar]
- 46.Griffith O. W., Bridges R. J., Meister A. (1978) Proc. Natl. Acad. Sci. U.S.A. 75, 5405–5408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu R. M., Shi M. M., Giulivi C., Forman H. J. (1998) Am. J. Physiol. 274, L330–L336 [DOI] [PubMed] [Google Scholar]
- 48.Schäfer C., Fels C., Brucke M., Holzhausen H. J., Bahn H., Wellman M., Visvikis A., Fischer P., Rainov N. G. (2001) Acta Oncol. 40, 529–535 [DOI] [PubMed] [Google Scholar]
- 49.Davis G. D., Masilamoni J. G., Arul V., Kumar M. S., Baraneedharan U., Paul S. F., Sakthivelu I. V., Jesudason E. P., Jayakumar R. (2009) Cell Biol. Toxicol. 25, 331–340 [DOI] [PubMed] [Google Scholar]
- 50.Bump E. A., Brown J. M. (1990) Pharmacol. Ther. 47, 117–136 [DOI] [PubMed] [Google Scholar]
- 51.Dirven H. A., van Ommen B., van Bladeren P. J. (1996) Chem. Res. Toxicol. 9, 351–360 [DOI] [PubMed] [Google Scholar]