

Abstract
Disruption of neurotoxic effects of amyloid β protein (Aβ) is one of the major, but as yet elusive, goals in the treatment of Alzheimer's disease (AD). The amylin receptor, activated by a pancreatic polypeptide isolated from diabetic patients, is a putative target for the actions of Aβ in the brain. Here we show that in primary cultures of human fetal neurons (HFNs), AC253, an amylin receptor antagonist, blocks electrophysiological effects of Aβ. Pharmacological blockade of the amylin receptor or its down-regulation using siRNA in HFNs confers neuroprotection against oligomeric Aβ-induced caspase-dependent and caspase-independent apoptotic cell death. In transgenic mice (TgCRND8) that overexpress amyloid precursor protein, amylin receptor is up-regulated in specific brain regions that also demonstrate an elevated amyloid burden. The expression of Aβ actions through the amylin receptor in human neurons and temporospatial interrelationship of Aβ and the amylin receptor in an in vivo model of AD together provide a persuasive rationale for this receptor as a novel therapeutic target in the treatment of AD.
Several lines of evidence support a role for amyloid β-protein (Aβ) in the pathogenesis of Alzheimer's disease (AD). The genetic data include the occurrence of AD with inherited amyloid precursor protein (APP) mutations adjacent to the β- and γ-secretase cleavage sites, trisomy 21 with the APP gene, and early-onset PS1 and PS2 mutations in the γ-secretase catalytic subunit.1 Other data include neurotoxicity of soluble oligomeric Aβ when applied to neurons2 and the generation of APP-overexpressing mice that recapitulate certain neuropathological and behavioral features of AD.3 Although Aβ exerts a wide range of biological effects and is potently neurotoxic, there is as yet no unequivocally identified receptor for Aβ. Several putative receptor candidates for Aβ have been reported (eg, RAGE receptors, the p75NTR receptor, scavenger receptors, neuronal nicotinic receptors, and the tachykinin and serpin-enzyme complex receptors), but the functional significance of Aβ interactions with such receptors in the brain has yet to be identified or remains controversial.4–8
Several epidemiological studies have attempted to link AD and diabetes mellitus, a disorder of glucose metabolism and insulin secretion.9,10,11 At a cellular level, human amylin (islet amyloid peptide, diabetes-associated peptide), a 37-amino-acid amyloidogenic peptide first isolated from protein deposits within the pancreatic islets of Langerhans of non-insulin-dependent diabetes mellitus patients, shares similar biophysical and physiological properties with Aβ.12,13,14,15 Electrophysiological data reveal that human amylin and Aβ affect the same suite of potassium conductances in rat cholinergic basal forebrain neurons and that each peptide is able to occlude the response of the other, suggesting a common mechanism of action.16,17 Furthermore, Aβ and human amylin not only induce apoptotic cell death in cultured neurons and pancreatic β-islet cells, but demonstrate a neurotoxicity profile that is identical, including time- and concentration-dependent induction of apoptotic genes.14,15,18 Recent data using quantitative iTRAC proteomics analysis (iTRAC stands for “isobaric tag for relative and absolute quantitation”) reveal that human amylin and Aβ deregulate identical mitochondrial proteins, further supporting the notion that both amyloidoses have common targets.19 Collectively, these observations suggest that the human amylin receptor, which serves as the endogenous receptor for the pancreatic amylin peptide, could also serves as a putative receptor for the expression of the biological effects of Aβ.
Dimerization of the calcitonin receptor (CTR) with RAMP3 yields a receptor that binds amylin with a significantly higher affinity than CGRP and adrenomedullin, two other peptides belonging to this family.20,21 Several peptides, typically analogs of salmon calcitonin, have been developed as amylin receptor antagonists, chiefly with a view to treating diabetes mellitus.22,23 Of these, AC187 and AC253 are highly selective and potent antagonists at the amylin receptor.21,23,24,25 We have identified a novel interaction of Aβ and human amylin with the amylin receptor in cholinergic neurons of the rat basal forebrain, where loss of such neurons is linked to the cognitive impairment observed in AD.17 We have shown that both the acute electrophysiological and neurotoxic effects of amylin and Aβ in the rat cholinergic basal forebrain neurons can be blocked using amylin receptor antagonists.17,26 An important question raised by our observations is whether blockade of the amylin receptor confers neuroprotection against Aβ toxicity in cultures of human neurons. This is a critical issue, because rodents (rats, mice, hamsters), the species in which the effects of Aβ have been most widely studied, do not develop an age-related human equivalent of AD.
In the present study, using whole-cell patch clamp recordings from primary cultures of human fetal neurons (HFNs), we found that acute applications of nanomolar concentrations of Aβ result in an activation of a suite of potassium conductances, which can be blocked by exposure to the amylin receptor antagonist AC253. Furthermore, the amyloid-induced toxicity mediated via caspase-dependent and -independent pathways in HFNs can be attenuated with pretreatment of cultures with AC253 or through down-regulation of the amylin receptor gene expression with small interfering RNA (siRNA). Finally, we demonstrate that in transgenic mice that overexpress APP (TgCRND8), amylin receptor expression in the brain is up-regulated in an age-dependent manner, but only within specific brain regions that demonstrate an increased amyloid burden.
Materials and Methods
All experiments were conducted in compliance with the relevant laws and the guidelines set by the Canadian Council for Animal Care and with the approval of the Human Research Ethics Board and Animal Care Use Committee (Health Sciences) at the University of Alberta.
Electrophysiological Recordings from HFNs
HFNs, grown on coverslips, were bathed with oxygenated artificial cerebrospinal fluid that contained (in mmol/L) 140 NaCl, 2.5 KCl, 1.5 CaCl2, 1.2 MgCl2, 10 HEPES, and 33 d-glucose (pH 7.4). Whole cell patch-clamp recordings were performed at room temperature (20°C–22°C) using an Axopatch-200B amplifier in combination with a 1200A interface (Axon Instruments, Foster City, CA). Patch electrodes (thin wall with filament, 1.5-mm diameter; World Precision Instruments, Sarasota, FL) were pulled (PP-83 electrode puller; Narishige Scientific Instrument Lab, Tokyo, Japan) to yield resistances of 3–6 MΩ. The internal patch pipette solution contained (in mM) 140 K-methylsulfate, 10 EGTA, 5 MgCl2, 1 CaCl2, 10 HEPES, 2.2 Na2-ATP, and 0.3 Na-GTP (pH 7.2). All whole-cell recordings were made in current-clamp and voltage-clamp mode, and bridge balance and capacitance compensation were used. After whole-cell configuration was established with voltage-clamp mode (holding potential = −80 mV), we waited at least 5 minutes for the cell to stabilize, then started either voltage-clamp studies or switched to current-clamp recording mode. The current and membrane voltages were recorded using a low-pass filter at 5 kHz and were digitized at 10 kHz. All data were acquired and analyzed using pClamp8 software v8.0 (Axon Instruments). Whole-cell currents were activated by a voltage-ramp protocol, where the cells were held at −80 mV and subjected to voltage ramps from −110 to +30 mV at the rate of 20 mV/s. A 1-second-long hyperpolarizing command to −110 mV was applied to remove inactivation of K+ channels so that maximum current could be activated during the subsequent slow voltage ramp to +30 mV. Ca2+-dependent K+ conductances were isolated using the BK channel-specific blocker iberiotoxin. Drugs were either bath-applied or delivered via a focal applicator.
Primary Cultures of HFNs
Neuronal cultures were prepared from 12- to 15-gestational week fetuses with approval of the Human Ethics Research Board at the University of Alberta. The meninges and blood vessels were removed, the brain tissue was washed in minimum essential medium and mechanically dissociated by repeated trituration through a 20-gauge needle. Cells were centrifuged at 1500 g for 10 minutes and resuspended in minimum essential medium with 10% heat-inactivated fetal bovine serum, 0.2% N2 supplement, and 1% antibiotic solution (104 U of penicillin G per milliliter, 10 mg streptomycin per milliliter, and 25 mg amphotericin B per milliliter in 0.9% NaCl).27 Subsequently, cultures were treated with arabinofuranosylcytosine (25 μmol/L) for 2 weeks and were plated (at a density of 5 × 10−5 per well) on 96-well plates. HFN cultures were grown in a 5% CO2 humidified incubator at 37°C. Sample wells were immunostained for the neuronal marker microtubule-associated protein 2, and only cultures in which >70% of the cells stained positive for the marker were used for experiments. All reagents were obtained from Invitrogen (Burlington, ON, Canada) and antibodies from Sigma-Aldrich (Oakville, ON, Canada).
Treatments and Cell Death Assays
Soluble oligomeric Aβ1-42 or the inverse sequence peptide Aβ42-1, human amylin, and AC253 were prepared according to published protocols.28,29 All peptides were purchased from American Peptide (Sunnyvale, CA). To determine the dose-dependent toxicity of Aβ peptides and human amylin. HFNs were treated with different concentrations of the peptides (0.5–50 μmol/L) (see Supplemental Figure S1 at http://ajp.amjpathol.org). In each experiment and in subsequent ones described below, two rows of eight wells each (of a 96-well plate) received the same treatment, and each experiment was repeated a minimum of four times. To evaluate the neuroprotective effects of the amylin receptor antagonist AC253 against Aβ toxicity, cultured HFNs were preexposed to AC253 (10 μmol/L) for 24 hours and then to soluble oligomeric 20 μmol/L Aβ1-42 or human amylin (2 μmol/L). Cells in adjacent rows of wells received applications of either 20 μmol/L Aβ1-42, 2 μmol/L human amylin, or 10 μmol/L or AC253. Controls included applications of the inverse sequence Aβ peptide Aβ42-1 or no drug. After 48 hours, the control and treated cultures were examined for neuronal survival using a 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Sigma-Aldrich).26,30
For experiments examining the effects of caspase inhibition on Aβ toxicity, the cell cultures were pretreated for 2 hours with individual caspase inhibitors (20 μmol/L; kit from RD Systems, Minneapolis, MN), followed by exposure to 20 μmol/L Aβ1-42 for 48 hours. At the end of this period, the cultures were processed for MTT assay. Each experiment was conducted four times.
Immunoblotting
Western blotting was performed as described previously.26,30 Briefly, samples of control and treated groups of cultured cells (with AC253, Aβ1-42, or AC253 pretreatment followed by Aβ1-42) with equal amounts of protein were separated by 12% polyacrylamide gel electrophoresis and the resolved proteins were transferred onto nitrocellulose membranes and probed with anti-caspase 3, 6, and 9 antibodies and also with antibodies to pro- and antiapoptotic intermediaries (cytochrome c, SMAC, PUMA, Bax, and Bcl-2; New England Biolabs, Ipswich, MA). Blots were also probed with anti-β actin (Abcam, Cambridge, MA) as loading control. To assess whether AC253 also blocked Aβ-induced caspase-independent cell death, we performed Western blot and immunohistochemistry to detect cleavage and translocation of apoptosis inducing factor (AIF) from the activated mitochondria using AIF antibody (Santa Cruz Biotechnology, Santa Cruz, CA) and MitoTracker Red dye (Molecular Devices, Sunnyvale, CA). Each experiment was repeated four times. For all quantitative analysis of Western blots, Image J software v1.44j (National Institutes of Health, Bethesda, MD) was used. Relative intensities were calculated by comparing the intensity of protein bands of interest to control β-actin loading bands.
siRNA
siRNA corresponding to the human CTR (NM_001742) and RAMP3 (NM_005856) genes, purchased from Open BioSystems (Huntsville, AL), were inserted into a vector (pGIPz) that produced green fluorescent protein. HFNs were plated at a density of 2 million cells/ml in a 6-well plate and cultured for 24 hours before transfection. On the day of transfection, culture media was replaced with OptiMEM (Invitrogen). Four micrograms of plasmid siRNA mixed with Lipofectamine 2000 (Invitrogen) was incubated for 30 minutes and then added to the cultures. Cultures were gently mixed and placed in an incubator at 37°C with 5% CO2 for 10 hours, after which normal culture media was replaced. Subsequently, transfected and non-transfected HFN cultures in 96-well plates were exposed to soluble oligomeric Aβ1-42 (20 μmol/L) or the inverse sequence peptide, Aβ42-1 (20 μmol/L), and cell survival was measured using the MTT assay.
Enzyme-Linked Immunosorbent Assay for Measurement of Aβ Levels in Brain Regions of TgCRND8 APP and Control Mice
Brains were quickly removed from 1-, 4-, and 6-month-old TgCRND8 APP mice (human APP695 transgene array incorporating Swedish K670M/N671L and Indiana V717F mutations superimposed on a C57BL6/C3H genetic background) and from control nontransgenic C57BL6/C3H littermate mice. The cortex, hippocampus, diagonal band of Broca, brainstem, and cerebellum were dissected and weighed before the preparation of normalized lysates. Proteins from each dissected region were isolated according to the protocol provided with the Aβ1-42 enzyme-linked immunosorbent assay kit (BioSource, Burlington, ON, Canada). In brief, in this protocol a monoclonal antibody specific to the NH-2 terminus of the Aβ1-42 is coated to the wells of microtiter strips. Proteins are incubated in the well. A secondary antibody specific to the COOH-terminus of the Aβ1-42 sequence is then incubated in the wells. Bound rabbit antibody is detected with a horseradish peroxidase-labeled anti-rabbit antibody. Excess anti-rabbit antibody is removed by washing, and then a substrate solution is added and is converted, producing a color. The color is measured at 450 nm and the intensity of color is directly proportional to the amount of Aβ1-42 in the tissue.
Expression of Amylin Receptor in Brain Regions of TgCRND8 APP-Overexpressing Mice
Under halothane anesthesia, TgCRND8 APP mice (1, 4, and 6 months of age, n = 5 for each age group) and age-matched control mice (n = 5 for each age group) were decapitated and brains were quickly removed. Brains were hemisected and one half of the brain was used for dissecting the cortex, hippocampus, diagonal band of Broca, cerebellum, and brainstem. Tissue was weighed and the placed in cold RIPA (radio-immunoprecipitation assay) buffer with protease inhibitors and proteins were isolated and measured using a Bio-Rad protein assay kit (Mississauga, ON, Canada). Proteins were diluted with SDS sample buffer (New England Biolabs) and were loaded at 30 μg per lane on a 12% agarose gel. Proteins were transferred to nitrocellulose blocked with nonfat milk. Nitrocellulose membranes were incubated with antibodies overnight at 4°C on a shaker. Blots were incubated in the following antibodies, one at a time, and were stripped between each application: CTR (1:1000 rabbit; kindly supplied by Professor P. Sexton, Monash University, VIC, Australia), RAMP3 (1:1000 goat; Santa Cruz Biotechnology), 6E10 for Aβ (1:5000 mouse; 6E10; Signet Laboratories, Dedham, MA), and β-actin (1:1000 mouse; Sigma-Aldrich). Blots were subsequently incubated with corresponding secondary antibodies labeled with horseradish peroxidase and were washed with Immobilon Western chemiluminescent substrate (Millipore, Billerica, MA). Blots were exposed to X-ray film (Kodak BioMax MR) for 10 seconds to 5 minutes. X-ray blots were then scanned and analyzed with Image J software. The other half of the brains from TgCRND8 and control mice was fixed for immunohistochemical analysis of brain sections from cortex and hippocampus to determine Aβ plaque deposition, and the same sections were also double-stained using the CTR antibody.
Statistical Analysis
Data are presented as means ± SEM. Unless otherwise indicated, group data were compared using one-way analysis of variance followed by Newman-Keuls post hoc test with the P-value set at 0.05.
Results
Electrophysiological Effects of Aβ Are Blocked by Amylin Receptor Antagonist
In HFNs, applications of oligomeric Aβ1-42 caused a time-independent and concentration-dependent reduction in whole-cell outward currents (WCCs) in the voltage range −30 to +30 mV (see Supplemental Figure S2 at http://ajp.amjpathol.org). WCCs were not affected by applications of the inverse sequence Aβ42-1 peptide (data not shown). AC253 is a selective peptidergic amylin receptor antagonist that, applied in the concentration range 10−5 to 10−6 mol/L, has been shown to block the actions of human amylin.17,31 We investigated whether the effects of Aβ could also be blocked using this amylin receptor antagonist. Application of 10 nmol/L AC253 blocked Aβ1-42-induced reduction in WCCs (20 nmol/L, n = 8, P < 0.01) (Figure 1, A and B). Peak WCC (at +30 mV) in the presence of Aβ1-42 was significantly reduced, compared with the control level of current (27.7%, n = 8, P < 0.01), but in the presence of AC253, the Aβ1-42-evoked response was abolished (Figure 1C). When the high conductance Ca2+-activated K+ channels Ic (BK channels) were inhibited with iberiotoxin (25 nmol/L), Aβ1-42-induced reduction in WCCs was significantly blocked (Figure 1D), indicating that Aβ effects are expressed via this ionic conductance.
Figure 1.
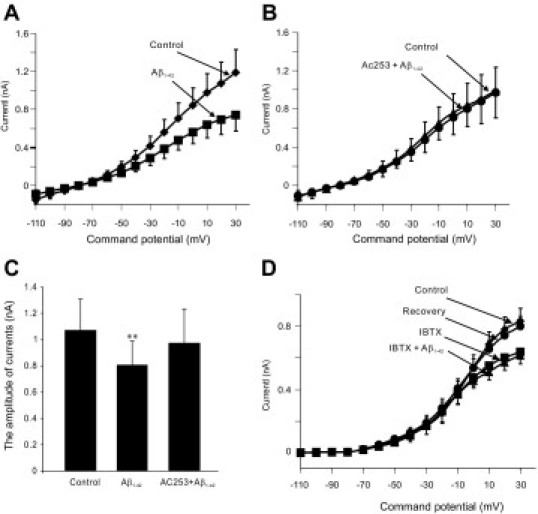
Electrophysiological effects of β-amyloid (Aβ) on whole-cell currents in human fetal neurons (HFNs) are expressed via the amylin receptor. A: Bath application of Aβ1-42 (20 nmol/L) decreases outward currents in the voltage range from −30 to +30 mV (n = 8). B: AC253, a selective amylin receptor antagonist, blocks the effect of Aβ1-42 on whole-cell currents in HFNs (n = 8). C: Histograms showing that Aβ1-42-evoked decrease in peak amplitude of the whole-cell currents (measured at +30 mV) is decreased in the presence of AC253 (n = 8, P < 0.01). D: Under voltage-clamp recording conditions, Ca2+-activated K+ currents are blocked in the presence of iberiotoxin (IBTX) (10 nmol/L), a selective inhibitor of Ic (BK channels) (n = 9, P < 0.01). Subsequent application of Aβ1-42 (10 nmol/L) in the presence of IBTX did not result in a further significant reduction in such currents. Error bars indicate ±SEM.
Under current-clamp conditions, the number of spikes generated by injection of a current pulse in HFNs was significantly increased in the presence of Aβ1-42 (10 nmol/L), compared with control conditions (Figure 2A). Application of AC253 (10 nmol/L) blocked the increase in excitability induced by Aβ1-42. The average number of spikes elicited by current injection was 1.2 ± 0.3 Hz under control conditions, which significantly increased to 16.6 ± 3.5 Hz in the presence of Aβ1-42 (Figure 2B) (n = 5, P < 0.01). In the presence of AC253, application of Aβ1-42 did not result in a significant increase in firing (n = 5, P > 0.05), compared with control. Under resting conditions, application of Aβ1-42 (10 nmol/L) to HFNs resulted in membrane depolarization and an increase in action potential firing, which were blocked in the presence of AC253 (Figure 2C).
Figure 2.

Aβ-evoked increase in excitability of human fetal neurons (HFNs) is blocked by amylin receptor antagonist. A: Under current-clamp conditions, action potential generation evoked by injecting depolarizing current pulses (5 pA, 1 second) in a HFN is significantly increased in the presence of the evoked Aβ1-42 (10 nmol/L). Application of AC253 (10 nmol/L), antagonist of amylin receptor, completely blocks the increase in firing frequency induced by Aβ1-42. B: Histograms depict summary of data obtained from five HFNs after application of Aβ1-42 alone and in the presence of Aβ1-42. *Significant difference from control, P < 0.05. C: Continous current-clamp recording from a HFN depicting membrane depolarization and increased action potential firing with application of Aβ1-42 (10 nmol/L), which is blocked in the presence of AC253. RMP of cell = −65 mV. Error bars indicate ±SEM.
Aβ Neurotoxicity Is Expressed through the Amylin Receptor
To determine whether Aβ toxicity is expressed via amylin receptors, we used primary cultures of HFNs to initially establish that application of oligomeric Aβ1-42 (see Supplemental Figure S3 at http://ajp.amjpathol.org and also the Materials and Methods section) or human amylin evokes concentration-dependent cell death in such neurons (see Supplemental Figure S1 at http://ajp.amjpathol.org). Pretreatment of HFNs for 24 hours with the amylin receptor antagonist AC253 (10 μmol/L) resulted in a significant improvement in neuronal survival of HFNs exposed to either Aβ1-42 (20 μmol/L) or human amylin (2 μmol/L) (Figure 3, A and B). Concentrations of extracellularly applied oligomeric Aβ1-42 used were based on our data (see Supplemental Figure S1 at http://ajp.amjpathol.org) and data reported in the literature for studies on human neurons.32,33,34
Figure 3.

Amylin receptor antagonist protects against Aβ and human amylin neurotoxicity in primary cultures of human fetal neurons (HFNs). A: Aβ1-42 induces cell death and loss of HFNs (identified with MAP2 staining), compared with control HFN cultures. Loss of HFNs is attenuated with pretreatment of cultures with the amylin receptor antagonist AC253. Applied alone, AC253 has no effect on HFNs. Scale bar = 10 μm. B: Quantification of the neurotoxic effects of soluble oligomeric Aβ1-42 and human amylin applications to cultures of HFNs. Control HFN cultures treated with the inverse sequence peptide, Aβ42-1, do not show evidence of cell death. Neuroprotective effects are apparent in HFN cultures pretreated with AC253 before Aβ1-42 or human amylin exposure. Cell survival measured with MTT assay. Pooled data from five experiments. Significant difference (*P < 0.05) in neuronal survival with AC253 pretreatment, compared with Aβ1-42 or human amylin applications, respectively. Error bars indicate ±SEM.
To corroborate our pharmacological observations concerning neuroprotective effects of AC253, we also examined whether down-regulation of the amylin receptor using an siRNA approach blunts Aβ1-42-induced neurotoxicity in HFNs. To suppress amylin receptor in HFNs, we generated siRNA corresponding to the two components of the amylin receptor, the human CTR (NM_001742) and RAMP3 (NM_005856) genes, and inserted these into a vector (pGIPz) that produces green fluorescent protein (Figure 4A). Controls were packaged with scrambled siRNA. We first confirmed amylin receptor suppression by the siRNAs in HFNs using separately validated antibodies (Figure 4B). HFN cultures were exposed to Aβ1-42 at 4 days after transfection and neuronal viability was assessed. Knockdown of the amylin receptor using siRNA rendered HFNs more resistant to Aβ1-42-induced cell death (Figure 4C).
Figure 4.

Aβ toxicity is blunted in cultures of human fetal neurons (HFNs) where expression of amylin receptors is down-regulated. A: HFNs transfected with siRNA directed at calcitonin receptor (CTR) and RAMP3, components of the amylin receptor. Fluorescent cells (green fluorescent protein-positive) identify those cells that express CTR (left) or RAMP3 (right). Scale bar = 10 μm. B: Western blot of control (nontransfected) and transfected HFNs, showing that CTR and RAMP3 protein are down-regulated with siRNA transfections. Lanes 1 and 3: transfected HFN cultures; lanes 2 and 4: control HFN cultures. β-actin was used as a loading control. C: Neuronal viability after Aβ1-42 exposure in HFNs transfected with vectors for shRNA RAMP3, shRNA CTR, or a combination of the two vectors. Aβ neurotoxicity is attenuated in HFN cultures transfected with shRNA RAMP3 and shRNA CTR. Significant difference compared with nontransfected HFNs exposed to Aβ1-42, *P < 0.05. Error bars indicate ±SEM.
Amylin Receptor Blockade Attenuates Aβ-Induced Caspase-Dependent and -Independent Apoptotic Cell Death
In animal in vitro models, Aβ may induce apoptosis by activation of a number of cell-death signaling pathways involving caspases, a family of cysteine proteases.35,36 Alternatively, Aβ-induced cell death may be caspase-independent, occurring through mitochondrial release, cleavage, and translocation of AIF to the nucleus with consequent DNA fragmentation and nuclear condensation.37 We initially identified caspases that are relevant in our HFN model of Aβ-induced apoptosis. HFNs pretreated with the pan-caspase inhibitor Z-VAD-FMK or with inhibitors of specific caspases (3, 6, 8, and 9) demonstrated significant resistance to oligomeric Aβ1-42 neurotoxicity (see Supplemental Figure S4 at http://ajp.amjpathol.org). Subsequently, we determined whether antagonism of the amylin receptor is able to block the Aβ-induced activation of specific caspase pathways. HFN cultures were initially treated with oligomeric Aβ1-42 and time-dependent activation of caspases 3, 6, and 9 was detected using antibodies to the cleaved forms of these caspases (Figure 5A). Pretreatment of HFNs with the amylin receptor antagonist AC253 for 24 hours effectively blocked the cleavage of caspases 3, 6, and 9 induced by oligomeric Aβ1-42 (Figure 5B). Quantitative analysis of pro-caspase 3 confirmed its reduction (due to conversion to cleaved caspase 3 product) after exposure of HFNs to Aβ1-42, but not Aβ42-1 (see Supplemental Figure S5A at http://ajp.amjpathol.org). In the presence of AC253, the Aβ1-42-induced decrease in the pro-caspase 3 substrate was significantly attenuated, indicating that less of the activated (cleaved) caspase 3 was generated to cause apoptotic cell death.
Figure 5.

AC253 inhibits Aβ activation of proapoptotic caspase pathways. A: HFN cultures exposed to soluble oligomeric Aβ1-42 (20 μmol/L) show a time-dependent increase in the proapoptotic cleaved caspases 3 and 6 over 48 hours. On Aβ1-42 exposure over the same time frame, pro-caspase 9, the mitochondrial associated caspase substrate, shows a steady decrease, indicating its cleavage to an active proapoptotic ∼37-kDa product. B: Alterations in caspases 3, 6, and 9 observed at 48 hours in HFN cultures exposed to Aβ1-42 (A) are attenuated in cells pretreated with AC253 (10 μmol/L). β-actin was used as a loading control in A and B.
A similar quantitative analysis revealed that the Aβ1-42-evoked increase in cleaved caspase 9 could be blocked by AC253 pretreatment (see Supplemental Figure S5B at http://ajp.amjpathol.org). Proapoptotic intermediaries in the caspase 9 pathway, including cytochrome c, Bax, SMAC, and PUMA, are also increased after exposure to Aβ1-42. Pretreatment with AC253 significantly attenuated or blocked not only the cleavage of caspases 9 but also activation of cytochrome c, BAX, SMAC, and PUMA (Figure 6). At the same time, Aβ1-42-evoked reduction in antiapoptotic mediators Bcl-2, Akt, and PI3-kinase was attenuated by AC253 (Figure 6).
Figure 6.

Temporal profile for mediators of apoptosis in cultures of human fetal neurons (HFNs) exposed to Aβ. A: Western blots identifying time-dependent alterations in proapoptotic (cytochrome c, BAX, SMAC, and PUMA) or antiapoptotic [Bcl-2, phospho-Akt (pAkt), PI-3 kinase] markers in HFN cultures exposed to oligomeric Aβ1-42 (20 μmol/L). Time-course for initial activation and subsequent decrease for pAkt and PI-3 kinase occurs earlier than that for proapoptotic mediators. B: Pretreatment of HFN cultures with amylin receptor antagonist AC253 (10 μmol/L) before Aβ1-42 exposure results in attenuation of proapoptotic markers and at the same time an up-regulation of antiapoptotic proteins shown in A. β-actin was used as a loading control in A and B.
To determine whether blockade of the amylin receptor also attenuated apoptotic cell death via a caspase-independent pathway, we examined the effects of AC253 pretreatment on Aβ1-42-induced cleavage and translocation of AIF from the mitochondria to the nucleus. Aβ1-42 (20 μmol/L), but not Aβ42-1 (20 μmol/L), caused a time-dependent increase in AIF from its 67-kDa form to the cleaved 57-kDa moiety (Figure 7A), which was significantly reduced in HFNs pretreated with AC253 (Figure 7, B and C). Exposure of HFNs to Aβ1-42 under the same conditions also resulted in activation of activation of caspase 9 through a caspase-dependent pathway (Figure 5). To confirm cellular localization of AIF within the mitochondria and its translocation to the nucleus, we used immunofluorescence methods in SK-N-SH neuroblastoma cells, which (by virtue of their larger size, relative to HFNs) provide better visualization of the cytoplasm and the spatial relationship of the mitochondria to the nucleus. Aβ1-42-treated SK-N-SH cells demonstrated a significant decrease in retention of AIF within the mitochondria (identified using MitoTracker Red dye), but a corresponding increase in AIF within the nuclei (identified using the nuclear stain 4′,6-diamidino-2-phenylindole), compared with control cultures (Figure 7D). This nuclear translocation of AIF was not apparent in SK-N-SH cells pretreated with AC253 and subsequently exposed to Aβ1-42. In such cells, AIF was retained within the cytoplasm and colocalized with the mitochondrial-specific marker.
Figure 7.

Time course exposure of Aβ on apoptosis-inducing factor (AIF) expression in cultures of human fetal neurons (HFNs). Western blot (A) shows the time course for AIF expression after exposure to either (1) Aβ1-42 (20 μmol/L) or (2) Aβ42-1 (20 μmol/L), the inactive, reverse sequence peptide. Lower bands are loading controls, β-actin. B: AC253 (10 μmol/L) pretreated HFNs exposed to Aβ1-42 (20 μmol/L) show AIF expression is attenuated. C: Quantification of Western blots for AC253 pretreated HFNs after exposure to Aβ1-42 (20 μmol/L). Relative intensities were calculated by comparing the intensity of AIF bands to β-actin bands. Significant increase in relative intensity with exposure to Aβ1-42, compared with β-actin loading control (n = 5, *P < 0.05). D: Photomicrographs revealing MitoTracker Red staining (red) to identify mitochondria, immunohistochemistry for AIF (green), and nuclear staining with DAPI (blue) in SK-N-SH neuroblastoma cell line. Overlays in the right column (DAP/AIF) highlight that Aβ1-42-induced translocation of AIF from mitochondria to the nucleus is significantly reduced in AC253 pretreated cells exposed to Aβ1-42 (compare photomicrographs in the overlay column on the right for the last two rows). Scale bar = 20 μm. Error bars indicate ±SEM.
Up-Regulation of Amylin Receptor in Brains of Transgenic Mice That Overexpress Aβ
We sought to determine the relationship between expression of two components of the amylin receptor, RAMP3 and CTR, and the amount of Aβ present in the brains of APP-overexpressing transgenic mice (TgCRND8 APP swe/APP+/−V717F), which serve as a surrogate in vivo model of AD.38 Western blot analyses of brains from TgCRND8 mice at 1, 4, and 6 months of age revealed an age-dependent up-regulation of CTR and RAMP3 (Figure 8). Brains of age-matched control mice did not reveal an increase in either amylin receptor components or Aβ. The age-dependent increase in Aβ in TgCRND8 mice relative to control age-matched mice was confirmed by enzyme-linked immunosorbent assay including measurement of soluble Aβ species (see Supplemental Figure S6 at http://ajp.amjpathol.org). Furthermore, the increases in CTR and RAMP3 levels in TgCRND8 were specific to brain regions (the cortex, hippocampus, and basal forebrain) that demonstrate a high amyloid burden, compared with other regions (the brainstem and cerebellum) that are relatively spared of amyloid deposition (Figure 8, see also Supplemental Figures S6 and S7 at http://ajp.amjpathol.org). Immunohistochemical localization CTR was observed in close vicinity of Aβ plaque deposition within the cortex, hippocampus, and basal forebrain of TgCRND8 mice (Figure 9).
Figure 8.

Quantitative analysis of RAMP3 and CTR expression in TgCRND8 mice. Quantification of Western blots for RAMP3, CTR expression in TgCRND8 and control mice at 1, 4, and 6 months of age. Specific brain regions [the cortex, hippocampus, and diagonal band of Broca (DBB)] in 4- and 6-month-old TgCRND8 mice demonstrate an increase in RAMP3 and CTR expression relative to age-matched control mice. Significant difference for transgenic mice compared with age-matched controls within specific brain regions, n = 5 animals, *P < 0.01.
Figure 9.

Immunohistochemical localization of Aβ and CTR in brains of TgCRND8 and control mice. Immunohistochemistry for Aβ (green, A, D, G, J, and M) and CTR (red, B, E, H, K, and N) and overlay of Aβ and CTR staining (C, F, I, L, and O) in sections from the basal forebrain nucleus, diagonal band of Broca (top two rows), and hippocampus and the overlying cortex (bottom two rows). Photomicrographs A, B, C, J, K, and L are from a 6-month-old control mouse. Photomicrographs D, E, F, M, N, and O are from a 6-month-old TgCNDR8 mouse. Higher magnification of Aβ deposits (G), CTR immunostaining (H), and overlay of Aβ and CTR (I) show the close apposition of amyloid to CTR-labeled profiles. Scale bar = 50 μm for G, H, I. Scale bar = 100 μm for remaining panels.
Discussion
Our data demonstrate that Aβ-induced depression of whole-cell currents in HFNs results in membrane depolarization and a consequent increase in excitability of these neurons. Such effects of Aβ appear to be mediated via an inhibition of the calcium-activated potassium currents (BK channels), because in the presence of iberiotoxin, a specific blocker of these channels, Aβ-induced reduction of whole-cell currents was abolished. Importantly, the actions of Aβ under both voltage- and current-clamp conditions could be blocked in the presence of the amylin receptor antagonist AC253. A sustained Aβ-evoked neuronal excitation would be expected to affect a number of cellular processes, including augmented calcium entry into the cell and apoptosis through pathways that are activated on elevation of intracellular levels of this ion. Indeed, in the present study a sustained exposure of HFNs to Aβ demonstrated concentration- and time-dependent apoptotic cell death that was attenuated by blockade of the human amylin receptor.
Aβ-generated neurotoxicity has been shown to be mediated through both caspase-dependent and caspase-independent pathways.35,36,39 As in other cell culture systems, exposure of HFNs to Aβ results in activation of caspases 3, 6, 8, and 9. Our data indicate that blockade of the amylin receptor by the antagonist AC253 attenuates cleavage of these caspases associated with apoptotic cell death. On the other hand, Aβ can activate a caspase-independent apoptotic pathway, where cleavage and translocation of mitochondrial AIF to the nucleus result in nuclear condensation and DNA fragmentation.37,40 In the present study, we observed a similar activation of a caspase-independent pathway in HFNs treated with Aβ. However, HFNs exposed to Aβ, but pretreated with AC253, revealed an attenuation of AIF cleavage and retention of AIF within the mitochondrial compartment. Aβ-induced down-regulation of antiapoptotic signaling pathways, which occurs in parallel with an up-regulation of proapoptotic mediators, was restored to basal levels after pretreatment of HFNs with AC253. This simultaneous attenuation of multiple proapoptotic pathways (and up-regulation of antiapoptotic mediators) by AC253 suggests that the neuroprotective actions of this compound result from blockade of the neurotoxic interaction of Aβ with human amylin receptors localized on the postsynaptic membrane.
A siRNA approach to down-regulate the expression of amylin receptor components (CTR and RAMP3) rendered transfected HFN cultures resistant to Aβ toxicity. Neuroprotection against Aβ toxicity with the siRNA approach was not as robust as with the use of the amylin receptor antagonist AC253. This observation is not surprising, however, considering that Lipofectamine-facilitated transfection rates of primary neuronal cell cultures under optimum conditions are at best in 30%–40% range, a level achieved in our experiments. Therefore, neuroprotection conferred by down-regulation of the amylin receptor in primary cultures of HFNs would be expected to be less effective against Aβ toxicity than is pharmacological blockade of the amylin receptor. Nonetheless, results from siRNA experiments complement our pharmacological data concerning ability of the amylin receptor antagonist AC253 to block both the electrophysiological and neurotoxic effects of Aβ. The precise nature of how AC253 blocks Aβ effects is unclear, as is the case for other receptors that have been postulated as targets for the actions of Aβ. However, the ability to blunt Aβ toxicity by down-regulating amylin receptors located on HFNs and electrophysiological data showing that direct Aβ-evoked responses could be blocked by the amylin receptor antagonist AC253 appear to suggest some form of a direct interaction between Aβ and the postsynaptically located amylin receptors on HFNs. Collectively, these studies point to a central role for the amylin receptor as a target for the characteristic lethal actions of Aβ on human neurons.
An important issue raised by the electrophysiological and cell culture experiments concerns the relationship of the amylin receptor to the overexpression of Aβ that is observed under in vivo conditions, such as in the brains of transgenic mouse models of AD. TgCRND8 APP mice carrying combined APP Swedish (K670M/N671L) and Indiana (V717F) mutations exhibit an aggressive pathology that by 6 months consists of large numbers of core plaques and peculiar floccular diffuse parenchymal deposits.38,41 The present study shows that there is a time-dependent increase in the two amylin receptor components (CTR and RAMP3) relative to age-matched controls that parallels the overexpression of Aβ observed in TgCRND8 APP mice. These findings suggest that the increased levels of Aβ (both in soluble and plaque form) observed in these mice cause deleterious effects through an overexpression of the putative target, the amylin receptor. Thus, an interaction between Aβ and amylin receptors over an extended period of time could result in synaptic disruption, neuronal loss, and the behavioral deficits that are a hallmark of AD.
For such a hypothesis to be tenable, the up-regulation of the amylin receptor would be have to demonstrate a regional specificity within the brain, such that the receptor is preferentially increased only in specific areas demonstrating a large burden of amyloid (ie, the cortex, hippocampus, and basal forebrain, but not the brainstem or cerebellum). Indeed, our observations support this notion, because the levels of amylin receptor markers (CTR and RAMP3) were significantly increased only in the aforementioned areas and, in addition, these correlated with the high levels of Aβ detected in the very same regions. These observations also suggest that the mechanism that elevates amyloid burden in transgenic mice may also signal up-regulation of the amylin receptor.
Numerous epidemiological studies have linked type 2 diabetes mellitus with an increased risk of developing AD.11,42 At a cellular level, the pathophysiological linkage between the two conditions has been based on several avenues of investigation that point to a potential role for insulin signaling and insulin-degrading enzyme in neurodegeneration and cognitive decline.43,44 Accelerated rate of formation and accumulation of advanced glycation end products are observed in both type 2 diabetes mellitus and AD.45,46 Furthermore, RAGE, the receptor for advanced glycation end products, has been shown to promote intraneuronal accumulation of Aβ (and the consequent toxicity attributed to this peptide) and is also implicated in the development of diabetic vascular complications.47 Although the involvement of amylin and its receptor in the development of diabetes has been known for some time,12 the present study provides novel evidence linking the amylin receptor to neurodegenerative mechanisms in human neurons and in a transgenic APP-overexpressing mouse model of AD.
Because Aβ evokes cell death through a number of apoptotic pathways (both caspase-dependent and caspase-independent), neuroprotective strategies aimed at attenuating the apoptotic actions of Aβ are more likely to be effective if directed upstream to activation of these multiple intracellular signaling pathways. A working model summarizing the observations from the present study is presented in Figure 10. Based on this model, AC253 and similar amylin-receptor-based compounds that block the expression of Aβ effects via human amylin may have considerable potential as a treatment approach for AD.
Figure 10.

Schematic summarizing Aβ-amylin receptor interactions. Aβ activation of amylin receptor (CTR and RAMP3 complex) results in depression of calcium-activated potassium conductance (gK,Ca), membrane depolarization, and consequent entry of calcium into the cell. This triggers an activation of caspase-dependent and -independent signaling pathways that converge to cause apoptotic cell death. Blockade of the amylin receptor with the antagonist AC253 results in an inhibition of Aβ-mediated membrane events and apoptotic cell death.
Acknowledgment
We thank Dr. Khem Jhamandas for useful comments and suggestions on the manuscript.
Footnotes
Supported by the Canadian Institutes of Health Research (MOP 93601) and the Canada Research Chairs program.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2010.11.022.
Supplementary Data
β-amyloid (Aβ) and human amylin neurotoxicity in primary cultures of human fetal neurons (HFNs). Histograms depict neuronal viability against varying concentrations of soluble oligomeric form of Aβ1-42 (A) and human amylin (B). Cell viability is measured using the MTT assay. C: Histograms depicting neuronal survival of HFNs, plated at different densities in 96-well plates, in response to application of varying concentrations of Aβ1-42. Error bars indicate ±SEM.
Electrophysiological effects of Aβ on human fetal neurons (HFNs). A: Aβ1-42-evoked reduction of whole-cell outward currents in HFNs is a time-independent response. Aβ1-42 (20 nmol/L) significantly reduced whole-cell outward currents, with applications 10 minutes apart (n = 5) indicating the Aβ-response does not desensitize with repeated applications of the peptide in the same neuron. B: Effects of Aβ1-42 on HFNs are dose-dependent. EC50 is 9 nmol/L. All points of the graph represented an average reduction of the outward current from four HFNs. Error bars indicate ±SEM.
Electron microscopic (EM) and Western blot analysis of Aβ species in solution. A: EM visualization of soluble oligomeric form of Aβ used in both the electrophysiological and the cell culture experiments. B: EM visualization of fibrillar form of Aβ. C: Western blot of soluble oligomeric Aβ probed with antiserum to Aβ shows the presence of trimers and tetramers in addition to monomers of Aβ (lane 1) and composition of the fibrillar Aβ species (lane 2). Note the presence of large fibrillar aggregates in this preparation.
Caspase inhibition protects against Aβ neurotoxicity in cultured human fetal neurons (HFNs). The pan-caspase inhibitor Z-VAD-FMK as well as inhibitors of caspases 3, 6, 8, and 9 increase cell viability. All caspase inhibitors (20 μmol/L) were applied to cultures for 2 hours and cells were then exposed to Aβ1-42 (20 μmol/L) for 48 hours. Data shown are pooled results from 14 wells (of a 96-well plate) for each inhibitor. *Significant difference in neuronal survival, compared with Aβ1-42 treatment, P < 0.05.
Quantitative analysis of amylin receptor inhibition of caspase 3 and 9 activation. A: Aβ1-42 (20 μmol/L) applied alone for 48 hours results in a reduction of pro-caspase 3 activity (on account of its cleavage to a form that induces apoptosis), whereas application of AC253 (10 μmol/L), Aβ42-1, or Aβ1-42 after AC253 pretreatment for 24 hours demonstrates persistence of pro-caspase 3 activity. Pooled data from four experiments. *Significant difference for HFNs treated with AC253 and Aβ1-42, compared with Aβ1-42 alone, P < 0.05. B: Amount of cleaved caspase 9 generated in HFNs exposed to 20 μmol/L oligomeric Aβ1-42 is significantly attenuated in presence of AC253. Pooled data from four experiments. *Significant difference for HFNs treated with AC253 and Aβ1-42, compared with Aβ1-42 alone, P < 0.05.
Aβ expression in TgCRND8 and age-matched control mice. Progressive increase in Aβ levels (measured by enzyme-linked immunosorbent assay) in TgCRND8 mice at 1, 4, and 6 months of age, compared with age-matched control mice. Aβ levels within the cortex, hippocampus and diagonal band of Broca (DBB), but not the brainstem or cerebellum, are significantly elevated in 4- and 6-month-old transgenic mice, compared with controls. *Significant difference for transgenic mice from age-matched controls within specific brain regions, P < 0.01.
Expression of amylin receptor levels in TgCRND8 mice. Western blot showing time-dependent and regional brain expression of protein levels for amylin receptor components, RAMP3 and CTR, in TgCRND8 and control mice. Increasing levels of protein for RAMP3 and CTR in TgCRND8 are detected in the cortex, hippocampus, and diagonal band of Broca (DBB) of 4- and 6-month-old mice, compared with age-matched control mice. Low levels of basal RAMP3 and CTR expression are noted in 1-month-old TgCRND8 and control mice. Note the relative lack of increase in RAMP3 and CTR protein levels within the brainstem and cerebellum. For all blots β-actin was used as a loading control.
References
- 1.Brouwers N., Sleegers K., Van Broeckhoven C. Molecular genetics of Alzheimer's disease: an update. Ann Med. 2008;40:562–583. doi: 10.1080/07853890802186905. [DOI] [PubMed] [Google Scholar]
- 2.Yankner B.A., Lu T. Amyloid beta-protein toxicity and the pathogenesis of Alzheimer disease. J Biol Chem. 2009;284:4755–4759. doi: 10.1074/jbc.R800018200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Price D.L., Wong P.C., Markowska A.L., Lee M.K., Thinakaren G., Cleveland D.W., Sisodia S.S., Borchelt D.R. The value of transgenic models for the study of neurodegenerative diseases. Ann N Y Acad Sci. 2000;920:179–191. doi: 10.1111/j.1749-6632.2000.tb06920.x. [DOI] [PubMed] [Google Scholar]
- 4.Chen X., Walker D.G., Schmidt A.M., Arancio O., Lue L.F., Yan S.D. RAGE: a potential target for Abeta-mediated cellular perturbation in Alzheimer's disease. Curr Mol Med. 2007;7:735–742. doi: 10.2174/156652407783220741. [DOI] [PubMed] [Google Scholar]
- 5.Salminen A., Ojala J., Kauppinen A., Kaarniranta K., Suuronen T. Inflammation in Alzheimer's disease: amyloid-beta oligomers trigger innate immunity defence via pattern recognition receptors. Prog Neurobiol. 2009;87:181–194. doi: 10.1016/j.pneurobio.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 6.Sotthibundhu A., Sykes A.M., Fox B., Underwood C.K., Thangnipon W., Coulson E.J. Beta-amyloid(1-42) induces neuronal death through the p75 neurotrophin receptor. J Neurosci. 2008;28:3941–3946. doi: 10.1523/JNEUROSCI.0350-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parri R.H., Dineley T.K. Nicotinic acetylcholine receptor interaction with beta-amyloid: molecular, cellular, and physiological consequences. Curr Alzheimer Res. 2010;7:27–39. doi: 10.2174/156720510790274464. [DOI] [PubMed] [Google Scholar]
- 8.Mehta T.K., Dougherty J.J., Wu J., Choi C.H., Khan G.M., Nichols R.A. Defining pre-synaptic nicotinic receptors regulated by beta amyloid in mouse cortex and hippocampus with receptor null mutants. J Neurochem. 2009;109:1452–1458. doi: 10.1111/j.1471-4159.2009.06070.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun M.K., Alkon D.L. Links between Alzheimer's disease and diabetes. Drugs Today (Barc) 2006;42:481–489. doi: 10.1358/dot.2006.42.7.973588. [DOI] [PubMed] [Google Scholar]
- 10.Craft S. Insulin resistance and Alzheimer's disease pathogenesis: potential mechanisms and implications for treatment. Curr Alzheimer Res. 2007;4:147–152. doi: 10.2174/156720507780362137. [DOI] [PubMed] [Google Scholar]
- 11.Lin L., Hölscher C. Common pathological processes in Alzheimer disease and type 2 diabetes: a review. Brain Res Rev. 2007;56:384–402. doi: 10.1016/j.brainresrev.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 12.Cooper G.J., Willis A.C., Clark A., Turner R.C., Sim R.B., Reid K.B. Purification and characterization of a peptide from amyloid-rich pancreases of type 2 diabetic patients. Proc Natl Acad Sci USA. 1987;84:8628–8632. doi: 10.1073/pnas.84.23.8628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arispe N., Pollard H.B., Rojas E. Giant multilevel cation channels formed by Alzheimer disease amyloid beta-protein [A beta P-(1-40)] in bilayer membrane. Proc Natl Acad Sci USA. 1993;90:10573–10577. doi: 10.1073/pnas.90.22.10573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.May P.C., Boggs L.N., Fuson K.S. Neurotoxicity of human amylin in rat primary hippocampal cultures: similarity to Alzheimer's disease amyloid-beta neurotoxicity. J Neurochem. 1993;61:2330–2333. doi: 10.1111/j.1471-4159.1993.tb07480.x. [DOI] [PubMed] [Google Scholar]
- 15.Tucker H.M., Rydel R.E., Wright S., Estus S. Human amylin induces “apoptotic” pattern of gene expression concomitant with cortical neuronal atrophy. J Neurochem. 1998;71:506–516. doi: 10.1046/j.1471-4159.1998.71020506.x. [DOI] [PubMed] [Google Scholar]
- 16.Jhamandas J.H., Cho C., Jassar B., Harris K., MacTavish D., Easaw J. Cellular mechanisms for amyloid-beta protein activation of rat cholinergic basal forebrain neurons. J Neurophysiol. 2001;86:1312–1320. doi: 10.1152/jn.2001.86.3.1312. [DOI] [PubMed] [Google Scholar]
- 17.Jhamandas J.H., Harris K.H., Cho C., Fu W., MacTavish D. Human amylin actions on rat cholinergic basal forebrain neurons: antagonism of beta-amyloid effects. J Neurophysiol. 2003;90:3130–3136. doi: 10.1152/jn.01138.2002. [DOI] [PubMed] [Google Scholar]
- 18.Lim Y.A., Ittner L.M., Lim Y.L., Götz J. Human but not rat amylin shares neurotoxic properties with Abeta42 in long-term hippocampal and cortical cultures. FEBS Lett. 2008;582:2188–2194. doi: 10.1016/j.febslet.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 19.Lim Y.A., Rhein V., Baysang G., Meier F., Poljak A., Raftery M.J., Guilhaus M., Ittner L.M., Eckert A., Götz J. Abeta and human amylin share a common toxicity pathway via mitochondrial dysfunction. Proteomics. 2010;10:1621–1633. doi: 10.1002/pmic.200900651. [DOI] [PubMed] [Google Scholar]
- 20.Poyner D.R., Sexton P.M., Marshall I., Smith D.M., Quirion R., Born W., Muff R., Fischer J.A., Foord S.M. International Union of Pharmacology. XXXII. The mammalian calcitonin gene-related peptides, adrenomedullin, amylin, and calcitonin receptors. Pharmacol Rev. 2002;54:233–246. doi: 10.1124/pr.54.2.233. [DOI] [PubMed] [Google Scholar]
- 21.Young A. Receptor pharmacology. Adv Pharmacol. 2005;52:47–65. doi: 10.1016/S1054-3589(05)52003-9. [DOI] [PubMed] [Google Scholar]
- 22.Hay D.L., Christopoulos G., Christopoulos A., Sexton P.M. Amylin receptors: molecular composition and pharmacology. Biochem Soc Trans. 2004;32:865–867. doi: 10.1042/BST0320865. [DOI] [PubMed] [Google Scholar]
- 23.Hay D.L., Christopoulos G., Christopoulos A., Poyner D.R., Sexton P.M. Pharmacological discrimination of calcitonin receptor: receptor activity-modifying protein complexes. Mol Pharmacol. 2005;67:1655–1665. doi: 10.1124/mol.104.008615. [DOI] [PubMed] [Google Scholar]
- 24.Cantarella G., Martinez G., Di Benedetto G., Loreto C., Musumeci G., Prato A., Lempereur L., Matera M., Amico-Roxas M., Bernardini R., Clementi G. Protective effects of amylin on reserpine-induced gastric damage in the rat. Pharmacol Res. 2007;56:27–34. doi: 10.1016/j.phrs.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 25.Morfis M., Tilakaratne N., Furness S.G., Christopoulos G., Werry T.D., Christopoulos A., Sexton P.M. Receptor activity-modifying proteins differentially modulate the G protein-coupling efficiency of amylin receptors. Endocrinology. 2008;149:5423–5431. doi: 10.1210/en.2007-1735. [DOI] [PubMed] [Google Scholar]
- 26.Jhamandas J.H., MacTavish D. Antagonist of the amylin receptor blocks beta-amyloid toxicity in rat cholinergic basal forebrain neurons. J Neurosci. 2004;24:5579–5584. doi: 10.1523/JNEUROSCI.1051-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Power C., McArthur J.C., Nath A., Wehrly K., Mayne M., Nishio J., Langelier T., Johnson R.T., Chesebro B. Neuronal death induced by brain-derived human immunodeficiency virus type 1 envelope genes differs between demented and nondemented AIDS patients. J Virol. 1998;72:9045–9053. doi: 10.1128/jvi.72.11.9045-9053.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stine W.B., Jr, Dahlgren K.N., Krafft G.A., LaDu M.J. In vitro characterization of conditions for amyloid-beta peptide oligomerization and fibrillogenesis. J Biol Chem. 2003;278:11612–11622. doi: 10.1074/jbc.M210207200. [DOI] [PubMed] [Google Scholar]
- 29.White J.A., Manelli A.M., Holmberg K.H., Van Eldik L.J., Ladu M.J. Differential effects of oligomeric and fibrillar amyloid-beta 1-42 on astrocyte-mediated inflammation. Neurobiol Dis. 2005;18:459–465. doi: 10.1016/j.nbd.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 30.Ding X., MacTavish D., Kar S., Jhamandas J.H. Galanin attenuates beta- amyloid (Abeta) toxicity in rat cholinergic basal forebrain neurons. Neurobiol Dis. 2006;21:413–420. doi: 10.1016/j.nbd.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 31.Riediger T., Rauch M., Schmid H.A. Actions of amylin on subfornical organ neurons and on drinking behaviour in rats. Am J Physiol. 1999;276:R514–R521. doi: 10.1152/ajpregu.1999.276.2.R514. [DOI] [PubMed] [Google Scholar]
- 32.Mattson M.P., Cheng B., Davis D., Bryant K., Lieberburg I., Rydel R.E. beta-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci. 1992;12:376–389. doi: 10.1523/JNEUROSCI.12-02-00376.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Afkhami-Goli A., Noorbakhsh F., Keller A.J., Vergnolle N., Westaway D., Jhamandas J.H., Andrade-Gordon P., Hollenberg M.D., Arab H., Dyck R.H., Power C. Proteinase-activated receptor-2 exerts protective and pathogenic cell type-specific effects in Alzheimer's disease. J Immunol. 2007;179:5493–5503. doi: 10.4049/jimmunol.179.8.5493. [DOI] [PubMed] [Google Scholar]
- 34.Ryan S.D., Whitehead S.N., Swayne L.A., Moffat T.C., Hou W., Ethier M., Bourgeois A.J., Rashidian J., Blanchard A.P., Fraser P.E., Park D.S., Figeys D., Bennett S.A. Amyloid-beta42 signals tau hyperphosphorylation and compromises neuronal viability by disrupting alkylacylglycerophosphocholine metabolism. Proc Natl Acad Sci USA. 2009;106:20936–20941. doi: 10.1073/pnas.0905654106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Villa P., Kaufmann S.H., Earnshaw W.C. Caspases and caspase inhibitors. Trends Biochem Sci. 1997;22:388–393. doi: 10.1016/s0968-0004(97)01107-9. [DOI] [PubMed] [Google Scholar]
- 36.Yuan J., Yankner B.A. Apoptosis in the nervous system. Nature. 2000;407:802–809. doi: 10.1038/35037739. [DOI] [PubMed] [Google Scholar]
- 37.Cregan S.P., Dawson V.L., Slack R.S. Role of AIF in caspase-dependent and caspase-independent cell death. Oncogene. 2004;23:2785–2796. doi: 10.1038/sj.onc.1207517. [DOI] [PubMed] [Google Scholar]
- 38.Chishti M.A., Yang D.S., Janus C., Phinney A.L., Horne P., Pearson J., Strome R., Zuker N., Loukides J., French J., Turner S., Lozza G., Grilli M., Kunicki S., Morissette C., Paquette J., Gervais F., Bergeron C., Fraser P.E., Carlson G.A., George-Hyslop P.S., Westaway D. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein. J Biol Chem. 2001;276:21562–21570. doi: 10.1074/jbc.M100710200. [DOI] [PubMed] [Google Scholar]
- 39.Bredesen D.E., Rao R.V., Mehlen P. Cell death in the nervous system. Nature. 2006;443:796–802. doi: 10.1038/nature05293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krantic S., Mechawar N., Reix S., Quirion R. Apoptosis-inducing factor: a matter of neuron life and death. Prog Neurobiol. 2007;81:179–196. doi: 10.1016/j.pneurobio.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 41.Van Vickle G.D., Esh C.L., Kalback W.M., Patton R.L., Luehrs D.C., Kokjohn T.A., Fifield F.G., Fraser P.E., Westaway D., McLaurin J., Lopez J., Brune D., Newel A.J., Poston M., Beach T.G., Roher A.E. TgCRND8 amyloid precursor protein transgenic mice exhibit an altered gamma-secretase processing and an aggressive, additive amyloid pathology subject to immunotherapeutic modulation. Biochemistry. 2007;46:10317–10327. doi: 10.1021/bi700951u. [DOI] [PubMed] [Google Scholar]
- 42.Götz J., Ittner L.M., Lim Y.A. Common features between diabetes mellitus and Alzheimer's disease. Cell Mol Life Sci. 2009;66:1321–1325. doi: 10.1007/s00018-009-9070-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Farris W., Mansourian S., Chang Y., Lindsley L., Eckman E.A., Frosch M.P., Eckman C.B., Tanzi R.E., Selkoe D.J., Guenette S. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci USA. 2003;100:4162–4167. doi: 10.1073/pnas.0230450100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gasparini L., Xu H. Potential roles of insulin and IGF-1 in Alzheimer's disease. Trends Neurosci. 2003;26:404–406. doi: 10.1016/S0166-2236(03)00163-2. [DOI] [PubMed] [Google Scholar]
- 45.Münch G., Schinzel R., Loske C., Wong A., Durany N., Li J.J., Vlassara H., Smith M.A., Perry G., Riederer P. Alzheimer's disease—synergistic effects of glucose deficit, oxidative stress and advanced glycation endproducts. J Neural Transm. 1998;105:439–461. doi: 10.1007/s007020050069. [DOI] [PubMed] [Google Scholar]
- 46.Yamagishi S., Takeuchi M., Inagaki Y., Nakamura K., Imaizumi T. Role of advanced glycation end products (AGEs) and their receptor (RAGE) in the pathogenesis of diabetic microangiopathy. Int J Clin Pharmacol Res. 2003;23:129–134. [PubMed] [Google Scholar]
- 47.Ledesma M.D., Bonay P., Colaço C., Avila J. Analysis of microtubule-associated protein tau glycation in paired helical filaments. J Biol Chem. 1994;269:21614–21619. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
β-amyloid (Aβ) and human amylin neurotoxicity in primary cultures of human fetal neurons (HFNs). Histograms depict neuronal viability against varying concentrations of soluble oligomeric form of Aβ1-42 (A) and human amylin (B). Cell viability is measured using the MTT assay. C: Histograms depicting neuronal survival of HFNs, plated at different densities in 96-well plates, in response to application of varying concentrations of Aβ1-42. Error bars indicate ±SEM.
Electrophysiological effects of Aβ on human fetal neurons (HFNs). A: Aβ1-42-evoked reduction of whole-cell outward currents in HFNs is a time-independent response. Aβ1-42 (20 nmol/L) significantly reduced whole-cell outward currents, with applications 10 minutes apart (n = 5) indicating the Aβ-response does not desensitize with repeated applications of the peptide in the same neuron. B: Effects of Aβ1-42 on HFNs are dose-dependent. EC50 is 9 nmol/L. All points of the graph represented an average reduction of the outward current from four HFNs. Error bars indicate ±SEM.
Electron microscopic (EM) and Western blot analysis of Aβ species in solution. A: EM visualization of soluble oligomeric form of Aβ used in both the electrophysiological and the cell culture experiments. B: EM visualization of fibrillar form of Aβ. C: Western blot of soluble oligomeric Aβ probed with antiserum to Aβ shows the presence of trimers and tetramers in addition to monomers of Aβ (lane 1) and composition of the fibrillar Aβ species (lane 2). Note the presence of large fibrillar aggregates in this preparation.
Caspase inhibition protects against Aβ neurotoxicity in cultured human fetal neurons (HFNs). The pan-caspase inhibitor Z-VAD-FMK as well as inhibitors of caspases 3, 6, 8, and 9 increase cell viability. All caspase inhibitors (20 μmol/L) were applied to cultures for 2 hours and cells were then exposed to Aβ1-42 (20 μmol/L) for 48 hours. Data shown are pooled results from 14 wells (of a 96-well plate) for each inhibitor. *Significant difference in neuronal survival, compared with Aβ1-42 treatment, P < 0.05.
Quantitative analysis of amylin receptor inhibition of caspase 3 and 9 activation. A: Aβ1-42 (20 μmol/L) applied alone for 48 hours results in a reduction of pro-caspase 3 activity (on account of its cleavage to a form that induces apoptosis), whereas application of AC253 (10 μmol/L), Aβ42-1, or Aβ1-42 after AC253 pretreatment for 24 hours demonstrates persistence of pro-caspase 3 activity. Pooled data from four experiments. *Significant difference for HFNs treated with AC253 and Aβ1-42, compared with Aβ1-42 alone, P < 0.05. B: Amount of cleaved caspase 9 generated in HFNs exposed to 20 μmol/L oligomeric Aβ1-42 is significantly attenuated in presence of AC253. Pooled data from four experiments. *Significant difference for HFNs treated with AC253 and Aβ1-42, compared with Aβ1-42 alone, P < 0.05.
Aβ expression in TgCRND8 and age-matched control mice. Progressive increase in Aβ levels (measured by enzyme-linked immunosorbent assay) in TgCRND8 mice at 1, 4, and 6 months of age, compared with age-matched control mice. Aβ levels within the cortex, hippocampus and diagonal band of Broca (DBB), but not the brainstem or cerebellum, are significantly elevated in 4- and 6-month-old transgenic mice, compared with controls. *Significant difference for transgenic mice from age-matched controls within specific brain regions, P < 0.01.
Expression of amylin receptor levels in TgCRND8 mice. Western blot showing time-dependent and regional brain expression of protein levels for amylin receptor components, RAMP3 and CTR, in TgCRND8 and control mice. Increasing levels of protein for RAMP3 and CTR in TgCRND8 are detected in the cortex, hippocampus, and diagonal band of Broca (DBB) of 4- and 6-month-old mice, compared with age-matched control mice. Low levels of basal RAMP3 and CTR expression are noted in 1-month-old TgCRND8 and control mice. Note the relative lack of increase in RAMP3 and CTR protein levels within the brainstem and cerebellum. For all blots β-actin was used as a loading control.