

Abstract
Chemokines (i.e. chemoattractant cytokines) are a family of small secreted molecules that mediate leukocyte migration. It is becoming increasingly more evident that chemokines play an integral role in the initiation of a specific immune response. With respect to cancer, chemokines are being studied for both their role in tumor biology and as promising immunotherapy candidates. We review several areas of chemokine importance in tumor immunity and discuss the experimental evidence that is leading to the clinical use of this cytokine family in new treatment approaches for patients with cancer.
Keywords: chemokines, chemokine receptors, antitumor immunity, immunotherapy, gene therapy, cancer vaccines, dendritic cells, infiltrating T cells
Over the past few years there has been growing interest in chemokines (i.e. chemoattractant cytokines), which are small protein molecules involved in immune and inflammatory responses. Chemokines are known to direct leukocyte trafficking to areas of injury as well as to locations where primary immune responses are initiated (e.g., secondary lymphoid tissues such as lymph nodes, spleen, Peyer’s patches and tonsils) (1, 2). There are four classes of chemokine molecules (C, CC, CXC, and CX3C) that are named for the number and location of cysteine residues on the amino terminus of the protein. To date, the total number of distinct molecules comprising these four classes is 48 (i.e. CCL1-28; CXCL1-17; XCL1 and 2; CX3CL1; Table 1).
Table 1.
List of Chemokines and Chemokine Receptors
| CC chemokines | ||||
|---|---|---|---|---|
| Name | Gene | Other name(s) | Receptor | Uniprot |
| CCL1 | Scya1 | I-309, TCA-3 | CCR8 | |
| CCL2 | Scya2 | MCP-1 | CCR2, CCR2 | P13500 |
| CCL3 | Scya3 | MIP-1a | CCR1 | P10147 |
| CCL4 | Scya4 | MIP-1β | CCR1, CCR5 | P13236 |
| CCL5 | Scya5 | RANTES | CCR5 | P13501 |
| CCL6 | Scya6 | C10, MRP-2 | CCR1 | P27784 |
| CCL7 | Scya7 | MARC, MCP-3 | CCR2 | P80098 |
| CCL8 | Scya8 | MCP-2 | CCR1, CCR2B, CCR5 | P80075 |
| CCL9/CCL10 | Scya9 | MRP-2, CCF18, MIP-1? | CCR1 | P51670 |
| CCL11 | Scya11 | Eotaxin | CCR2, CCR3, CCR5 | P51671 |
| CCL12 | Scya12 | MCP-5 | Q62401 | |
| CCL13 | Scya13 | MCP-4, NCC-1, Ckβ10 | CCR2, CCR3, CCR5 | Q99616 |
| CCL14 | Scya14 | HCC-1, MCIF, Ckβ1, NCC-2, CCL | CCR1 | Q16627 |
| CCL15 | Scya15 | Leukotactin-1, MIP-5, HCC-2, NCC-3 | CCR1, CCR3 | Q16663 |
| CCL16 | Scya16 | LEC, NCC-4, LMC, Ckβ12 | CCR1, CCR2, CCR5, CCR8 | O15467 |
| CCL17 | Scya17 | TARC, dendrokine, ABCD-2 | CCR4 | Q92583 |
| CCL18 | Scya18 | PARC, DC-CK1, AMAC-1, Ckβ7, MIP-4 | P55774 | |
| CCL19 | Scya19 | ELC, Exodus-3, Ckβ11 | CCR7 | Q99731 |
| CCL20 | Scya20 | LARC, Exodus-1, Ckβ4 | CCR6 | P78556 |
| CCL21 | Scya21 | SLC, 6Ckine, Exodus-2, Ckβ9, TCA-4 | CCR7 | O00585 |
| CCL22 | Scya22 | MDC, DC/β-CK | CCR4 | O00626 |
| CCL23 | Scya23 | MPIF-1, Ckβ8, MIP-3, MPIF-1 | CCR1 | P55773 |
| CCL24 | Scya24 | Eotaxin-2, MPIF-2, Ckβ6 | CCR3 | O00175 |
| CCL25 | Scya25 | TECK, Ckβ15 | CCR9 | O15444 |
| CCL26 | Scya26 | Eotaxin-3, MIP-4a, IMAC, TSC-1 | CCR3 | Q9Y258 |
| CCL27 | Scya27 | CTACK, ILC, Eskine, PESKY, skinkine | CCR10 | Q9Y4X3 |
| CCL28 | Scya28 | MEC | CCR3, CCR10 | Q9NRJ3 |
| CXC chemokines | ||||
|---|---|---|---|---|
| Name | Gene | Other name(s) | Receptor | Uniprot |
| CXCL1 | Scyb1 | Gro-a, GRO1, NAP-3, KC | CXCR2 | P09341 |
| CXCL2 | Scyb2 | Gro-β, GRO2, MIP-2a | CXCR2 | P19875 |
| CXCL3 | Scyb3 | Gro-?, GRO3, MIP-2β | CXCR2 | P19876 |
| CXCL4 | Scyb4 | PF-4 | CXCR3B | P02776 |
| CXCL5 | Scyb5 | ENA-78 | CXCR2 | P42830 |
| CXCL6 | Scyb6 | GCP-2 | CXCR1, CXCR2 | P80162 |
| CXCL7 | Scyb7 | NAP-2, CTAPIII, β-Ta, PEP | P02775 | |
| CXCL8 | Scyb8 | IL-8, NAP-1, MDNCF, GCP-1 | CXCR1, CXCR2 | P10145 |
| CXCL9 | Scyb9 | MIG, CRG-10 | CXCR3 | Q07325 |
| CXCL10 | Scyb10 | IP-10, CRG-2 | CXCR3 | P02778 |
| CXCL11 | Scyb11 | I-TAC, β-R1, IP-9 | CXCR3 | O14625 |
| CXCL12 | Scyb12 | SDF-1, PBSF | CXCR4 | P48061 |
| CXCL13 | Scyb13 | BCA-1, BLC | CXCR5 | O43927 |
| CXCL14 | Scyb14 | BRAK, bolekine | O95715 | |
| CXCL15 | Scyb15 | Lungkine, WECHE | Q9WVL7 | |
| CXCL16 | Scyb16 | SRPSOX | Q9H2A7 | |
| CXCL17 | VCC-1 | DMC, VCC-1 | Q6UXB2 | |
| C chemokines | ||||
|---|---|---|---|---|
| Name | Gene | Other name(s) | Receptor | Uniprot |
| XCL1 | Scyc1 | Lymphotactin a, SCM-1a, ATAC | XCR1 | P47992 |
| XCL2 | Scyc2 | Lymphotactin β, SCM-1β | XCR1 | Q9UBD3 |
| CX3C chemokines | ||||
|---|---|---|---|---|
| Name | Gene | Other name(s) | Receptor | Uniprot |
| CX3CL1 | Scyd1 | Fractalkine, Neurotactin, ABCD-3 | CX3CR1 | P78423 |
These molecules communicate with their target cells via G-protein coupled receptors that are pertussis toxin sensitive. Different chemokines act on different leukocyte populations, thereby modulating the influx of immune effector cells to the area in question based on the needs of the particular situation. In vivo, secreted chemokines form a concentration gradient, and attracted immune cells move through the gradient towards the higher concentration of chemokine.
CHEMOKINES AND THE ANTITUMOR IMUNE RESPONSE
Various chemokines have been identified that have profound activities on the antitumor immune response. Forced expression of the tumor necrosis factor superfamily member LIGHT in the tumor environment can induce substantial infiltration of naïve T cells that correlates with an upregulation of both chemokine (notably, CCL21) production and expression of adhesion molecules (3). The arriving T cells become primed inside the LIGHT-expressing tumors, leading to eradication of established tumors at both local and distal sites. As other examples, tumor-derived, heat shock protein 70 can initiate antitumor immunity by inducing chemokine production (4), IL-12 produced by dendritic cells (DC) can augment CD8+ T cell activation through the production of CCL1 and CCL17 (5), CCL20 can regulate the migration of inflammatory and regulatory T cells (6), CCL2 can mediate tumor tropism of adoptively transferred T cells (7), CXCL9 induced by IFN-γ production by tumor cells is critical for T cell-mediated suppression of cutaneous tumors (8), CXCL14 expression in tumor cells can cause attraction of DC in vivo (9), CXCL12 can mediate the migration of cytotoxic T cells (CTL) toward melanoma resulting in tumor regression (10), CXCL9 combined with systemic IL-2 can inhibit tumor growth by increased intratumoral infiltration of CXCR3+ mononuclear cells (11), radiation-induced CXCL16 release by breast cancer cells can attract effector T cells and elicit tumor regression (12), and therapeutic T cells can induce tumor-directed chemotaxis of innate immune cells through tumor-specific secretion of CCL3, CCL4, and CCL5 (13). Injection of CCL16 and CCL20 into established murine tumors can inhibit their growth and prolong survival of treated animals by eliciting T cell specific immunity (14). In addition, administration of the immunostimulatory cytokines IL-2 and IL-12 can enhance antitumor effects generated by XCL1 and CXCL10, respectively (14).
Early work investigating the idea of using chemokines to induce antitumor immunity involved stably transducing tumors to produce them. The first report of this approach is one by us in 1996 in which antitumor immunity was successfully generated when tumors were transduced to produce RANTES (Table 2), a CC chemokine denoted CCL5 (Table 1; 15). This effect was attributable, at least partially, to the recruitment of monocytes and T cells to the tumor site. Similar effects of lymphocyte effector recruitment have been observed subsequently by other investigators using Mig (CXCL9), and Lymphotactin (XCL1) among others. At least two shortcomings of this approach, however, are that it is dependent on the creation of a gene-modified tumor in order to initiate an antitumor response, which first requires removal and manipulation of an often limited number of viable tumor cells ex vivo, and relies entirely on the inherent immunogenicity of that tumor for successful immune priming.
Table 2.
Rantes-Secreting MCA-205 (WP4) Tumors Fail to Grow Progressively
| Tumor transfectantsa | RANTES-secreting | Tumor incidence |
|
|---|---|---|---|
| Day 21 | Day 51 | ||
| WP4-1 | + | 0/6b (0 ± 0)c | 0/6 (0 ± 0) |
| WP4-2 | − | 6/6 (9 ± 1) | 6/6 (16 ± 2) |
| WP4-3 | − | 6/6 (7 ± 1) | 6/6 (11 ± 1) |
| WP4-12 | + | 0/6 (0 ± 0) | 0/6 (0 ± 0) |
| WP4-NEO | − | 6/6 (8 ± 1) | 6/6 (14 ± 1) |
Injected SQ at 1 × 107 cells/site.
Number with tumor/total.
Mean tumor diameter (mm ± SEM).
CHEMOKINES AND DENDRITIC CELL-BASED VACCINES
Ex vivo generated DC in both mouse and humans have very limited movement from subcutaneous or intradermal injection sites to locally draining lymph node(s) and essentially none to spleen (16, 17). This limitation is considered one of the significant weaknesses in the use of DC-based vaccines to date. It is also clear that the intravenous route of administration of DC has proven ineffective to target multiple peripheral lymphoid organs. Most DC administered by this route appear to be trapped rapidly in the capillaries of the lungs, in the spleen, and in the liver where the DC tend to be cleared. Immunization by this route is generally inadequate and some investigators have abandoned the intravenous delivery of DC both in animal studies and in human clinical trials. Recently, the direct intranodal delivery of antigen-loaded DC has gained much favor, as this route appears to be somewhat superior for inducing immune responses compared to the subcutaneous or intradermal route (18–20). However, it is logistically and technically impractical to deliver a large number of DC to a single lymph node, especially in the setting of chemotherapy-induced shrinkage, as well as to target multiple lymph nodes by the current methodology.
It has been shown that limited infiltration of exogenous DC and naïve T cells restricts immune responses in peripheral lymph nodes (21). Rather than focusing on attempts to deliver greater numbers of ex vivo generated DC to lymph nodes, new strategies are being employed that deliver immune cells to the site(s) of administered DC through the use of chemokines.
We became interested in CCL21 (also denoted secondary lymphoid tissue chemokine or SLC, Exodus-2, thymus-derived chemotactic agent 4, 6CKine), which is a CC chemokine found in the high endothelial venules of the lymph node for the treatment of solid tumors, particularly melanoma and lung cancer (22–26). It was initially reported to be a chemoattractant specifically for naïve T lymphocytes and DC. In 2000, we reported for the first time that intratumoral injection of recombinant CCL21 could lead to potent immune-dependent antitumor responses in preclinical models of lung cancer (27). In two murine lung cancer models, the intratumoral administration of recombinant CCL21 mediated T cell-dependent antitumor responses. CCL21-mediated antitumor responses were lymphocyte dependent as evidenced by the fact that the therapy did not alter tumor growth in SCID mice. In immunocompetent mice, intratumoral CCL21 injection led to a significant increase in CD4+ and CD8+ T lymphocytes and DC infiltrating both the tumor and draining lymph nodes. Studies performed in CD4 and CD8 gene knockout mice revealed a direct therapeutic requirement for both CD4+ and CD8+ T cell subsets for CCL21-mediated tumor regression (27).
By genetically modifying mouse B16 melanoma lysate-pulsed DC (TL-DC) to produce CCL21 (Figure 1), we were able to specifically recruit naïve T cells to the site of TL-DC injection in the skin by creating a new “lymph node-like” structure that was, importantly, functional (i.e. the TL-DC served as antigen presenting cells to “educate” the large numbers of arriving naïve T cells that were recruited to the site by the local production of CCL21) (Figures 2, 3, 4, 5; 22, 23). In this case, “structure” is defined histologically by the dense accumulation of immune cells with the presence of DC as APC, without a definable capsule and without HEVs (high endothelial venules). When naïve T cells encountered TL-DC at the vaccination site, the TL-DC interacted with the naïve T cells through costimulatory and MHC Class I and II molecules, thus initiating a primary immune response that then created a powerful systemic antitumor immunity (as the “educated” T cells egressed from the local site and systemically disseminated through the body’s peripheral lymphoid system and blood) that caused regression of local tumor at the skin site in addition to metastatic disease at distant, visceral sites (Figures 6, 7). Thus, CCL21 could both induce antitumor responses and enhance the antitumor immunity elicited by TL-DC in vivo. Also, direct administration of DC genetically modified to express CCL21 into growing tumors themselves could result in a substantial, sustained influx of T cells within the mass with only a transient increase in T cell numbers in the draining lymph node (DLN). TL-DC were retained at the tumor site with only a very small percentage trafficking to the DLN. The T cells infiltrating the tumor mass expressed the activation marker CD25 within 24 hours and developed IFN-gamma-secreting function specifically to the tumor within 7 days as tumor growth became inhibited. Importantly, similar results were obtained in lymphotoxin-alpha gene knock out (Ltα −/−) mice, which completely lacked peripheral lymph nodes (Figure 8). These data demonstrated for the first time that effective T cell priming could occur extranodally and result in measurable, enhanced antitumor effects in vivo, through creation of new, functional “lymph node-like” structures. This latter finding has relevance to the recent, intriguing finding of ectopic lymph nodes present within human solid tumor masses that appear to correlate with better patient prognosis, which is further mentioned below (Figures 9, 10; 28–30).
Figure 1.

Genetic modification of DC to produce SLC/CCL21. DC were harvested from 4-to 6-day-old bone marrow cultures and infected with adenoviral vectors encoding for either CCL21 or green fluorescent protein (GFP). Supernatants were harvested 18 hr after infection and were used on the bottom chamber of a 24-well plate microchemotaxis assay; CD4+ T cells from splenocytes served as responders. Recombinant CCL21 (10–5000 ng/ml) was used to generate a standard curve from which the effective concentration of CCL21 in the cultured supernatants was determined. Duplicate to quadruplicate samples were run for each supernatant tested. Data are presented as the means of the amount (in ng) of CCL21 produced in 18 hr by 1× 106 cells and is cumulative of nine separate infections; bars, SE.
Figure 2.

CD4+ and CD8+ T cells migrate to the injection sites containing SLC/CCL21-expressing DC. Mice received injections subcutaneously (s.c.) with 1 × 106 tumor lysate-pulsed DC expressing either GFP (□) or SLC/CCL21 ( ). Three days after injection, 1.5 × 1.5-cm sections of skin were harvested, minced, and digested in collagenase, DNase I, and hyaluronidase to obtain a single cell suspension (1 × 106 cells/ml). Polystyrene beads (Cf = 5 × 105 beads/ml) were added to enumerate migrating T cells. T cell subsets were analyzed by FACS. Data are presented as the mean numbers of migrating cells from four (GFP) and five (SLC/CCL21) mice; bars, SE. *, P < 0.05 by Student’s t test.
). Three days after injection, 1.5 × 1.5-cm sections of skin were harvested, minced, and digested in collagenase, DNase I, and hyaluronidase to obtain a single cell suspension (1 × 106 cells/ml). Polystyrene beads (Cf = 5 × 105 beads/ml) were added to enumerate migrating T cells. T cell subsets were analyzed by FACS. Data are presented as the mean numbers of migrating cells from four (GFP) and five (SLC/CCL21) mice; bars, SE. *, P < 0.05 by Student’s t test.
Figure 3.

Retention of gene-modified DC in treated tumors. Mice bearing 6-day s.c. B16 tumors were injected with 1 × 106 genetically modified DC that had been labeled with PKH26 dye prior to injection. Tumors were harvested 24 hr after injection, stained with FITC-conjugated anti-CD11c, and analyzed for the presence of labeled cells by flow cytometry. Cell percentages were quantified. Bars, SE.
Figure 4.

Detection of recently activated T cells within treated tumors. Tumors were enzymatically digested and analyzed for presence of CD25 in the CD4+ (A) and CD8+ (B) T cell subsets with PE-conjugated anti-CD25- and Cy-Chrome-conjugated antibodies. Analysis was carried out in the CD11b−/B220− population, and cell numbers were normalized to the tumor volume. *, **, and ***, at least P < 0.05, P < 0.01, and P < 0.001 for DC-SLC/CCL21 versus all others groups, respectively. n = 4–6 mice/time points; bars, SE.
Figure 5.

Enumeration of the IFN-γ-producing T cells within treated tumors. Intracellular IFN-γ cytokine staining in the CD4+ (A) and CD8+ (B) T cell subsets was performed on tumor samples. Cell numbers were normalized to tumor volume and presented as cells/mm3. ** and ***, at least P < 0.01 and P < 0.001 for DC-SLC/CCL21 versus all other groups, respectively. n = 4 mice/time point. Numbers above columns for DC-SLC/CCL21 represent the percentage (bars, SE) of CD4+(A) or CD8+(B) T cells producing IFN-γ.
Figure 6.

Immunization with tumor lysate-pulsed, SLC/CCL21 gene-modified DC elicit antitumor effect. Mice bearing 6-day s.c. B16-BL6 tumors in the right flank were immunized in the left flank twice on days 6 and 13 with 5 × 105 DC that had been infected with adenovirus encoding SLC/CCL21 (●) or GFP (○) and pulsed for 18 hr with B16 cell lysate. Control mice received injections of HBSS alone (□). Tumor size was measured. Bars, SE.
Figure 7.

Treatment of established B16 melanoma with rSLC/CCL21 and gene-modified DC. Tumors were established in B6 mice by s.c. injection with 1 × 105 viable cells. Six days after tumor challenge, mice were treated with daily injections of rSLC/CCL21 (3 μg/dose) from days 6–10 or 1 × 106 gene-modified DC on days 6 and 9. Tumors were monitored for growth by perpendicular diameter measurement. Tumor volumes (means; bars, SE) are presented in mm3. *, P < 0.05 for DC-βgal and rSLC versus PBS; **, P < 0.01 for DC-SLC/CCL21 versus all other groups. N = 6–10 mice/time point.
Figure 8.

Priming and differentiation of effector T cells within treated tumors in the absence of LN. B16 tumors were established in either B6 or Ltα−/− mice after injection of 1 × 105 tumor cells. Gene-modified DC were given on days 6 and 9, and tumors were harvested on day 14 after tumor challenge. Intracellular staining in IFN-γ was performed for CD4+ (A) and CD8+ (B) T cells. The numbers in the upper right-hand quadrant of each dot plot represent the average percentage of T cells producing IFN-γ (n = 3 mice).
Figure 9.

Ectopic lymph node structures in lung adenocarcinoma demonstrating B cell follicles with T cell marginal zones by immunohistochemistry.
Figure 10.

Ectopic lymph node structures in colorectal cancer demonstrating a B cell follicle with T cell marginal zone by immunohistochemistry.
Because the levels of CCL21 required for immune modulation of the tumor microenvironment for tumor rejection can be achieved by using CCL21 transduced DC as the transfer vehicle, we also evaluated intratumoral injection of DC overexpressing CCL21 in a transplantable and a spontaneous bronchoalveolar cell carcinoma model of lung cancer (31, 32). We found that in both these models, CCL21 gene-modified DC could generate systemic antitumor responses and confer tumor immunity. Intratumoral administration of DC expressing CCL21 orchestrated the recruitment and activation of T cell effectors that mediated antitumor responses as those seen in our B16 melanoma work. Moreover, these studies demonstrated that elaboration of CCL21 in the tumors by DC promotes the CXCR3/CXCR3 ligand efferent arm of the immune response for the modulation of antitumor activity; i.e. neutralization of the CXCR3 ligands CXCL9 or CXCL10 inhibited the antitumor responses (Figure 11; 31). The potent antitumor properties demonstrated by administered DC overexpressing CCL21 have provided the rationale for prompting us to evaluate this strategy in the regulation of tumor immunity and its clinical use in patients with advanced lung cancer and melanoma, as mentioned below.
Figure 11.
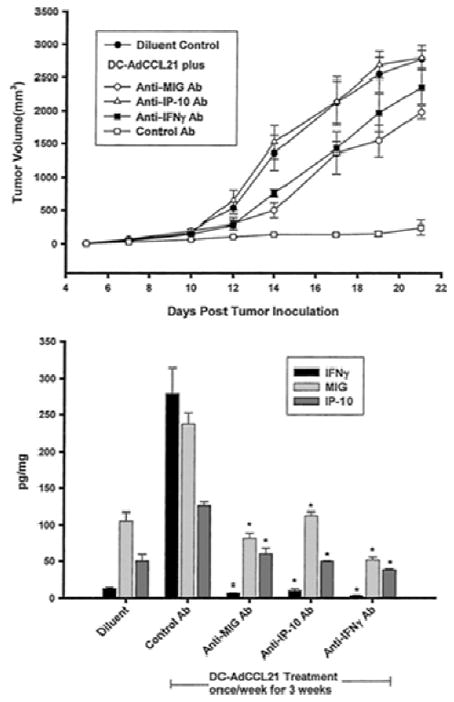
DC-AdCCL21-mediated antitumor responses require IFN-γ, MIG/CXCL9, and IP-10/CXCL10. Upper, L1C2 tumors (1.5 × 105 cells) were implanted in BALB/c mice. Five days after tumor implantation, mice were treated intratumorally with DC-AdCCL21 once a week for 3 weeks. One day before DC-AdCCL21 administration, mice were given the respective cytokine antibody by i.p. injection. The antibodies were administered three times per week. Antibodies to IP-10/CXCL10, MIG/CXCL9, and IFN-γ inhibited the antitumor efficacy of DC-AdCCL21 (P < 0.01 for anti-IFN-γ and anti-MIG/CXCL9; P < 0.001 for anti-IP-10/CXCL10 compared with the control antibody treated group; n = 8 mice group). Lower, DC-AdCCL21-treated mice had a significant induction in IFN-γ, MIG/CXCL9, and IP-10/CXCL10 at the tumor site compared with diluent-treated control tumor-bearing mice (P < 0.001). Assessment of cytokine production at the tumor site of DC-AdCCL21-treated mice receiving anti-IFN-γ, anti-MIG/CXCL9, and anti-IP-10/CXCL10 showed an interdependence of IFN-γ, MIG/CXCL9, and IP-10/CXCL10; neutralization of any one of these cytokines in vivo caused a concomitant decrease in all three cytokines (*, P < 0.01 compared with the control antibody treated group). Results are expressed as pg/mg of total protein. Total protein was determined by the Bradford assay (n = 8 mice per group); bars, ±SE.
Other investigators have embarked on studies to expand the value of CCL21 in tumor immunotherapy applications. Solheim’s group used CCL21 as an effective surgical neoadjuvant for treatment of murine mammary tumors (33). Expression of CCL21 by gene-modified murine TRAMPC2 could result in inhibition of primary prostate tumor growth and metastases (34). Similar results were reported in a B16-F10 melanoma model (35). In all these cases, the enhanced antitumor effects observed were correlated with CCL21’s chemotactic activity on lymphocytes and APC. Other investigators have recently published on biologic activities of CCL21, outside of chemoattraction, for enhanced antitumor effects, which include inducing apoptosis resistance in DC (36), improving efficacy of adoptive immunotherapy by promoting the survival and cytotoxic activity of transferred T cells (37), and enhancing tumor sensitivity to subsequent chemotherapy (38). Curiously, a recent report has suggested that B16-F10 tumor expression of CCL21 can, in contrast, induce both lymphoid-like stroma and immune escape by tumors that express this chemokine (39). In our opinion, the authors did not adequately reconcile the mounting literature consistently showing that CCL21 can cause substantial tumor regression and enhance antitumor immunity. Moreover, in this study, control tumors and CCL21-overexpressors curiously resulted in similar chemotaxis in vitro of cells that should be CCL21 responsive. And, in our hands, we have not found B16-F10 melanoma to make constitutive CCL21 or have mRNA expression using highly sensitive detection assays nor have we observed appreciable influx/accumulation of CD4+ CD25+ FoxP3+ T regulatory cells in vivo into B16 melanomas gene-modified to secrete CCL21, as reported in the Shields and colleagues’ study. Given these discordant results, it is conceivable that the selected and manipulated tumors in the Shields et al. work may be attributed to multiple modifications introduced in addition to just the overexpression of CCL21.
HUMAN CHEMOKINE STUDIES
In melanoma, we have genetically modified human TL-DC to secrete human CCL21 that, similar to our murine studies, could potently recruit naïve human CD4+ and CD8+ T cells (in this instance in vitro, Figure 12; 24, 40). Importantly, we also showed for the first time that TL-DC secreting the CCL21 could significantly enhance the level/number of tumor antigen-specific T cells to at least two, specific melanoma peptides [i.e. MART-1 (Figure 13) and gp100]. Thus, TL-DC producing CCL21 served as a vehicle for both recruiting naïve T cells and enhancing the production of tumor-specific T cells. These in vitro data in human have provided the feasibility for an ongoing clinical trial (in collaboration with the NCI-RAID program) in melanoma patients that we are conducting at the Moffitt Cancer Center. In this regard, we have embarked on a phase I clinical trial to assess the toxicity, immune responses and anti-tumor clinical responses in HLA-A*0201 positive patients with chemotherapy-naïve metastatic melanoma receiving escalating doses of adenoviral CCL21-transduced DC matured ex vivo with a cytokine cocktail and pulsed with MART-1/gp100/NY-ESO-1 class I peptides and KLH. In this study, patients are receiving the vaccine intradermally at multiple sites. To date, 12 patients (the first two of three dose cohorts) have been treated and early results show -indicative of the known chemotactic selectivity of CCL21 -the accumulation of CD3+ T cells, but not CD56+ NK cells or CD19+ B cells in biopsies taken of one of several injection sites (Figure 14).
Figure 12.

Functional SLC/CCL21 is produced by AdSLC/CCL21-transduced human DC. Supernatants from 1 × 106 AdSLC/CCL21-transduced DC at 48 hr following infection at varying numbers of viral particles per cell (MOI) were placed in the bottom chamber of a microchemotaxis assay, and human T cells were allowed to migrate through a porous membrane in response to SLC/CCL21 present in the lower chamber. SLC/CCL21 concentrations in the samples were extrapolated from a standard curve generated by the migration of human T cells in response to known concentrations of human rSLC/CCL21. T cells migrate in response to supernatants taken from AdSLC/CCL21-transduced DC (at a wide range of MOI), but not uninfected DC. Values represent the average SLC/CCL21 concentration ± the SEM from separate experiments as determined by nonlinear regression from the standard curve.
Figure 13.

AdSLC/CCL21-transduced DC enhance the priming of naïve human T cells in vitro. Uninfected and AdSLC/CCL21-transduced, HLA-A201-positive DC were pulsed with MART-1 peptide, and used to stimulate nylon-wool-purified, autologous human T cells. Following two or three stimulations, MART-1 tetramer staining was performed. Results are representative of three experiments.
Figure 14.

CD3+ T cells but not NK cells or B cells are observed by immunohistochemistry in a biopsy taken of an injection site of CCL21-producing human DC compared to normal skin from a patient with melanoma.
With respect to lung cancer, the concept of intratumoral administration of agents designed to enhance immune responses is supported by previous reports of induction of systemic antitumor reactivity following intratumoral administration of cytokines or chemokine genes in murine models and patients (31, 41–45). Our research group has demonstrated the clinical feasibility and safety of local injection of pulmonary tumors with immune stimulators (45). In fact, the first clinical gene therapy trials for lung cancer evaluated the intratumoral route for gene delivery and included patients who experienced significant tumor reduction in response to the therapy (46, 47). We have developed an alternative strategy for lung cancer in which intratumoral administration of DC is utilized to activate specific immune responses within the tumor microenvironment and, in addition, to generate systemic immunity (31–48). Our studies (41) and those of others (49) suggest that intratumoral DC administration may be particularly effective. Lung cancer patients have decreased numbers of circulating competent DC (50). Thus, injecting DC within the lung tumor site may be a particularly effective approach. A correlation exists between the number of tumor-infiltrating DC and survival (51–53). In fact, there is a relationship between tumor-infiltrating DC aggregation and apoptosis in situ in human non-small cell lung cancer (NSCLC) (54). This is consistent with recent studies indicating that attraction and activation of DC at the site of tumor elicits potent antitumor immunity (55).
Dieu-Nosjean et al (53) have identified ectopic lymph node or tertiary lymphoid structures within human NSCLC specimens and demonstrated a correlation of their cellular content with clinical outcome. These structures have been referred to as tumor-induced bronchus-associated lymphoid tissue (Ti-BALT), which are follicle-like and contain germinal centers, similar to those in secondary lymphoid follicles of lymph nodes. The density of DC-Lamp+, mature DC within these structures is a predictor of long-term survival in lung cancer patients (53). These findings suggest that Ti-BALT have clinical relevance and participate in the host’s antitumor immune response, and they are consistent with previously reported pre-clinical and clinical data (51, 52, 56). For example, in murine tumor models we (56) reported that DC genetically modified to secrete CCL21 can produce lymphoid cell aggregates and, importantly, prime naıve T cells extranodally within a tumor mass, resulting in the generation of tumor-specific T cells and subsequent tumor regression (22, 23). Thus, the intratumoral approach may achieve tumor antigen presentation by utilizing the tumor as an in vivo source of antigen for DC. In contrast to in vitro immunization with purified peptide antigen(s), autologous tumor has the capacity to provide the activated DC administered at the tumor site access to the entire repertoire of available antigens in situ. This may increase the likelihood of a response and reduce the potential for tumor resistance due to phenotypic modulation. Based on the pre-clinical data described here we have begun a phase I clinical evaluation at UCLA (in collaboration with the NCI-RAID program) in patients with advanced, stage IIIb/IV NSCLC of the safety, maximum tolerated dose, biologic and clinical activities of the intratumoral administration of autologous DC, transduced with a replication deficient adenoviral vector to express the CCL21 (57).
OTHER AREAS OF CHEMOKINE INVOLVEMENT IN IMMUNOTHERAPY
Recently, chemokine genes have been identified as key elements of molecular signatures that appear to be predictive of clinical efficacy of immunotherapy in melanoma (58–61) and NSCLC (62). In addition, more recent work (28–30) suggests that in situ expression of certain chemokines can be linked to the appearance of ectopic “lymph node-like” structures within solid tumor masses, which appear to correlate with better prognosis. Tumor immune escape by the loss of homeostatic chemokine expression has been reported recently.
Muller et al. (63) have shown that human keratinocyte-derived skin tumors may evade T cell immunity by down-regulating the expression of CCL27. Neutralization of CCL27 in mice could lead to decreased immune cell recruitment to tumor masses and could significantly increase primary tumor growth in vivo. Another reported form of tumor immune escape involving chemokines encompasses so-called “chemorepulsion”. Vianello and colleagues (64) have reported that engineered expression of CXCL12 by B16 melanoma cells can result in repulsion of tumor antigen-specific T cells, allowing the tumor to evade immune control. To date, however, it remains unreported whether or not a similar mechanism exists in human tumors in any clinically relevant setting.
Chemokine-chemokine receptor engagement is another promising strategy that is being employed in cancer immunotherapy applications. In this regard, Kershaw et al. (65) have developed a strategy to direct T cells toward chemokines expressed by tumors. T cells were retrovirally transduced to express CXCR2, which recognizes Gro-α. These gene-modified T cells became endowed with the capacity to potently and specifically react (by production of interferon-gamma and microchemotaxis) to both recombinant Gro-α as well as the tumor-derived chemokine. It will be valuable to ascertain whether or not this strategy of redirecting T cells to tumors will be useful in vivo in adoptive transfer studies to elicit tumor regression. Stasi and associates (66) have recently shown in a Hodgkin’s tumor model that T cells coexpressing CCR4 (by forced expression) and a chimeric antigen receptor targeting CD30 have sustained cytotoxic function and cytokine secretion in vitro, as well as improved homing and antitumor activity in vivo. However, chemokine-chemokine receptor engagement may not be a universally promising strategy to enhance cancer immunotherapy. For example, Liu et al. (67) have shown that CXCR3 chemokine ligands secreted by hepatocellular carcinomas can elicit functional desensitization of CXCR3 in lymphocytes from these patients. This interaction can cause lymphocyte dysfunction and subsequently impaired immune defense against tumors.
CONCLUSION
It is becoming increasingly clear that chemokines can have a profound role in the biology and progression or rejection of tumors. With respect to tumor immunity, chemokines can potently re-direct the trafficking of immune cells to sites of tumor as well as enhance their activation. Dendritic cells are appearing to serve as an ideal vehicle for delivering chemokines in vivo for therapeutic applications to concentrate immune cells at vaccine sites to enhance the antitumor response. T cells are being engineered to express chemokine receptors to re-direct their trafficking to and accumulation in tumors that express known endogenous chemokine ligands. In addition, chemokine genes have been identified as key elements of molecular signatures that appear to be predictive of better prognosis (survival) and clinical efficacy of immunotherapy for solid tumors. Overall, chemokines are holding much promise in the future, and the expanding knowledge of their unique biologic properties will be no doubt be important to further exploit their use in cancer therapeutics.
References
- 1.Schall TJ, Bacon KB. Chemokines, leukocyte trafficking, and inflammation. Curr Opin Immunol. 1995;6:86573. doi: 10.1016/0952-7915(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 2.Balkwill F. Cancer and the chemokine network. Nature Rev Cancer. 2004;4:540–50. doi: 10.1038/nrc1388. [DOI] [PubMed] [Google Scholar]
- 3.Yu P, Lee Y, Liu W, Chin RK, Wang J, Wang W, Schietinger A, Philip M, Schreiber H, Fu Y-X. Priming of naïve T cells inside tumors leads to eradication of established tumors. Nature Immunol. 2004;5:141–49. doi: 10.1038/ni1029. [DOI] [PubMed] [Google Scholar]
- 4.Chen T, Guo J, Han C, Yang M, Cao X. Heat shock protein 70, released from heat-stressed tumor cells, initiates antitumor immunity by inducing tumor cell chemokine production and activating dendritic cells via TLR4 pathway. J Immunol. 2009;182(3):1449–59. doi: 10.4049/jimmunol.182.3.1449. [DOI] [PubMed] [Google Scholar]
- 5.Henry CJ, Ornelles DA, Mitchell LM, Brzoza-Lewis KL, Hiltbold EM. IL-12 produced by dendriticcells augments CD8+ T cell activation through the production of the chemokines CCL1 and CCL17. J Immunol. 2008;181(12):8576–84. doi: 10.4049/jimmunol.181.12.8576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamazaki T, Yang XO, Chung Y, Fukunaga A, Nurieva R, Pappu B, Martin-Orozco N, Kang HS, Ma L, Panopoulos AD, Craig S, Watowich SS, Jetten AM, Tian Q, Dong C. CCR6 regulates the migration of inflammatory and regulatory T cells. J Immunol. 2008;181(12):8391–401. doi: 10.4049/jimmunol.181.12.8391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown CE, Vishwanath RP, Aguilar B, Starr R, Najbauer J, Aboody KS, Jensen MC. Tumor-derived chemokine MCP-1/CCL2 is sufficient for mediating tumor tropism of adoptively transferred T cells. J Immunol. 2007;179(5):3332–41. doi: 10.4049/jimmunol.179.5.3332. [DOI] [PubMed] [Google Scholar]
- 8.Gorbachev AV, Kobayashi H, Kudo D, Tannenbaum CS, Finke JH, Shu S, Farber JM, Fairchild RL. CXC chemokine ligand 9/monokine induced by IFN-gamma production by tumor cells is critical for T cell-mediated suppression of cutaneous tumors. J Immunol. 2007;178(4):2278–86. doi: 10.4049/jimmunol.178.4.2278. [DOI] [PubMed] [Google Scholar]
- 9.Shurin GV, Ferris RL, Tourkova IL, Perez L, Lokshin A, Balkir L, Collins B, Chatta GS, Shurin MR. Loss of new chemokine CXCL14 in tumor tissue is associated with low infiltration by dendritic cells (DC), while restoration of human CXCL14 expression in tumor cells causes attraction of DC both in vitro and in vivo. J Immunol. 2005;174(9):5490–8. doi: 10.4049/jimmunol.174.9.5490. [DOI] [PubMed] [Google Scholar]
- 10.Zhang T, Somasundaram R, Berencsi K, Caputo L, Rani P, Guerry D, Furth E, Rollins BJ, Putt M, Gimotty P, Swoboda R, Herlyn M, Herlyn D. CXC chemokine ligand 12 (stromal cell-derived factor 1 alpha) and CXCR4-dependent migration of CTLs toward melanoma cells in organotypic culture. J Immunol. 2005;174(9):5856–63. doi: 10.4049/jimmunol.174.9.5856. [DOI] [PubMed] [Google Scholar]
- 11.Pan J, Burdick MD, Belperio JA, Xue YY, Gerard C, Sharma S, Dubinett SM, Strieter RM. CXCR3/CXCR3 ligand biological axis impairs RENCA tumor growth by a mechanism of immunoangiostasis. J Immunol. 2006;176(3):1456–64. doi: 10.4049/jimmunol.176.3.1456. [DOI] [PubMed] [Google Scholar]
- 12.Matsumura S, Wang B, Kawashima N, Braunstein S, Badura M, Cameron TO, Babb JS, Schneider RJ, Formenti SC, Dustin ML, Demaria S. Radiation-induced CXCL16 release by breast cancer cells attracts effector T cells. J Immunol. 2008;181(5):3099–107. doi: 10.4049/jimmunol.181.5.3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Winter H, van den Engel NK, Rüttinger D, Schmidt J, Schiller M, Poehlein CH, Löhe F, Fox BA, Jauch KW, Hatz RA, Hu HM. Therapeutic T cells induce tumor-directed chemotaxis of innate immune cells through tumor-specific secretion of chemokines and stimulation of B16BL6 melanoma to secrete chemokines. J Transl Med. 2007;5:56. doi: 10.1186/1479-5876-5-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ruffini PA, Morandi P, Cabioglu N, Altundag K, Cristofanilli M. Manipulating the chemokinechemokine receptor network to treat cancer. Cancer. 2007;109(12):2392–404. doi: 10.1002/cncr.22706. [DOI] [PubMed] [Google Scholar]
- 15.Mulé JJ, Custer M, Averbook B, Yang JC, Weber JS, Goeddel DV, Rosenberg SA, Schall TJ. RANTES secretion by gene-modified tumor cells results in loss of tumorigenicity in vivo: role of immune cell subpopulations. Hum Gene Ther. 1996;7(13):1545–53. doi: 10.1089/hum.1996.7.13-1545. [DOI] [PubMed] [Google Scholar]
- 16.Chang AE, Redman BG, Whitfield JR, Nickoloff BJ, Braun TM, Lee PP, Geiger JD, Mulé JJ. A phase I trial of tumor lysate-pulsed dendritic cells in the treatment of advanced cancer. Clin Cancer Res. 2002;8(4):1021–32. [PubMed] [Google Scholar]
- 17.Geiger JD, Hutchinson RJ, Hohenkirk LF, McKenna EA, Yanik GA, Levine JE, Chang AE, Braun TM, Mulé JJ. Vaccination of pediatric solid tumor patients with tumor lysate-pulsed dendritic cells can expand specific T cells and mediate tumor regression. Cancer Res. 2001;61(23):8513–9. [PubMed] [Google Scholar]
- 18.Adema GJ, de Vries IJ, Punt CJ, Figdor CG. Migration of dendritic cell based cancer vaccines: in vivo veritas? Curr Opin Immunol. 2005;17(2):170–4. doi: 10.1016/j.coi.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 19.Verdijk P, Aarntzen EH, Punt CJ, de Vries IJ, Figdor CG. Maximizing dendritic cell migration in cancer immunotherapy. Expert Opin Biol Ther. 2008;8(7):865–74. doi: 10.1517/14712598.8.7.865. [DOI] [PubMed] [Google Scholar]
- 20.Lambert LA, Gibson GR, Maloney M, Durell B, Noelle RJ, Barth RJ., Jr Intranodal immunization with tumor lysate-pulsed dendritic cells enhances protective antitumor immunity. Cancer Res. 2001;61(2):641–6. [PubMed] [Google Scholar]
- 21.Mullins DW, Engelhard VH. Limited infiltration of exogenous dendritic cells and naive T cells restricts immune responses in peripheral lymph nodes. J Immunol. 2006;176(8):4535–42. doi: 10.4049/jimmunol.176.8.4535. [DOI] [PubMed] [Google Scholar]
- 22.Kirk CJ, Hartigan-O’Connor D, Nickoloff BJ, Chamberlain JS, Giedlin M, Aukerman L, Mule JJ. T cell dependent antitumor immunity mediated by secondary lymphoid tissue chemokine: augmentation of dendritic cell-based immunotherapy. Cancer Res. 2001;61(5):2062–70. [PubMed] [Google Scholar]
- 23.Kirk CJ, Hartigan-O’Connor D, Mulé JJ. The dynamics of the T-cell antitumor response: chemokine secreting dendritic cells can prime tumor-reactive T cells extranodally. Cancer Res. 2001;61(24):8794–802. [PubMed] [Google Scholar]
- 24.Terando A, Roessler B, Mulé JJ. Chemokine gene modification of human dendritic cell-based tumor vaccines using a recombinant adenoviral vector. Cancer Gene Ther. 2004;11(3):165–73. doi: 10.1038/sj.cgt.7700671. [DOI] [PubMed] [Google Scholar]
- 25.Kirk CJ, Mulé JJ. Gene-modified dendritic cells for use in tumor vaccines. Hum Gene Ther. 2000;11(6):797806. doi: 10.1089/10430340050015419. [DOI] [PubMed] [Google Scholar]
- 26.Yang SC, Batra RK, Hillinger S, Reckamp KL, Strieter RM, Dubinett SM, Sharma S. Intrapulmonary administration of CCL21 gene-modified dendritic cells reduces tumor burden in spontaneous murine bronchoalveolar cell carcinoma. Cancer Res. 2006;66(6):3205–13. doi: 10.1158/0008-5472.CAN-05-3619. [DOI] [PubMed] [Google Scholar]
- 27.Sharma S, Stolina M, Luo J, et al. Secondary lymphoid tissue chemokine mediates T cell-dependent antitumor responses in vivo. J Immunol. 2000;164(9):4558–63. doi: 10.4049/jimmunol.164.9.4558. [DOI] [PubMed] [Google Scholar]
- 28.Coppola D, Khalil F, Tjoelker L, Moyle M, Mulé J. Ecotpic lymphoid tissue in colonic adenocarcinoma. [abstract]. Proceedings of the 100th Annual Meeting of the American Association for Cancer Research; 2009 April 18–22. Denver, CO. Philadelphia (PA): AACR; 2009. Late Breaking Abstract nr 9047. [Google Scholar]
- 29.Coppola D, Mulé JJ. Ectopic lymph nodes within human solid tumors. J Clin Oncol. 2008;26(27):4369–70. doi: 10.1200/JCO.2008.17.6149. [DOI] [PubMed] [Google Scholar]
- 30.Coppola D, Nebozhyn M, Khalil F, Loboda A, Yeatman T, Mulé JJ. Ectopic lymph node-like structure presence and composition in colorectal carcinoma are predicted by immune gene array profiles: potential for cancer patient selection for vaccines and other immunotherapies. Proc Amer Soc Gene Cell Ther. in press. [Google Scholar]
- 31.Yang SC, Hillinger S, Riedl K, et al. Intratumoral administration of dendritic cells overexpressing CCL21 generates systemic antitumor responses and confers tumor immunity. Clin Cancer Res. 2004;10(8):2891–901. doi: 10.1158/1078-0432.ccr-03-0380. [DOI] [PubMed] [Google Scholar]
- 32.Yang SC, Batra RK, Hillinger S, et al. Intrapulmonary administration of CCL21 gene-modified dendritic cells reduces tumor burden in spontaneous murine bronchoalveolar cell carcinoma. Cancer Res. 2006;66(6):3205–13. doi: 10.1158/0008-5472.CAN-05-3619. [DOI] [PubMed] [Google Scholar]
- 33.Ashour AE, Lin X, Wang X, Turnquist HR, Burns NM, Tuli A, Sadanandam A, Suleiman K, Singh RK, Talmadge JE, Solheim JC. Cancer Biol Ther. 2007;6(8):1206–10. doi: 10.4161/cbt.6.8.4405. [DOI] [PubMed] [Google Scholar]
- 34.Yousefieh N, Hahto S, Ciavarra RP. Gene Therapy: CCL21 (SLC) Inhibits Primary Prostate Tumor Growth and Metastases. The FASEB Journal. 2008;22:1076.15. [Google Scholar]
- 35.Novak L, Igoucheva O, Cho S, Alexeev V. Characterization of the CCL21-mediated melanoma-specific immune responses and in situ melanoma eradication. Mol Cancer Ther. 2007;6(6):1755–64. doi: 10.1158/1535-7163.MCT-06-0709. [DOI] [PubMed] [Google Scholar]
- 36.Wu S, Xing W, Peng J, Yuan X, Zhao X, Lei P, Li W, Wang M, Zhu H, Huang B, Huang L, Shen G. Tumor transfected with CCL21 enhanced reactivity and apoptosis resistance of human monocyte derived dendritic cells. Immunobiology. 2008;213(5):417–26. doi: 10.1016/j.imbio.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 37.Thanarajasingam U, Sanz L, Diaz R, Qiao J, Sanchez-Perez L, Kottke T, Thompson J, Chester J, Vile RG. Delivery of CCL21 to metastatic disease improves the efficacy of adoptive T-cell therapy. Cancer Res. 2007;67(1):300–8. doi: 10.1158/0008-5472.CAN-06-1017. [DOI] [PubMed] [Google Scholar]
- 38.Selvakumaran M, Biade S, Schick J, Marinucci M, Morgan M, Livolsi V, O’Dwyer PJ, Johnson SW. Role of CCL19 and CCL21 chemokines in the response of ovarian tumors to platinum-based chemotherapy (abstract) Proc Amer Assoc Cancer Res. 1472;45:2004. [Google Scholar]
- 39.Shields JD, Kourtis IC, Tomei AA, Roberts JM, Swartz MA. Induction of lymphoidlike stroma and immune escape by tumors that express the chemokine CCL21. Science. 2010;328(5979):749–52. doi: 10.1126/science.1185837. [DOI] [PubMed] [Google Scholar]
- 40.Riedl K, Baratelli F, Batra RK, Yang SC, Luo J, Escuadro B, Figlin R, Strieter R, Sharma S, Dubinett S. Overexpression of CCL-21/secondary lymphoid tissue chemokine in human dendritic cells augments chemotactic activities for lymphocytes and antigen presenting cells. Mol Cancer. 2003;2:35. doi: 10.1186/1476-4598-2-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sharma S, Miller PW, Stolina M, et al. Multicomponent gene therapy vaccines for lung cancer: effective eradication of established murine tumors in vivo with interleukin-7/herpes simplex thymidine kinasetransduced autologous tumor and ex vivo activated dendritic cells. Gene Ther. 1997;4(12):1361–70. doi: 10.1038/sj.gt.3300531. [DOI] [PubMed] [Google Scholar]
- 42.Miller PW, Sharma S, Stolina M, et al. Dendritic cells augment granulocyte-macrophage colony-stimulating factor (GM-CSF)/herpes simplex virus thymidine kinase-mediated gene therapy of lung cancer. Cancer Gene Ther. 1998;5(6):380–9. [PubMed] [Google Scholar]
- 43.Sharma S, Yang SC, Batra RK, Dubinett SM. Intratumoral therapy with cytokine gene-modified dendritic cells in murine lung cancer models. Methods Mol Med. 2003;75:711–22. doi: 10.1385/1-59259-324-0:711. [DOI] [PubMed] [Google Scholar]
- 44.Dubinett SM, Patrone L, Tobias J, Cochran AJ, Wen DR, McBride WH. Intratumoral interleukin-2 immunotherapy: activation of tumor-infiltrating and splenic lymphocytes in vivo. Cancer Immunol Immunother. 1993;36(3):156–62. doi: 10.1007/BF01741086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Suh RD, Goldin JG, Wallace AB, et al. Metastatic renal cell carcinoma: CT-guided immunotherapy as a technically feasible and safe approach to delivery of gene therapy for treatment. Radiology. 2004;231(2):35964. doi: 10.1148/radiol.2312021754. [DOI] [PubMed] [Google Scholar]
- 46.Gahery-Segard H, Molinier-Frenkel V, Le Boulaire C, et al. Phase I trial of recombinant adenovirus gene transfer in lung cancer. Longitudinal study of the immune responses to transgene and viral products. J Clin Invest. 1997;100(9):2218–26. doi: 10.1172/JCI119759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roth JA, Nguyen D, Lawrence DD, et al. Retrovirus-mediated wild-type p53 gene transfer to tumors of patients with lung cancer. Nat Med. 1996;2(9):985–91. doi: 10.1038/nm0996-985. [DOI] [PubMed] [Google Scholar]
- 48.Miller PW, Sharma S, Stolina M, et al. Intratumoral administration of adenoviral interleukin 7 gene-modified dendritic cells augments specific antitumor immunity and achieves tumor eradication. Hum Gene Ther. 2000;11(1):53–65. doi: 10.1089/10430340050016157. [DOI] [PubMed] [Google Scholar]
- 49.Tatsumi T, Huang J, Gooding WE, et al. Intratumoral delivery of dendritic cells engineered to secrete both interleukin (IL)-12 and IL-18 effectively treats local and distant disease in association with broadly reactive Tc1-type immunity. Cancer Res. 2003;63(19):6378–86. [PubMed] [Google Scholar]
- 50.Almand B, Resser JR, Lindman B, et al. Clinical significance of defective dendritic cell differentiation in cancer. Clin Cancer Res. 2000;6(5):1755–66. [PubMed] [Google Scholar]
- 51.Zeid NA, Muller HK. S100 positive dendritic cells in human lung tumors associated with cell differentiation and enhanced survival. Pathology. 1993;25(4):338–43. doi: 10.3109/00313029309090853. [DOI] [PubMed] [Google Scholar]
- 52.Coppola D, Mule JJ. Ectopic lymph nodes within human solid tumors. J Clin Oncol. 2008;26(27):4369–70. doi: 10.1200/JCO.2008.17.6149. [DOI] [PubMed] [Google Scholar]
- 53.Dieu-Nosjean MC, Antoine M, Danel C, et al. Long-term survival for patients with non-small-cell lung cancer with intratumoral lymphoid structures. J Clin Oncol. 2008;26(27):4410–7. doi: 10.1200/JCO.2007.15.0284. [DOI] [PubMed] [Google Scholar]
- 54.Kurabayashi A, Furihata M, Matsumoto M, Hayashi H, Ohtsuki Y. Distribution of tumor-infiltrating dendritic cells in human non-small cell lung carcinoma in relation to apoptosis. Pathol Int. 2004;54(5):302–10. doi: 10.1111/j.1440-1827.2004.01624.x. [DOI] [PubMed] [Google Scholar]
- 55.Lapteva N, Aldrich M, Rollins L, et al. Attraction and Activation of Dendritic Cells at the Site of Tumor Elicits Potent Antitumor Immunity. Mol Ther. 2009;17(9):1626–36. doi: 10.1038/mt.2009.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kirk CJ, Hartigan-O’Connor D, Nickoloff BJ, et al. T cell-dependent antitumor immunity mediated by secondary lymphoid tissue chemokine: augmentation of dendritic cell-based immunotherapy. Cancer Res. 2001;61(5):2062–70. [PubMed] [Google Scholar]
- 57.Lee JM, Garon EB, Baratelli F, Schaue D, Pak PS, Wallace WD, Suh R, Abtin F, Zeng G, Dubinett SM. Phase I Trial of CCL21 gene modified dendritic cells in non-small cell lung cancer. ASCO Trials in Progress 2010. 2010 abstract # 51761. [Google Scholar]
- 58.Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, McKee M, Gajewski TF. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009;69(7):3077–85. doi: 10.1158/0008-5472.CAN-08-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gajewski TF. Insights into Mechanisms of Immune Resistance in the Tumor Microenvironment through Molecular Profiling. In: Yefenof E, editor. Innate and Adaptive Immunity in the Tumor Microenvironment. Vol. 1. Netherlands: Springer; 2008. pp. 77–89. [Google Scholar]
- 60.Lehmann F, Louahed J, Gaulis S, Gruselle O, Brichard V. Clinical response to the MAGE-A3 immunotherapeutic in metastatic melanoma patients is associated with a specific gene profile present prior to treatment. Cancer Immunity. 2008;8(suppl 2):27. [Google Scholar]
- 61.Louahed J, Gruselle O, Gaulis S, Coche T, Eggermont AM, Kruit W, Dreno B, Chiarion Sileni V, Lehmann F, Brichard VG. Expression of defined genes identified by pretreatment tumor profiling: Association with clinical responses to the GSK MAGE-A3 immunotherapeutic in metastatic melanoma patients (EORTC 16032-18031) J Clin Oncol. 2008;26:9045. [Google Scholar]
- 62.Vansteenkiste JF, Zielinski M, Dahabreh IJ, Linder A, Lehmann F, Gruselle O, Therasse P, Louahed J, Brichard VG. Association of gene expression signature and clinical efficacy of MAGE-A3 antigen specific cancer immunotherapeutic (ASCI) as adjuvant therapy in resected stage IB/II non-small cell lung cancer (NSCLC) J Clin Oncol. 2008;26:7501. [Google Scholar]
- 63.Pivarcsi A, Müller A, Hippe A, Rieker J, van Lierop A, Steinhoff M, Seeliger S, Kubitza R, Pippirs U, Meller S, Gerber PA, Liersch R, Buenemann E, Sonkoly E, Wiesner U, Hoffmann TK, Schneider L, Piekorz R, Enderlein E, Reifenberger J, Rohr UP, Haas R, Boukamp P, Haase I, Nürnberg B, Ruzicka T, Zlotnik A, Homey B. Tumor immune escape by the loss of homeostatic chemokine expression. Proc Natl Acad Sci U S A. 2007;104(48):19055–60. doi: 10.1073/pnas.0705673104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vianello F, Papeta N, Chen T, Kraft P, White N, Hart WK, Kircher MF, Swart E, Rhee S, Palu G, Irimia D, Toner M, Weissleder R, Poznansky MC. Murine B16 melanomas expressing high levels of the chemokine stromal-derived factor-1/CXCL12 induce tumor-sepecific T cell chemorepulsion and escape from immune control. J Immunol. 2006;176:2902–14. doi: 10.4049/jimmunol.176.5.2902. [DOI] [PubMed] [Google Scholar]
- 65.Kershaw MH, Wang G, Westwood JA, Pachynski RK, Tiffany HL, Marincola FM, Wang E, Young HA, Murphy PM, Hwu P. Redirecting migration of T cells to chemokine secreted from tumors by genetic modification with CXCR2. Hum Gene Ther. 2002;13(16):1971–80. doi: 10.1089/10430340260355374. [DOI] [PubMed] [Google Scholar]
- 66.Di Stasi A, De Angelis B, Rooney CM, Zhang L, Mahendravada A, Foster AE, Heslop HE, Brenner MK, Dotti G, Savoldo B. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood. 2009;113(25):6392–402. doi: 10.1182/blood-2009-03-209650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu Y-Q, Poon RT, Hughes J, Li Q-Y, Yu W-C, Fan S-T. Desensitization of T lymphocyte function by CXCR3 ligands in human hepatocellular carcinoma. World J Gastroenterol. 2005;11(2):164–170. doi: 10.3748/wjg.v11.i2.164. [DOI] [PMC free article] [PubMed] [Google Scholar]