

Abstract
The human α7 neuronal nicotinic acetylcholine receptor gene (CHRNA7) is a candidate gene for schizophrenia and an important drug target for cognitive deficits in the disorder. Activation of the α7*nAChR, results in opening of the channel and entry of mono- and divalent cations, including Ca++, that presynaptically participates to neurotransmitter release and postsynaptically to down-stream changes in gene expression. Schizophrenic patients have low levels of α7*nAChR, as measured by binding of the ligand [125I]-α-bungarotoxin (I-BTX). The structure of the gene, CHRNA7, is complex. During evolution, CHRNA7 was partially duplicated as a chimeric gene (CHRFAM7A), which is expressed in the human brain and elsewhere in the body. The association between a 2bp deletion in CHRFAM7A and schizophrenia suggested that this duplicate gene might contribute to cognitive impairment. To examine the putative contribution of CHRFAM7A on receptor function, co-expression of α7 and the duplicate genes was carried out in cell lines and Xenopus oocytes. Expression of the duplicate alone yielded protein expression but no functional receptor and co-expression with α7 caused a significant reduction of the amplitude of the ACh-evoked currents. Reduced current amplitude was not correlated with a reduction of I-BTX binding, suggesting the presence of non-functional (ACh-silent) receptors. This hypothesis is supported by a larger increase of the ACh-evoked current by the allosteric modulator 1-(5-chloro-2,4-dimethoxy-phenyl)-3-(5-methyl-isoxazol-3-yl)-urea (PNU-120596) in cells expressing the duplicate than in the control. These results suggest that CHRFAM7A acts as a dominant negative modulator of CHRNA7 function and is critical for receptor regulation in humans.
1. Introduction
The human α7 neuronal nicotinic receptor gene (CHRNA7) is one of approximately 8 genes consistently reported to be associated with schizophrenia [1]. Expression of α*7nAChRs in schizophrenic postmortem brain is decreased [2], but the mechanism for this reduction has not been definitively determined. In the current report, we show that a chimeric gene, CHRFAM7A formed as a partial duplication of CHRNA7, is a dominant negative regulator of function when coexpressed with the CHRNA7 gene product.
Genetic linkage to the CHRNA7 gene at 15q13.3 was first found to an endophenotype in schizophrenia, the P50 deficit (LOD = 5.30) [3] and then to the disease itself. Replicated linkage has been found in multiple ethnic populations suggesting that this candidate gene is associated with schizophrenia in most all populations [4-12]. Genetic data also implicates the CHRNA7 gene in smoking in the disorder. A specific dinucleotide repeat allele (D15S1360) in intron 2 of the CHRNA7 gene was found to be associated with smoking in schizophrenia [13]. Recent genetic linkage studies showed that three nicotinic receptors are associated with smoking in schizophrenia, the α2 nicotinic receptor subunit gene (CHRNA2; 8p21), the β2 nicotinic receptor subunit gene (CHRNB2;1q21), and the α7 nicotinic receptor subunit gene (CHRNA7; 15q14) [14].
Behaviorally, smoking normalizes auditory sensory deficits, found in schizophrenics and 50% of their first-degree relatives [15], and improves cognition in human subjects [16]. It has been suggested that smoking may be a form of self-medication for these patients [17; 18]. Smoking cessation in control subjects shows improvement in visuospatial working memory tasks, but in schizophrenics, withdrawal from smoking leads to decreased cognitive performance [19]. These results may be due to differences in neuroadaptive responses to nicotine resulting from decreased levels of nicotinic receptors [20].
The α7 receptor subunit gene, CHRNA7, is a member of a large gene family coding for neuronal nicotinic acetylcholine receptor subunits. Unlike high-affinity nicotinic receptors in which multiple subunits assemble to form α4β2*nAChRs, the α7*nAChR is homomeric in most tissues and binds nicotine with low affinity [21]. The antagonist, α-bungarotoxin, usually used as the iodinated product [125I]-α-bungarotoxin specifically labels α7*nAChRs showing very broad expression throughout the brain [22-24]. Recently, [3H]-CHIBA-1001 a molecule derived from the specific α7 agonist SSR-180711 was used for α7 labeling in humans, confirming the high level and general expression of this receptor subtype [25;26]. Such promising ligands are expected to advance our understanding of the cholinergic system in brain impairment. α7*nAChRs are located both pre- and postsynaptically [27;28]. Presynaptically, they are involved in neurotransmitter release, including release of GABA, glutamate, and dopamine from specific terminals [28-33]. Postsynaptically, α7*nAChRs are localized in or near the postsynaptic density (PSD) [34], where the Ca++ flux increases phosphorylation and affects gene expression [27;35]. The α7*nAChR is also found in the periphery where it is involved in neuropeptide release and protects against inflammatory processes [36;37].
The low-affinity nicotine binding site in the α7*nAChR is specifically measured by binding of [I125]-α-bungarotoxin (I-BTX) [22;24]. In postmortem hippocampus, frontal cortex, dorsolateral prefrontal cortex, cingulate gyrus, and in the reticular nucleus of the thalamus of schizophrenic subjects, α7*nAChR levels are decreased, as determined by I-BTX binding and western blot [2;38-40], suggesting that functional surface receptors are decreased in all brain regions examined to date. I-BTX, which binds to the low-affinity nicotine site, can bind to a partially assembled muscle receptor of three subunits [41]. It is assumed, but not known whether this is also true of the α7*nAChR. Thus, schizophrenic subjects with low levels of I-BTX binding, may have defects early in the assembly process. The structure and regulation of α7*nAChR expression is, however, complex. Decreased receptor expression can occur at one of many levels, including transcription [42], translation [43], or copy number variation [44].
1.1 Structure of the CHRNA7 gene
The CHRNA7 gene on chromosome 15q13.3 has 10 exons, differing from other nicotinic receptor subunits, which only have six. We cloned the human gene from postmortem brain (Genbank U40583) and discovered that the gene is partially duplicated [45]. Exons 5-10 are duplicated, along with a large cassette of DNA (∼300kb). The duplication interrupted a second partial duplication of another gene, and maps centromeric to CHRNA7 by 1.6Mb [45-47]. The chimeric gene CHRFAM7A is complex (Figure 1). Upstream of Exons 5-10 of CHRNA7 (red), are three exons from a partial duplication of ULK4 (C,B,A;blue), a serine/threonine kinase gene mapping to 3p22.1. 5′ of these three exons is an additional exon of unknown provenance (D; pink). CHRNA7 DNA sequence in CHRFAM7A is 99.9% conserved [45]. CHRFAM7A is expressed in human brain at low levels (approximately one order of magnitude less than CHRNA7 in hippocampus), but is expressed abundantly in peripheral lymphocytic cells and in multiple other tissues. While a full phylogenetic study has not yet been done, CHRFAM7A is not found in closely related primates [48] and does not appear to be found in rodents (unpublished data). Thus, the duplication is recent. This may not be surprising as CHRNA7 is one of the oldest ligand-gated ion channel genes and phylogenetic duplication may have contributed to the origin of this large gene family [49-51]. CHRFAM7A was recently shown to have a regulatory role that, thus, is unique to humans [52].
Figure 1. Structure of the CHRNA7/CHRFAM7A gene cluster on chromosome 15q13.3.
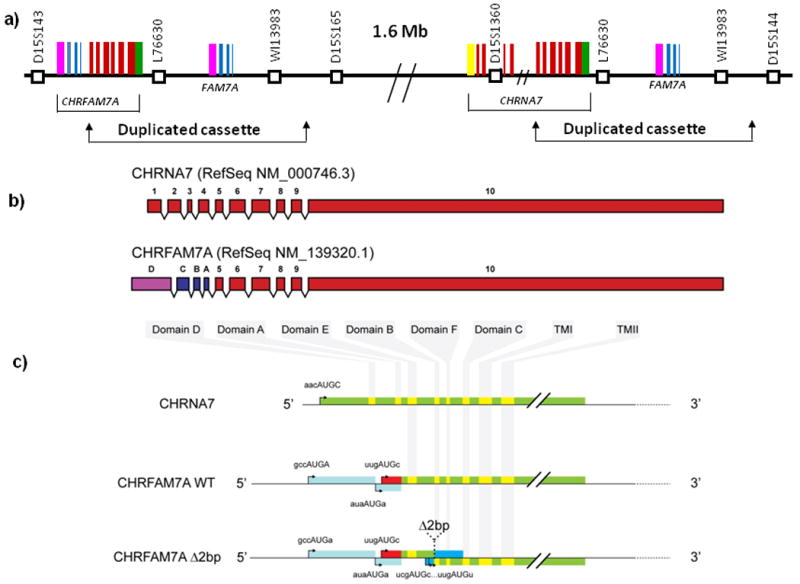
Fig. 1a, Map of the partial duplication of CHRNA7 on 15q13.3. Exons 5-10 of CHRNA7 were duplicated in a duplicon of ∼300kb, mapping centromeric by 1.6 Mb. The duplicon interrupted a partial duplication of a second gene, ULK4. CHRNA7 exons, red; ULK4 exons, blue; exon D, pink. Fig. 1b, Schematic representation of the exon organisation of the transcripts coding for CHRNA7, CHRFAM7A, CHRFAM7A based on the RefSeq NM_000746.3 and NM_139320.1. Fig 1c, Putative translation products from CHRNA7, CHRFAM7A, and CHRFAM7AΔ2bp mRNAs. Amino acid sequence of α7 is represented in green and yellow (for the different domains). Alternative amino acids from CHRFAM7A are indicated in red and alternative amino acids from CHRFAM7AΔ2bp in blue. The start codons in Kozak context are indicated, as are the stop codons.
1.2 Copy number variation in the chromosome 15q13.3 region
Reports of large copy number variations (CNV) on 15q13.3 that include the CHRNA7 gene suggest that CNV in this gene cluster is associated with schizophrenia [44;53]. Examination of these reports of duplications and deletions at 15q13.3 show that in many cases CHRNA7 has been deleted, but the partial duplication, CHRFAM7A is present. It has not been known whether copy number of CHRFAM7A could be important. There are also smaller insertions and deletions within the CHRNA7/CHRFAM7A gene cluster. Approximately 10% of individuals have only one copy of CHRFAM7A. This CNV has an allele frequency of 0.08 in Caucasians and 0.12 in African Americans (not significantly different). Rare individuals are missing both copies [45;54]. Again, a common CNV for CHRFAM7A would not be unusual if it is a new duplication.
1.3 Mutations in the CHRNA7 and CHRFAM7A genes in schizophrenia
Mutation screening in exons and splice junctions of the CHRNA7 gene and its partial duplication, CHRFAM7A revealed no mutations in the coding region of the full-length gene, CHRNA7, that were associated with schizophrenia [45;55]. Therefore, the receptors from CHRNA7 that are expressed in the disorder have a normal amino acid sequence. However, we did find mutations in the proximal promoter region of the CHRNA7 gene that functionally reduce transcription and are associated with both schizophrenia and the P50 deficit [42]. The promoter mutations are more prevalent in schizophrenic non-smokers and may account for the low levels of CHRNA7 mRNA in that group [56]. One of these mutations, rs3087454, has recently been associated with a pharmacogenomic response to DMXB-A, an α7*nAChR partial agonist [57].
We also found a 2bp deletion in exon 6 that is found only in the CHRFAM7A gene [55]. The overall allele frequency of this mutation, CHRFAM7AΔ2bp is 0.22. To evaluate the 2bp deletion in exon 6 of CHRFAM7A, the CNV must be measured in addition to screening for the polymorphism, since ∼10% of individuals have only one copy. We have recently completed such a study and find that the 2bp deletion in exon 6 of CHRFAM7A is associated with schizophrenia [54]. Others have found association of the 2bp deletion to schizophrenia [58] or to sensory processing deficits such as the P50 deficit and antisaccade performance [59-61].
The 2bp deletion in CHRFAM7A is also associated with a gene inversion [47]. The wild type allele is in a head-to-head orientation with respect to the full-length CHRNA7 gene, but CHRFAM7AΔ2bp is oriented in the same direction. CHRFAM7AΔ2bp is more common in Caucasians than in African Americans, suggesting that the inversion of the gene occurred simultaneously or shortly prior to the 2bp deletion [54]. The functions of these two versions of the gene are not known.
There is a common, but synonymous, mutation in CHRFAM7A at position 654bp. Of the Caucasian individuals that have been genotyped for both the 2bp deletion and the SNP at 654bp, the two mutations appear to be in linkage disequilibrium. However, more subjects need to be genotyped to confirm this finding. An additional common mutation is non-synonymous, 1466bp C→T, resulting in an amino acid change of a serine to a leucine at aa 489. This amino acid would be in the cytosolic tail. Although a function for this S489L mutation has not yet been defined, a similar mutation that changed a glycine to a serine in CHRNA7 resulted in phosphorylation differences and alterations in receptor function [62].
1.4 The CHRNA7 gene is regulated differently in schizophrenic smokers, compared to control smokers
A conundrum concerning the expression of the CHRNA7 gene product, α7*nAChR, exists in schizophrenia. In a microarray comparison of gene expression in postmortem hippocampus of control and schizophrenic smokers and non-smokers, we found that the CHRNA7 gene is differentially regulated at the mRNA and protein level in schizophrenic smokers [35;56]. Schizophrenic non-smokers have low levels of CHRNA7 mRNA and protein, compared to controls, but schizophrenic smokers have mRNA and protein levels for CHRNA7 similar to control smokers. Thus, in schizophrenic smokers there is adequate mRNA and protein [56]. These results are not consistent with the low levels of [125I]-α-bungarotoxin and [3H]-methyllycaconitine binding seen in human postmortem brain of schizophrenic subjects [2;39;40]. The data are consistent, however, with an assembly, trafficking, or binding site defect. In the current report we investigated the possibility that the gene duplication, CHRFAM7A regulates the expression and function of CHRNA7.
2. Materials and Methods
2.1 Cloning of CHRFAM7A and CHRFAM7AΔ2bp
Generation of a full-length cDNA clone of the duplicated gene, CHRFAM7A, was performed by PCR cloning. The exon D –3′ UTR cDNA was generated by PCR amplification of reverse transcribed mRNA derived from human primary lymphocytes, which abundantly express CHRFAM7A. Primer sequences were 5′-CTCGGTGCCCCTTGCCATTT-3′ (Sense) and 5′-CCTTGCCCATCTGTGAGTTTTCCAC-3′ (antisense) and were located at position 140 and 1850 from the start of exon D. PCR was performed using taq/pfu polymerase mix with supplied buffers and reagents (Stratagene, La Jolla, CA). After TA cloning, the fragment was moved to pcDNA3.1 (Invitrogen, Carlsbad, CA), a mammalian expression vector.
The 2bp deletion in exon 6 of CHRFAM7A was introduced into pcDNA3.1 CHRFAM7A by PCR with the QuikChange II XL Site-Directed Mutagenesis Kit, according to the manufacturer's protocols (Stratagene, La Jolly, CA). All clones were sequenced bidirectionally using internal primers and dRhodamine dye terminators on an ABI 3100 Avant DNA sequencer (Applied Biosytems, Inc., Foster City, CA) for confirmation of correct sequence. Clones were designated as pcDNA3.1-CHRFAM7A and pcDNA3.1-CHRFAM7AΔ2bp.
An α7 full-length clone was also constructed as a control. Primer sequences were 5′-CGCTGCAGCTCCGGGACTCAACATG-3′ (sense) and 5-TGCCCATCTGTGAGTTTTCCA CATG-3′ (anti-sense) and were located at position (from start ATG) (-22) and +1638 respectively. PCR fragments were gel purified and TA cloned into the pcDNA3.1 mammalian expression vector (Invitrogen)(pcDNA3.1-CHRNA7).
From the original exon D-10/pcDNA3.1 and α7/pcDNA3.1 cDNA constructs, PCR primers were designed to amplify α7-in frame sequences for expression with a V5 epitope; the 5′ end of the antisense primer terminated just prior to the normal stop codon in exon 10. PCR fragments were purified from 1% agarose and TA cloned into the pcDNA3 Topo/V5 vector (Invitrogen, Carlsbad, CA), transformed, isolated, prepped and sequenced as described above. The clone is designated as pcDNA3.1-CHRFAM7A-V5.
2.2 Cell Culture and Transfections
SH-EP1 cells were used for heterologous expression studies. SH-EP1 are a non-neuronal epithelial cell line derived from the neuroblastoma cell line SH-SY5Y [63]. Wild-type SH-EP1 cells do not express endogenously detectable α7 mRNA or protein. Cells were cultured in DMEM media supplemented with 10% heat-inactivated horse serum, 5% fetal bovine serum, 1 mM sodium pyruvate, 4 mM L-glutamine, and 100 U/ml penicillin/streptomycin (Gibco Life Technologies/ Invitrogen, Rockville, MD) in 5% CO2 at 37°C.
Optimal transfection conditions were determined empirically using Green Fluorescent Protein (Invitrogen). Cells (150,000) were seeded in 12-well plates containing 12 mm poly-L-lysine-coated glass coverslips (Becton-Dickinson, Franklin Lakes, NJ). Transfections were performed 24 hours after plating using Superfect reagent (Qiagen, Valencia, CA). Briefly, 5-10 μg of DNA was added to an unsupplemented D-MEM in final volume of 150 μl. Superfect (60 μl) was then added and the mixture was allowed to incubate for 10 minutes at room temperature before the addition of 1 ml of complete media. During DNA complex formation, cells were washed once with phosphate-buffered saline (PBS) and the DNA/Superfect mix was added to the cells. Cells were then incubated for 2 hours before the transfection reagent was removed. Cells were washed twice with PBS and returned to the incubator with complete media for 48 hours. Forty-eight hours post-transfection, cells were fixed in 4% paraformaldehyde, washed with PBS and permeabilized in successive washes of 10, 95 and 100% ethanol. After washing, coverslips were blocked for 20 minutes in complete media. Cells were then blocked using 4% goat serum (containing 2% bovine gamma globulin and 0.3% Triton-X100) at room temperature for 1 hour. Primary antibodies were mouse anti-V5 (Invitrogen) diluted 1:100 and rabbit anti-α7 (kind gift of Cecilia Gotti, University of Milan) diluted 1:1000. Cells were incubated for 1 hour, washed 3× with PBS and secondary antibodies applied. Secondary antibodies were donkey-anti-mouse Cy2 (1:1000) and donkey anti-rabbit tetramethyl rhodamine (TRITC) (1:1000) (Jackson Immunochemicals, West Grove, PA) and were incubated for 1 hour at room temperature. All coverslips were then washed 3× with PBS, mounted and visualized using appropriate filters on a Microphot-FXA fluorescence microscope (Nikon, Melville, NY).
2.3 Oocyte expression
Oocytes were isolated and selected as previously described [64]. Briefly, oocytes were harvested from mature Xenopus laevis females, anesthetized with tricaine. Frogs were sacrificed following ethical animal protocols in Geneva, Switzerland. Oocytes were isolated by mechanical and enzymatic action using type I collagenase (Sigma, Basel Switzerland) at 0.2% in a medium deprived of calcium. Stage V and VI oocytes were placed in 96 wells microtiter plates (NUNC) in BARTH medium that contained 88 mM NaCl, 1 mM KCl, 2.4 mM NaHCO3, 10 mM HEPES, 0.82 mM MgSO4·7H2O, 0.33 mM Ca(NO3)2·4H2O, 0.41 mM CaCl2·6H2O, adjusted to pH 7.4 with NaOH and supplemented with kanamycin (20 mg/ml), penicillin (100 U/ml) and streptomycin (100 U/ml) (Sigma). Nuclear injections of 2 ng of cDNA expression vectors, containing equal molar ratios of the genes of interest were performed with an automatic injection system [65]. Cells were subsequently maintained in Barth's medium at 18 °C.
2.4 Recording of α7* receptor properties
Electrophysiological properties of oocytes were determined 2 to 5 days after injection with an automated system equipped with a two electrode voltage clamp (Geneclamp Axon Instrument, Molecular Devices, Sunnyvale, CA). Oocytes were continuously superfused with OR2 (control medium) that contained 82.5 mM NaCl, 2.5 mM KCl, 2.5 mM CaCl2, 1 mM MgCl2, 5 mM HEPES, adjusted to pH 7.4 with NaOH. Current was generated by 5s pulses of 200 μM acetylcholine (ACh). Data were filtered at 10 Hz and digitized at 100 Hz with an analog to digital converter (National Instruments, Austin, TX) and stored on a personal computer. Data acquisition, treatment and statistics were performed using Matlab (MathWorks Inc., Natick, MA).
2.5 [I125]-α-Bungarotoxin assay
Injected oocytes generating a current were selected and were pre-incubated in a solution of OR2 with 2%BSA for 10-20 min. Oocytes were then incubated in 50 μl of OR2-2%BSA with 10nM [I125]-α-bungarotoxin (Institute of Isotopes Co., Budapest, Hungary) for 1h at 18°c. After 3 washes with OR2-2%BSA, the radioactivity of each oocyte was measured with a scintillation counter and normalized to non-injected oocytes.
2.6 RNA isolation from injected oocytes and QRT-PCR quantification of CHRFAM7A and CHRNA7
Oocytes were placed in 1 ml RNAlater (Ambion, Austin, TX) at 4°C for >24 hour. Ten eggs were homogenized for 30 seconds in 0.5ml of Trizol (Invitrogen, Carlsbad, CA) using a Pestle Grinder (Fisher Scientific, Pittsburgh, PA). The homogenate was placed on ice for one minute, then room temperature for 5 minutes before extraction with chloroform. Following centrifugation at 12,000rpm for 15 minutes at 4°C, the aqueous layer was removed to a new tube. Purification of RNA was done using an RNeasy mini column (Qiagen, Valencia, CA) using manufacturer's protocol. Yield was 0.5-1 ug total RNA per oocyte.
cDNA was synthesized using Superscript 3 (Invitrogen) according to manufacturer's directions. Real-time PCR was performed using human CHRNA7 primer sets (Forward 5′-TTTACAGTGGAATGTGTCAGA-3′, Reverse 5′-TGTGGAATGTGGCGTCAAG-3′), CHRFAM7A (Forward 5′-TGGATAGCTGCAAACTGCGA-3′, Reverse 5′-TACTGGCAATGCCCAGAAGA-3′) and GAPDH (5′-Forward GGTATCGTGGAAGGACTC-3′, Reverse 5′-GGATGATGTTCTGGAGAGC-3′), using iQ SYBR Green (Biorad, Hercules, CA). All three real-time assays were run for 40 cycles; 94°C for 15 sec, 58°C for 30 sec, 72°C for 30 sec.
3. Results
3.1 Structure of the CHRNA7/CHRFAM7A gene cluster
The partial duplication of the CHRNA7 gene is recent, not being found in primates or rodents [48]. The relationship of the two genes is shown in Figure 1A. They are approximately 1.6 Mb apart. The region between the CHRFAM7A and CHRNA7 contains two other genes TRPM1 and KLF13. A detailed study, utilizing tagged SNPs across the linkage region at 15q13.3 shows that there is broad association with schizophrenia at this locus [66].
The chimeric gene, CHRFAM7A, contains exons 5-10 of the full-length CHRNA7 gene, exons A, B, and C duplicated from ULK4, and exon D of unknown provenance (Figure 1A). Exon sizes were determined: A, 47bp; B, 63bp; C, 125bp; D, 297bp [45] and the gene was registered in GenBank (AF029837). Mutation screening of the coding regions and exon/intron borders in both CHRNA7 and CHRFAM7A was previously reported [55]. Non-synonymous mutations were rare in CHRNA7 and were not associated with schizophrenia. Therefore, the receptors from CHRNA7 that are expressed in the disorder have a normal amino acid sequence.
Several mutations were mapped specifically to CHRFAM7A (Gault et al., 2003). A 2bp deletion in exon six of CHRFAM7A (CHRFAM7AΔ2bp) was found. The mutation is common; the overall allele frequency of this mutation is 0.22, and it is more prevalent in Caucasian individuals than in African Americans [55]. The CHRFAM7A gene also varies in copy number. Approximately 30% of individuals have only one copy and 5% have no copies. Thus to evaluate the 2bp deletion in exon 6 of CHRFAM7A, copy number variation must be measured in addition to screening for the polymorphism. We have recently completed such a study and find that the 2bp deletion in exon 6 of CHRFAM7A is associated with schizophrenia in both Caucasians and African Americans [54]. Others have found association of the 2bp deletion to schizophrenia [58] or to sensory processing deficits such as the P50 deficit and antisaccade performance [59;60]. The 2bp deletion may change the structure of the protein translated from the mRNA.
3.2 CHRFAM7A is expressed in transfected cells
A cDNA clone of CHRFAM7A with a V5 peptide tag (pcDNA3.1 CHRFAM7A-V5) was transfected into SH-EP cells, plated on cover slips. An anti-V5 antibody (Invitrogen) was utilized to detect protein product. Figure 2A shows a positive V5 signal, indicating that the peptide product was transcribed and translated, although it is not abundantly expressed in the transfected cells. An α7 polyclonal antibody to the large cytoplasmic loop of the protein was used in 2B, labeled with rhodamine. A positive signal and overlap in 2C shows that the translated peptide contains sequence for the α7 subunit.
Figure 2. Translation of CHRFAM7A.
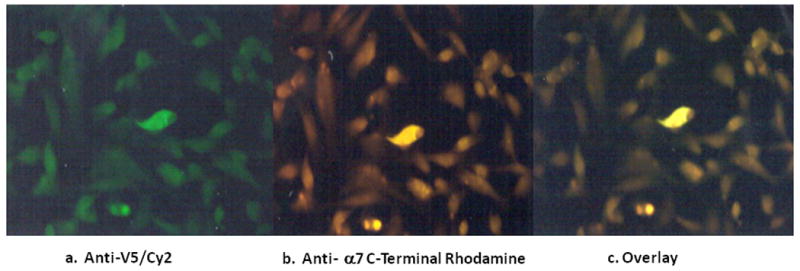
Cloned CHRFAM7A-V5 was transfected into SHEP cells. A V5 antibody labeled with Cy2 (green), in 2a indicates that CHRFAM7A-V5 protein is present. The anti-α7 antibody and the overlay (2b and 2c) confirm that the peptide has amino acid sequence in-frame with α7 amino acid sequence.
3.3 Translation of CHRFAM7A mRNA
Several initiator methionines in exon D could result in truncated transcripts that do not contain CHRNA7 coding sequence. Use of an initiator methionine in exon B of CHRFAM7A would produce a protein including amino acids coded by part of exon B, exon A, and exons 5-10 of CHRNA7. The peptide would include the disulfide bridge and vicinal cysteines contained in the amino terminus of the α7 subunit. A putative glycosylation site would be lost and part of the agonist binding site (Figure 1C). If the 2bp deletion in exon 6 is present, use of the same initiating methionine would code for a peptide with exon 6 amino acid sequence to the deletion. At that point a frame shift would result in 40 unique amino acids before a stop codon was reached. There are, however, two ATG codons in exon 6. A translation start at one of these would terminate within a few amino acids unless the 2bp deletion was present. Deletion of these two base pairs would allow translation of a peptide with considerable α7 sequence. The peptide would be out of frame until the 2bp were reached, coding for either 6 or 13 amino acids, depending on which ATG was used. At that point, the frame shift caused by the 2bp deletion would return the amino acid sequence to that of the α7 subunit. The peptide resulting from translation of mRNA without the 2bp, initiating in exon 6, would be missing the signal peptide and nearly the entire binding site, but would contain all of the CHRNA7 membrane-spanning regions.
3.4 The CHRFAM7A gene product is a dominant negative regulator of α7*nAChR function
The CHRFAM7A gene is expressed in both human brain and in the periphery. To study function of this chimeric gene product and effects of the 2bp deletion in exon 6, we expressed clones of the genes alone and together with a full length CHRNA7 cDNA in oocytes. Xenopus laevis oocytes were isolated and injected as described. A total of 2ng of cDNA, containing molar equivalent amounts for each gene were injected. Three injection groups were utilized: pcDNA3.1-CHRNA7 + vector, pcDNA3.1-CHRNA7 + pcDNA3.1-CHRFAM7A, and pcDNA3.1-CHRNA7 + pcDNA3.1-CHRFAM7AΔ2bp. Two to five days after injection, oocytes were superfused with control medium and current, generated by 5s pulses of 200 μM acetylcholine (ACh), was measured.
Determination of the ACh-evoked current in a large population of oocytes obtained from different batches revealed that expression of the duplicate gene caused a reduction of the response amplitude (Figure 3A). Currents were reduced by 53% when pcDNA3.1-CHRFAM7A was included. The presence of the 2bp deletion in exon 6, pcDNA3.1-CHRFAM7AΔ2bp, further reduced the current amplitude of the full-length gene product by an additional 10%. The difference in current amplitude is best evidenced by plotting the histogram of current distribution for pcDNA3.1-CHRNA7 + pcDNA3.1, pcDNA3.1-CHRNA7 + pcDNA3.1-CHRFAM7A, and pcDNA3.1-CHRNA7 + pcDNA3.1-CHRFAM7AΔ2bp as shown in Figures B, C and D, respectively.
Figure 3. The CHRFAM7A gene product is a dominant-negative regulator of α7*nAChR function.
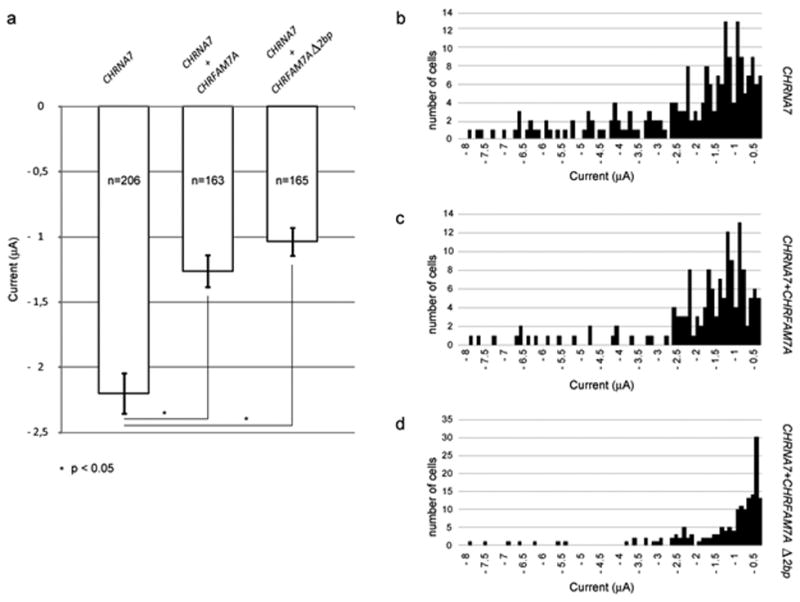
Oocytes were harvested from mature Xenopus females and isolated by mechanical and enzymatic action. Stage V and VI oocytes were placed in 96 wells microtiter plates (NUNC) in Barth medium. Nuclear injections of 1 ng CHRNA7 and 1 ng of pcDNA3 or CHRFAM7A or CHRFAM7AΔ2bp were made and cells maintained at 18 °C. Electrophysiological properties of oocytes were determined 3 days later with an automated two electrode voltage clamp. Oocytes were continuously superfused with OR2 (control medium) and tested by the application of 200 mM of ACh. Fig. 3a, Mean of the currents obtained for pcDNA3.1-CHRNA7+pcDNA3.1, pcDNA3.1-CHRNA7+ pcDNA3.1-CHRFAM7A, or pcDNA3.1-CHRNA7+ pcDNA3.1-CHRFAM7AΔ2bp; Fig. 3b, Current distribution for pcDNA3.1-CHRNA7+ pcDNA3.1; Fig. 3c, for pcDNA3.1-CHRNA7+pcDNA3.1-CHRFAM7A, and Fig. 3d, for pcDNA3.1-CHRNA7+ pcDNA3.1-CHRFAM7AΔ2bp.
3.5 Coexpression of CHRNA7 and CHRFAM7A does not alter transcription from either gene
Total RNA was isolated from oocytes injected with cDNA coding for CHRNA7 and vector or cDNA for CHRNA7 and either CHRFAM7A or CHRFAM7AΔ2bp. Real-time QRT-PCR was utilized to quantify message for CHRNA7 and CHRFAM7A in RNA isolated from each preparation, with primers specific for each gene. Mean normalized expression was calculated from the control values for the gene of interest and the housekeeping gene, GAPDH, and the amplification efficiencies [67]. As shown in Table 1, expression of the gene duplication, CHRFAM7A did not alter the transcription of CHRNA7.
Table 1. No effect of CHRFAM7A on transcription of CHRNA7. RNA was isolated from oocytes injected with molar equivalents of either CHRNA7 and empty vector, or CHRNA7 and CHRFAM7A with or without the 2bp deletion. MNE, mean normalized expression to GAPDH.
| OOCYTE | MNE | Fold Change |
|---|---|---|
| CHRNA7 + pcDNA3.1 | 243.9 | 1.0 |
| CHRNA7 + CHRFAM7A | 245.6 | 1.0 |
| CHRNA7 + CHRFAM7AΔ2bp | 261.4 | 1.1 |
3.6 Presence of the duplicated gene product from CHRFAM7A, changes the [125I]-α-bungarotoxin binding properties of α7*nAChR
Oocytes were injected with cDNA for CHRNA7+pcDNA3.1, CHRNA7+CHRFAM7A, or CHRNA7+CHRFAM7AΔ2bp at a final concentration of 2ng/μl. After 5-7 days, the amplitude of the current elicited by brief pulses (5 s) of ACh (200 μM) was measured as described in Methods. Oocytes responding to the ACh test pulse were incubated in OR2-BSA with 10nM [125I]-α-bungarotoxin (I-BTX) for 1h. Cells were then thoroughly washed to remove unspecific binding and the radioactivity in each oocyte was measured in a scintillation counter. Non-specific binding was determined by measuring the average I-BTX binding on non-injected oocytes. The histograms of I-BTX binding obtained after subtraction of the non-specific binding obtained for CHRNA7, CHRNA7+CHRFAM7A and CHRNA7+CHRFAM7AΔ2bp are shown in Figure 4A and suggest that cells expressing the duplicate might have a smaller number of binding sites at the cell surface. As there was less current for equivalent binding this is consistent with the hypothesis that the assembly of a truncated subunit, such as the chimeric CHRFAM7A or CHRFAM7AΔ2bp with the CHRNA7 gene product produces a receptor that is less functional. A plot of the peak current versus counts (CPM) yielded linear relationships that were readily fitted by linear regression with high correlation coefficients (CHRNA7+pcDNA3.1, R2=0.995; CHRNA7+CHRFAM7A, R2=0.867; CHRNA7+CHRFAM7AΔ2bp, R2=0.915 (Figure 4B).
Figure 4. Plot of the current registered versus the [125I]-α-bungarotoxin binding activity for oocytes injected with plasmid expressing CHRNA7 with different variants of CHRFAM7A.

Oocytes were injected with pcDNA3.1-CHRNA7+pcDNA3.1, pcDNA3.1-CHRNA7+pcDNA3.1-CHRFAM7A, or pcDNA3.1-CHRNA7+pcDNA3.1-CHRFAM7AΔ2bp at a final concentration of 2ng/μl. On 5 to 7 days after the injection, the current generated by an application of 200 μM of ACh for 5s was registered by an homemade automated two electrode voltage clamp. Oocytes generating a current were selected and incubated into OR2-BSA with 10nM [125I]-α-bungarotoxin for 1h. After 3 washes with OR2-BSA, the radioactivity of each oocyte was measured with a scintillation counter and normalized to non-injected oocytes. Fig. 4a, Means of binding in each group. Fig. 4b, Lines represent a linear regression. CHRNA7+pcDNA3 (Y=-225.93X+249,89 R2=0.995), CHRNA7+CHRFAM7A (Y=-555.45X+207.02 R2=0.867), CHRNA7+CHRFAM7AΔ2bp (Y=-805.79X+244.72 R2=0.915).
3.7 Potentiation of CHRNA7 and CHRFAM7A by the allosteric modulator PNU-120596
PNU-120596 is a potent allosteric modulator of the α7*nAChR causing an increase of the maximal amplitude of the ACh-evoked current, reduction of the EC50 and reduced desensitization [68]. Previous work suggested that co-expression of the duplicate gene could alter the allosteric modulation of the receptor [52]. To understand the possible relevance of such an observation it must be recalled that the absence of potentiation by PNU-120596 observed in the α7-5HT3 chimera suggested that this molecule must interact with the transmembrane domain [69]. As the duplicate is thought to have a different N-terminal domain from the α7 subunit, but has the same amino acid sequence for the transmembrane domains, PNU-120596 potentiation in the presence of the duplicate in the receptor complex might be expected. To assess the hypothesis, PNU-120596 effects were examined at CHRNA7, CHRNA7+CHRFAM7A and CHRNA7+CHRFAM7AΔ2bp, injected oocytes. Cells injected with an equivalent concentration of the cDNAs encoding the two genes showed non-significant differences in the magnitude of potentiation measured as the ratio of the ACh-evoked current observed after PNU-120596 versus the current evoked by the same ACh test pulse (1280 μM, 5 s) in control (Figure 5). As shown in Figure 5A, exposure to 10 μM PNU-120596 for 30 s causes a dramatic increase in the amplitude of the ACh-evoked current that is accompanied by a significant increase in the response duration that was interpreted as a reduction of desensitization [68]. It could be argued that only receptors composed of CHRNA7 are potentiated by PNU-120596 and this effect might mask the fraction of receptors that include the duplicate. To address this hypothesis additional experiments were designed in which the ratio of CHRNA7 versus the duplicate gene was reduced to 1:10. Currents evoked by a brief pulse of 1280 μM ACh for 5 seconds were measured in control and following 30 seconds exposure to 10 μM PNU-12056. Average currents measured on the same day in sibling oocytes were respectively 5.8 ± 0.66 μA for CHRNA7, 0.9 ± 0.33 μA for CHRNA7+CHRFAM7A and 1.1 ± 0.19 μA for cells injected with CHRNA7+CHRFAM7AΔ2bp. These data are in agreement with previous experiments indicating that expression of the duplicate gene causes a reduction of the average ACh-evoked current [52]. However, a different picture emerged when examining the amplitude of the ACh-evoked current after PNU-120596 exposure. Peak inward currents were of 28.0 ± 3.3 μA for CHRNA7, 12.8 ± 1.23 μA for CHRNA7+CHRFAM7A and 22.0 ± 1.77 μA for cells injected with CHRNA7+CHRFAM7AΔ2bp. The potentiation caused by PNU-120596 is best observed when plotting the ratio of these values as shown in Figure 6A.
Figure 5. Potentiation of acetylcholine stimulated currents by PNU-120596.

Fig. 5a. Typical current recorded in an oocyte injected with CHRNA7 + CHRFAM7A (in a 1:1 ratio) evoked by 1280 μM ACh in control and following exposure to 10 μM PNU-120596. Note the large increase in current amplitude and reduction of desensitization. Fig. 5b. Histogram of the ratio of currents measured following exposure to the positive allosteric modulator versus control (1:1 ratio). Fig. 5c. Potentiation by PNU-120596 in cells injected with a 1:10 ratio in favor of the duplicate gene. Histogram of the ratio of currents measured following exposure to the positive allosteric modulator versus control. Recordings were performed on the same day in sibling oocytes.
Figure 6. Putative incorporation of the CHRFAM7A gene product into α7*nAChR.
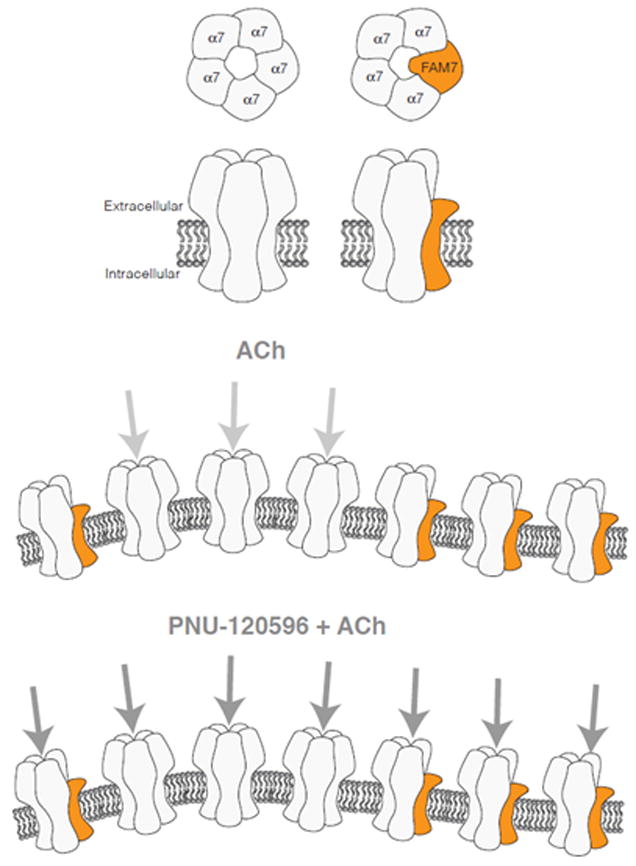
Fig. 6a. The gene product from CHRFAM7A would be missing the signal peptide and part of the amino terminal binding site for ligands. A CHRFAM7AΔ2bp product would be missing the entire binding site. As suggested in this figure, there may be steric effects on the other subunits that would alter the ligand binding site.
Fig 6b. Assuming that a fraction of the receptors comprise α7, whereas another fraction includes the duplicate protein, it can be proposed that ACh activates only the α7 containing receptors. Exposure to PNU-120596 might facilitate the opening of the receptor causing larger inward currents and increase in the PNU-120596 potentiation.
Determination of the concentration activation curves in control and following PNU-120596 exposure revealed no major difference in sensitivity between the α7 cells and those expressing the duplicate gene. In control conditions the EC50s and Hill Coefficients were: 38 ± 3 μM, nH = 1 for CHRNA7; 105 ± 18 μM, nH = 0.8 for CHRNA7+CHRFAM7A; 47 ± 8 μM, nH = 0.9 for CHRNA7+CHRFAM7AΔ2bp. Following exposure to PNU-120596 the values were: 3.6 ± 0.1 μM, nH = 5 for CHRNA7; 5 ± 0.9 μM, nH = 3 for CHRNA7+CHRFAM7A; 3.6 ± 0.3 μM, nH = 3 for CHRNA7+CHRFAM7AΔ2bp. As expected, exposure to the positive allosteric modulator caused a shift to the left of the ACh sensitivity accompanied by an increase in the Hill coefficient. This suggests that while expression of the duplicate gene causes a reduction of the ACh current amplitude it does not significantly affect the sensitivity of the receptor to its natural ligand.
4. Discussion
The α7*nAChR gene, CHRNA7, has been consistently associated with schizophrenia [1;4]. Expression of α7*nAChR, as measured by [125I]-α-bungarotoxin binding, is decreased in postmortem hippocampus, cortex, and the reticular nucleus of the thalamus in schizophrenic subjects [2;39;40]. We recently found that CHRNA7 mRNA and protein are differentially regulated in postmortem hippocampus of schizophrenics [56]. In schizophrenic non-smokers, the CHRNA7 mRNA and protein are both decreased compared to controls. However, in schizophrenic smokers, both mRNA and protein return to control levels. Thus, in schizophrenic smokers, there is adequate α7 protein, but low bungarotoxin binding, suggesting a problem with assembly or trafficking of the receptor.
There are multiple mechanisms for regulation of α7*nAChR expression. Mutations in the proximal promoter decrease transcription of CHRNA7 and are associated with both schizophrenia and with the P50 deficit in the disorder [42]. Another mutation 1,831bp from exon 1 is associated with schizophrenia [70], and with an improvement in the fMRI default network after treatment with DMXB-A, an α7*nAChR agonist [57]. Additional complexity is introduced by the partial duplication of CHRNA7, which results in the formation of a chimeric gene, CHRFAM7A [45-47]. Large deletions of 15q13.3, the map locus of both CHRNA7 and CHRFAM7A have been associated with schizophrenia [44;53]. In many of these patients, the CHRNA7 gene is deleted, but the CHRFAM7A gene remains. We also found that a 2bp deletion in exon 6 of the duplication CHRFAM7A, but not in the full-length gene, CHRNA7, is associated with schizophrenia [54]. This deletion is associated with the P50 deficit [60] and with a chromosomal inversion of the CHRFAM7A gene [47]. There is a copy number variation for CHRFAM7A; approximately 10% of individuals have only one copy and 5% have no copies [45;54]. A recent study suggested that the CHRFAM7A gene may be a dominant-negative regulator of α7*nAChR function [52]. These investigators found that co-expression of α7 subunits and the CHRFAM7A gene product dupα7 in oocytes resulted in decreased ACh stimulated current.
In the current study, we have examined the effects of both the CHRFAM7A gene and the gene copy with the 2bp deletion in exon 6, CHRFAM7AΔ2bp, on function of the full-length gene product of CHRNA7 in an oocyte expression system. Our results confirm the de Lucas-Cerrillo et al. study [52] and show that the 2bp deletion in exon 6 further decreases function when co-expressed with the α7 subunit. Thus copy number variation in CHRFAM7A and presence of the 2bp deletion are likely to be factors when considering the overall activity of α7*nAChR in human subjects. Rodents and primates apparently do not have such a chimeric gene, suggesting that it is a relatively new duplication [47;54;71].
The mechanism by which CHRFAM7A acts as a dominant negative regulator of α7*nAChR function is not clear. CHRFAM7A is missing CHRNA7 exons 1-4, including the signal peptide, and does not appear to assemble alone (not shown). Leukocytes express a protein, identified with an α7 antibody, but are not labeled by I-BTX, suggesting that the peptide expressed is from CHRFAM7A and that functional receptors are not assembled nor do they migrate to the surface [72]. Thus, co-expression of the CHRFAM7A and CHRNA7 gene products could result in receptor sequestration in the endoplasmic reticulum. Further, CHRFAM7A mRNA is down regulated by nicotine [52] and by bacterial infection [73].
Our [125I]-α-bungarotoxin experiments also suggest another alternative; the binding sites for bungarotoxin and other ligands may be altered. When only the α7 gene product was expressed, current increased commensurate with bungarotoxin binding, a measure of receptor number. Moreover, since labeling was carried out on intact oocytes, labeling corresponds to receptors expressed in the plasma membrane. When the CHRFAM7A gene product was present, bungarotoxin binding increases showed a significantly steeper correlation slope. The increase in bungarotoxin binding for an equivalent current suggests the presence in the membrane of receptors that cannot be activated by ACh. Putative translation start sites for CHRFAM7A and CHRFAM7AΔ2bp and possible structures of the assembled receptor are illustrated in Figure 1 and Figure 7. Translation of CHRFAM7A mRNA likely starts in exon B and is in frame through the CHRNA7 sequence. The peptide is missing the signal peptide and two of the glycosylation sites, retaining the cysteine bridge and vicinal cysteines. It is, thus, missing domains A and D of the binding site [74]. The 2bp deletion in exon 6 would result in a truncated receptor, not likely to assemble with α7 subunits, if translation of CHRFAM7AΔ2bp mRNA begins in exon B. A translation start from either of the two AUG codons in exon 6 would result in an open reading frame with CHRNA7 sequence after the 2bp deletion through exon 10, but the amino terminus would be missing domains E and B in addition to domains A and D. The vicinal cysteines and the cysteine bridge would be absent. We speculate that this could result in an assembled receptor resembling that in Figure 7. The lack of steric association in the upstream amino termini of the assembled subunits might lead to some instability, changing the receptor properties.
Interestingly, the de Lucas-Cerrillo et al. study [52] indicates that expression of the duplicate does not modify the receptor sensitivity to ACh. As it is known that the ligand binding site lies at the interface between two adjacent subunits, reviewed in [74;75], the incorporation of the duplicate should not contribute to the formation of distinct ACh binding sites. The stoichiometry of wild type α7 versus the duplicate in a single receptor is unknown; some receptor complexes might, thus, incorporate more than one duplicate subunit. Such receptors may display distinct properties and be less activatable by ACh. To evaluate this hypothesis, experiments were carried out using the positive allosteric modulator PNU-120596 [68]. Evaluation of the potentiation caused by brief exposure to 10 μM PNU carried out in sibling oocytes injected with CHRNA7 and CHRFAM7A genes showed no detectable differences when genes were injected in a 1:1 ratio. However, a marked difference was observed when the gene ratio was changed to 1:10 in favor of the duplicate. Cells expressing this gene ratio showed a significantly higher potentiation by PNU-120596 suggesting the presence of otherwise silent receptors. Although our observations differ from those reported by de Lucas-Cerrillo et al [52], these authors restricted their study at 1 μM PNU-120596 which, as shown in Figure 1 of the original work published by Hurst and collaborators [68], is in the lowest part of the concentration activity curve for this compound.
Experimental evidence indicates that PNU-120596 interacts with transmembrane domains of the receptors [69]. As the duplicate gene encodes an equivalent amino acid sequence in the transmembrane segments, it can be postulated that receptors containing the duplicate must have an equivalent pore formation and transmembrane domain organization. It is therefore likely that an allosteric modulator interacting through the transmembrane domain must remain efficacious in receptors incorporating the duplicate gene product.
The use of an allosteric modulator interacting with the transmembrane domains presents several advantages versus orthosteric ligands that are known to bind at the interfaces between N-terminal domains of the subunits. Namely, as it was shown that expression of α7 with the duplicate causes no changes in the ACh sensitivity, it can be speculated that some α7-α7 interfaces are conserved with complexes comprising the duplicate. This hypothesis is further supported by the fact that these receptors are still able to bind α-bungarotoxin and, in view of the marked difference observed in the amino-acid sequence of the duplicate N-terminal domain, it is unlikely that this subunit can contribute an identical principal or complementary binding site. The most likely explanation is therefore that receptors incorporating the duplicate maintain enough α7-α7 interfaces to keep most of the pharmacological properties of the receptors. Our transfection results in Figure 2 suggest that CHRFAM7A is not abundantly expressed, which may result in more α7-α7 interfaces than α7-duplicate interfaces. Furthermore, it is important to recall some of the unique features of the α7*nAChR and their modulation by endogenous proteins such as Lynx-1 or SLURP-1 [76-79]. Although it can be speculated that such modulation is likely to remain present in receptors containing the duplicate, further investigation into the pharmacological properties of CHRFAM7A is needed. It is not known whether CHRFAM7A expression is altered in schizophrenia or by the use of tobacco products in these patients. Based on the data presented here, one hypothesis for the decreased I-BTX binding in schizophrenic postmortem brain could be overexpression of CHRFAM7A or CHRFAM7AΔ2bp.
As a strong regulator of α7*nAChR function, CHRFAM7A holds some promise as a drug target. The CHRNA7 gene is not only associated with mental illness, but plays a role in inflammation where it is protective and seems to prevent cytokine expression following an inflammatory event [37;80-82]. Maternal infection is a known risk factor for development of neuropsychiatric disorders, including schizophrenia [83-85]. Agonists, specific for α7*nAChR protect against inflammation [86]. Recently lipopolysaccharide, a surrogate for bacterial inflammation was shown to down regulate CHRFAM7A [73]. Nicotine also down regulates CHRFAM7A expression [52]. Since the CHRFAM7A gene product is a dominant negative regulator, decreasing its expression may be beneficial for treatment of both mental illness and inflammation. Additionally, the development of new Type II positive allosteric modulators for the α7*nAChR might open new therapeutic avenues.
Acknowledgments
An antibody for the α7 subunit was kindly provided by Dr. Cecilia Gotti, University of Milan. The authors thank Margaret Short for technical help with this project. The work was supported by National Institutes of Health grants MH81174, DA09457, and the Veterans Affairs Medical Research Service to SL, and EEC grant Neurocypres to DB.
Abbreviations
- CHRNA7
human α7 nicotinic acetylcholine receptor gene
- Chrna7
mouse α7 nicotinic acetylcholine receptor gene
- α7
α7 receptor subunit protein
- α7*nAChR
α7 assembled surface pentameric receptor
- CHRFAM7A
duplicated CHRNA7 gene
- CHRFAM7AΔ2bp
duplicated gene containing a 2bp deletion in exon 6
- I-BTX
[125I]-α-bungarotoxin
- α-BTX
α-bungarotoxin
- PNU-120596
1-(5-chloro-2,4- dimethoxy-phenyl)-3-(5-methyl-isoxazol-3-yl)-urea
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psych. 2005;10:40–68. doi: 10.1038/sj.mp.4001558. [DOI] [PubMed] [Google Scholar]
- 2.Freedman R, Hall M, Adler LE, Leonard S. Evidence in postmortem brain tissue for decreased numbers of hippocampal nicotinic receptors in schizophrenia. Biol Psych. 1995;38:22–33. doi: 10.1016/0006-3223(94)00252-X. [DOI] [PubMed] [Google Scholar]
- 3.Freedman R, Coon H, MylesWorsley M, OrrUrtreger A, Olincy A, Davis A, Polymeropoulos M, Holik J, Hopkins J, Hoff M, Rosenthal J, Waldo MC, Reimherr F, Wender P, Yaw J, Young DA, Breese CR, Adams C, Patterson D, Adler LE, Kruglyak L, Leonard S, Byerley W. Linkage of a neurophysiological deficit in schizophrenia to a chromosome 15 locus. Proc Nat Acad Sci. 1997;94:587–92. doi: 10.1073/pnas.94.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Freedman R, Leonard S, Olincy A, Kaufmann CA, Malaspina D, Cloninger CR, Svrakic D, Faraone SV, Tsuang MT. Evidence for the multigenic inheritance of schizophrenia. Am J Med Gen. 2001;105:794–800. doi: 10.1002/ajmg.10100. [DOI] [PubMed] [Google Scholar]
- 5.Leonard S, Freedman R. Genetics of chromosome 15q13-q14 in schizophrenia. Biol Psych. 2006;60:115–22. doi: 10.1016/j.biopsych.2006.03.054. [DOI] [PubMed] [Google Scholar]
- 6.Kaufmann CA, Suarez B, Malaspina D, Pepple J, Svrakic D, Markel PD, Meyer J, Zambuto CT, Schmitt K, Matise TC, Harkavy-Friedman JM, Hampe C, Lee H, Shore D, Wynne D, Faraone SV, Tsuang MT, Cloninger CR. NIMH Genetics Initiative Millenium Schizophrenia Consortium: linkage analysis of African-American pedigrees. Am J Med Genet. 1998;81:282–9. [PubMed] [Google Scholar]
- 7.Liu CM, Hwu HG, Lin MW, Ou-Yang WC, Lee SFC, Fann CSJ, Wong SH, Hsieh SH. Suggestive evidence for linkage of schizophrenia to markers at chromosome 15q13-14 in Taiwanese families. Am J Med Gen. 2001;105:658–61. doi: 10.1002/ajmg.1547. [DOI] [PubMed] [Google Scholar]
- 8.Riley BP, Makoff AM, Magudi-Carter M, Jenkins TJ, Williamson R, Collier DA, Murray RM. Haplotype transmission disequilibrium and evidence for linkage of the CHRNA7 gene region to schizophrenia in Southern African Bantu families. Am J Med Gen. 2000;96:196–201. doi: 10.1002/(sici)1096-8628(20000403)96:2<196::aid-ajmg15>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 9.Gejman PV, Sanders AR, Badner JA, Cao QH, Zhang J. Linkage analysis of schizophrenia to chromosome 15. Am J Med Gen. 2001;105:789–93. doi: 10.1002/ajmg.1552. [DOI] [PubMed] [Google Scholar]
- 10.Xu JZ, Pato MT, Dalla Torre C, Medeiros H, Carvalho C, Basile VS, Bauer A, Dourado A, Valente J, Soares MJ, Macedo AA, Coelho I, Ferreira CP, Azevedo MH, Macciardi F, Kennedy JL, Pato CN. Evidence for linkage disequilibrium between the alpha 7- nicotinic receptor gene (CHRNA7) locus and schizophrenia in Azorean families. Am J Med Gen. 2001;105:669–74. doi: 10.1002/ajmg.1549. [DOI] [PubMed] [Google Scholar]
- 11.Fallin MD, Lasseter VK, Wolyniec PS, McGrath JA, Nestadt G, Valle D, Liang KY, Pulver AE. Genomewide linkage scan for schizophrenia susceptibility loci among Ashkenazi Jewish families shows evidence of linkage on chromosome 10q22. Am J Hum Gen. 2003;73:601–11. doi: 10.1086/378158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsuang DW, Skol AD, Faraone SV, Bingham S, Young KA, Prabhudesai S, Haverstock SL, Mena F, Menon AS, Bisset D, Pepple J, Sauter F, Baldwin C, Weiss D, Collins J, Boehnke M, Schellenberg GD, Tsuang MT. Examination of genetic linkage of chromosome 15 to schizophrenia in a large veterans affairs cooperative study sample. Am J Med Gen. 2001;105:662–8. [PubMed] [Google Scholar]
- 13.De Luca V, Wong AHC, Muller DJ, Wong GWH, Tyndale RF, Kennedy JL. Evidence of association between smoking and alpha 7 nicotinic receptor subunit gene in schizophrenia patients. Neuropsychopharmacol. 2004;29:1522–6. doi: 10.1038/sj.npp.1300466. [DOI] [PubMed] [Google Scholar]
- 14.Faraone SV, Su J, Taylor L, Wilcox M. A novel permutation testing method implicates sixteen nicotinic acetylcholine receptor genes as risk factors for smoking in schizophrenia families. Hum Hered. 2004;57:59–68. doi: 10.1159/000077543. [DOI] [PubMed] [Google Scholar]
- 15.Adler LE, Hoffer LD, Wiser A, Freedman R. Normalization of auditory physiology by cigarette smoking in schizophrenic patients. Am J Psych. 1993;150:1856–61. doi: 10.1176/ajp.150.12.1856. [DOI] [PubMed] [Google Scholar]
- 16.Rezvani AH, Levin ED. Nicotinic-glutamatergic interactions and attentional performance on an operant visual signal detection task in female rats. Eur J Pharmacol. 2003;465:83–90. doi: 10.1016/s0014-2999(03)01439-0. [DOI] [PubMed] [Google Scholar]
- 17.Leonard S, Mexal S, Freedman R. Smoking, Genetics and Schizophrenia: Evidence for Self Medication. J Dual Diag. 2007;3:43–59. doi: 10.1300/J374v03n03_05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kumari V, Postma P. Nicotine use in schizophrenia: The self medication hypotheses. Neurosci Biobehav Rev. 2005;29:1021–34. doi: 10.1016/j.neubiorev.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 19.George TP, Vessicchio JC, Termine A, Sahady DM, Head CA, Pepper WT, Kosten TR, Wexler BE. Effects of smoking abstinence on visuospatial working memory function in schizophrenia. Neuropsychopharmacol. 2002;26:75–85. doi: 10.1016/S0893-133X(01)00296-2. [DOI] [PubMed] [Google Scholar]
- 20.Leonard S. Consequences of low levels of nicotinic acetylcholine receptors in schizophrenia for drug development. Drug Dev Res. 2003;60:127–36. [Google Scholar]
- 21.Hogg RC, Raggenbass M, Bertrand D. Nicotinic acetylcholine receptors: from structure to brain function. Rev Physiol Biochem Pharmacol. 2003;147:1–46. doi: 10.1007/s10254-003-0005-1. [DOI] [PubMed] [Google Scholar]
- 22.Marks MJ, Stitzel JA, Romm E, Wehner JM, Collins AC. Nicotinic binding sites in rat and mouse brain: Comparison of acetylcholine, nicotine and α-bungarotoxin binding. Mol Pharmacol. 1986;30:427–36. [PubMed] [Google Scholar]
- 23.Quik M, Choremis J, Komourian J, Lukas RJ, Puchacz E. Similarity between rat brain nicotinic alpha-bungarotoxin receptors and stably expressed alpha-bungarotoxin binding sites. J Neurochem. 1996;67:145–54. doi: 10.1046/j.1471-4159.1996.67010145.x. [DOI] [PubMed] [Google Scholar]
- 24.OrrUrtreger A, Goldner FM, Saeki M, Lorenzo I, Goldberg L, DeBiasi M, Dani JA, Patrick JW, Beaudet AL. Mice deficient in the alpha 7 neuronal nicotinic acetylcholine receptor lack alpha-bungarotoxin binding sites and hippocampal fast nicotinic currents. J Neurosci. 1997;17:9165–71. doi: 10.1523/JNEUROSCI.17-23-09165.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tanibuchi Y, Wu J, Toyohara J, Fujita Y, Iyo M, Hashimoto K. Characterization of [H-3]CHIBA-1001 binding to alpha 7 nicotinic acetylcholine receptors in the brain from rat, monkey, and human. Brain Res. 2010;1348:200–8. doi: 10.1016/j.brainres.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 26.Toyohara J, Sakata M, Wu J, Ishikawa M, Oda K, Ishii K, Iyo M, Hashimoto K, Ishiwata K. Preclinical and the first clinical studies on [C-11]CHIBA-1001 for mapping alpha 7 nicotinic receptors by positron emission tomography. Ann Nuc Med. 2009;23:301–9. doi: 10.1007/s12149-009-0240-x. [DOI] [PubMed] [Google Scholar]
- 27.Conroy WG, Liu ZP, Nai Q, Coggan JS, Berg DK. PDZ-containing proteins provide a functional postsynaptic scaffold for nicotinic receptors in neurons. Neuron. 2003;38:759–71. doi: 10.1016/s0896-6273(03)00324-6. [DOI] [PubMed] [Google Scholar]
- 28.Jones IW, Wonnacott S. Precise localization of alpha 7 nicotinic acetylcholine receptors on glutamatergic axon terminals in the rat ventral tegmental area. J Neurosci. 2004;24:11244–52. doi: 10.1523/JNEUROSCI.3009-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bertolino M, Kellar KJ, Vicini S, Gillis RA. Nicotinic receptor mediates spontaneous GABA release in the rat dorsal motor nucleus of the vagus. Neurosci. 1997;79:671–81. doi: 10.1016/s0306-4522(97)00026-2. [DOI] [PubMed] [Google Scholar]
- 30.Fisher JL, Dani JA. Nicotinic receptors on hippocampal cultures can increase synaptic glutamate currents while decreasing the NMDA-receptor component. Neuropharmacol. 2000;39:2756–69. doi: 10.1016/s0028-3908(00)00102-7. [DOI] [PubMed] [Google Scholar]
- 31.McGehee DS, Role LW. Presynaptic ionotropic receptors. Curr Opin Neurobiol. 1996;6:342–9. doi: 10.1016/s0959-4388(96)80118-8. [DOI] [PubMed] [Google Scholar]
- 32.Rousseau SJ, Jones IW, Pullar IA, Wonnacott S. Presynaptic alpha 7 and non-alpha 7 nicotinic acetylcholine receptors modulate [H-3]D-aspartate release from rat frontal cortex in vitro. Neuropharmacol. 2005;49:59–72. doi: 10.1016/j.neuropharm.2005.01.030. [DOI] [PubMed] [Google Scholar]
- 33.Schilstrom B, Fagerquist MV, Zhang X, Hertel P, Panagis G, Nomikos GG, Svensson TH. Putative role of presynaptic alpha 7*nicotinic receptors in nicotine stimulated increases of extracellular levels of glutamate and aspartate in the ventral tegmental area. Synapse. 2000;38:375–83. doi: 10.1002/1098-2396(20001215)38:4<375::AID-SYN2>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 34.Levy RB, Aoki C. alpha 7 nicotinic acetylcholine receptors occur at postsynaptic densities of AMPA receptor-positive and -negative excitatory synapses in rat sensory cortex. J Neurosci. 2002;22:5001–15. doi: 10.1523/JNEUROSCI.22-12-05001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mexal S, Frank M, Berger R, Adams CE, Ross RG, Freedman R, Leonard S. Differential modulation of gene expression in the NMDA postsynaptic density of schizophrenic and control smokers. Mol Brain Res. 2005;139:317–32. doi: 10.1016/j.molbrainres.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 36.Ulloa L, Huston J, Liao H, Rosas-Ballina M, Ochani M, Ocham K, Tracey KJ. The cholinergic anti-inflammatory pathway. J Neuroimmunol. 2006;178:70. [Google Scholar]
- 37.Rosas-Ballina M, Tracey KJ. Cholinergic control of inflammation. J Internal Med. 2009;265:663–79. doi: 10.1111/j.1365-2796.2009.02098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guan ZZ, Zhang X, Blennow K, Nordberg A. Decreased protein level of nicotinic receptor alpha7 subunit in the frontal cortex from schizophrenic brain. Neuroreport. 1999;10:1779–82. doi: 10.1097/00001756-199906030-00028. [DOI] [PubMed] [Google Scholar]
- 39.Marutle A, Zhang X, Court J, Piggott M, Johnson M, Perry R, Perry E, Nordberg A. Laminar distribution of nicotinic receptor subtypes in cortical regions in schizophrenia. J Chem Neuroanat. 2001;22:115–26. doi: 10.1016/s0891-0618(01)00117-x. [DOI] [PubMed] [Google Scholar]
- 40.Court J, Spurden D, Lloyd S, McKeith I, Ballard C, Cairns N, Kerwin R, Perry R, Perry E. Neuronal nicotinic receptors in dementia with Lewy bodies and schizophrenia: alpha-bungarotoxin and nicotine binding in the thalamus. J Neurochem. 1999;73:1590–7. doi: 10.1046/j.1471-4159.1999.0731590.x. [DOI] [PubMed] [Google Scholar]
- 41.Green WN, Claudio T. Acetylcholine receptor assembly: subunit folding and oligomerization occur sequentially. Cell. 1993;74:57–69. doi: 10.1016/0092-8674(93)90294-z. [DOI] [PubMed] [Google Scholar]
- 42.Leonard S, Gault J, Hopkins J, Logel J, Vianzon R, Short M, Drebing C, Berger R, Venn D, Sirota P, Zerbe G, Olincy A, Ross RG, Adler LE, Freedman R. Association of promoter variants in the alpha 7 nicotinic acetylcholine receptor subunit gene with an inhibitory deficit found in schizophrenia. Arch Gen Psych. 2002;59:1085–96. doi: 10.1001/archpsyc.59.12.1085. [DOI] [PubMed] [Google Scholar]
- 43.Marutle A, Warpman U, Bogdanovic N, Nordberg A. Regional distribution of subtypes of nicotinic receptors in human brain and effect of aging studied by (+/-)-[H- 3]epibatidine. Brain Res. 1998;801:143–9. doi: 10.1016/s0006-8993(98)00558-7. [DOI] [PubMed] [Google Scholar]
- 44.Stone JL, O'Donovan MC, Gurling H, Kirov GK, Blackwood DHR, Corvin A, Craddock NJ, Gill M, Hultman CM, Lichtenstein P, McQuillin A, Pato CN, Ruderfer DM, Owen MJ, St Clair D, Sullivan PF, Sklar P, Purcell SM. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–41. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gault J, Robinson M, Berger R, Drebing C, Logel J, Hopkins J, Moore T, Jacobs S, Meriwether J, Choi MJ, Kim EJ, Walton K, Buiting K, Davis A, Breese CR, Freedman R, Leonard S. Genomic organization and partial duplication of the human α7 neuronal nicotinic acetylcholine receptor gene. Genomics. 1998;52:173–85. doi: 10.1006/geno.1998.5363. [DOI] [PubMed] [Google Scholar]
- 46.Riley B, Williamson M, Collier D, Wilkie H, Makoff A. A 3-Mb map of a large segmental duplication overlapping the alpha 7-nicotinic acetylcholine receptor gene (CHRNA7) at human 15q13-q14. Genomics. 2002;79:197–209. doi: 10.1006/geno.2002.6694. [DOI] [PubMed] [Google Scholar]
- 47.Flomen RH, Davies AF, Di Forti M, La Cascia C, Mackie-Ogilvie C, Murray R, Makoff AJ. The copy number variant involving part of the alpha 7 nicotinic receptor gene contains a polymorphic inversion. Eur J Hum Gen. 2008;16:1364–71. doi: 10.1038/ejhg.2008.112. [DOI] [PubMed] [Google Scholar]
- 48.Locke DP, Archidiacono N, Misceo D, Cardone MF, Deschamps S, Roe B, Rocchi M, Eichler EE. Refinement of a chimpanzee pericentric inversion breakpoint to a segmental duplication cluster. Genome Biol. 2003;4 doi: 10.1186/gb-2003-4-8-r50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Le Novere N, Corringer PJ, Changeux JP. The diversity of subunit composition in nAChRs: Evolutionary origins, physiologic and pharmacologic consequences. J Neurobiol. 2002;53:447–56. doi: 10.1002/neu.10153. [DOI] [PubMed] [Google Scholar]
- 50.Ortells MO, Lunt GG. Evolutionary history of the ligand-gated ion-channel superfamily of receptors. Trends Neurosci. 1995;18:121–7. doi: 10.1016/0166-2236(95)93887-4. [DOI] [PubMed] [Google Scholar]
- 51.Tsunoyama K, Gojobori T. Evolution of nicotinic acetylcholine receptor subunits. Mol Biol Evol. 1998;15:518–27. doi: 10.1093/oxfordjournals.molbev.a025951. [DOI] [PubMed] [Google Scholar]
- 52.de Lucas-Cerrillo AM, Maldifassi MC, Arnalich F, Renart J, Atienza G, Serantes R, Cruces J, Sanchez-Pacheco A, Andres-Mateos E, Montiel C. Function of Partially Duplicated Human alpha 7 Nicotinic Receptor Subunit CHRFAM7A Gene. J Biol Chem. 2011;286:594–606. doi: 10.1074/jbc.M110.180067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stefansson H, Rujescu D, Cichon S, Pietilainen OPH, Ingason A, Steinberg S, Fossdal R, Sigurdsson E, Sigmundsson T, Buizer-Voskamp JE, Hansen T, Jakobsen KD, Muglia P, Francks C, Matthews PM, Gylfason A, Halldorsson BV, Gudbjartsson D, Thorgeirsson TE, Sigurdsson A, Jonasdottir A, Jonasdottir A, Bjornsson A, Mattiasdottir S, Blondal T, Haraldsson M, Magnusdottir BB, Giegling I, Moller HJ, Hartmann A, Shianna KV, Ge DL, Need AC, Crombie C, Fraser G, Walker N, Lonnqvist J, Suvisaari J, Tuulio-Henriksson A, Paunio T, Toulopoulou T, Bramon E, Di Forti M, Murray R, Ruggeri M, Vassos E, Tosato S, Walshe M, Li T, Vasilescu C, Muhleisen TW, Wang AG, Ullum H, Djurovic S, Melle I, Olesen J, Kiemeney LA, Franke B, Sabatti C, Freimer NB, Gulcher JR, Thorsteinsdottir U, Kong A, Andreassen OA, Ophoff RA, Georgi A, Rietschel M, Werge T, Petursson H, Goldstein DB, Nothen MM, Peltonen L, Collier DA, St Clair D, Stefansson K. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–7. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sinkus ML, Lee MJ, Gault J, Logel J, Short M, Freedman R, Christian SL, Lyon J, Leonard S. A 2-base pair deletion polymorphism in the partial duplication of the alpha 7 nicotinic acetylcholine gene (CHRFAM7A) on chromosome 15q14 is associated with schizophrenia. Brain Res. 2009;1291:1–11. doi: 10.1016/j.brainres.2009.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gault J, Hopkins J, Berger R, Drebing C, Logel J, Walton K, Short M, Vianzon R, Olincy A, Ross RG, Adler LE, Freedman R, Leonard S. Comparison of polymorphisms in the α7 nicotinic receptor gene and its partial duplication in schizophrenic and control subjects. Am J Med Gen. 2003;123B:39–49. doi: 10.1002/ajmg.b.20061. [DOI] [PubMed] [Google Scholar]
- 56.Mexal S, Berger R, Logel J, Ross RG, Freedman R, Leonard S. Differential regulation of α7 nicotinic receptor gene (CHRNA7) expression in schizophrenic smokers. J Mol Neurosci. 2010;40:185–195. doi: 10.1007/s12031-009-9233-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tregellas JR, Tanabe J, Rojas DC, Shatti S, Olincy A, Johnson L, Martin LF, Soti F, Kem WR, Leonard S, Freedman R. Effects of an alpha 7-nicotinic agonist on default network activity in schizophrenia. Biol Psych. 2010;69:7–11. doi: 10.1016/j.biopsych.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Flomen RH, Collier DA, Osborne S, Munro J, Breen G, St Clair D, Makoff AJ. Association study of CHRFAM7A copy number and 2bp deletion polymorphisms with schizophrenia and bipolar affective disorder. Am J Med Gen Part B. 2006;141B:571–5. doi: 10.1002/ajmg.b.30306. [DOI] [PubMed] [Google Scholar]
- 59.Petrovsky N, Schmechtig A, Flomen RH, Kumari V, Collier D, Makoff A, Wagner M, Ettinger U. CHRFAM7A copy number and 2-bp deletion polymorphisms and antisaccade performance. Int J Neuropsychopharmacol. 2009;12:267–73. doi: 10.1017/S1461145708009784. [DOI] [PubMed] [Google Scholar]
- 60.Raux G, Bonnet-Brilhault F, Louchart S, Houy E, Gantier R, Levillain D, Allio G, Haouzir S, Petit M, Martinez M, Frebourg T, Thibaut F, Campion D. The-2 bp deletion in exon 6 of the ‘alpha 7-like’ nicotinic receptor subunit gene is a risk factor for the P50 sensory gating deficit. Mol Psych. 2002;7:1006–11. doi: 10.1038/sj.mp.4001140. [DOI] [PubMed] [Google Scholar]
- 61.Dempster EL, Toulopoulou T, McDonald C, Bramon E, Walshe M, Wickham H, Sham PC, Murray RM, Collier DA. Episodic memory performance predicted by the 2bp deletion in exon 6 of the “alpha 7-like” nicotinic receptor subunit gene. Am J Psychiatry. 2006;163:1832–U2. doi: 10.1176/ajp.2006.163.10.1832. [DOI] [PubMed] [Google Scholar]
- 62.Tsuneki H, Kobayashi S, Takagi K, Kagawa S, Tsunoda M, Murata M, Matsuoka T, Wada T, Kurachi M, Kimura I, Sasaoka T. Novel G423S mutation of human alpha 7 nicotinic receptor promotes agonist-induced desensitization by a protein kinase C-dependent mechanism. Mol Pharmacol. 2007;71:777–86. doi: 10.1124/mol.106.030866. [DOI] [PubMed] [Google Scholar]
- 63.Biedler JL, Helson L, Spengler BA. Morphology and growth, tumorigenicity, and cytogenetics of human neuroblastoma cells in continuous culture. Cancer Res. 1973;33:2643–52. [PubMed] [Google Scholar]
- 64.Hoda JC, Gu W, Friedli M, Phillips HA, Bertrand S, Antonarakis SE, Goudie D, Roberts R, Scheffer IE, Marini C, Patel J, Berkovic SF, Mulley JC, Steinlein OK, Bertrand D. Human nocturnal frontal lobe epilepsy: Pharmocogenomic profiles of pathogenic nicotinic acetylcholine receptor beta-subunit mutations outside the ion channel pore. Mol Pharmacol. 2008;74:379–91. doi: 10.1124/mol.107.044545. [DOI] [PubMed] [Google Scholar]
- 65.Hogg RC, Bandelier F, Benoit A, Dosch R, Bertrand D. An automated system for intracellular and intranuclear injection. J Neurosci Meth. 2008;169:65–75. doi: 10.1016/j.jneumeth.2007.11.028. [DOI] [PubMed] [Google Scholar]
- 66.Stephens SH, Franks A, Berger R, Palionyte M, Fingerlin TE, Wagner B, Logel J, Olincy A, Ross RG, Freedman R, Leonard S. Association in the 15q13-14 schizophrenia linkage region. Psych Gen. 2010 doi: 10.1097/YPG.0b013e32834c0c33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Simon P. Q-Gene: processing quantitative real-time RT-PCR data. Bioinformatics. 2003;19:1439–40. doi: 10.1093/bioinformatics/btg157. [DOI] [PubMed] [Google Scholar]
- 68.Hurst RS, Hajos M, Raggenbass M, Wall TM, Higdon NR, Lawson JA, Rutherford-Root KL, Berkenpas MB, Hoffmann WE, Piotrowski DW, Groppi VE, Allaman G, Ogier R, Bertrand S, Bertrand D, Arneric SP. A novel positive allosteric modulator of the alpha 7 neuronal nicotinic acetylcholine receptor: In vitro and in vivo characterization. J Neurosci. 2005;25:4396–405. doi: 10.1523/JNEUROSCI.5269-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bertrand D, Bertrand S, Cassar S, Gubbins E, Li JH, Gopalakrishnan M. Positive Allosteric Modulation of the alpha 7 Nicotinic Acetylcholine Receptor: Ligand Interactions with Distinct Binding Sites and Evidence for a Prominent Role of the M2-M3 Segment. Mol Pharmacol. 2008;74:1407–16. doi: 10.1124/mol.107.042820. [DOI] [PubMed] [Google Scholar]
- 70.Stephens SH, Logel J, Barton A, Franks A, Schultz J, Short M, Dickenson J, James B, Fingerlin T, Wagner B, Hodgkinson CA, Graw S, Ross RG, Freedman R, Leonard S. Association of the 5′-upstream regulatory region of the α7 nicotinic acetylcholine receptor subunit gene (CHRNA7) with schizophrenia. Schizo Res. 2009;109:102–12. doi: 10.1016/j.schres.2008.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Locke DP, Jiang Z, Pertz LM, Misceo D, Archidiacono N, Eichler EE. Molecular evolution of the human chromosome 15 pericentromeric region. Cytogen Genome Res. 2005;108:73–82. doi: 10.1159/000080804. [DOI] [PubMed] [Google Scholar]
- 72.Villiger Y, Szanto I, Jaconi S, Blanchet C, Buisson B, Krause KH, Bertrand D, Romand JA. Expression of an alpha 7 duplicate nicotinic acetylcholine receptor-related protein in human leukocytes. J Neuroimmunol. 2002;126:86–98. doi: 10.1016/s0165-5728(02)00057-7. [DOI] [PubMed] [Google Scholar]
- 73.Benfante R, Antonini RA, De Pizzol M, Gotti C, Clementi F, Locati M, Fornasari D. Expresssion of the α7 nAChR subunit duplicate form (CHRFAM7A) is down-regulated in the monocytic cell line THP-1 on treatment with LPS. J Immunol. 2011;230:74–84. doi: 10.1016/j.jneuroim.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 74.Taly A, Corringer PJ, Guedin D, Lestage P, Changeux JP. Nicotinic receptors: allosteric transitions and therapeutic targets in the nervous system. Nature Rev Drug Discov. 2009;8:733–50. doi: 10.1038/nrd2927. [DOI] [PubMed] [Google Scholar]
- 75.Dani JA, Bertrand D. Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Ann Rev Pharmacol Toxicol. 2007;47:699–729. doi: 10.1146/annurev.pharmtox.47.120505.105214. [DOI] [PubMed] [Google Scholar]
- 76.Chimienti F, Hogg RC, Plantard L, Lehmann C, Brakch N, Fischer J, Huber M, Bertrand D, Hohl D. Identification of SLURP-1 as an epidermal neuromodulator explains the clinical phenotype of Mal de Meleda. Hum Mol Genet. 2003;12:3017–24. doi: 10.1093/hmg/ddg320. [DOI] [PubMed] [Google Scholar]
- 77.Miwa JM, Stevens TR, King SL, Caldarone BJ, Ibanez-Tallon I, Cheng X, Fitzsimonds RM, Pavildes C, Lester HA, Picciotto MR, Heintz N. The prototoxin lynx1 acts on nicotinic acetylcholine receptors to balance neuronal activity and survival in vivo. Neuron. 2006;51:587–600. doi: 10.1016/j.neuron.2006.07.025. [DOI] [PubMed] [Google Scholar]
- 78.Miwa JM, Ibanez-Tallon I, Crabtree GW, Sanchez R, Sali A, Role LW, Heintz N. lynx1, an endogenous toxin-like modulator of nicotinic acetylcholine receptors in the mammalian CNS. Neuron. 1999;23:105–14. doi: 10.1016/s0896-6273(00)80757-6. [DOI] [PubMed] [Google Scholar]
- 79.Morishita H, Miwa JM, Heintz N, Hensch TK. Lynx1, a Cholinergic Brake, Limits Plasticity in Adult Visual Cortex. Science. 2010;330:1238–40. doi: 10.1126/science.1195320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gallowitsch-Puerta M, Tracey KJ. Immunologic role of the cholinergic anti-inflammatory pathway and the nicotinic acetylcholine alpha 7 receptor. Hum Immunol: Patient-Based Res. 2005;1062:209–19. doi: 10.1196/annals.1358.024. [DOI] [PubMed] [Google Scholar]
- 81.Pavlov VA, Ochani M, Yang LH, Gallowitsch-Puerta M, Ochani K, Lin XC, Czura CJ, LaRosa G, Miller EJ, Tracey KJ, Al-Abed Y. Selective alpha7 nicotinic receptor activation improves survival in murine sepsis. FASEB J. 2006;20:A1383. [Google Scholar]
- 82.Pavlov VA, Parrish WR, Rosas-Ballina M, Ochani M, Puerta M, Qchani K, Chavan S, Al-Abed Y, Tracey KJ. Brain acetylcholinesterase activity controls systemic cytokine levels through the cholinergic anti-inflammatory pathway. Brain Behav Immun. 2009;23:41–5. doi: 10.1016/j.bbi.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hsiao EY, Patterson PH. Activation of the maternal immune system induces endocrine changes in the placenta via IL-6. Brain Behav Immun. 2011;25:604–15. doi: 10.1016/j.bbi.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Patterson PH. Immune involvement in schizophrenia and autism: Etiology, pathology and animal models. Behav Brain Res. 2009;204:313–21. doi: 10.1016/j.bbr.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 85.Meyer U, Feldon J. Neural basis of psychosis-related behaviour in the infection model of schizophrenia. Behav Brain Res. 2009;204:322–34. doi: 10.1016/j.bbr.2008.12.022. [DOI] [PubMed] [Google Scholar]
- 86.Rosas-Ballina M, Goldstein RS, Gallowitsch-Puerta M, Yang LH, Valdes-Ferrer SI, Patel NB, Chavan S, Al-Abed Y, Yang H, Tracey KJ. The Selective alpha 7 Agonist GTS-21 Attenuates Cytokine Production in Human Whole Blood and Human Monocytes Activated by Ligands for TLR2, TLR3, TLR4, TLR9, and RAGE. Mol Med. 2009;15:195–202. doi: 10.2119/molmed.2009.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]