

Abstract
Autism spectrum disorder (ASD) is a devastating neurodevelopmental disorder. Over the last decade, evidence has emerged that some children with ASD suffer from undiagnosed co-morbid medical conditions. One of the medical disorders that has been consistently associated with ASD is mitochondrial dysfunction. Individuals with mitochondrial disorders without concomitant ASD manifest dysfunction in multiple high energy organ systems, such as the central nervous, muscular and gastrointestinal systems. Interestingly, these are the identical organ systems affected in a significant number of children with ASD. This finding raises the possibility that mitochondrial dysfunction may be one of the keys that explains the many diverse symptoms observed in some children with ASD. This manuscript will review the importance of mitochondria in human health and disease, the evidence for mitochondrial dysfunction in ASD, the potential role of mitochondrial dysfunction in the co-morbid medical conditions associated with ASD, and how mitochondrial dysfunction can bridge the gap for understanding how these seemingly disparate medical conditions are related. We also review the limitation of this evidence and other possible explanations for these findings. This new understanding of ASD should provide researchers a pathway for understanding the etiopathogenesis of ASD and clinicians the potential to develop medical therapies.
Autism spectrum disorder (ASD) is an umbrella term that describes a heterogeneous group of neurodevelopmental disorders that share a common set of behavioral and cognitive impairments. ASD includes the more specific diagnoses of autistic disorder, Asperger syndrome, and pervasive developmental disorder-not otherwise specified (PDD-NOS). These disorders are defined behaviorally by impairments in communication and social interaction along with restrictive and/or repetitive behaviors. Recent estimates suggest that up to 1 in 110 individuals in the United States are affected with ASD (1).
The etiology and biological basis of ASD is unclear at this time. Although several genetic syndromes, such as Fragile X and Rett syndromes, have been associated with ASD, empirical studies have estimated that genetic syndromes only account for 6–15% of the cases of ASD (2). Thus, the majority of ASD cases are non-syndromic with a clear etiology yet to be defined. The abnormal behavioral and cognitive features that define ASD are believed to arise from dysfunction of the central nervous system (CNS). However, evidence from many fields of medicine has documented multiple non-CNS biological abnormalities associated with ASD.
Over the last decade, we have started to understand that some children with ASD suffer from undiagnosed co-morbid medical conditions such as abnormalities in the peripheral nervous, musculoskeletal, endocrine, gastrointestinal, immune, detoxification, redox regulation and energy generation systems (3). This has changed our view of ASD from a primary CNS disorder to a disorder that affects multiple physiological systems. This recognition has resulted in the evaluation of children with ASD by a wide variety of medical practitioners and leads to the justification for a wide variety of medical treatments for ASD. Although certain novel and complementary medical treatments may prove to be effective, the lack of objective, well controlled clinical studies has limited the recommendations of such treatments (4). One of the difficulties with treating particular physiological (e.g., detoxification) or organ specific (e.g., gastrointestinal) abnormalities is that it is not clear whether or not such abnormalities are primary to the disease process associated with ASD or are secondary to a more basic abnormality of cellular function. Thus, it is not clear whether many ASD treatments address the physiological cause of ASD or whether they merely address specific symptoms. This can be attributed to the fact that the biological mechanisms that underlie the cause of ASD have not been fully elucidated. In fact, few theories have been formulated to connect the seemingly disparate abnormalities in multiple organ and physiologic systems found in ASD. Connecting all of these seemingly disparate abnormalities to a dysfunction in one particular aspect of cellular, physiologic or metabolic function would help researchers better understand the underlying cause of ASD and develop treatments to more effectively treat, or even prevent, ASD.
There is growing evidence that mitochondrial dysfunction may be important in non-syndromic ASD (5), and limited evidence suggests that mitochondrial dysfunction may also be associated with genetic syndromes that include ASD features, such as Rett (6) and Angelman’s (7) syndromes. Mitochondrial dysfunction is an excellent candidate for a basic cellular abnormality that could cause disturbances in a wide range of physiological and organ systems since mitochondria are essential for many basic cellular functions throughout the body (8). Evidence for mitochondrial dysfunction in ASD was first suggested over 20 years ago. Indeed, Coleman and Blass hypothesized that individuals with ASD may have an abnormality in carbohydrate metabolism in 1985 (9) and Lombard proposed that ASD may be a disorder of impaired mitochondrial function and abnormal brain bioenergetics in 1998 (10). Over the past decade, evidence has accumulated that some individuals with ASD have concomitant mitochondrial dysfunction. Estimates of the prevalence of mitochondrial dysfunction in ASD vary widely. While a large population-based study estimated the prevalence of mitochondrial disease in ASD as 7.2% (11), a more recent controlled study in the Journal of the American Medical Association suggested that mitochondrial dysfunction may be present in up to 80% of children with ASD (12). In addition, while some investigators have proposed a “mitochondrial autism” subgroup (13). In a systematic review and meta-analysis, we recently demonstrated that there is significant evidence for both mitochondrial disease and dysfunction in children with ASD (5).
In this review we discuss some of the evidence that supports the notion that mitochondrial dysfunction may contribute to many of the clinical findings associated with ASD as well as explain some of the evidence linking physiological abnormalities to ASD and animal models of ASD that have been linked to mitochondrial dysfunction. We also discuss the limitations of these studies.
Mitochondrial Function and Dysfunction
Mitochondrial medicine is a relatively new and rapidly evolving field of medicine. Mitochondrial disease was once thought to be uncommon but is now considered the most recognized cause of metabolic disease (14), with the minimum birth prevalence of an electron transport chain defect estimated at 1 in 7,634 individuals (15). Mitochondrial disorders have been recently implicated in many neurological and psychiatric diseases, including neurodegenerative diseases such as Huntington’s disease and Friedreich’s ataxia (16) as well as Parkinson’s disease and amyotrophic lateral sclerosis (17).
Mitochondria are distinct cellular organelles that oxidize glucose and fatty acids to generate adenosine triphosphate, the energy carrier in most mammalian cells (8) (Figure 1). Mitochondria contain two plasma membranes, an inner and an outer membrane. One of the key energy-associated pathways, the tricarboxylic acid cycle, is located in the mitochondrial matrix, while the final common pathway for energy production, the electron transport chain, is located on the inner mitochondrial membrane. The number of mitochondria in each cell depends on the cellular energy demands. Low energy cells, such as skin cells, have fewer mitochondria, while cells that require high energy demands, such as muscle, brain, and gastrointestinal (GI) cells possess many mitochondria. Mitochondria are the only organelle in mammalian cells with their own genome. Electron transport chain enzymes are coded by both mitochondrial DNA and nuclear DNA while other mitochondrial enzymes are coded by nuclear DNA (14). In addition to energy production, the mitochondria are intimately involved in programmed cell death (apoptosis), calcium homeostasis, synaptic plasticity and neurotransmitter release (18,19).
Figure 1.
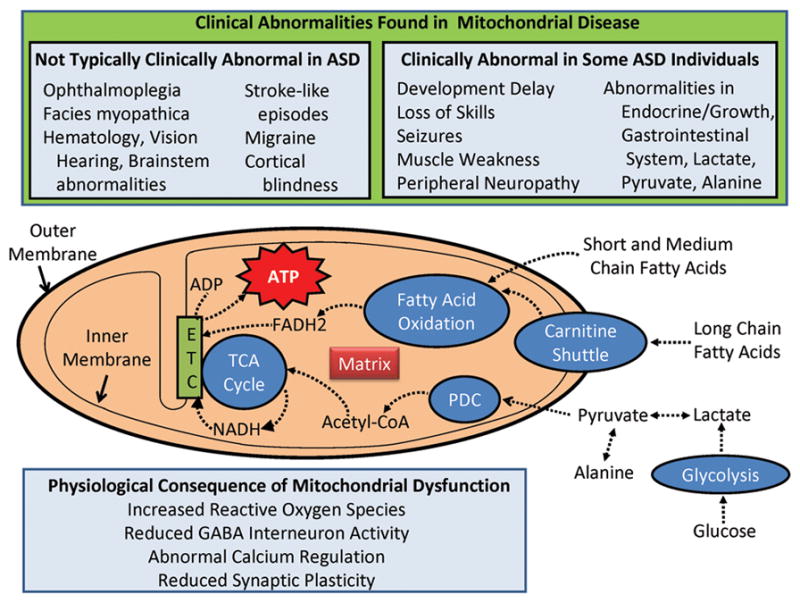
Mitochondrial function, features associated with mitochondrial disease and the features and consequences of mitochondrial dysfunction in autism spectrum disorder (ASD). The top panel outlines some of the features and metabolic markers used to diagnose mitochondrial disease. The features are divided up into those that are not typically clinically abnormal in ASD and those found to be clinically abnormal in some individuals with ASD. Features in which the association with ASD is unclear are not listed in this panel. The mitochondrion is represented diagrammatically in the middle of the figure. Glucose and fatty acids are processed through separate but overlapping pathways to create adenosine triphosphate (ATP). The fatty acid oxidation pathway, tricarboxylic acid (TCA) cycle and the pyruvate dehydrogenase complex are located within the mitochondrion matrix which is located inside the inner mitochondrial membrane. Long chain fatty acids require the carnitine shuttle to enter the mitochondrial matrix while medium and short chain fatty acids do not require the carnitine shuttle. One of the enzymes for the TCA cycle, succinate dehydrogenase, is complex II of the electron transport chain (ETC). Electron carriers, NADH and FADH2, transfer energy from the TCA cycle and fatty acid oxidation pathway to the ETC. The ETC is the final common pathway for the generation of ATP from adenosine diphosphate (ADP). The bottom panel lists some of the physiological consequences of mitochondrial dysfunction.
The diagnosis of mitochondrial disease can be challenging, and is based on several objective clinical, histological, biochemical, molecular, neuroimaging, and enzymatic findings. Several major diagnostic criteria can be used; Table 1 provides the outline of a widely used criterion for diagnosing children with mitochondria disease, the Morava et al. criterion (20). This criterion assigns point values to clinical, metabolic, imaging and morphological features. A mitochondrial disease criteria (MDC) score is calculated by summing the points associated with these features. The MDC score predicts mitochondrial disease as: not likely (<=1), possible (2–4 points), probable (5–7 points), or definite (>=8 points). Notably, some of the laboratory evidence utilized in this criterion requires an invasive muscle biopsy procedure in order to obtain tissue for histology, electron microscopy and/or functional enzymatic assays. Morava et al. (20) outlined a MDC score of >=3 as criterion for performing a further workup, including a muscle biopsy, in order to confirm a diagnosis of mitochondrial disease. The need for a muscle biopsy to obtain additional evidence of mitochondrial disease is a confounding factor for all of the criteria used to diagnose mitochondrial disease since not all families agree to this invasive procedure. This is not an uncommon problem in clinical practice and probably biases the confidence of diagnosing mitochondrial disease in both clinical practice and research studies.
Table 1.
One clinical criteria used for the diagnosis of mitochondria disease
| Probably associated with ASD (% in ASD) | Might be associated with ASD | Probably not associated with ASD | ||
|---|---|---|---|---|
| I. Clinical signs and symptoms, 1 point/symptom (max. 4 points) | A. Muscular presentation (max. 2 points) | Muscle weakness (myopathies) | Abnormal EMG Exercise intolerance Rhabdomyolysis |
Ophthalmoplegia† Facies myopathica |
| B. CNS presentation (max. 2 points) | Developmental delay (100%) Loss of skills (33%) Seizures (25%) |
Extrapyramidal signs Myoclonus Pyramidal signs |
Stroke-like episode Migraine Cortical Blindness Brainstem involvement |
|
| C. Multisystem disease (max. 3 points) | GI tract (7–91.4%) Endocrine/growth Recurrent/familial (10.9%) Neuropathy |
Heart Kidney |
Vision Hearing Hematology |
|
| II. Metabolic/imaging studies (max. 4 points) | Elevated lactate† (17.1–76.6%) Elevated L/P ratio (27.6%) Elevated alanine† (36.0%) Elevated lactate/MRS (11.1%) |
Elevated CSF lactate†, protein and alanine Urinary TA excretion† Stroke-like picture/MRI |
Ethylmalonic aciduria Leigh syndrome/MRI† (0%) |
|
| III. Morphology (max. 4 points) | Abnormal mitochondria/EM† Reduced COX staining‡ Ragged red/blue fibers‡ |
COX-negative fibers‡ Reduced SDH staining SDH positive blood vessels† |
||
Scores 2 points.
Scores 4 points.
GI = gastrointestinal; L/P = lactate/pyruvate; COX = cytochrome c oxidase; SDH = succinate dehydrogenase; EM = electron microscopy; EMG = electromyography; TA = tricarbon acid.; CSF = Cerebrospinal fluid
Clinical features associated with ASD are also associated with mitochondrial disease
Table 1 divides the Morava et al. criterion into the symptoms and laboratory findings that have been typically associated with ASD, those that might be associated with ASD, and those that are probably not associated with ASD. The association between these features and ASD was determined by a review of the literature and the best estimates of the prevalence of these features in ASD, if one exists, are provided in parentheses.
First, we review clinical features shared by both ASD and mitochondrial disease. Some clinical features are very common in ASD. For example, developmental delay is part of the definition of both autistic disorder and PDD-NOS. Developmental regression (i.e., loss of skills) is also relatively common in these disorders. It is generally believed that approximately one-third of children with ASD have a history of regression (21). Since children with regression will acquire developmental delays as a result of the regression, and some studies have suggested that children with regression have subtle developmental delays prior to the regression (22), many children with regressive-type ASD will have a higher probability of a mitochondrial disorder than children with non-regressive children with ASD.
Some clinical features of mitochondrial dysfunction are not commonly recognized as being associated with ASD. In fact, Weissman et al. (13) suggested that children with ASD and mitochondrial disease appeared to have unusual non-neurologic medical disorders and symptoms not typically associated with idiopathic ASD, particularly constitutional symptoms, such as excessive fatigability. Many of the clinical features reported in Table 1 have been well studied in ASD while others have not. A recent large study from Taiwan of over 3,400 children with ASD demonstrated a significant increase in the prevalence of neurological disorders, including epilepsy, disorders of the peripheral nervous system (i.e., neuropathy) and myopathies, as well as endocrine disorders, but did not provide prevalence (23). Others have reported that up to 25% of individuals with ASD eventually develop clinical seizures (24, 25). The reported prevalence of GI abnormalities in ASD varies widely from 7% to 91.4%. A recent consensus statement published in Pediatrics concluded that it is likely that the prevalence of GI disorders in ASD is high (26, 27). Accelerated growth in head circumference, height and length within the first year of life has been documented in some ASD children in several studies, although the prevalence of this overgrowth has not been studied (28). Finally, a recent large study reported the prevalence of recurrent risk of ASD in a sibling as 10.9% (29).
Some of the features of mitochondrial disease listed in Table 1 might be associated with ASD, but these features have not been well studied in the general ASD population. Given that peripheral neuropathies are associated with abnormal electromyography studies, and that there is an increased incidence of peripheral neuropathy in ASD (23), an increased prevalence of abnormal electromyography is likely in ASD. Exercise intolerance has been found to be a common symptom of children with ASD and concomitant mitochondrial disease (13). Rhabdomyolysis and myoclonus have been reported in ASD associated with mitochondrial disease (30), extrapyramidal signs may be seen in children with ASD and creatine deficiency syndromes (31) and rhabdomyolysis and extrapyramidal signs have been reported in individuals with ASD taking anti-psychotic medications (32, 33). Pyramidal signs are typically seen in neurological disorders, such as cerebral palsy, which may also be associated with ASD (34). Heart and kidney defects are not uncommonly seen in genetic syndrome associated with ASD (2, 35). Other symptoms listed in Table 1 as probably not associated with ASD have not been reported to have a specific association with ASD but also have not been specifically studied in ASD.
The estimation of the prevalence of features listed in Table 1 in ASD is complicated and studies that have provided estimates have notable limitations. One of the most important limitations is the probability of referral bias. For example, children with GI symptoms are more likely to be referred to a GI clinic than those without such symptoms, resulting in prevalence estimates of GI abnormalities derived from GI clinics biased to higher prevalence rates than found in the general ASD population. The other major limitation when considering clinical features of ASD children is the fact that children with ASD often have other co-morbid conditions (e.g., cerebral palsy, genetic syndromes) other than mitochondrial disease that might or might not account for these features. Of course, this is further complicated by the fact that many co-morbid conditions may also be associated with mitochondrial disease (e.g., seizures, cerebral palsy, genetic syndromes). However, from this brief review, we see that some clinical features that define mitochondrial disease are rather prevalent in ASD while other clinical features that are not uncommon in mitochondrial disease are not typically observed in ASD. If a subgroup of children with ASD and mitochondrial disease exists, it would be expected that this ASD subgroup would manifest the multiple clinical features associated with mitochondrial disease. However, the overlap of these various symptoms in ASD has not been well studied. Further research will be needed to examine the prevalence of these clinical features in more detail in children with ASD and document the co-occurrence of these features.
Now we review the prevalence of metabolic and imaging abnormalities characteristic of mitochondrial disease in ASD as defined in Table 1. The prevalence of an elevated lactate in ASD has been examined in six studies varying in sample sizes from 30 to 210 individuals and ranging in prevalence from 17.1% to 76.6% (11, 36–39). One study examining 192 ASD participants reported a prevalence of 27.6% for an elevated lactate-to-pyruvate ratio (36). Another study examining 25 ASD children reported the prevalence of an elevated alanine as 36.0% (40). The prevalence of elevations in cerebrospinal fluid lactate, protein and alanine as well as urinary organic acids has not been well studied in children with ASD, but given the fact that other biomarkers for mitochondrial disease are elevated, it is possible that future studies will reveal an association between these markers of mitochondrial disease and ASD. Only one magnetic resonance spectroscopy study examined lactate levels in 9 children with ASD and demonstrated elevated lactate in 11.1% (41). Additionally, only one large clinical study examined clinical magnetic resonance imaging findings in non-syndromic ASD (42) and reported that 31.9% of participants had T2-weighted white matter abnormalities. It is possible that some of these white matter abnormalities could be described as stroke-like but there is insufficient information in the study to determine whether or not a subset of such children had stroke-like findings. Furthermore, this large study did not describe any cases with Leigh syndrome abnormalities.
This review of the metabolic and imaging abnormalities associated with ASD suggests that approximately one-third of children with ASD have metabolic abnormalities that would contribute to the definition of mitochondrial disease while substantially less have imaging scans consistent with mitochondrial disease. One significant limitation of these studies is that they do not report whether different abnormalities are shared by the same patients since, for the most part, individual studies examined specific metabolic or neuroimaging abnormalities instead of a wide range of metabolic and neuroimaging abnormalities. Further studies will need to examine the overlap in metabolic and neuroimaging abnormalities in ASD to determine the true percentage of children with mitochondrial-type metabolic and neuroimaging abnormalities. These studies also are limited by the factors discussed above, such as potential sampling bias. In addition, metabolic studies are particularly difficult to interpret as improper collection, handing and storage of biological samples can distort the results. For example, it is well known that the use of a tourniquet during collection of blood lactate may artificially increase the value. Some studies have used control patients in order to partially control for blood collection techniques, but for the most part, most studies have relatively small numbers of subjects and/or are uncontrolled. One large population-based study (discussed below) provides more reassuring evidence that the prevalence of an abnormal lactate is truly elevated in ASD (11).
Only a few studies have reported morphological mitochondrial abnormalities in ASD and such reports described children with clear mitochondrial disease. For example, these reports have found reduced cytochrome oxidase staining (13), ragged-red fibers (13, 43, 44), and electron microscopy abnormalities (13, 44–49). However, it is difficult to determine the prevalence of such morphological abnormalities in children with ASD since most individuals with ASD do not undergo muscle biopsy unless mitochondrial disease is strongly considered.
Given the fact that children with ASD have been reported to have features suggestive of a possible mitochondrial disease, it is easy to see why some have suggested that mitochondrial and other metabolic disease should be carefully considered in children with ASD (50–53). However, the limitations of prevalence studies, and the lack of knowledge of whether there is a subgroup that manifests multiple abnormalities, makes it difficult to determine the true prevalence of a subgroup of ASD children with concomitant mitochondrial disease.
Studies examining the prevalence of mitochondrial disease in ASD
Although several studies have reported the prevalence of mitochondrial disease in ASD, one study was particularly well designed to prevent sampling bias. This population-based study screened 67,795 children in Portugal and the Azores Islands for ASD (11). Of the 120 children with ASD identified, 69 were screened for mitochondrial disease by examining plasma lactate. Of the 14 patients (20%) with an elevated lactate level, 11 (79%) underwent a deltoid muscle biopsy for confirmation of mitochondrial disease. Five of the 11 (46%) who underwent muscle biopsy met criterion for “definite” mitochondrial disease, yielding a mitochondrial disease prevalence in the general ASD population of 7.2%. This prevalence estimate is markedly higher than the prevalence of mitochondrial disease in the general population (~0.01%) (15).
Although the above study was particularly robust in its design, it is likely that the prevalence of mitochondrial disease was underestimated for several reasons. First, individuals with a normal lactate did not undergo a thorough evaluation for mitochondrial disease even though some individuals with ASD and concomitant mitochondrial disease are known to have normal lactate levels. An example of this is provided a the recent controlled study which found a prevalence of mitochondrial dysfunction in 80% of children with ASD despite only 20% of such children demonstrating elevated lactate levels and only 10% meeting criteria for definite mitochondrial disease (12). Of course this recent study was limited in sample size and examined mitochondrial function in lymphocytes instead of more typical examinations of muscle and/or skin. The examination of mitochondrial function in lymphocytes in children with ASD is particularly problematic as these children may have immune system abnormalities which might adversely affect mitochondrial function. For example, children with ASD, as compared to controls, have been shown to have elevations in tumor necrosis factor-α in lymphocytes (54) and in the CNS (55,56). Tumor necrosis factor-α is a pro-inflammatory cytokine that can inhibit mitochondrial function (57,58).
Secondly, lactate alone was used to screen for mitochondrial disease in the population study despite the fact that mitochondrial markers other than lactate may have identified ASD children with mitochondrial disease. For example, abnormalities in pyruvate (12, 39), ubiquinone (59), and acyl-carnitines (60), all markers of mitochondrial dysfunction, have been reported in children with ASD. In addition, children with ASD and concomitant mitochondrial disease have been reported to have elevations in ammonia (49) and the alanine-to-lysine ratio (61), two markers not typically considered when conducting a workup for mitochondrial disorders in ASD. Thus, additional studies will be needed with a more comprehensive set of mitochondrial markers to consider the true prevalence of mitochondrial disease in children with ASD.
Physiological abnormalities associated with ASD can be explained by mitochondrial dysfunction
Mitochondria are a predominant source of reactive oxygen species (ROS), especially in the context of abnormal electron transport chain function (16, 62). Individuals with ASD have been shown, as a group, to be under higher oxidative stress and have reduced levels of antioxidants as compared to controls (63–65). It is likely that mitochondrial dysfunction could be the cause of abnormally high levels of oxidative stress found in ASD individuals. This notion is consistent with two studies reporting in vitro mitochondrial function in lymphoblasts obtained from the Autism Genetic Resource Exchange. Higher concentrations of ROS and reactive nitrogen species was found in ASD mitochondria compared to controls in one study (Chauhan A et al., Mitochondrial abnormalities in lymphoblasts from autism, International Meeting for Autism Research, May15–17, 2008, London, England, Abstract 146.17) and a lower mean concentration of reduced mitochondrial glutathione, a key antioxidant that protects the mitochondria, as compared to controls was found in the other study (65). The depletion of reduced glutathione in mitochondria has been associated with impaired mitochondrial function (66) and additional ROS production (67). This may initiate a vicious cycle as increased ROS can further impair mitochondrial function (67, 68).
Mitochondrial function can also contribute to abnormalities in neuronal functional that have been reported in ASD. For example, an imbalance in the excitatory (glutamate) and inhibitory (gamma-aminobutyric acid) neurotransmitter systems has been implicated in the pathogenesis of ASD, with a relative increase in the excitatory-to-inhibitory ratio (69). Mitochondrial dysfunction can lead to reduced synaptic neurotransmitter release in neurons that have high firing rates, such as inhibitory gamma-aminobutyric acid interneurons (19). Thus, mitochondrial dysfunction can result in a reduction in inhibition and the relative increase in the excitatory-to-inhibitory ratio observed in ASD. Furthermore, calcium signaling is essential for the integrity of synaptic function and abnormal calcium signaling has also been implicated in ASD (70, 71). Mitochondria are well known to have a pivotal role in the regulation of plasma membrane calcium channels and transporter activities (72).
Animal models of ASD that demonstrate mitochondrial dysfunction
The maternal ubiquitin protein ligase E3A (UBE3A) deficiency mouse is a model of Angelman’s syndrome, a syndrome that frequently includes ASD features. Recently this mouse model was reported to have abnormal mitochondrial morphology and a partial oxidative phosphorylation defect in complex III in the hippocampus (7). Additionally, the methyl CpG binding protein 2-null mouse is a model of Rett syndrome, a neurodegenerative disorder that includes ASD features. This mouse demonstrates decoupling of the respiratory complexes and overexpression of the nuclear gene for ubiquinol-cytochrome c reductase core protein 1, a gene that codes for a subunit of the electron transport chain complex III (73). Recently one research group has demonstrated that intraventricular injection of propionic acid into the rat results in some behavioral features of ASD such as repetitive behaviors, social interaction problems and hyperactivity. This rat model has been demonstrated to have mitochondrial dysfunction (74–77). However, the biological validity of intraventricular propionic acid injection as an etiological mechanism for causing ASD is rather unclear. Other animal models of ASD exist, but many of them have not been examined for mitochondrial dysfunction. Closer examination of other animal models of ASD will help investigators understand whether or not mitochondrial dysfunction is a common link that helps explain the animal model link to ASD.
Conclusion: Mitochondrial disease and ASD: Is there a connection?
Mitochondrial dysfunction is the most common metabolic abnormality associated with ASD (5, 78). A large population-based study estimated the prevalence of mitochondrial disease in ASD to be approximately 7.2% while a more recent but small controlled study has suggested that the prevalence of mitochondrial dysfunction (at least as measured in lymphocytes) in ASD may be as high as 80% (12). Many clinical features as well as biochemical and physiological abnormalities associated with ASD could be related to mitochondrial disease and/or dysfunction. In addition, the immune, detoxification, and redox regulation systems are highly energy dependent systems and have been shown to be dysfunctional in ASD. Lastly, some animal models of ASD have been reported to have mitochondrial dysfunction. It is important to recognize that many studies examining ASD patients have small sample sizes and that the clinical diagnosis of ASD is quite heterogeneous, perhaps because of the variety of psychological instruments used to diagnose ASD; this finding adds to variability across studies. However, despite the limitations of many studies, there is clear evidence that suggests that mitochondrial dysfunction is important in at least a subset of individuals with ASD. As laboratory tests advance to allow clinicians to more accurately detect genetic and metabolic disorders, investigators will be able to increase the diagnostic yield of children with ASD to determine which individuals have genetic, metabolic, mitochondrial and other underlying disorders.
Mitochondrial dysfunction can unify the seemingly disparate clinical findings and physiological abnormalities associated with ASD. However, it should be noted that even if mitochondrial dysfunction underlies the abnormalities in multiple organs and physiological systems, mitochondrial dysfunction could still be secondary to other biological processes such as elevated ROS or chronic immune dysfunction, both of which have been implicated in ASD (54–56,63–65).
Acknowledgments
Statement of Financial Support: This study was funded in part by the National Institute of Neurological Disorders and Stroke (NINDS), grant no. K23NS046565, and the Jane Botsford Johnson Foundation.
Abbreviation list
- ASD
Autism spectrum disorder
- GI
Gastrointestinal
- MDC
Mitochondrial disease criteria
- PDD-NOS
Pervasive developmental disorder-not otherwise specified
- ROS
Reactive oxygen species
Footnotes
Publisher's Disclaimer: Pediatric Research Articles Ahead of Print contains articles in unedited manuscript form that have been peer-reviewed and accepted for publication. As a service to our readers, we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting and review of the resulting proof before it is published in its final definitive form. Please note that during the production process errors may be discovered, which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rice C for the Autism and Developmental Disabilities Monitoring Network Surveillance Year. 2006. Principal Investigators, Centers for Disease Control and Prevention (CDC) Prevalence of autism spectrum disorders - Autism and Developmental Disabilities Monitoring Network, United States, 2006. MMWR Surveill Summ. 2009;58:1–20. [PubMed] [Google Scholar]
- 2.Schaefer GB, Mendelsohn NJ. Genetics evaluation for the etiologic diagnosis of autism spectrum disorders. Genet Med. 2008;10:4–12. doi: 10.1097/GIM.0b013e31815efdd7. [DOI] [PubMed] [Google Scholar]
- 3.Ming Xue, Brimacombe M, Chaaban J, Zimmerman-Bier B, Wagner GC. Autism spectrum disorders: concurrent clinical disorders. J Child Neurol. 2008;23:6–13. doi: 10.1177/0883073807307102. [DOI] [PubMed] [Google Scholar]
- 4.Rossignol DA. Novel and emerging treatments for autism spectrum disorders: a systematic review. Ann Clin Psychiatry. 2009;21:213–236. [PubMed] [Google Scholar]
- 5.Rossignol DA, Frye RE. Mitochondrial dysfunction in autism spectrum disorders: a systematic review and meta-analysis. Mol Psychiatry. 2011 doi: 10.1038/mp.2010.136. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cornford ME, Philippart M, Jacobs B, Scheibel AB, Vinters HV. Neuropathology of Rett syndrome: case report with neuronal and mitochondrial abnormalities in the brain. J Child Neurol. 1994;9:424–431. doi: 10.1177/088307389400900419. [DOI] [PubMed] [Google Scholar]
- 7.Su H, Fan W, Coskun PE, Vesa J, Gold JA, Jiang YH, Potluri P, Procaccio V, Acab A, Weiss JH, Wallace DC, Kimonis VE. Mitochondrial dysfunction in CA1 hippocampal neurons of the UBE3A deficient mouse model for Angelman syndrome. Neurosci Lett. 2011;487:129–133. doi: 10.1016/j.neulet.2009.06.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haas RH, Parikh S, Falk MJ, Saneto RP, Wolf NI, Darin N, Cohen BH. Mitochondrial disease: a practical approach for primary care physicians. Pediatrics. 2007;120:1326–1333. doi: 10.1542/peds.2007-0391. [DOI] [PubMed] [Google Scholar]
- 9.Coleman M, Blass JP. Autism and lactic acidosis. J Autism Dev Disord. 1985;15:1–8. doi: 10.1007/BF01837894. [DOI] [PubMed] [Google Scholar]
- 10.Lombard J. Autism: a mitochondrial disorder? Med Hypotheses. 1998;50:497–500. doi: 10.1016/s0306-9877(98)90270-5. [DOI] [PubMed] [Google Scholar]
- 11.Oliveira G, Diogo L, Grazina M, Garcia P, Ataide A, Marques C, Miguel T, Borges L, Vicente AM, Oliveira CR. Mitochondrial dysfunction in autism spectrum disorders: a population-based study. Dev Med Child Neurol. 2005;47:185–189. doi: 10.1017/s0012162205000332. [DOI] [PubMed] [Google Scholar]
- 12.Giulivi C, Zhang YF, Omanska-Klusek A, Ross-Inta C, Wong S, Hertz-Picciotto I, Tassone F, Pessah IN. Mitochondrial dysfunction in autism. JAMA. 2010;304:2389–2396. doi: 10.1001/jama.2010.1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weissman JR, Kelley RI, Bauman ML, Cohen BH, Murray KF, Mitchell RL, Kern RL, Natowicz MR. Mitochondrial disease in autism spectrum disorder patients: a cohort analysis. PLoS ONE. 2008;3:e3815. doi: 10.1371/journal.pone.0003815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zeviani M, Bertagnolio B, Uziel G. Neurological presentations of mitochondrial diseases. J Inherit Metab Dis. 1996;19:504–520. doi: 10.1007/BF01799111. [DOI] [PubMed] [Google Scholar]
- 15.Skladal D, Halliday J, Thorburn DR. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain. 2003;126:1905–1912. doi: 10.1093/brain/awg170. [DOI] [PubMed] [Google Scholar]
- 16.Trushina E, McMurray CT. Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience. 2007;145:1233–1248. doi: 10.1016/j.neuroscience.2006.10.056. [DOI] [PubMed] [Google Scholar]
- 17.Martin LJ. Mitochondriopathy in Parkinson disease and amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2006;65:1103–1110. doi: 10.1097/01.jnen.0000248541.05552.c4. [DOI] [PubMed] [Google Scholar]
- 18.Roberts RA, Laskin DL, Smith CV, Robertson FM, Allen EM, Doorn JA, Slikker W. Nitrative and oxidative stress in toxicology and disease. Toxicol Sci. 2009;112:4–16. doi: 10.1093/toxsci/kfp179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anderson MP, Hooker BS, Herbert MR. Bridging from cells to cognition in autism pathophysiology: biological pathways to defective brain function and plasticity. Am J Biochem Biotechnol. 2008;4:167–176. [Google Scholar]
- 20.Morava E, van den Heuvel L, Hol F, de Vries MC, Hogeveen M, Rodenburg RJ, Smeitink JA. Mitochondrial disease criteria: diagnostic applications in children. Neurology. 2006;67:1823–1826. doi: 10.1212/01.wnl.0000244435.27645.54. [DOI] [PubMed] [Google Scholar]
- 21.Stefanatos GA. Regression in autistic spectrum disorders. Neuropsychol Rev. 2008;18:305–319. doi: 10.1007/s11065-008-9073-y. [DOI] [PubMed] [Google Scholar]
- 22.Fernell E, Hedvall A, Norrelgen F, Eriksson M, Hoglund-Carlsson L, Barnevik-Olsson M, Svensson L, Holm A, Westerlund J, Gillberg C. Developmental profiles in preschool children with autism spectrum disorders referred for intervention. Res Dev Disabil. 2010;31:790–799. doi: 10.1016/j.ridd.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 23.Chen CY, Chen KH, Liu CY, Huang SL, Lin KM. Increased risks of congenital, neurologic, and endocrine disorders associated with autism in preschool children: cognitive ability differences. J Pediatr. 2009;154:345–350. doi: 10.1016/j.jpeds.2008.09.043. [DOI] [PubMed] [Google Scholar]
- 24.Volkmar FR, Nelson DS. Seizure disorders in autism. J Am Acad Child Adolesc Psychiatry. 1990;29:127–129. doi: 10.1097/00004583-199001000-00020. [DOI] [PubMed] [Google Scholar]
- 25.Hara H. Autism and epilepsy: a retrospective follow-up study. Brain Dev. 2007;29:486–490. doi: 10.1016/j.braindev.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 26.Buie T, Campbell DB, Fuchs GJ, 3rd, Furuta GT, Levy J, Vandewater J, Whitaker AH, Atkins D, Bauman ML, Beaudet AL, Carr EG, Gershon MD, Hyman SL, Jirapinyo P, Jyonouchi H, Kooros K, Kushak R, Levitt P, Levy SE, Lewis JD, Murray KF, Natowicz MR, Sabra A, Wershil BK, Weston SC, Zeltzer L, Winter H. Evaluation, diagnosis, and treatment of gastrointestinal disorders in individuals with ASDs: a consensus report. Pediatrics. 2010;125:S1–S18. doi: 10.1542/peds.2009-1878C. [DOI] [PubMed] [Google Scholar]
- 27.Buie T, Fuchs GJ, 3rd, Furuta GT, Kooros K, Levy J, Lewis JD, Wershil BK, Winter H. Recommendations for evaluation and treatment of common gastrointestinal problems in children with ASDs. Pediatrics. 2010;125:S19–S29. doi: 10.1542/peds.2009-1878D. [DOI] [PubMed] [Google Scholar]
- 28.Fukumoto A, Hashimoto T, Mori K, Tsuda Y, Arisawa K, Kagami S. Head circumference and body growth in autism spectrum disorders. Brain Dev. 2010 doi: 10.1016/j.braindev.2010.09.004. in press. [DOI] [PubMed] [Google Scholar]
- 29.Constantino JN, Zhang Y, Frazier T, Abbacchi AM, Law P. Sibling recurrence and the genetic epidemiology of autism. Am J Psychiatry. 2010;167:1349–1356. doi: 10.1176/appi.ajp.2010.09101470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Connolly BS, Feigenbaum AS, Robinson BH, Dipchand AI, Simon DK, Tarnopolsky MA. MELAS syndrome, cardiomyopathy, rhabdomyolysis, and autism associated with the A3260G mitochondrial DNA mutation. Biochem Biophys Res Commun. 2010;402:443–447. doi: 10.1016/j.bbrc.2010.10.060. [DOI] [PubMed] [Google Scholar]
- 31.Nasrallah F, Feki M, Kaabachi N. Creatine and creatine deficiency syndromes: biochemical and clinical aspects. Pediatr Neurol. 2010;42:163–171. doi: 10.1016/j.pediatrneurol.2009.07.015. [DOI] [PubMed] [Google Scholar]
- 32.Karakaya P, Yis U, Kurul SH, Turkmen MA. Rhabdomyolysis associated with olanzapine treatment in a child with Autism. Pediatr Emerg Care. 2010;26:41–42. doi: 10.1097/PEC.0b013e3181c39a22. [DOI] [PubMed] [Google Scholar]
- 33.Correia CT, Almeida JP, Santos PE, Sequeira AF, Marques CE, Miguel TS, Abreu RL, Oliveira GG, Vicente AM. Pharmacogenetics of risperidone therapy in autism: association analysis of eight candidate genes with drug efficacy and adverse drug reactions. Pharmacogenomics J. 2010;10:418–430. doi: 10.1038/tpj.2009.63. [DOI] [PubMed] [Google Scholar]
- 34.Mouridsen SE, Rich B, Isager T. A longitudinal study of epilepsy and other central nervous system diseases in individuals with and without a history of infantile autism. Brain Dev. 2010 doi: 10.1016/j.braindev.2010.07.002. in press. [DOI] [PubMed] [Google Scholar]
- 35.Johansson M, Gillberg C, Rastam M. Autism spectrum conditions in individuals with Mobius sequence, CHARGE syndrome and oculo-auriculo-vertebral spectrum: diagnostic aspects. Res Dev Disabil. 2010;31:9–24. doi: 10.1016/j.ridd.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 36.Correia C, Coutinho AM, Diogo L, Grazina M, Marques C, Miguel T, Ataide A, Almeida J, Borges L, Oliveira C, Oliveira G, Vicente AM. Brief report: High frequency of biochemical markers for mitochondrial dysfunction in autism: no association with the mitochondrial aspartate/glutamate carrier SLC25A12 gene. J Autism Dev Disord. 2006;36:1137–1140. doi: 10.1007/s10803-006-0138-6. [DOI] [PubMed] [Google Scholar]
- 37.Germanò E, Gagliano A, Magazù A, Calarese T, Calabrò ME, Bonsignore M, Tortorella G, Calamoneri F. Neurobiology of autism: Study of a sample of autistic children. Minerva Pediatr. 2006;58:109–120. [PubMed] [Google Scholar]
- 38.László A, Horváth E, Eck E, Fekete M. Serum serotonin, lactate and pyruvate levels in infantile autistic children. Clin Chim Acta. 1994;229:205–207. doi: 10.1016/0009-8981(94)90243-7. [DOI] [PubMed] [Google Scholar]
- 39.Moreno H, Borjas L, Arrieta A, Sáez L, Prassad A, Estévez J, Bonilla E. Clinical heterogeneity of the autistic syndrome: a study of 60 families. Invest Clin. 1992;33:13–31. [PubMed] [Google Scholar]
- 40.Arnold GL, Hyman SL, Mooney RA, Kirby RS. Plasma amino acids profiles in children with autism: potential risk of nutritional deficiencies. J Autism Dev Disord. 2003;33:449–454. doi: 10.1023/a:1025071014191. [DOI] [PubMed] [Google Scholar]
- 41.Chugani DC, Sundram BS, Behen M, Lee ML, Moore GJ. Evidence of altered energy metabolism in autistic children. Prog Neuropsychopharmacol Biol Psychiatry. 1999;23:635–641. doi: 10.1016/s0278-5846(99)00022-6. [DOI] [PubMed] [Google Scholar]
- 42.Boddaert N, Zilbovicius M, Philipe A, Robel L, Bourgeois M, Barthélemy C, Seidenwurm D, Meresse I, Laurier L, Desguerre I, Bahi-Buisson N, Brunelle F, Munnich A, Samson Y, Mouren MC, Chabane N. MRI findings in 77 children with non-syndromic autistic disorder. PLoS ONE. 2009;4:e4415. doi: 10.1371/journal.pone.0004415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pancrudo J, Shanske S, Coku J, Lu J, Mardach R, Akman O, Krishna S, Bonilla E, DiMauro S. Mitochondrial myopathy associated with a novel mutation in mtDNA. Neuromuscul Disord. 2007;17:651–654. doi: 10.1016/j.nmd.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fillano JJ, Goldenthal MJ, Rhodes CH, Marin-Garcia J. Mitochondrial dysfunction in patients with hypotonia, epilepsy, autism, and developmental delay: HEADD syndrome. J Child Neurol. 2002;17:435–439. doi: 10.1177/088307380201700607. [DOI] [PubMed] [Google Scholar]
- 45.Shoffner J, Hyams L, Langley GN, Cossette S, Mylacraine L, Dale J, Ollis L, Kuoch S, Bennett K, Aliberti A, Hyland K. Fever plus mitochondrial disease could be risk factors for autistic regression. J Child Neurol. 2010;25:429–434. doi: 10.1177/0883073809342128. [DOI] [PubMed] [Google Scholar]
- 46.Pons R, Andreu AL, Checcarelli N, Vila MR, Engelstad K, Sue CM, Shungu D, Haggerty R, de Vivo DC, DiMauro S. Mitochondrial DNA abnormalities and autistic spectrum disorders. J Pediatr. 2004;144:81–85. doi: 10.1016/j.jpeds.2003.10.023. [DOI] [PubMed] [Google Scholar]
- 47.Gargus JJ, Imtiaz F. Mitochondrial energy-deficient endophenotype in autism. Am J Biochem Biotechnol. 2008;4:198–207. [Google Scholar]
- 48.Chauhan A, Chauhan V, Brown WT. Autism: Oxidative Stress, Inflammation, and Immune Abnormalities. CRC Press; Boca Raton: 2010. [Google Scholar]
- 49.Filipek PA, Juranek J, Smith M, Mays LZ, Ramos ER, Bocian M, Masser-Frye D, Laulhere TM, Modahl C, Spence MA, Gargus JJ. Mitochondrial dysfunction in autistic patients with 15q inverted duplication. Ann Neurol. 2003;53:801–804. doi: 10.1002/ana.10596. [DOI] [PubMed] [Google Scholar]
- 50.Benvenuto A, Moavero R, Alessandrelli R, Manzi B, Curatolo P. Syndromic autism: causes and pathogenetic pathways. World J Pediatr. 2009;5:169–176. doi: 10.1007/s12519-009-0033-2. [DOI] [PubMed] [Google Scholar]
- 51.Manzi B, Loizzo AL, Giana G, Curatolo P. Autism and metabolic diseases. J Child Neurol. 2008;23:307–314. doi: 10.1177/0883073807308698. [DOI] [PubMed] [Google Scholar]
- 52.Zecavati N, Spence SJ. Neurometabolic disorders and dysfunction in autism spectrum disorders. Curr Neurol Neurosci Rep. 2009;9:129–136. doi: 10.1007/s11910-009-0021-x. [DOI] [PubMed] [Google Scholar]
- 53.Frye RE. Autism. In: Carney PR, Geyer JD, editors. Pediatric Practice: Neurology. McGraw-Hill; New York, NY: 2010. pp. 348–361. [Google Scholar]
- 54.Malik M, Sheikh AM, Wen G, Spivack W, Brown WT, Li X. Expression of inflammatory cytokines, Bcl2 and cathepsin D are altered in lymphoblasts of autistic subjects. Immunobiology. 2011;216:80–85. doi: 10.1016/j.imbio.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 55.Chez MG, Dowling T, Patel PB, Khanna P, Kominsky M. Elevation of tumor necrosis factor-alpha in cerebrospinal fluid of autistic children. Pediatr Neurol. 2007;36:361–365. doi: 10.1016/j.pediatrneurol.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 56.Li X, Chauhan A, Sheikh AM, Patil S, Chauhan V, Li XM, Ji L, Brown T, Malik M. Elevated immune response in the brain of autistic patients. J Neuroimmunol. 2009;207:111–116. doi: 10.1016/j.jneuroim.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Samavati L, Lee I, Mathes I, Lottspeich F, Huttemann M. Tumor necrosis factor alpha inhibits oxidative phosphorylation through tyrosine phosphorylation at subunit I of cytochrome c oxidase. J Biol Chem. 2008;283:21134–21144. doi: 10.1074/jbc.M801954200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vempati UD, Diaz F, Barrientos A, Narisawa S, Mian AM, Millan JL, Boise LH, Moraes CT. Role of cytochrome C in apoptosis: increased sensitivity to tumor necrosis factor alpha is associated with respiratory defects but not with lack of cytochrome C release. Mol Cell Biol. 2007;27:1771–1783. doi: 10.1128/MCB.00287-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kurup RK, Kurup PA. A hypothalamic digoxin-mediated model for autism. Int J Neurosci. 2003;113:1537–1559. doi: 10.1080/00207450390231482. [DOI] [PubMed] [Google Scholar]
- 60.Clark-Taylor T, Clark-Taylor BE. Is autism a disorder of fatty acid metabolism? Possible dysfunction of mitochondrial beta-oxidation by long chain acyl-CoA dehydrogenase. Med Hypotheses. 2004;62:970–975. doi: 10.1016/j.mehy.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 61.Poling JS, Frye RE, Shoffner J, Zimmerman AW. Developmental regression and mitochondrial dysfunction in a child with autism. J Child Neurol. 2006;21:170–172. doi: 10.2310/7010.2006.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fernández-Checa JC, Garcia-Ruiz C, Colell A, Morales A, Mari M, Miranda M, Ardite E. Oxidative stress: role of mitochondria and protection by glutathione. Biofactors. 1998;8:7–11. doi: 10.1002/biof.5520080102. [DOI] [PubMed] [Google Scholar]
- 63.James SJ, Melnyk S, Fuchs G, Reid T, Jernigan S, Pavliv O, Hubanks A, Gaylor DW. Efficacy of methylcobalamin and folinic acid treatment on glutathione redox status in children with autism. Am J Clin Nutr. 2009;89:425–430. doi: 10.3945/ajcn.2008.26615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.James SJ, Melnyk S, Jernigan S, Cleves MA, Halsted CH, Wong DH, Cutler P, Bock K, Boris M, Bradstreet JJ, Baker SM, Gaylor DW. Metabolic endophenotype and related genotypes are associated with oxidative stress in children with autism. Am J Med Genet B Neuropsychiatr Genet. 2006;141B:947–956. doi: 10.1002/ajmg.b.30366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.James SJ, Rose S, Melnyk S, Jernigan S, Blossom S, Pavliv O, Gaylor DW. Cellular and mitochondrial glutathione redox imbalance in lymphoblastoid cells derived from children with autism. FASEB J. 2009;23:2374–2383. doi: 10.1096/fj.08-128926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vali S, Mythri RB, Jagatha B, Padiadpu J, Ramanujan KS, Andersen JK, Gorin F, Bharath MM. Integrating glutathione metabolism and mitochondrial dysfunction with implications for Parkinson’s disease: a dynamic model. Neuroscience. 2007;149:917–930. doi: 10.1016/j.neuroscience.2007.08.028. [DOI] [PubMed] [Google Scholar]
- 67.Fernández-Checa JC, Kaplowitz N, Garcia-Ruiz C, Colell A, Miranda M, Mari M, Ardite E, Morales A. GSH transport in mitochondria: defense against TNF-induced oxidative stress and alcohol-induced defect. Am J Physiol. 1997;273:G7–G17. doi: 10.1152/ajpgi.1997.273.1.G7. [DOI] [PubMed] [Google Scholar]
- 68.Wallace DC. Mitochondrial diseases in man and mouse. Science. 1999;283:1482–1488. doi: 10.1126/science.283.5407.1482. [DOI] [PubMed] [Google Scholar]
- 69.Rubenstein JL, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003;2:255–267. doi: 10.1034/j.1601-183x.2003.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K, Tager-Flusberg H, Priori SG, Sanguinetti MC, Keating MT. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119:19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 71.Krey JF, Dolmetsch RE. Molecular mechanisms of autism: a possible role for Ca2+ signaling. Curr Opin Neurobiol. 2007;17:112–119. doi: 10.1016/j.conb.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 72.Demaurex N, Poburko D, Frieden M. Regulation of plasma membrane calcium fluxes by mitochondria. Biochim Biophys Acta. 2009;1787:1383–1394. doi: 10.1016/j.bbabio.2008.12.012. [DOI] [PubMed] [Google Scholar]
- 73.Kriaucionis S, Paterson A, Curtis J, Guy J, Macleod N, Bird A. Gene expression analysis exposes mitochondrial abnormalities in a mouse model of Rett syndrome. Mol Cell Biol. 2006;26:5033–5042. doi: 10.1128/MCB.01665-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.MacFabe DF, Cain DP, Rodriguez-Capote K, Franklin AE, Hoffman JE, Boon F, Taylor AR, Kavaliers M, Ossenkopp KP. Neurobiological effects of intraventricular propionic acid in rats: possible role of short chain fatty acids on the pathogenesis and characteristics of autism spectrum disorders. Behav Brain Res. 2007;176:149–169. doi: 10.1016/j.bbr.2006.07.025. [DOI] [PubMed] [Google Scholar]
- 75.MacFabe DF, Rodríguez-Capote K, Hoffman JE, Franklin AE, Mohammad-Asef Y, Taylor AR, Boon F, Cain DP, Kavaliers M, Possmayer F, Ossenkopp K-P. A novel rodent model of autism: intraventricular infusions of propionic acid increase locomotor activity and induce neuroinflammation and oxidative stress in discrete regions of adult rat brain. Am J Biochem Biotechnol. 2008;4:146–166. [Google Scholar]
- 76.Shultz SR, MacFabe DF, Ossenkopp KP, Scratch S, Whelan J, Taylor R, Cain DP. Intracerebroventricular injection of propionic acid, an enteric bacterial metabolic end-product, impairs social behavior in the rat: implications for an animal model of autism. Neuropharmacology. 2008;54:901–911. doi: 10.1016/j.neuropharm.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 77.Thomas RH, Foley KA, Mepham JR, Tichenoff LJ, Possmayer F, MacFabe DF. Altered brain phospholipid and acylcarnitine profiles in propionic acid infused rodents: further development of a potential model of autism spectrum disorders. J Neurochem. 2010;113:515–529. doi: 10.1111/j.1471-4159.2010.06614.x. [DOI] [PubMed] [Google Scholar]
- 78.Oliveira G, Ataide A, Marques C, Miguel TS, Coutinho AM, Mota-Vieira L, Goncalves E, Lopes NM, Rodrigues V, Carmona da Mota H, Vicente AM. Epidemiology of autism spectrum disorder in Portugal: prevalence, clinical characterization, and medical conditions. Dev Med Child Neurol. 2007;49:726–733. doi: 10.1111/j.1469-8749.2007.00726.x. [DOI] [PubMed] [Google Scholar]