

Abstract
Epithelial-mesenchymal transition (EMT) is a key process in tumor metastatic cascade that is characterized by the loss of cell-cell junctions and cell polarity, resulting in the acquisition of migratory and invasive properties. However, the precise molecular events that initiate this complex EMT process in head and neck cancers are poorly understood. Increasing evidence suggests that tumor microenvironment plays an important role in promoting EMT in tumor cells. We have previously shown that head and neck tumors exhibit significantly higher Bcl-2 expression in tumor-associated endothelial cells and overexpression of Bcl-2 alone in tumor-associated endothelial cells was sufficient to enhance tumor metastasis of oral squamous cell carcinoma in a SCID mouse model. In this study, we show that endothelial cells expressing Bcl-2 (EC-Bcl-2), when co-cultured with head and neck tumor cells (CAL27), significantly enhance EMT-related changes in tumor cells predominantly by the secretion of IL-6. Treatment with recombinant IL-6 or stable IL-6 overexpression in CAL27 cells or immortalized oral epithelial cells (IOE) significantly induced the expression of mesenchymal marker, vimentin, while repressing E-cadherin expression via the JAK/STAT3/Snail signaling pathway. These EMT-related changes were further associated with enhanced tumor and IOE cell scattering and motility. STAT3 knock-down significantly reversed IL-6-mediated tumor and IOE cell motility by inhibiting FAK activation. Furthermore, tumor cells overexpressing IL-6 showed marked increase in lymph node and lung metastasis in a SCID mouse xenograft model. Taken together, these results demonstrate a novel function for IL-6 in mediating EMT in head and neck tumor cells and increasing their metastatic potential.
Keywords: IL-6, head and neck cancer, EMT, tumor metastasis, STAT3
Background
Tumor metastases in head and neck cancer patients almost invariably heralds a poor prognosis with an average survival of 6 months and treatment of the patients is usually palliative (1). Five year survival rates for patients with early stage localized head and neck cancers are over 80% but drop to 40% when the disease has spread to the neck nodes, and to below 20% for patients with distant metastatic disease (2). Uncontrolled cell proliferation and angiogenesis are key characteristics of initiation and early growth of epithelial origin cancers (3). However, it is the subsequent acquisition of motility and invasiveness that leads to the metastatic dissemination of these tumor cells (4). Tumor metastasis is a complex process consisting of multiple individual steps (5). A key process in this metastatic cascade that converts an adherent epithelial cell into a migratory cell, which can invade through the extracellular matrix, is known as epithelial-mesenchymal transition (EMT) (6). Many in vitro and in vivo studies have demonstrated that tumor cells of epithelial origin can acquire mesenchymal phenotype (7, 8) and that these cells are typically seen at the invasive front of primary tumors (4, 9, 10). The role of EMT in tumor metastasis is further highlighted by the observations that acquisition of mesenchymal markers such as vimentin or S100A4 by epithelial cells is associated with increased metastatic potential (11, 12).
Functional loss of E-cadherin in epithelial cell has been considered a hallmark of EMT (13). During tumor progression, E-cadherin can be functionally inactivated or silenced by a number of different mechanisms including somatic mutations (14), down regulation of gene expression through promoter hypermethylation (15), histone deacetylation (16), or transcriptional repression (17). Several EMT-inducing regulators repress E-cadherin transcription via interaction with specific E-boxes of the proximal E-cadherin promoter (7). Most prominent E-cadherin regulators are the snail-related zinc-finger transcription factors (snail and slug), (17, 18) SIP1/ZEB2, (19) δEF-1/ZEB1 (20) and Twist (21). Several studies have shown that snail-related transcriptional factors play a transcriptional and regulatory role in invasion, metastasis, and poor outcome for several epithelial malignancies, including head and neck squamous cell carcinoma (HNSCC) (16, 22). Medelsohn et al have recently reported that Snail is an independent marker of tumor metastasis in patients with HNSCC (23).
A number of studies have highlighted the role of tumor microenvironment in promoting tumor metastasis (24-26). We have recently shown that tumor-associated endothelial cells exhibit significantly higher Bcl-2 expression which is directly correlates with metastatic status of head and neck cancer patients (25, 27). In addition, overexpression of Bcl-2 alone in tumor-associated endothelial cells was sufficient to promote tumor metastasis in a SCID mouse model (25). We designed this study to understand how these tumor-associated endothelial cells may be promoting EMT in head and neck tumor cells. Our results suggest that endothelial cells expressing Bcl-2 induce EMT-related changes in tumor cells predominantly via the secretion of IL-6. IL-6 is a pleiotropic cytokine that is involved in acute phase of inflammation and is a major inducer of C-reactive protein (28). Recently, Lederle et al demonstrated that IL-6 promotes malignant growth of skin squamous cell carcinoma by regulating a complex cytokine and protease network (29). IL-6 is one of main chemokines present in serum samples of head and neck cancer patients and elevated IL-6 levels independently predict tumor recurrence, poor survival and tumor metastasis (30, 31). However, very little is known about the role of IL-6 in head and neck tumor metastasis and the process of EMT.
In this study, we demonstrate that treatment of head neck tumor cells or immortalized oral epithelial cells with recombinant IL-6 or stable IL-6 overexpression significantly induced the expression of vimentin and snail proteins while repressing E-cadherin expression. In addition, overexpression of IL-6 in tumor and normal oral epithelial cells significantly enhanced cell scattering and motility. STAT3 knock-down significantly reversed IL-6-mediated EMT phenotype of tumor and immortalized oral epithelial cells. Furthermore, tumor cells overexpressing IL-6, when implanted in SCID mice, showed marked increase in EMT and metastasis to lymph node and lungs. These results suggest an important role of IL-6 in mediating EMT and metastasis of HNSCC tumor cells.
Methods
Cell culture and reagents
Primary human dermal microvascular endothelial cells (ECs) were purchased from Lonza (Walkersville, MD). ECs were maintained in Endothelial Cell Basal Medium-2 (EBM-2) containing 5% FBS and growth supplements. Head and neck squamous carcinoma cell (HNSCC) line CAL27 (ATCC) was maintained in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% FBS. Immortalized (HPV E6/E7) oral epithelial cells (IOE) were provided by Drs. William Foulkes and Ala-Eddin Al Moustafa (McGill University, Montreal, Quebec, Canada) (32) and cultured in keratinocyte serum-free medium (K-SFM, Invitrogen, Carlsbad, CA). Primary antibodies against E-Cadherin (Cat # 3195), Akt (Cat # 9272), pAkt (Cat # 9271), STAT3 (Cat # 9132), STAT3 (pY705) (Cat # 9131), STAT3 (pS727) (Cat # 9134), p44/42 MAPK (ERK1/2, Cat # 9102), pERK1/2 (Cat # 4377), FAK (Cat # 3285) and Tubulin (Cat # 2148) were purchased from Cell Signaling (Danvers, MA); Snail (Cat # AB70983), pFAK (Cat # AB4803), and Vimentin (Cat # AB20346) were from Abcam; pJAK1 (Cat # 44422G) and pJAK2 (Cat # 44426G) were from Invitrogen (Carlsbad, CA) and IL-6 (Cat # AF-206-NA) was from R&D Systems (Minneapolis, MN). Recombinant IL-6, IL-8 and CXCL1/GRO-alpha proteins were purchased from PeproTech Inc (Rocky Hill, NJ).
Transduction of endothelial cells with Bcl-2 and CAL27 and IOE cells with IL-6
Bcl-2 was introduced into human microvascular endothelial cells (ECs) as described previously (33). Interleukin-6 (IL-6) expression plasmid IL6- pTargeT (Promega, Madison, WI; a kind gift from Dr. Tushar Patel, The Ohio State University) containing full-length IL-6 (34) was amplified using Vent polymerase (New England Biolabs, Beverly, MA). To generate retroviral expression vector containing IL-6, the cDNA for full-length IL-6 was amplified using the forward: 5′-cggaattcTCCAGGAGCCCAGCTATG-3′ and the reverse: 5′-taggatccAATCTGAGGTGCCC ATG-3′ primers and the PCR product was sub cloned at the EcoRI/BamHI site of pLXSN vector (Clontech, Palo Alto, CA). We verified the authenticity of the expression vector by sequencing the plasmid DNA. IL-6 was introduced into CAL27 and IOE cells by using the same protocol as described above for Bcl-2 transduction.
Tumor and endothelial cell co-culture
1 × 105 Endothelial cells overexpressing Bcl-2 (EC-Bcl-2) or endothelial cells transduced with vector alone (EC-VC cells) were layered on top of collagen coated 6-well transwell inserts (3 μM, Greiner Bio-One, Monroe, NC) and these transwell inserts containing endothelial cells were then carefully placed on top of 6-well plates containing tumor cells. After 72 hrs of co-culture, transwell inserts were removed and CAL27 cell lysate was prepared for Western Blotting. In the immunofluorescence staining experiments, CAL27 cells were cultured on top of cover slips in 6-well plates while the rest of the protocol was same as described above. For IL-6 neutralizing experiments, CAL27 cells were co-cultured with EC-Bcl-2 in presence of neutralizing anti-IL-6 antibody (50 ng/ml)) or isotype control antibody.
ELISA
Endothelial cells, CAL27 or IOE cells were cultured in 6-well plates till they were 80% confluent. Cells were washed and fresh media was added. After 24 hours, culture supernatants were collected from 3 separate experiments and cell number in each well was counted. Chemokines levels (IL-8, IL-6 and CXCL1/GRO-α) in culture supernatants were measured using Quantikine human ELISA kits (R&D Systems, Minneapolis, MN) as per manufacturer's instructions and normalized to 1×105 cells.
Transient transfection with siRNA
CAL27 overexpressing IL-6 (CAL27-IL-6) and IOE cells overexpressing IL-6 (IOE-IL-6) cells were transfected with siRNA for Akt, STAT3 or p42/42 MAPK (ERK1/2) using Signal Silence siRNA's from Cell Signaling according to the manufacturer's instructions. Seventy two hours post transfection, cells were either used for migration experiments or whole cell lysates were prepared for Western blotting.
Western Blot Analysis
Whole cell lysates were separated by 4-12% NuPAGE Bis-Tris gels (Invitrogen, Carlsbad, CA) and transferred onto PVDF (GE Healthcare, Waukesha, WI) membranes. Nonspecific binding was blocked by incubating the blots with 3% BSA in Tris buffered saline containing 0.1% Tween-20 (TBST) for 1 hr at room temperature (RT). The blots were then incubated with primary antibody in TBST + 3% BSA at 4°C overnight. After washing with TBST, the blots were incubated with horseradish peroxidase-conjugated sheep anti-mouse IgG (1:10,000) or with donkey anti-rabbit IgG (1:10,000) for 1 hr at RT. An ECL-plus detection system (Amersham Life Sciences, Piscataway, NJ) was used to detect specific protein bands. Protein loading in all the experiments was normalized by stripping the blots and then re-probing with anti-tubulin antibody. Alpha Innotech (San Leandro, CA) imaging software was used to quantify Western blot bands.
Immunofluorescent staining
Endothelial cells, CAL27 cells or IOE cells were cultured in labtech chambers. CAL27 and IOE cells were treated with recombinant IL-6 (50 ng/ml) for different time points. At the end of incubation, cells were fixed with 4% paraformaldehyde for 15 minutes at RT and permeablized by treating with 100% methanol for 10 minutes at -20°C. Next, slides were washed with PBS, blocked with normal goat IgG for 1 hr at room temperature and incubated overnight at 4°C with rabbit anti-E-cadherin and mouse anti-vimentin antibodies. After washing with PBS, chamber slides were incubated with secondary antibodies (goat anti-mouse-IgG-Alexa Fluor 488 and goat anti-rabbit-IgG-Alexa Fluor 594). Chamber slides were then mounted with Prolong gold antifade Reagent with DAPI (Invitrogen). The fluorescent images were captured using Nikon Eclipse 80i microscope with DS-Ri1 camera at 600× magnification and overlaid using NIS-Elements-Basic Research software (Nikon, Melville, NY).
Tumor cell motility assay
Tumor cell motility was examined by Xcelligence system using the RTCA DP instrument (Roche, Mannheim, Germany) as per manufacturer's instructions. In brief, 160 μl of media (DMEM for CAL27 cells and K-SFM for IOE) containing 10% FBS was added to the lower chambers. The upper chamber (sensor surface facing down) was then carefully assembled on top of lower chamber and 50 μl of serum free media was added to the wells. After 1 hr of equilibration with media, 100 μl of cell suspension (50,000 cells/well in serum free media) was added to each well. Cell migration to lower chamber was monitored and expressed as cell migration index at 24 hrs.
Tumor metastasis models
CAL27 transduced with vector alone (CAL27-VC) or CAL27-IL-6 (1×106) and endothelial cells (1×106) were mixed with 100 μl of Matrigel and injected subcutaneously in the flanks of SCID mice as described previously (25). Tumor volume measurements began on day 6 and continued twice a week until the end of the study. The length and width of the tumors were measured using a digital caliper and tumor volumes were calculated using the formula, volume (mm3) = L × W2/2 (length L, mm; width W, mm). After 40 days, primary tumors, regional lymph nodes, and lungs were carefully removed. To label and identify flank draining lymph nodes, 25 μl of 5% Evans Blue dye (Sigma, St. Louis, MO) was injected in the foot pads of animals 30 minutes before euthanizing (35). Primary tumors, lymph nodes and lungs were fixed with 4% paraformaldehyde and then processed for immunohistochemistry. Tumor tissue sections were deparaffinized and antigen retrieval was achieved by pressure cooking in a Decloaking chamber at 120°C for 20 minutes (36). Slides were then washed with TBST, blocked with normal goat IgG for 1 hr, and incubated overnight at 4°C with rabbit anti-pSTAT3 or Rabbit anti-snail or rabbit anti-E-cadherin and mouse anti-vimentin antibodies. After washing with PBS, slides were incubated with secondary antibodies (goat anti-mouse-IgG-Alexa Fluor 488 and goat anti-rabbit-IgG-Alexa Fluor 594). Slides were then mounted with Prolong gold antifade reagent with DAPI (Invitrogen, Carlsbad, CA). The fluorescent images were captured using Nikon Eclipse 80i microscope with DS-Ri1 camera at 400× or 600× magnification and overlaid using NIS-Elements-Basic Research software (Nikon, Melville, NY). Lymph nodes were stained with mouse anti-pan cytokeratin antibody (ab7753, Abcam).
Statistical analysis
Data from all the experiments are expressed as mean ± SEM. Statistical differences were determined by two-way analysis of variance and Student's t test. A p value of <0.05 was considered significant.
Results
Bcl-2 expressing endothelial cells (EC-Bcl-2) promote EMT changes via the secretion of IL-6
We have previously shown that Bcl-2 expression is significantly elevated in tumor-associated blood vessels of head and neck cancer patients as compared to matched control samples (27). Recently, we demonstrated that up-regulation of Bcl-2 in tumor-associated endothelial cells is sufficient to enhance tumor metastasis, in vivo (25). However, the molecular mechanism by which EC-Bcl-2 promotes tumor metastasis is poorly understood. In this study, we examined if secreted factors from EC-Bcl-2 could induce EMT-related changes in head and neck tumor cells. Indeed, co-culture of EC-Bcl-2 along with tumor cells (CAL27) markedly enhanced vimentin expression (76% increase) while significantly decreasing E-cadherin expression (52% decrease) in CAL27 cells (Fig. 1A-B). In addition, co-culture of EC-Bcl-2 with CAL27 also upregulated snail expression, a key repressor of E-cadherin (Fig. 1B). We next examined the chemokine profile of EC-Bcl-2 cells. Culture supernatants from EC-Bcl-2 cells contained significantly higher levels of IL-8, IL-6 and Gro-α as compared to endothelial cells transduced with vector alone (EC-VC) (Fig. 1C). To further examine the role of these chemokines in EMT process, CAL27 cells were treated with recombinant IL-8, IL-6, or Gro-α for 3 days and E-cadherin and vimentin levels were analyzed by Western blotting. Interestingly, only IL-6 was able to significantly induce the expression of vimentin (63%) while repressing E-cadherin expression in CAL27 (51%, Fig. 1D). Similarly, co-culture of CAL27 cells and EC-Bcl-2 in the presence of neutralizing IL-6 antibody significantly reversed IL-6-mediated EMT changes (Fig. 1E-F).
Figure 1. Endothelial cells overexpressing Bcl-2 promotes EMT changes in head and neck cancer cells via the secretion of IL-6.
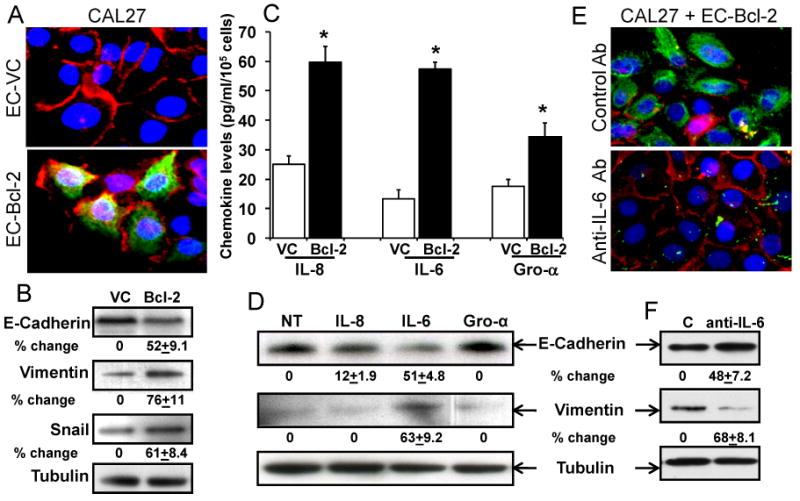
A: CAL27 cells were co-cultured with EC-Bcl-2 or EC-VC cells for 72 hours and stained for E-cadherin (red), vimentin (green) or DNA/nucleus (blue). Photomicrographs were taken at 600×. B: CAL27 cells were co-cultured with EC-Bcl-2 or EC-VC cells for 72 hours and whole cell lysate from each group was Western blotted for E-cadherin, vimentin and snail. Equal protein loading was verified by stripping the blots and reprobing with tubulin antibody. Band density of each protein was normalized with tubulin and expressed as % increase or decrease ± SE as compared to controls. C: Culture supernatants from EC-Bcl-2 or EC-VC cells were examined for IL-8, IL-6 or Gro-α level by ELISA. *, represents a significant increase (p<0.05) in the respective chemokines in EC-Bcl-2 cells as compared to EC-VC. D: E-cadherin and vimentin levels were examined in CAL27 cells treated with different chemokines (IL-8, IL-6 or CXCL1/GRO-α) for 72 hours by Western blotting. E: CAL27 cells were co-cultured with EC-Bcl-2 in presence of neutralizing anti-IL-6 antibody or isotype control antibody and stained for E-cadherin (red), vimentin (green) or DNA/nucleus (blue). Photomicrographs were taken at 600×. F: CAL27 cells were co-cultured with EC-Bcl-2 in presence of neutralizing anti-IL-6 antibody or isotype control antibody (C) and whole cell lysate from each group was Western blotted for E-cadherin, vimentin or tubulin.
IL-6 promotes EMT changes in head and neck squamous cells and immortalized oral epithelial cells
To further examine the role of IL-6 in mediating EMT changes in head and neck squamous carcinoma cells, we cultured CAL27 cells for 3 days in the presence or absence of IL-6. IL-6 treatment of CAL27 cells markedly increased vimentin expression (62%) while at the same time decreasing the expression of E-cadherin (61%) (Fig. 2A-B). IL-6 treatment also significantly increased snail expression (51%, Fig. 2B). Similarly, IL-6 treatment of IOE cells significantly upregulated the expression of vimentin (95%) and snail (64%) while reducing the E-cadherin expression (Fig 2C-D). Next, we examined the IL-6 mediated signaling cascade in CAL27 cells by treating these cells with IL-6 for different time points (0-240 min). IL-6 predominantly activated JAK/STAT3, MAPK, and PI3K/Akt signaling pathways. JAK1/2 activation peaked 10 minutes post IL-6 treatment, whereas STAT3 (pY705 and pS727), ERK1/2 and AKT activation peaked 30 minutes post IL-6 treatment (Fig. 2E).
Figure 2. IL-6 induces the expression of vimentin and snail in CAL27 and IOE cells while downregulating E-Cadherin expression.

A: CAL27 cells were treated with IL-6 for 72 hours and then stained for E-cadherin (red), vimentin (green), or DNA/nucleus (blue). Photomicrographs were taken at 600×. B: E-cadherin, vimentin, and snail expression levels were examined in CAL27 cells treated with IL-6 by Western blotting. Equal protein loading in Western blotting was verified by stripping the blots and reprobing with tubulin antibody. Band density of each protein was normalized with tubulin and expressed as % increase or decrease ± SE as compared to controls from three independent experiments. C: IOE cells treated with IL-6 were stained for E-cadherin (red), vimentin (green) or DNA/nucleus (blue). Photomicrographs were taken at 600×. D: E-cadherin, vimentin, and snail expression levels were examined in IOE cells by Western blotting. E: CAL27 cells were treated with IL-6 for different time points and expression levels of pJAK1, pJAK2, pSTAT3 (Y705), pSTAT3 (S727), pERK1/2, and pAKT were examined by Western blotting. Equal protein loading was verified by stripping the blots and reprobing with tubulin antibody.
IL-6 overexpression in head and neck squamous cells and normal oral epithelial cells induces EMT changes and enhances tumor cell scattering and motility
To further understand the role of IL-6 in the EMT process, we stably overexpressed IL-6 in CAL27 cells (CAL27-IL-6) and IOE cells (IOE-IL-6) using a retroviral vector. IL-6 expression levels in CAL27 and IOE cells were verified by ELISA and Western blotting. CAL27-IL-6 produced more than 8 fold IL-6 as compared to CAL27 cells transduced by vector alone (CAL27-VC) (Supplementary Fig. 1A). CAL27-IL-6 cells also showed significantly higher STAT3 phosphorylation (pY705) as compared to CAL27-VC cells (Supplementary Fig. 1B) thereby suggesting that IL-6 produced by CAL27-IL-6 cells is biologically active. Interestingly, overexpression of IL-6 in CAL27 was also associated with scattering effect (Fig. 3Aa), a hallmark of EMT process. In addition, CAL27-IL-6 cells showed significantly higher vimentin (54%) and snail expression (51%) whereas E-cadherin expression was significantly reduced (53%, Fig. 3B). Furthermore, IL-6 overexpression in CAL27 cells significantly enhanced tumor cell migration (Fig. 3C).
Figure 3. IL-6 overexpression in CAL27 and IOE cells induces EMT phenotype characterized by cell scattering and enhanced motility.

A: CAL-IL-6 or CAL-VC were cultured on labtech chambers. Aa: Cells were photographed using phase contrast microscope (400×) or Ab: stained with E-cadherin (red), vimentin (green), and DNA/nucleus (blue) then photographed using fluorescent microscope (600×). B: Expression levels of E-cadherin, vimentin and snail were examined by Western blotting. Equal protein loading was verified by stripping the blots and reprobing with tubulin antibody. Band density of each protein was normalized with tubulin and expressed as % increase or decrease ± SE as compared to controls. C: CAL27 cell migration was examined by Xcelligence system and expressed as cell migration index. *, represents a significant increase (p<0.05). Da: IOE cells were photographed using phase contrast microscope (400×) or Db: stained with E-cadherin (red), vimentin (green), and DNA/nucleus (blue) and photographed using fluorescent microscope (600×). E: Expression levels of E-cadherin, vimentin, and snail were examined by Western blotting. F: IOE cell migration was examined by Xcelligence system and expressed as cell migration index.
IOE cells produced very low baseline levels of IL-6 and overexpression of IL-6 markedly increased IL-6 production (>30 fold) as compared to IOE-VC (Supplementary Fig. 2A). Similar to CAL27, IL-6 overexpression induced scattering effect in IOE cells (Fig. 3Da) and significantly increased vimentin (63%) and snail expression (70%) while downregulating E-cadherin levels (77%, Fig. 3E). In addition, IL-6 overexpression significantly enhanced IOE cell motility (Fig. 3F).
IL-6 mediates EMT via the activation of JAK-STAT3-snail pathway
Our signaling experiments showed that IL-6 activates 3 distinct signaling pathways: JAK/STAT3, MAPK, and PI3K/Akt (Fig. 2E). To examine which of these pathways is used by IL-6 to mediate its EMT-related effects, we knocked down Akt, STAT3, and ERK1/2 in CAL27 and IOE cells by siRNA treatment. Down regulation of Akt, STAT3, and ERK1/2 protein levels were verified by Western blotting (Fig. 4A-B). Knocking down of STAT3 in CAL27-IL-6 (Fig. 4A and C) and IOE-IL-6 cells (Fig. 4B and D) significantly reversed IL-6 mediated upregulation of vimentin (52% and 48% in CAL27-IL-6 and IOE-IL-6 respectively) and snail protein levels (58% and 64%), whereas Akt and ERK knock down did not significantly alter vimentin and snail levels. Similarly, STAT3 knock down significantly reversed IL-6-mediated loss of E-cadherin expression (67% and 45%), while Akt knock down had no effect and ERK knock down had only partial effect on E-cadherin expression (Fig. 4).
Figure 4. IL-6 mediates EMT changes via the JAK/STAT3 signaling pathway.

Akt, STAT3, or ERK1/2 proteins were knocked down in CAL-IL6 and IOE-IL-6 cells by respective siRNAs. A-B: Expression level of different proteins (Akt, STAT3, ERK1/2, E-cadherin, vimentin, and snail) was examined in CAL-IL-6 (A) or IOE-IL-6 (B) cells by Western blotting. Equal protein loading was verified by stripping the blots and reprobing with tubulin antibody. Band density of each protein was normalized with tubulin and expressed as % increase or decrease ± SE as compared to controls. C-D: Expression levels of E-cadherin (red) and vimentin (green) was examined in CAL-IL-6 cells (C) or IOE-IL-6 cells (D) by fluorescent microscope (600×).
STAT3 knock-down significantly reverses IL-6 mediated CAL27 and IOE motility by inhibiting FAK activation
We further examined the role of different signaling pathways in IL-6 mediated tumor cell and IOE cell migration. As observed with EMT changes, knocking down of STAT3 significantly decreased CAL27-IL-6 and IOE-IL-6 cell migration (Fig. 5A and C). Knocking down of ERK had partial effect while Akt knock down had the least effect on CAL27 and IOE cell migration (Fig. 5A and C). To further understand how STAT3 knock down may be inhibiting CAL27 and IOE motility, we examined the activation profile of focal adhesion kinase (FAK), a key player in cell migration. Interestingly, STAT3 knockdown in CAL27-IL-6 and IOE-IL-6 cells markedly reduced FAK phosphorylation whereas Akt and ERK knockdown had no effect on FAK activation (Fig. 5B and D).
Figure 5. STAT3 knockdown significantly reverses IL-6 mediated cell migration by inhibiting FAK activation.

Akt, STAT3, or ERK1/2 proteins were knocked down in CAL-IL6 and IOE-IL-6 cells by respective siRNAs. A: CAL-IL-6 cell migration was examined by Xcelligence system and expressed as cell migration index. *, represents a significant decrease (p<0.05) in the cell migration as compared to scramble control group. B: Expression level of pFAK in CAL27 was examined by Western blotting. Equal protein loading was verified by stripping the blots and reprobing with total FAK antibody. C: IOE-IL-6 cell migration was examined by Xcelligence system and expressed as cell migration index. D: Expression level of pFAK in IOE cells was examined by Western blotting. Equal protein loading was verified by stripping the blots and reprobing with total FAK antibody.
IL-6 promotes tumor growth, EMT and tumor metastasis, in vivo
To further investigate the role of IL-6 in EMT process and tumor metastasis, we used a SCID mouse xenograft model. CAL27-IL-6 tumors showed a moderate but significant increase in tumor growth as compared to CAL27-VC cells (Fig. 6A). CAL27-IL-6 tumor cells exhibited fibroblastic morphology (spindle shape) in contrast to cobblestone morphology of CAL27-VC cells (Fig. 6B). In addition, CAL27-IL-6 tumors showed markedly elevated levels of activated STAT3 (pY705, Fig. 6C) and snail proteins (Fig. 6D) that were predominantly localized in the nucleus. CAL27-IL-6 tumors also showed decreased E-cadherin and increased vimentin expression (Fig. 6E-F).
Figure 6. IL-6 overexpression in tumor cells promotes tumor growth and EMT changes.
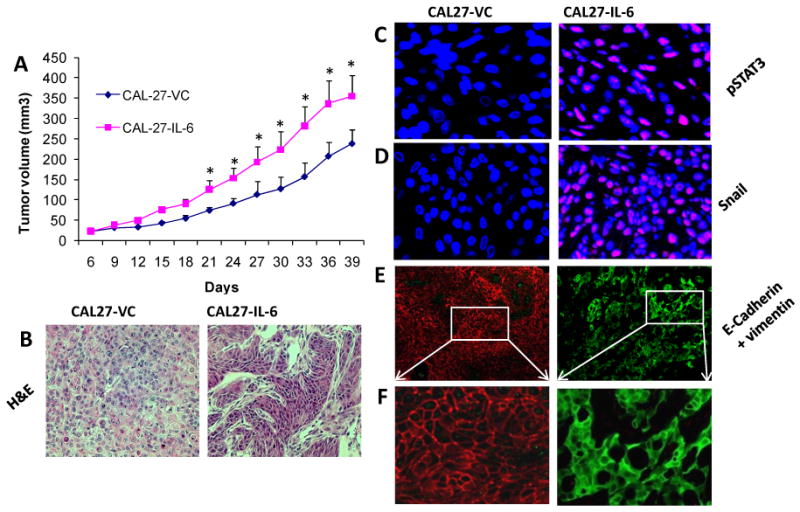
A: Tumor growth curves for CAL-27 cells overexpressing IL-6 (CAL-27-IL-6) or CAL-27 cells expressing vector alone (CAL-27-VC). *, represents a significant difference (p<0.05, n=6). B: H&E stained sections of CAL27-IL-6 and CAL27-VC tumors (400×). C: Tumor samples stained with p-STAT3 (pY705, red) and DAPI (blue) then photographed at 600×. D: Tumor samples stained with Snail (red) and DAPI (blue) then photographed at 600×. E: Tumor samples stained with E-cadherin (red) and vimentin (green) then photographed at 200×. F: Magnified area (600×).
Lymph nodes from animals carrying CAL27-VC tumors were negative for metastatic disease whereas 60% of lymph nodes from animals carrying CAL27-IL-6 tumors were positive for metastatic disease (Fig. 7A-C). Similarly, lungs from animals with CAL27-IL-6 tumors showed marked increase in metastatic nodes (Fig. 7D-E).
Figure 7. IL-6 promotes tumor metastasis in a SCID mouse model.

A: Lymph nodes stained with pan cytokeratin (brown) then photographed at 100×. B: Magnified area from lymph nodes (400×). C: Percentage of lymph nodes positive for tumor cell metastasis. D: H&E stained sections of lungs (200×). The presence of metastatic nodes in lungs is marked with green arrow. E: The number of metastasis nodules present in these sections was counted under microscope. *p, represents a significant difference (p<0.05).
Discussion
EMT is an important biological process that plays a critical role in tumor cell metastasis and is commonly observed in tumor samples from head and neck cancer patients (37, 38). However, the precise molecular events that initiate this complex process of EMT in head and neck cancers are poorly understood. There is increasing evidence that suggests a dynamic interaction between cancer cells and the host microenvironment to support tumor growth and spread (39). Stromal fibroblasts represent a major component of tumor microenvironment and soluble factors from mammary carcinoma-associated fibroblasts were shown to induce an EMT phenotype in PMC42-LA human breast carcinoma cells (40). We have previously shown that head and neck tumors exhibit significantly higher levels of Bcl-2 expression in tumor associated endothelial cells (27) and overexpression of Bcl-2 alone in tumor-associated endothelial cells was sufficient to enhance tumor metastasis of oral squamous cell carcinoma in a SCID mouse model (25).
In this study, we show that endothelial cells expressing Bcl-2 (EC-Bcl-2), when co-cultured with head and neck tumor cells, significantly enhance EMT-related changes in tumor cells. In addition, EC-Bcl-2 cells produced significantly higher levels of IL-8, IL-6, and Gro-α chemokines. Out of these different chemokines, only IL-6 was able to induce EMT-related changes in head and neck cancer cells. Cross-talk between endothelial cells and tumor cells via chemokines has been shown to promote transendothelial migration of cancer cells (41). Such observations reveal how cancer cells might utilize the chemokinetic network to modulate host microenvironment for their own progression. It is possible that multiple cytokines/chemokines may interact in a synergistic way to facilitate tumor cell release. In an elegant study, Lederle et al, showed that IL-6 induces a complex reciprocally regulated cytokine network in tumor cells which leads to the development of malignant and invasive tumors in a human skin carcinoma model (29). Interestingly, we also have previously observed that multiple Nuclear Factor-κB related serum cytokines (IL-6, IL-8, VEGF, HGF and Gro-α) are strongly associated with poor prognosis in HNSCC patients (42). Out of these cytokines/chemokines, IL-6 was an independent predictor of tumor recurrence, poor survival and tumor metastasis (30, 31). In addition to IL-6, HGF has also been shown to regulate cell scattering and tumor metastasis in HNSCC (43).
Therefore, we designed this study to further understand the role and mechanism(s) of IL-6-mediated EMT changes in human head and neck cancer. Our results demonstrate that IL-6 induces EMT changes in head and neck tumor cells via the activation of STAT3/Snail signaling pathway and STAT3 knock down significantly reversed IL-6-mediated EMT changes. These results provides a mechanistic explanation for the prognostic studies that have directly linked IL-6 (30, 31), STAT3 signaling (44), and Snail (23, 45) with tumor recurrence, tumor metastasis, and poor survival in head and neck cancer patients. Recently, Sullivan et al have shown that IL-6 promotes EMT changes in breast cancer by upregulating the expression of Twist (46). IL-6 is also shown to enhance human skin carcinoma cell invasiveness by inducing the overexpression of MMP-1 (29). Furthermore, as tumor cells often contain multiple genetic alterations and some of these genetic alterations may indirectly influence the IL-6 effect, we therefore examined the effect of IL-6 using immortalized oral epithelial cells (IOE). Similar to tumor cells, IL-6 treatment transformed these IOE cells from typical epithelial cell phenotype to more scattered mesenchymal phenotype, a hallmark of EMT process. To our knowledge, this is the first study that has demonstrated that IL-6 can induce EMT changes in immortalized oral epithelial cells and head and neck tumor cells.
We validated our in vitro results by implanting IL-6 overexpressing tumor cells (CAL27-IL-6) in SCID mice. Interestingly, IL-6 overexpression alone was enough to transform a non-metastatic cell line (CAL27) into a metastatic cell line. It is possible that IL-6 mediated EMT changes may also contribute to subsequent steps of the metastatic cascade (e.g. anoikis resistance), as EMT-specific genes are known to play an important role in these metastatic steps (47, 48). Taken together, our results provide novel insight into the role of IL-6 in HNSCC that could explain the direct correlation observed between high serum IL-6 levels and tumor metastasis in head and neck cancer patients.
Supplementary Material
Acknowledgments
Financial support: NIH/NCI-CA133250 (PK) and Joan's fund Research Grant (BK and PK).
Abbreviations
- HNSCC
head and neck squamous cell carcinoma
- EMT
epithelial-mesenchymal transition
- SCID
severe combined immunodeficiency disease
- EC
endothelial cells
- IOE cells
HPV E6/E7 immortalized oral epithelial cells
- STAT3
signal transducers and activators of transcription protein 3
- FAK
focal adhesion kinase
- HGF
hepatocyte growth factor
Footnotes
Conflict of interest: The authors declare that they have no competing interests.
References
- 1.Ferlito A, Rinaldo A, Buckley JG, Mondin V. General Considerations on Distant Metastases from Head and Neck Cancer. ORL. 2001;63:189–91. doi: 10.1159/000055738. [DOI] [PubMed] [Google Scholar]
- 2.Kalavrezos N, Bhandari R. Current trends and future perspectives in the surgical management of oral cancer. Oral Oncol. 2010;46:429–32. doi: 10.1016/j.oraloncology.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 4.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–54. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 5.Ahmad A, Hart IR. Mechanisms of metastasis. Crit Rev Oncol Hematol. 1997;26:163–73. doi: 10.1016/s1040-8428(97)10002-6. [DOI] [PubMed] [Google Scholar]
- 6.Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14:818–29. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 7.Giroldi LA, Bringuier PP, de Weijert M, Jansen C, van Bokhoven A, Schalken JA. Role of E boxes in the repression of E-cadherin expression. Biochem Biophys Res Commun. 1997;241:453–8. doi: 10.1006/bbrc.1997.7831. [DOI] [PubMed] [Google Scholar]
- 8.Hajra KM, Chen DY, Fearon ER. The SLUG zinc-finger protein represses E-cadherin in breast cancer. Cancer Res. 2002;62:1613–8. [PubMed] [Google Scholar]
- 9.Brabletz T, Jung A, Reu S, Porzner M, Hlubek F, Kunz-Schughart LA, et al. Variable beta-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc Natl Acad Sci U S A. 2001;98:10356–61. doi: 10.1073/pnas.171610498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fidler IJ, Poste G. The “seed and soil” hypothesis revisited. Lancet Oncol. 2008;9:808. doi: 10.1016/S1470-2045(08)70201-8. [DOI] [PubMed] [Google Scholar]
- 11.Hu L, Lau SH, Tzang CH, Wen JM, Wang W, Xie D, et al. Association of Vimentin overexpression and hepatocellular carcinoma metastasis. Oncogene. 2004;23:298–302. doi: 10.1038/sj.onc.1206483. [DOI] [PubMed] [Google Scholar]
- 12.Garrett SC, Varney KM, Weber DJ, Bresnick AR. S100A4, a Mediator of Metastasis. Journal of Biological Chemistry. 2006;281:677–80. doi: 10.1074/jbc.R500017200. [DOI] [PubMed] [Google Scholar]
- 13.Cavallaro U, Christofori G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat Rev Cancer. 2004;4:118–32. doi: 10.1038/nrc1276. [DOI] [PubMed] [Google Scholar]
- 14.Berx G, Cleton-Jansen AM, Nollet F, de Leeuw WJ, van de Vijver M, Cornelisse C, et al. E-cadherin is a tumour/invasion suppressor gene mutated in human lobular breast cancers. Embo J. 1995;14:6107–15. doi: 10.1002/j.1460-2075.1995.tb00301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hasegawa M, Nelson HH, Peters E, Ringstrom E, Posner M, Kelsey KT. Patterns of gene promoter methylation in squamous cell cancer of the head and neck. Oncogene. 2002;21:4231–6. doi: 10.1038/sj.onc.1205528. [DOI] [PubMed] [Google Scholar]
- 16.Peinado H, Ballestar E, Esteller M, Cano A. Snail Mediates E-Cadherin Repression by the Recruitment of the Sin3A/Histone Deacetylase 1 (HDAC1)/HDAC2 Complex. Mol Cell Biol. 2004;24:306–19. doi: 10.1128/MCB.24.1.306-319.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bolos V, Peinado H, Perez-Moreno MA, Fraga MF, Esteller M, Cano A. The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: a comparison with Snail and E47 repressors. J Cell Sci. 2003;116:499–511. doi: 10.1242/jcs.00224. [DOI] [PubMed] [Google Scholar]
- 18.Batlle E, Sancho E, Franci C, Dominguez D, Monfar M, Baulida J, et al. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol. 2000;2:84–9. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- 19.Vandewalle C, Comijn J, De Craene B, Vermassen P, Bruyneel E, Andersen H, et al. SIP1/ZEB2 induces EMT by repressing genes of different epithelial cell-cell junctions. Nucl Acids Res. 2005;33:6566–78. doi: 10.1093/nar/gki965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dohadwala M, Wang G, Heinrich E, Luo J, Lau O, Shih H, et al. The role of ZEB1 in the inflammation-induced promotion of EMT in HNSCC. Otolaryngology -- Head and Neck Surgery. 2010;142:753–9. doi: 10.1016/j.otohns.2010.01.034. [DOI] [PubMed] [Google Scholar]
- 21.Vesuna F, van Diest P, Chen JH, Raman V. Twist is a transcriptional repressor of E-cadherin gene expression in breast cancer. Biochem Biophys Res Commun. 2008;367:235–41. doi: 10.1016/j.bbrc.2007.11.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yokoyama K, Kamata N, Hayashi E, Hoteiya T, Ueda N, Fujimoto R, et al. Reverse correlation of E-cadherin and snail expression in oral squamous cell carcinoma cells in vitro. Oral Oncol. 2001;37:65–71. doi: 10.1016/s1368-8375(00)00059-2. [DOI] [PubMed] [Google Scholar]
- 23.Mendelsohn AH, Lai CK, Shintaku IP, Fishbein MC, Brugman K, Elashoff DA, et al. Snail as a novel marker for regional metastasis in head and neck squamous cell carcinoma. American Journal of Otolaryngology. 2011 doi: 10.1016/j.amjoto.2010.11.018. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Padua D, Zhang XHF, Wang Q, Nadal C, Gerald WL, Gomis RR, et al. TGF2 Primes Breast Tumors for Lung Metastasis Seeding through Angiopoietin-like 4. Cell. 2008;133:66–77. doi: 10.1016/j.cell.2008.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumar P, Ning Y, Polverini PJ. Endothelial cells expressing Bcl-2 promotes tumor metastasis by enhancing tumor angiogenesis, blood vessel leakiness and tumor invasion. Lab Invest. 2008;88:740–9. doi: 10.1038/labinvest.2008.46. [DOI] [PubMed] [Google Scholar]
- 26.Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9:239–52. doi: 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumar P, Coltas IK, Kumar B, Chepeha DB, Bradford CR, Polverini PJ. Bcl-2 protects endothelial cells against gamma-radiation via a Raf-MEK-ERK-survivin signaling pathway that is independent of cytochrome c release. Cancer Res. 2007;67:1193–202. doi: 10.1158/0008-5472.CAN-06-2265. [DOI] [PubMed] [Google Scholar]
- 28.Kishimoto T. Interleukin-6: discovery of a pleiotropic cytokine. Arthritis Res Ther. 2006;8 2:S2. doi: 10.1186/ar1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lederle W, Depner S, Schnur S, Obermueller E, Catone N, Just A, et al. IL-6 promotes malignant growth of skin SCCs by regulating a network of autocrine and paracrine cytokines. Int J Cancer. 2011;128:2803–14. doi: 10.1002/ijc.25621. [DOI] [PubMed] [Google Scholar]
- 30.Duffy SA, Taylor JM, Terrell JE, Islam M, Li Y, Fowler KE, et al. Interleukin-6 predicts recurrence and survival among head and neck cancer patients. Cancer. 2008;113:750–7. doi: 10.1002/cncr.23615. [DOI] [PubMed] [Google Scholar]
- 31.Riedel F, Zaiss I, Herzog D, Gotte K, Naim R, Hormann K. Serum levels of interleukin-6 in patients with primary head and neck squamous cell carcinoma. Anticancer research. 2005;25:2761–5. [PubMed] [Google Scholar]
- 32.Al Moustafa AE, Foulkes WD, Benlimame N, Wong A, Yen L, Bergeron J, et al. E6//E7 proteins of HPV type 16 and ErbB-2 cooperate to induce neoplastic transformation of primary normal oral epithelial cells. Oncogene. 2004;23:350–8. doi: 10.1038/sj.onc.1207148. [DOI] [PubMed] [Google Scholar]
- 33.Nor JE, Christensen J, Liu J, Peters M, Mooney DJ, Strieter RM, et al. Up-Regulation of Bcl-2 in microvascular endothelial cells enhances intratumoral angiogenesis and accelerates tumor growth. Cancer Res. 2001;61:2183–8. [PubMed] [Google Scholar]
- 34.Meng F, Yamagiwa Y, Ueno Y, Patel T. Over-expression of interleukin-6 enhances cell survival and transformed cell growth in human malignant cholangiocytes. J Hepatol. 2006;44:1055–65. doi: 10.1016/j.jhep.2005.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harrell MI, Iritani BM, Ruddell A. Lymph node mapping in the mouse. J Immunol Methods. 2008;332:170–4. doi: 10.1016/j.jim.2007.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kumar P, Benedict R, Urzua F, Fischbach C, Mooney D, Polverini P. Combination treatment significantly enhances the efficacy of antitumor therapy by preferentially targeting angiogenesis. Lab Invest. 2005;85:756–67. doi: 10.1038/labinvest.3700272. [DOI] [PubMed] [Google Scholar]
- 37.Chung CH, Parker JS, Ely K, Carter J, Yi Y, Murphy BA, et al. Gene expression profiles identify epithelial-to-mesenchymal transition and activation of nuclear factor-kappaB signaling as characteristics of a high-risk head and neck squamous cell carcinoma. Cancer Res. 2006;66:8210–8. doi: 10.1158/0008-5472.CAN-06-1213. [DOI] [PubMed] [Google Scholar]
- 38.Schipper JH, Frixen UH, Behrens J, Unger A, Jahnke K, Birchmeier W. E-cadherin expression in squamous cell carcinomas of head and neck: inverse correlation with tumor dedifferentiation and lymph node metastasis. Cancer Res. 1991;51:6328–37. [PubMed] [Google Scholar]
- 39.Tse JC, Kalluri R. Mechanisms of metastasis: Epithelial-to-mesenchymal transition and contribution of tumor microenvironment. Journal of Cellular Biochemistry. 2007;101:816–29. doi: 10.1002/jcb.21215. [DOI] [PubMed] [Google Scholar]
- 40.Lebret SC, Newgreen DF, Thompson EW, Ackland ML. Induction of epithelial to mesenchymal transition in PMC42-LA human breast carcinoma cells by carcinoma-associated fibroblast secreted factors. Breast Cancer Res. 2007;9:R19. doi: 10.1186/bcr1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kukreja P, Abdel-Mageed AB, Mondal D, Liu K, Agrawal KC. Up-regulation of CXCR4 Expression in PC-3 Cells by Stromal-Derived Factor-1α (CXCL12) Increases Endothelial Adhesion and Transendothelial Migration: Role of MEK/ERK Signaling Pathway–Dependent NF-ΰB Activation. Cancer Research. 2005;65:9891–8. doi: 10.1158/0008-5472.CAN-05-1293. [DOI] [PubMed] [Google Scholar]
- 42.Allen C, Duffy S, Teknos T, Islam M, Chen Z, Albert PS, et al. Nuclear Factor-kB-Related Serum Factors as Longitudinal Biomarkers of Response and Survival in Advanced Oropharyngeal Carcinoma. Clinical Cancer Research. 2007;13:3182–90. doi: 10.1158/1078-0432.CCR-06-3047. [DOI] [PubMed] [Google Scholar]
- 43.Dong G, Lee TL, Yeh NT, Geoghegan J, Waes CV, Chen Z. Metastatic squamous cell carcinoma cells that overexpress c-Met exhibit enhanced angiogenesis factor expression, scattering and metastasis in response to hepatocyte growth factor. Oncogene. 2004;23:6199–208. doi: 10.1038/sj.onc.1207851. [DOI] [PubMed] [Google Scholar]
- 44.Song JI, Grandis JR. STAT signaling in head and neck cancer. Oncogene. 2000;19:2489–95. doi: 10.1038/sj.onc.1203483. [DOI] [PubMed] [Google Scholar]
- 45.Yang MH, Chang SY, Chiou SH, Liu CJ, Chi CW, Chen PM, et al. Overexpression of NBS1 induces epithelial-mesenchymal transition and co-expression of NBS1 and Snail predicts metastasis of head and neck cancer. Oncogene. 2007;26:1459–67. doi: 10.1038/sj.onc.1209929. [DOI] [PubMed] [Google Scholar]
- 46.Sullivan NJ, Sasser AK, Axel AE, Vesuna F, Raman V, Ramirez N, et al. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene. 2009;28:2940–7. doi: 10.1038/onc.2009.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schulze A, Lehmann K, Jefferies HB, McMahon M, Downward J. Analysis of the transcriptional program induced by Raf in epithelial cells. Genes Dev. 2001;15:981–94. doi: 10.1101/gad.191101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hemavathy K, Ashraf SI, Ip YT. Snail/slug family of repressors: slowly going into the fast lane of development and cancer. Gene. 2000;257:1–12. doi: 10.1016/s0378-1119(00)00371-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.