

Abstract
Fibroblast growth factor (FGF) receptor 1 (FGFR1) protein was expressed as the long and short as well as some truncated forms in ovine fetoplacental artery ex vivo and in vitro. Upon FGF2 stimulation, both the long and short FGFR1s were tyrosine phosphorylated and the PI3K/AKT1 and ERK1/2 pathways were activated in a concentration- and time- dependent manner in ovine fetoplacental artery endothelial (oFPAE) cells. Blockade of the PI3K/AKT1 pathway attenuated FGF2-stimulated cell proliferation and migration as well as tube formation; blockade of the ERK1/2 pathway abolished FGF2-stimulated tube formation and partially inhibited cell proliferation and did not alter cell migration. Both AKT1 and ERK1/2 were co-fractionated with caveolin-1 and activated by FGF2 in the caveolae. Disruption of caveolae by methyl-β-cyclodextrin inhibited FGF2 activation of AKT1 and ERK1/2. FGFR1 was found in the caveolae where it physically binds to caveolin-1. FGF2 stimulated dissociation of FGFR1 from caveolin-1. Downregulation of caveolin-1 significantly attenuated the FGF2-induced activation of AKT1 and ERK1/2 and inhibited FGF2-induced cell proliferation, migration and tube formation in oFPAE cells. Pretreatment with a caveolin-1 scaffolding domain peptide to mimic caveolin-1 overexpression also inhibited these FGF2-induced angiogenic responses. These data demonstrate that caveolae function as a platform for regulating FGF2-induced angiogenesis through spatiotemporally compartmentalizing FGFR1 and the AKT1 and ERK1/2 signaling modules; the major caveolar structural protein caveolin-1 interacts with FGFR1 and paradoxically regulates FGF2-induced activation of PI3K/AKT1 and ERK1/2 pathways that coordinately regulate placental angiogenesis.
Keywords: FGF2, caveolin-1, caveolae, cell signaling, endothelial cells, placenta
INTRODUCTON
Fibroblast growth factors (FGFs) are a large family of multifunctional peptide growth factors, including 28 distinct members encoded by 22 distinct genes in human. They play pivotal roles in many cellular processes including proliferation, differentiation, migration, and cell survival during all stages of prenatal and postnatal life (Beenken and Mohammadi, 2009). FGFs are major growth factors of the placenta, with FGF2 as the dominant form (Arany and Hill, 1998; Biswas et al., 1988; Borowicz et al., 2007). FGF2 are expressed by the trophoblast and endothelial cells in the uteri and placentas in ruminants (Borowicz et al., 2007; Cooke et al., 2009; Pfarrer et al., 2006) and human (Arany and Hill, 1998). Ovine fetoplacental artery endothelial (oFPAE) cells express FGF2 both in vivo (Borowicz et al., 2007; Zheng et al., 1997) and in vitro (Zheng et al., 1999). Fetoplacental FGF2 mRNA expression in vivo (Borowicz et al., 2007) and protein secretion ex vivo are developmentally regulated, greatest at ~day 120-130 of ovine pregnancy (Borowicz et al., 2007; Zheng et al., 1997). Expression of FGF2 changes little in uteroplacental but increases exponentially in fetoplacental tissues in late ovine gestation, implicating that FGF2 functions as a fetal angiogenic factor for branching angiogenesis that occurs mainly in the fetal cotyledonary tissues (Borowicz et al., 2007).
The pleiotropic activities of FGFs are mediated by a family of FGFRs composed of at least 4 distinct receptor tyrosine kinase (RTK) receptors (FGFR1-4) that are encoded by distinct genes (Beenken and Mohammadi, 2009). All FGFRs are expressed in the villous stroma and Hofbauer cells in human placentas across gestation and FGFRs 2-3 in these cells in the 2nd and 3rd trimesters (Anteby et al., 2005a; Anteby et al., 2005b). FGFR4 also expressed in the trophoblast cells across gestation. However, only FGFR1 was found in human placental capillaries in vivo in the 2nd and 3rd trimesters (Anteby et al., 2005b). In ovine placentas, FGFR1 is expressed in fetoplacental throughout gestation in vivo (Borowicz et al., 2007). Endothelial cells of different origins such as oFPAE cells express FGFR1 (Zheng et al., 1999), and in some cases FGFR2; FGFR3-4 are not expressed in any endothelial cells (Presta et al., 2005). Historically, the FGF/FGFR system occupies the central stage of angiogenesis field. The FGF/FGFR system is critical for endothelial functions and angiogenesis (Beenken and Mohammadi, 2009; Makarenkova et al., 2009). It regulates all aspects of angiogenesis including extracellular matrix degradation, endothelial cell migration and proliferation as well as tube formation (Presta et al., 2005), and maintains the integrity of mature vessels (Murakami et al., 2008). In oFPAE cells, FGF2 stimulates multiple common and distinct signaling pathways, including extracellular signal-regulated kinase (ERK1/2), phosphotidylinositol-3-kinase (PI3K)-protein kinase B/Akt1 and endothelial nitric oxide (NO) synthase (eNOS); all are highly relevant to placental endothelial proliferation and NO production (Mata-Greenwood et al., 2010; Mata-Greenwood et al., 2008; Zheng et al., 1999; Zheng et al., 2006). These findings implicate that FGF2 plays a key role in regulating placental angiogenesis and vasodilatation, i.e. two rate-limiting mechanisms for implementing increases in uterine and placental blood flows (Reynolds and Redmer, 2001) for the bidirectional mother-fetus exchanges of nutrients and respiratory gases essential for fetal growth/survival during pregnancy (Magness, 1999). However, how FGF2 signaling control of placental angiogenesis is regulated is poorly understood.
Caveolin-1 (Cav-1) is the principal structural protein (Rothberg et al., 1992) of the Ω-shape plasma membrane microdomains termed caveolae (Bruns and Palade, 1968a; Bruns and Palade, 1968b; Palade and Bruns, 1968) that are abundantly present in terminally differentiated cells (Predescu and Palade, 1993), including ECs (Minshall et al., 2002). Cav-1 is essential for the formation of caveolae as evidenced by the facts that ectopic expression of caveolin-1 leads to caveolae formation (Glenney and Soppet, 1992) and the loss of caveolae in cav-1−/− mice (Drab et al., 2001). Cav-1−/− mice display impaired NO signaling, uncontrolled proliferation of pulmonary ECs, and dramatic changes in vascular permeability (Drab et al., 2001), suggesting a critical role of cav-1/caveolae in angiogenesis. Cav-1 functions as a “scaffolding” protein directly interacting with various signaling molecules, and integrates specific transmembrane signaling pathways activated by various stimuli in the caveolae (Okamoto et al., 1998). Thus, it has been postulated that caveolae functions as a platform for signaling control of cell activity and reactivity (Isshiki et al., 2002a; Isshiki et al., 2002b). For example, we have shown that vascular endothelial growth factor (VEGF)-induced ERK1/2 activation is compartmentalized in the placental endothelial cell caveolae, which is important for endothelial cell migration, proliferation and differentiation (Liao et al., 2009; Liao et al., 2010). However, whether cav-1 plays a role in regulating FGF2-induced placental endothelial cell signaling towards angiogenesis has not been determined.
We hypothesized herein that cav-1/caveolae play a crucial role in regulating the FGF2 activated cell signaling control of placental angiogenesis. We specifically investigated the roles that cav-1 and caveolae play in FGF2-induced cell signaling pathways (i.e., PI3K/AKT1 and ERK1/2) as well as in vitro angiogenesis. Our data show that these pathways activated by FGF2 via FGFR1 are compartmentalized in the caveolae via interactions with cav-1, which are critical for FGF2-induced placental endothelial cell migration, proliferation and differentiation, e.g., angiogenesis.
MATERIALS AND METHODS
Antibodies and Chemicals
Recombinant human FGF2 (157aa) was purchased from R&D systems (Minneapolis, MN). Rabbit polyclonal antibody (pAb) of FGFR1 was from Zymed (South San Francisco, CA). pAbs against phospho-AKT1Ser473(pAKT1), phospho-ERK1/2Thr202/Tyr204(pERK1/2), AKT1, and ERK1/2, were from Cell Signaling (Beverly, MA). Mouse monoclonal antibody (mAb) against human eNOS was from Santa Cruz Biotechnology (Santa Cruz, CA). β-actin mAb was from Ambion (Austin, TX). Horseradish peroxidase-conjugated goat anti-mouse and anti-rabbit immunoglubins (IgG) were obtained from Pierce (Rockford, IL). Cav-1 scaffolding domain (Cav-SD, amino acids 82-101) fused with the N-terminus to the antennapedia internalization sequence (amino acids 43-58) and its negative control peptides (Cav-SDX) were from EMD Calbiochem (Gibbstown, NJ). Methyl-β-cyclodextrin (MβCD) and cholesterol and wortmannin were from Sigma-Aldrich (St. Louis, MO). PD98059 was from Cell Signaling (Danvers, MA); they were dissolved in DMSO with a final concentration less than 0.1%. Cell culture supplies were from Invitrogen/GIBCO (San Diego, CA). All other reagents were from Sigma unless indicated.
Animals and Tissue Sample Collection
The animal (sheep) use protocol was approved by the University of California San Diego Animal Subjects Committee. Secondary fetoplacental artery (PA) segments were collected from late pregnant (D120-130; gestation ~145 days) ewes immediately after sacrifice and fixed in 3.7% paraformaldehyde for preparing paraffin-embedded tissue blocks. A segment of PA (~1~2 cm) per ewe was homogenized in 4 volumes/wet weight of 20 mM Tris-HCl, pH 7.6, 1% Triton X-100, and 20% glycerol with a Pro200 tissue homogenizer (3 × 20s bursts) on ice. The homogenates were cleared by centrifugation and protein content was determined a Bio-Rad procedure using BSA as the standard.
Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
RT-PCR was performed to determine which FGFR isoform(s) were expressed in the ovine placental artery endothelial cells. Since the sequences of ovine fgfr1-4 genes are not available, the specific primer pairs for amplifying the ovine fgfr1-4 mRNAs were designed according to the highly divergent N-terminal sequences of bovine fgfr1-4 genes deposited in NCBI nucleotide database (supplemental table 1). Total RNAs were isolated by using TRIzol® Reagent (Invitrogen, Carlsbad, CA). The RNA samples (2 μg) were reverse transcribed as described previously (Liao et al., 2005). PCR reactions for all fgfr genes were performed with 36 cycles of denaturation at 94°C for 30 sec, annealing at 55°C for 30 sec, and extension at 72°C for 30 sec. The final PCR products were analyzed by agarose gel electrophoresized, stained and photographed. The partial ovine fgfr1 and fgfr3 cDNAs were confirmed by sequencing and deposited into GenBank with accession #JN129424 and #JN129425, respectively. These primers were also designed to match with human fgfr1-4 genes such that human cells and tissues can be used as positive controls. Human umbilical cord vein endothelial cells (HUVEC) and human placental tissues (HP) were used as controls. Collection of human umbilical cords and placental tissues were approved by institutional review board at the University of California, Irvine.
Immunofluorescence Microscopy
Sections (6 μm) of paraffin embedded fetoplacental arterial rings were cut and mounted onto slides. After deparaffinization and rehydration, the tissue sections were blocked in PBS + 1% BSA containing 2% goat serum for 30 min at room temperature, followed by incubation with rabbit anti-human FGFR1 pAb (2 μg/ml) in PBS + 1% BSA overnight at 4°C. Anti-eNOS mAb (2μg/ml) was applied to the samples for 1 hr. After washing (3 × 5 min) in PBS + 0.3% Triton X-100, the samples were incubated with Alexa Fluor488-labeled goat anti-mouse IgG (8 μg/ml, Invitrogen) and rhodamine-labeled donkey anti-rabbit IgG (0.8 μg/ml, Invitrogen) for 1 h at room temperature. After washing, the samples were mounted with ProLong Gold antifade reagent containing 4′-6-Diamidino-2-phenylindole (DAPI, Invitrogen). The samples were analyzed under a Leica fluorescence microscope. Digitalized images were captured and processed by using a high-resolution charge-coupled device (CCD) camera using the Simple PCI software (Compix Inc., Cranberry Township, PA). Omitting first antibody and replacing the first antibodies with rabbit and mouse IgG were used as controls.
For immunocytochemical analysis, cells were fixed with 3.7% paraformaldehyde in PBS on ice for 20 min. After rinsing with ice-cold PBS for three times, cells were permeabilized with 0.1% Triton X-100 in PBS for 20 min on ice. Nonspecific binding was blocked with 5% BSA in PBST (PBS with 0.1% Tween 20) for 1 h at room temperature. The cells were incubated with the primary antibodies overnight and washed (3×10 min) with PBST. Followed by incubation with Rhodamine Red-X conjugated goat anti-rabbit IgG or Alexa Fluor 488 conjugated goat anti-mouse IgG (1:1,000 dilution) for 1 h at room temperature and washing with PBST, cells were mounted in Prolong Gold antifade reagent with DAPI. The samples were analyzed same as the tissue sections.
Cell Culture and Preparation of Total Cell Extracts
oFPAE cells were isolated as previously described (Zheng et al., 1999). Cells were subcultured in MCDB-131 (GIBICO) containing 10% fetal bovine serum (Lonza, Walkersville, MD) and used at passages 7-11. HUVEC was isolated from healthy term placentas by collagenase digestion as described previously (Liao et al., 2009). Cord collection was from University of California Irvine Medical Center Hospital and approved by the Institutional Review Boards. Cells were cultured on gelatin-coated dishes in endothelial cell medium (ScienCell, Carlsbad, CA) containing 5% fetal bovine serum and supplemented with 1% antibiotics and 1% endothelial cell growth supplement and used within five passages. Prior to each experiment, subconfluent (~70-80%) cells were serum starved in M-199 containing 1% FBS, 0.1% BSA, 25 mM HEPES overnight. Following stimulation, total cellular proteins were harvested in a nondenaturing lysis buffer (Chen et al., 2005) and protein content was measured by the BioRad assay kit.
Stable cav-1 knockdown (KD) oFPAE cell line
Stable cav-1 KD oFPAE cell line was established as described (Liao et al., 2009). The cell line was established by transfection with the SureSilencing shRNA plasmid (SuperArray Bioscience Corp., Washington, DC) carrying ovine cav-1 RNA interference sequences: 5′-GGCAGTTGTACCATGCATTAA-3′ or a negative control sequence: 5′-GGAATCTCATTCGATGCATAC-3′. Stable oFPAE cells harboring shRNA plasmids were selected in complete MCDB-131 medium containing 600 μg/ml of Geneticin (G418, Invitrogen). The cells were maintained in complete MCDB131 medium containing 200 μg/ml of G418.
Isolation of caveolae membranes
Cell fractionation for detergent-free isolation of caveolae membranes was performed exactly as previously described (Liao et al., 2009).
Immunoprecipitation (IP), SDS-PAGE and immunoblotting
Equal amounts of proteins (1 mg/sample) were immunoprecipitated with 2 μg pY99 pAb (Santa Cruz Biotechology, Inc., Santa Cruz, CA) to pull down total tyrosine-phosphorylated proteins as described previously (Chen et al., 2005). To verify FGFR1 and caveolin-1 interaction, total oFPAE cell protein extracts (500 μg) were incubated with or without Cav-SD or Cav-SDX peptides (5 μM) for 6 h at 4°C. After pre-clear with protein-G agarose beads, rabbit anti-cav-1 pAb (1 μg) was added to immunoprecipitate cav-1. IP samples or total protein extracts were dissolved in SDS sample buffer and separated by SDS-PAGE. The proteins were transferred on PVDF membranes and analyzed by immunoblotting as described (Liao et al., 2005). The integrated relative densities of bands (proteins) were quantified by multiplying the absorbance of the surface areas using the NIH ImageJ software.
Cell Proliferation
Cell proliferation was measured by CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI) exactly following the manufacturer’s instructions.
Cell Migration
A scratch wound assay was performed to measure unidirectional cell migration as described elsewhere (Liao et al., 2010; Li et al., 2010).
Tube formation
Endothelial cell differentiation was measured by an in vitro tube formation assay as described elsewhere (Liao et al., 2009; Li et al., 2010).
Experimental replication and statistical analysis
All experiments were repeated at least four times. Data were presented as Means ± SD, and analyzed by one-way or two-way analysis of variance using SigmaStat 3.5 (Systat Software Inc., San Jose, CA). For comparison of data between two groups, we used paired Student t-test. Significance was defined as P < 0.05.
RESULTS
FGFR1 is the dominant FGFR isoform expressed in oFPAE cells and activated by FGF2
To determine which fgfr genes are expressed in the oFPAE cells, we measured mRNA levels of all fgfr isoforms in oFPAE cells and ovine placental artery (oPA) tissue samples as well as HUVEC and human healthy term placentas by using RT-PCR. The fgfr1 mRNA was detected in all the samples. The fgfr2 mRNA was only detected in human placentas. The fgfr3 mRNA was weakly expressed in the human placentas, oFPAE cells and oPA. The fgfr4 mRNA was only weakly expressed in HUVEC and human placentas (Fig. 1A). Thus, all four FGFRs are expressed in term human placenta as previously reported (Anteby et al., 2005b); however, among them, only FGFR1 was strongly expressed in ECs including oFPAE cells, HUVEC and oPA. Although FGFR4 is also expressed in HUVEC and FGFR3 in oFPAE cells and oPA, their expression levels are relatively low compared with FGFR1. These findings consist with previous reports that ECs of different origins express FGFR1, other FGFRs are not, or weakly expressed in ECs (Presta et al., 2005; Zheng et al., 1999).
Fig. 1. Fibroblast growth factor receptor 1 (FGFR1) is the dominant FGFR isoform expressed in ovine fetoplacental arteries and is activated by basic FGF (FGF2).

A: mRNA expression of fgfr isoforms in ovine placental artery endothelial oFPAE cells. Total RNAwas subjected to RT-PCR analysis for all fgfr mRNA expressions in oFPAE cells and ovine placental artery (oPA) and human umbilical cord vein endothelial cells (HUVEC) and human term placentas. B: Protein expression and localization of FGFR1 in ovine fetoplacental arteries ex vivo. Paraffin-embedded sections (6 μm) of ovine fetoplacental artery rings were incubated with specific antibodies of FGFR1 and endothelial nitric oxide synthase (eNOS) and followed by rhodamine-labeled donkey anti-rabbit IgG and Alexa Fluor488-labeled goat anti-mouse IgG. Immunofluorescence labeling of FGFR1 (red) and eNOS (green) were detected by immunofluorescence microscopy (200x). Cell nuclei were labeled with DAPI (blue). B: Expression of FGFR1 in oFPAE and HUVE cells. Total cell lysates from oFPAE and HUVE cells were immunoblotted with an anti-FGFR1 antibody. C: Effects of FGF2 on tyrosine phosphorylation of FGFR1. Serum-starved oFPAE cells were treated with FGF2 (10 ng/ml) for up to 1h. Total cell extracts were immunoprecipitated with anti-p-Tyr (PY99) antibody and the immunoprecipitates were analyzed by immunoblotting with anti-FGFR1 antibody. Experiments were repeated three times and the band intensities were quantified and expressed as means ± SD (n=3). *P <0.05 vs. time zero. FGFR1-L/S: FGFR1 long/short form; FGFR1-T: FGFR1 truncated forms; IgG-H, immunoglobin heavy chain.
We then determined FGFR1 protein expression in sections of late pregnant oPA by immunofluorescence microscopy with a specific anti-FGFR1 antibody. Immunofluorescence labeling of FGFR1 protein (red) was strongly observed in the endothelium that was specifically co-labeled with eNOS protein (green). Weaker immunoreactive FGFR1 labeling also was evident in the smooth muscle that was eNOS negative (Fig. 1B). The expression of FGFR1 in oFPAE cells and HUVEC was confirmed by immunoblotting with a specific anti-FGFR1 antibody that was used previously as reported in oFPAE cells (Xu et al., 1992; Zheng et al., 1999). Endothelial FGFR1 protein expressed as multiple bands; these included two major bands corresponding to a long-form (FGFR1-L, ~110 kDa) and a shortform (FGFR1-S, ~75 kDa) and, several truncated forms (<55 kDa) of FGFR1 (Fig. 1C). The multiple bands could be derived from splice variants of FGFR1 and/or post-translational modification (Furdui et al., 2006; Johnson et al., 1990).
Ligand binding renders FGFR1 autophosphorylated on tyrosine residues critical for initiating downstream signaling (Furdui et al., 2006). Treatment with FGF2 (10 ng/ml) stimulated tyrosine phosphorylation of FGFR1-L/S in oFPAE cells in a time-dependent manner. FGF2 stimulated FGFR1 tyrosine phosphorylation within 2 min, maximized at 5 min, and then declined and remained higher up to 60 min (Fig. 1D).
FGF2 promotes angiogenesis in oFPAE cells: role of the PI3K/AKT1 and ERK1/2 pathways
We have previously shown that activation of ERK1/2 and PI3K-AKT1 pathways is important for FGF2-induced oFPAE cell proliferation (Zheng et al., 2008). However, the effects of FGF2 on oFPAE cell migration and tube formation have yet to be determined. We investigated herein whether FGF2 stimulates oFPAE cell migration and tube formation and if yes, what are the roles of ERK1/2 and PI3K/AKT1 in these responses. First, we confirmed that FGF2 activated these two pathways in oFPAE cells. Both AKT1 and ERK1/2 were activated by FGF2 at 5 min in a concentration dependent manner; as little as 0.1 ng/ml of FGF2 stimulated both pathways and phosphorylated levels of AKT1 and ERK1/2 were increased 3 to 4 -fold (p < 0.05) to these of controls (Fig. S1A). Treatment with FGF2 (10 ng/ml) for various times also activated AKT1 and ERK1/2 in a time-dependent manner, beginning at 2 min, maximized at 5-10 min, and declined gradually but remained at significantly higher levels at 1 h (Fig. S1B). When cells were pretreated with the specific inhibitor wortmannin (100 nM, AKT1) or PD98059 (20 μM, ERK1/2), FGF2-induced phosphorylation of AKT1 and ERK1/2 were inhibited completely. Of note, inhibition of ERK1/2 by PD98059 increased the levels of Ser473 phosphorylated AKT1 without affecting the level of total AKT1 (Fig. 2A), suggesting a crosstalk existing between the two pathways in oFPAE cells.
Fig. 2. FGF2 stimulates oFPAE cell proliferation, migration and tube formation – role of PI3K/AKT1 and ERK1/2.
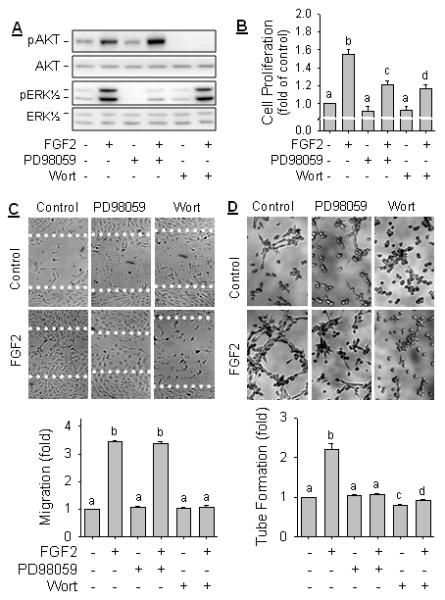
A: oFPAE cells were serum-starved and pretreated with or without 20 μM of PD98059 or 100 nM of wortmannin (Wort) for 1 h, followed by treatment with FGF2 (10 ng/ml) for 5 min. Activation of AKT1 and ERK1/2 were analyzed by immunoblotting with specific antibodies of their phosphorylated and non-phosphorylated forms. Images of a typical experiment shown represents one of three similar studies using cells from different ewes. B: Effects of blockade of ERK1/2 and PI3K/AKT1 on FGF2-induced oFPAE cell proliferation. Cells were treated with or without FGF2 (10 ng/ml) in the presence or absence of 20 μM of PD98059 or 100 nM of wortmannin (wort) for 48h. Untreated cells were used as controls. Experiments were repeated five times and summarized as means ± SD. Bars with different letters differ significantly (p<0.05). C: Effects of blockade of ERK1/2 and PI3K/AKT1 on FGF2-induced oFPAE cell migration determined. Confluent cells were scratch wounded and pretreated with 20 μM PD98059 or 100 nM Wort for 1 h, followed by treatment with or without FGF2 (10 ng/ml) for 20 h for cell migration. Images (200x) of a typical experiment are shown. Data are summarized as bar graphs (means ± SD, n=6) and bars with different letters differ significantly (p<0.05). D: Effects of blockade of ERK1/2 and PI3K/AKT1 on FGF2-induced oFPAE cell tube formation. Cells were seeded on Matrigel and treated with or without FGF2 (10ng/ml) in the presence or absence of 20 μM of PD98059 or 100 nM of Wort for 6h. Tube formation of three randomly chosen fields of each treatment was photographed. Images (200 x) of a typical experiment were shown. Tube-like structures were qualified by counting length and branches and summarized as bar graphs (means ± SD, n = 6). Bars with different letters differ significantly (p<0.05).
Next, we determined the roles of ERK1/2 and PI3K/AKT1 in FGF2-induced angiogenesis in oFPAE cells in vitro. FGF2 potently stimulated cell proliferation, cell migration, and differentiation (tube formation) in oFPAE cells in vitro. Pretreatment with 100 nM wortmannin partially but significantly inhibited FGF2-induced cell proliferation (Fig. 2B). It also suppressed (P < 0.05) FGF2-induced cell migration (Fig. 2B), and decelerated the process of tube formation. Pretreatment with 20 μM PD98059 completely blocked FGF2 induced tube formation (Fig. 2D), and partially inhibited (P < 0.05) cell proliferation (Fig. 2B) without altering cell migration (Fig. 2C).
FGF2-induced AKT1 and ERK1/2 activation in endothelial caveolae: role of cav-1
In purified caveolae membranes of oFPAE cells, AKT1 and ERK1/2 were co-fractionated with cav-1 (Fig. 3A). Treatment with FGF2 (10 ng/ml, 5 min) activated both AKT1 and ERK1/2 in the caveolae similarly as it did in the whole well extracts. These data confirmed our recent findings that ERK1/2 are compartmentalized in the caveolae (Liao et al., 2009) and suggested that AKT1 signaling may also be activated in the caveolae. Pre-treatment with a cholesterol depleting drug MβCD to destroy caveolae, FGF2 no longer activated AKT1 and ERK1/2 (Fig. 3B). These data suggest that integral caveolae are required for FGF2 signaling, similar to the role that they play in the VEGF-regulated angiogenesis in oFPAE cells (Liao et al., 2009; Liao et al., 2010).
Fig. 3. FGF2 activates AKT1 and ERK1/2 in oFPAE cell caveolae.

A: AKT1 and ERK1/2 co-fractionation with caveolin-1 in caveolae. oFPAE cells (~2 × 107) were lysed and subjected to fractionation by sucrose density gradient ultracentrifugation. Fractions were collected and AKT1 and ERK1/2 were measured by immunoblotting. B: Activation of AKT1 and ERK1/2 by FGF2 in the caveolae - effects of methyl-beta-cyclodextrin (MβCD). Cells were pretreated with or without 10 mM MβCD of for 1 h, followed by treatment with FGF2 (10 ng/ml) for 5 min. The cells were subjected to subcellular fractionation by sucrose density gradient ultracentrifugation. Equal amounts (10 μg) of samples from fraction 5 (caveolae membranes) and 9 were analyzed for AKT1 and ERK1/2 activation by immunoblotting with specific antibodies. Images of a typical experiment shown represents one of three similar studies using cells from different ewes.
Cav-1 is the major structural caveolar protein that directly interacts with various signaling molecules via caveolin scaffold domain (aa 82-101), thereby targeting various signaling molecules in the caveolae (Liao et al., 2009; Liao et al., 2010; Park and Han, 2009; Vihanto et al., 2006). We then investigated the role of cav-1 in FGF2-induced AKT1 and ERK1/2 activation in oFPAE cells. Pretreatment with Cav-SD peptide to mimic cav-1 overexpression significantly suppressed the FGF2-induced activations of AKT1 and ERK1/2. Treatment with the control peptide Cav-SDX (5 μM) did not alter FGF2-induced activations of AKT1 and ERK1/2 (Fig. 4A). Treatment with Cav-SD, but not control Cav-SDX, peptide significantly reduced basal and FGF2-stimulated ERK1/2 and AKT1 activation. We also investigated the effects of KD of endogenous cav-1 by its specific shRNA using our established cav-1 KD oFPAE cell model in which endogenous cav-1 protein were reduced by ~85% and caveolae abundance was significantly reduced. In the cav-1 KD cells, FGF2-induced AKT1 and ERK1/2 activation was also significantly inhibited (Fig. 4B).
Fig. 4. Role of caveolin-1 in FGF2-induced activation of AKT1 and ERK1/2 in oFPAE cells.
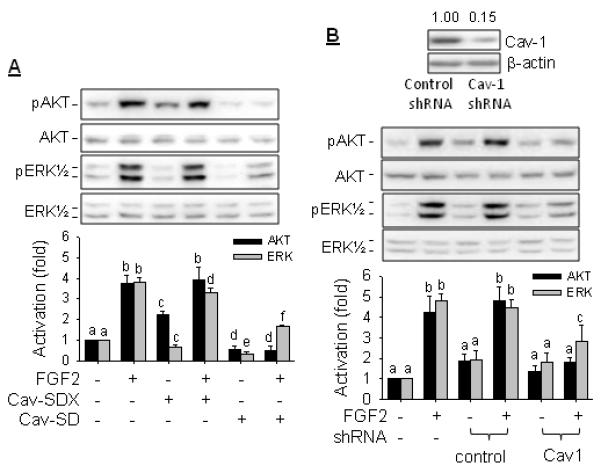
A: Effects of caveolin scaffold domain (Cav-SD) peptides. Cells pretreated with or without 5 μM of Cav-SD or Cav-SDX for 2 h, followed by treatment with FGF2 (10 ng/ml) for 5 min. B: Effects of knockdown of endogenous caveolin-1. oFPAE cells of caveolin-1 specific or control (CTL) shRNA transfected or nontransfected were treated with or without FGF2 (10 ng/ml) for 5 min. Total cell lysates were extracted and were analyzed for cav-1 knockdown and AKT1 and ERK1/2 activation by immunoblotting with specific antibodies. Images of a typical experiment were shown. Data from three independent experiments were summarized as bar graphs (means ± SD). Bars with different letters differ significantly (p<0.05).
Cav-1 regulates FGF2-induced angiogenesis
In the presence of the Cav-SDX peptide (5 μM), FGF2 was able to promote oFPAE cell proliferation, migration and tube formation; however, when the cells were pre-treated with 5 μM Cav-SD peptide, FGF2 no longer stimulated cell proliferation (Fig. 5A), migration (Fig. 5B) and tube formation (Fig. 5C). Of note, treatment with Cav-SD, but not Cav-SDX, peptides even lowered basal and FGF-stimulated cell migration to levels lesser than controls. We further tested the effects of KD of endogenous cav-1 on FGF2-induced in vitro angiogenesis in oFPAE cells. In control shRNA transfected cells, FGF2 can stimulates angiogenesis in vitro; however, in cav-1 KD oFPAE cells FGF2 did not stimulate cell proliferation (Fig 6A) and migration (Fig. 6B) as well as tube formation (Fig. 6C). Cav-1 KD also lowered basal cell migration in comparison to the control cells (Fig. 6B).
Fig. 5. Effects of caveolin-1 scaffolding domain (Cav-SD) peptides on FGF2-induced angiogenesis in oFPAE cells.

The serum-starved oFPAE cells were treated with 10 ng/ml of FGF2 with/without 5 μM of Cav-SD or Cav-SDX and subjected to assays for cell proliferation (A), migration (B), and tube formation (C) as described in Fig. 2. Images (200 x) of a typical experiments for A and B are shown. Data from six (A) and three (B&C) independent experiments were summarized as bar graphs (means ± SD). Bars with different letters differ significantly (p<0.05).
Fig. 6. Effects of downregulation of endogenous caveolin-1 on FGF2-induced angiogenesis in oFPAE cells.

Caveolin-1 (-) cells were serum-starved and treated with 10 ng/ml of FGF2. Cells proliferation assay (A), cell migration assay (B), and cell differentiation assay (C) were performed as described in Fig.2. Bars with different letters differ significantly (p<0.05).
FGFR1 is localized in the caveolae and binds to cav-1
Both long (FGFR1-L) and short (FGFR1-S) forms of FGFR1 were detected in the caveolar membranes by immunoblotting of the caveolae membranes isolated from resting oFPAE cells (Fig. 7A), although the majority of FGFR1 was present in non-caveolar fractions, similar to a previous report in other cells (Song et al., 1996). Double immunofluorescence labeling was used to assess the subcellular localization of cav-1 and FGFR1 in oFPAE cells in situ, co-localization of the two was clearly demonstrated at the plasma membrane (Fig. 7B). We tested the physical association of FGFR1 and cav-1 by co-immunoprecipitation experiments. Both long and short forms of FGFR1 were detected in the cav-1 immunoprecipitates. Moreover, the levels of cav-1 bound FGFR1-L and FGFR1-S were reduced by FGF2 treatment (Fig. 7C). Furthermore, pre-incubation with the synthetic Cav-SD, but not the control Cav-SDX peptides, inhibited the amounts of FGFR1-S immunoprecipitated by the anti-Cav-1 antibody from oFPAE cell lysates (Fig. 7D).
Fig. 7. FGFR1 is localized in the caveolae and interacts with caveolin-1 in oFPAE cells.

A. Localization of FGFR1 in caveolae. oFPAE cells were subjected to sucrose gradient centrifugation and fractions were analyzed by Immunoblotting with anti-FGFR1 or anti-caveolin-1 antibody. B. Immunocytochemical analysis of FGFR1 co-localization with caveolin-1. oFPAE cells were seeded on coverslips. After labeling FGFR1 (green) and caveolin-1 (red) by immunocytochemistry as described in Materials and Methods, cell images were captured by fluorescence microscopy. Triangles point to plasma membrane colocalization of FGFR1 and caveolin-1. C & D. FGFR1 association with caveolin-1 and displacement with Cav-SD peptide. Cell lysate were immunoprecipiated with rabbit anti-caveolin-1 antibody. Ant-rabbit IgG (rIgG) was used as a control. The immunoprecipitates were analyzed by immunoblotting with antibodies against FGFR1 and caveolin-1, respectively. Cell lysates used in C were from serum-starved oFPAE cells treated with or without 10 ng/ml of FGF2 for 5 min; cell lysates used in D were from untreated cells pre-incubated with or without 5 μM Cav-SD or Cav-SDX. Blots shown represent a typical experiment. Lower graphs summarize data from three independent experiments using cells from different pregnant ewes. TCL: total cell lysate; FGFR-L/FGFR-S: FGFR long/short from; IgG-H: IgG heavy chain; IgG-L: IgG light chain. *p <0.05.
DISCUSSION
We have demonstrated in this study that the FGFR1 is the dominant FGFR isoform that is expressed in the placenta artery endothelium ex vivo and in vitro. These findings implicate that FGF2 actions is primary mediated via FGFR1 in placental endothelial cells. Our in vitro angiogenesis studies have shown that this system is fully functional in regulating oFPAE cell proliferation, migration and tube formation. FGF2 stimulation of oFPAE cell angiogenesis requires activation of both AKT1 and ERK1/2 pathways. We have shown that the principal caveolar structural protein cav-1 plays a paradoxical role in regulating the FGF2/FGFR1 signaling control of placental angiogenesis. ERK1/2 and AKT1 are co-fractionated with cav-1 in the oFPAE cell caveolae. FGFR1 is also compartmentalized in the caveolae where it physically binds to cav-1. Disruption of caveolae completely inhibits FGF2 activation of both signaling pathways. Thus, these data demonstrate that integral caveolae are required for orchestrating signaling control of placental angiogenesis stimulated by the FGF2/FGFR1 system.
Multiple cell signaling pathways, especially PI3K/AKT1 pathway and MAPK/ERK1/2 pathway are responsible for FGF2-induced cell proliferation in oFPAE cells (Zheng et al., 2008). Here, we have further comprehensively investigated the roles of these two pathways in placental angiogenesis, including cell proliferation, migration and differentiation. Our data have shown that blocking ERK1/2 pathway with its specific inhibitor PD98059 only inhibits FGF2-induced tube formation and partially inhibits cell proliferation, but failed to inhibit cell migration. Compared with ERK1/2 pathway, blocking PI3K/AKT1 pathway with its specific inhibitor wortmannin inhibits all of these angiogenic cellular processes (cell proliferation, migration and differentiation) with much greater potency. Of note, FGF2-induced AKT1 phosphorylation on Ser473 necessary for its full activation (Osaki et al., 2004), is enhanced by inhibition of ERK1/2 activation. It has been reported that AKT1 phosphorylation could be negatively modulated by ERK via the Sos/Ras/PI3K pathway in response to FGF2 or EGF stimulation in NIH3T3 cells (Hayashi et al., 2008). Thus, the ERK1/2 pathway could be functionally upstream of the AKT1 pathway upon FGF2 stimulation and negatively regulates AKT1 phosphorylation. Meanwhile, AKT1 can also negatively regulate the ERK1/2 signaling pathway via phosphorylation and inactivation of the MAPK kinase kinase Raf-1 at Ser259 (Zimmermann and Moelling, 1999). Although our current study does not affirm that AKT1 inhibition arguments FGF2-induced ERK1/2 phosphorylation, we cannot exclude the possibility that the PI3K/AKT1 and ERK1/2 pathways crosstalk with each other in oFPAE cells. It is possible that the cross talk between ERK1/2 pathway and PI3K/AKT1 pathways is important for proper angiogenic responses and eNOS expression to FGF2 and/or other angiogenic growth factors as suggested previously (Mata-Greenwood et al., 2008; Zheng et al., 2008).
There is solid evidence suggesting that FGFR1 is possibly the major FGFR isoform important for mediating the roles of FGFs in placental angiogenesis and trophoblast functions (Anteby et al., 2005a; Arany and Hill, 1998 ; Zheng et al., 1999). However, little is known how its function is regulated in the placenta. The full-length of FGFR1 is membrane-bound protein with two or three extracellular Ig-like domain and two intracellular tyrosine kinase domains (Johnson et al., 1990); it may be secreted as a truncated form only with the extracellular Ig-like domains (Duan et al., 1992; Johnson et al., 1990; Root and Shipley, 2000). Our data suggest that FGFR1 expresses as multiple isoforms in the placental artery endothelial cells both ex vivo and in vitro. These include the full-length long form FGFR1 and the short as well as various truncated forms. The various immunoreactive FGFR1 proteins seem to be generated by alternative mRNA splicing (Bernard et al., 1991). In oFPAE cells, two major FGFR1 variants, FGFR1-L (~120 kDa) and FGFR1-S (~80 kDa) were detected by using an antibody specific for an N-terminal Ig loop. The phosphotyrosine antibody pull-down studies suggested that both of the long and short FGFR1 forms could be transiently tyrosine phosphorylated by FGF2, indicating that both FGFR1-L and FGFR1-S forms bear with tyrosine kinase domains and are potentially functional in mediating FGF2 signaling in oFPAE cells.
We have found both the long and short FGFR1 forms are present in oFPAE cell caveolae. There are a plethora of different classes of molecules that are concentrated within the caveolae. Most receptor tyrosine kinases, including receptors for epidermal growth factor, platelet-derived growth factor, nerve growth factor, VEGFR2, have one or more of the so-called “caveolin binding motif” within their conserved kinase domains (Couet et al., 1997). The binding motifs are with signature sequences of φXφXXXXφ or φXXXXφXXφ; where φ is one of the aromatic amino acids Trp, Phe or Tyr. They can directly interact with the caveolin scaffold domain, thereby providing a structural base for targeting these proteins into the caveolae (Couet et al., 1997; Frank et al., 2003; Labrecque et al., 2003; Liao et al., 2009; Vihanto et al., 2006). Interactions of the binding motifs with cav-1 could positively or negatively regulate receptor functions upon ligand stimulation (Labrecque et al., 2003; Vihanto et al., 2006). Sequence analysis shows that FGFR1 possesses a putative cav-1 binding motif (676WSFGVLLFEIF686, aromatic amino acids underlined). In this study, we have found: 1) FGFR1 co-fractionates with cav-1 in the caveolae membranes; 2) FGFR1 is co-localized with cav-1 as evidenced by fluorescence microscopy; 3) FGFR1 is detected in the cav-1 immunoprecipitates and; 4) pre-incubation with the Cav-SD peptide competes off cav-1 binding to FGFR1-S (Fig. 7D). These data support that FGFR1 is targeted into the caveolae via binding to cav-1, although how the interaction affects FGFR1 function is waiting to be determined. In addition, our data show that treatment with FGF2 stimulates FGFR1 dissociation from cav-1. This may indicate a mechanism potentially linked to increased FGFR1 activity as previous studies have shown that dissociation from cav-1 increase the activity of many caveolar proteins such as VEGF receptors (Labrecque et al., 2003) and eNOS (Garcia-Cardena et al., 1997), etc.
Cavolin-1 has multiple functions in different cells and generally acts as an inhibitor in cell growth and proliferation. For example, cav-1 inhibits HUVEC cell proliferation through arrest of cell cycle progression (Fang et al., 2007). In NIH 3T3 cells, downregulation of cav-1 with its antisense prevents cells from staurosporine-induced apoptosis. In contrast, overexpression of cav-1 sensitizes carcinoma cells to apoptotic stimuli, suggesting cav-1 is also pro-apoptotic (Liu et al., 2001). In addition, overexpression of cav-1 inhibits tumor cell growth and metastasis (Sloan et al., 2004). In vivo, Cav1−/ mice develop vascular abnormalities with up-regulated eNOS activity and hyperproliferative phenotypes (Razani et al., 2001). Yet, the precise regulatory roles of cav-1 and caveolae in angiogenesis have not fully elucidated. Formation of new vessels from existing ones (angiogenesis) is a complex three dimensional process in which endothelial cells play a central role (Liao et al., 2010). Cav-1 expression levels change during different phases of angiogenesis, i.e., proliferation, migration and differentiation, lowest with highest proliferative activity and greatest prior to the formation of capillary-like tubules in human microvascular endothelial cells (Liu et al., 2002). Overexpression of cav-1 or treatment with its SD peptide accelerated tubule formation (Liu et al., 2002). During cell migration, cav-1 exhibits an inhibitory role and appears to accumulate in the rear of cells in two-dimensional process; yet it shows a promoting role and distribute mainly in the front of cells in three-dimensional migration(Parat et al., 2003). Moreover, this polarized redistribution during migration is significantly directed by substrate topology (Santilman et al., 2006).
With regard to the role of cav-1/caveolae in placental angiogenesis, we have recently reported that both FGF2 and VEGF have no effects on oFPAE cell cav-1 expression (Liao et al., 2009). However, cav-1 and caveolae play a critical role in placental angiogenesis by regulating the angiogenic responses (proliferation, migration and tube formation) to VEGF stimulation. This is largely achieved by regulating various cell signaling pathways, including ERK1/2 and AKT1 and eNOS/NO, activated by VEGF in oFPAE cells. In these studies, we have shown that both overexpression and downregulation of cav-1 as well as disruption of caveolae are detrimental for VEGF stimulation of ERK1/2 and AKT1 pathways and oFPAE cell angiogenesis. We have also shown that caveolae functions as a platform for compartmentalizing VEGF signaling to angiogenesis (Liao et al., 2009; Liao et al., 2010). Our current study shows that FGF2 signals towards placental angiogenesis via FGFR1 and ERK1/2 and AKT1 pathways that are also compartmentalized in oFPAE cell caveolae. FGF2 activates both ERK1/2 and AKT1 in the caveolae (Fig. 3A) and disruption of caveolae completely causes loss of FGF2 stimulation of these signaling pathways. Activation of both pathways by FGF2 is regulated by cav-1 in a reciprocal manner similar to its role in the regulation of VEGF signaling in oFPAE cells (Liao et al., 2009; Liao et al., 2010). This is apparent because overexpression of exogenous cav-1 and downregulation of endogenous cav-1 cause similar inhibitory effects of FGF2 activation of ERK1/2 and AKT1 pathways. Further, both overexpression and downregulation of cav-1 inhibited FGF2 stimulation of FGF2-induced oFPAE cell proliferation, migration and tube formation. Obviously, a “paradox” exists as to the role of caveolin-1 in FGF2 signaling control of placental artery endothelial angiogenesis because both downregulation and overexpression of caveolin-1 result in similar effects on FGF2-induced ERK1/2 Akt signaling as well as cell proliferation, migration and tube formation in oFPAE cells. This phenomenon may be explained by the following facts. On one hand, caveolae represent a structural platform to facilitate and to amplify signaling cascades through compartmentalization of membrane receptors with their effectors/mediators for activation of various signaling pathways including ERK1/2 (Pike, 2005) and Akt (current study). On the other hand, the major residual protein of caveolae caveolin-1 contains the so-called “caveolin scaffold domain” (aa 82-101). This domain signifies a structural basis for caveolae compartmentalizing of many lipid modifying signaling molecules through direct interactions with their so called “caveolin-binding motifs” (Couet et al., 1997), which normally suppresses the catalytic activity of these signaling molecules. This “paradox” phenomenon is common to many scaffold proteins that, as suggested by Ferrell (2000), too much or too little of such component may decrease the output of the pathway. In other word, the optimal level of a scaffold protein is important to the optimal activation of signaling cascade, and indicating its check and balance effect on the control of signaling pathways (Ferrell, 2000). Thus, our findings consistently suggest that either too much or too little cav-1 or loss of caveolae impairs cell signaling towards placental angiogenesis upon stimulation by both FGF2 (current study) and VEGF (Liao et al., 2009; Liao et al., 2010). These findings also highlight that maintaining endogenous cav-1 at a proper level is important for the optimal output for cell signaling and placental angiogenesis.
Altogether, the plasma membrane microdomain caveolae functions as a platform for organizing FGF2-induced angiogenesis through spatiotemporally compartmentalizing FGFR1 and the ERK1/2 and PI3K/AKT1 signaling pathways in oFPAE cells. As one of major caveolar structural proteins, cav-1 interacts with FGFR1 directly and paradoxically regulates FGF2-induced activation of PI3K/AKT1 and ERK1/2 pathways that regulate placental angiogenesis coordinately (Fig. 8).
Fig. 8. Caveolin-1 orchestrates FGF2 signaling control of placental angiogenesis in placental artery endothelial cell caveolae.

Caveolae function as a platform for regulating FGF2-induced angiogenesis through spatiotemporally compartmentalizing FGFR1 and the AKT1 and ERK1/2 signaling modules; the major caveolar structural protein caveolin-1 interacts with FGFR1 physically and paradoxically regulates FGF2-induced activation of PI3K/AKT1 and ERK1/2 pathways that coordinately regulate placental endothelial cell proliferation, migration and tube formation, e.g., angiogenesis.
Supplementary Material
Acknowledgments
Source of Funding: The present study was supported in part by National Institutes of Health grants RO1 HL74947, RO1 HL70562 and R21 HL98746 (to DB Chen) and RO1 HL64703 (to J Zheng).
Abbreviations used
- FGF2
Fibroblast growth factor 2
- FGFR
FGF receptor
- VEGF
vascular endothelial growth factor
- VEGFR
VEGF receptor
- oFPAE
ovine fetal placental artery endothelial
- NO
nitric oxide
- eNOS
endothelial NO synthase
- MAPK
mitogen-activated protein kinase
- ERK1/2
extracellular signal-regulated protein kinase 1/2
- PI3K
phosphotidylinositol-3-kinase
- AKT1
v-Akt1 murine thymoma viral oncogene homolog 1
- pAb
polyclonal antibody
- mAb
monoclonal antibody
- IgG
immunoglobin
- Cav-1
caveolin-1
- SD
scaffold domain
Footnotes
Disclosure: The authors have nothing to disclose.
REFERENCES
- Anteby EY, Natanson-Yaron S, Greenfield C, Goldman-Wohl D, Haimov-Kochman R, Holzer H, Yagel S. Human placental Hofbauer cells express sprouty proteins: a possible modulating mechanism of villous branching. Placenta. 2005a;26(6):476–483. doi: 10.1016/j.placenta.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Anteby EY, Natanson-Yaron S, Hamani Y, Sciaki Y, Goldman-Wohl D, Greenfield C, Ariel I, Yagel S. Fibroblast growth factor-10 and fibroblast growth factor receptors 1-4: expression and peptide localization in human decidua and placenta. Eur J Obstet Gynecol Reprod Biol. 2005b;119(1):27–35. doi: 10.1016/j.ejogrb.2004.05.014. [DOI] [PubMed] [Google Scholar]
- Arany E, Hill DJ. Fibroblast growth factor-2 and fibroblast growth factor receptor-1 mRNA expression and peptide localization in placentae from normal and diabetic pregnancies. Placenta. 1998;19(2-3):133–142. doi: 10.1016/s0143-4004(98)90001-7. [DOI] [PubMed] [Google Scholar]
- Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and therapy. Nat Rev Drug Discov. 2009;8(3):235–253. doi: 10.1038/nrd2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard O, Li M, Reid HH. Expression of two different forms of fibroblast growth factor receptor 1 in different mouse tissues and cell lines. Proc Natl Acad Sci U S A. 1991;88(17):7625–7629. doi: 10.1073/pnas.88.17.7625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas SB, Hammond RW, Anderson LD. Fibroblast growth factors from bovine pituitary and human placenta and their functions in the maturation of porcine granulosa cells in vitro. Endocrinology. 1988;123(1):559–566. doi: 10.1210/endo-123-1-559. [DOI] [PubMed] [Google Scholar]
- Borowicz PP, Arnold DR, Johnson ML, Grazul-Bilska AT, Redmer DA, Reynolds LP. Placental growth throughout the last two thirds of pregnancy in sheep: vascular development and angiogenic factor expression. Biol Reprod. 2007;76(2):259–267. doi: 10.1095/biolreprod.106.054684. [DOI] [PubMed] [Google Scholar]
- Bruns RR, Palade GE. Studies on blood capillaries. I. General organization of blood capillaries in muscle. J Cell Biol. 1968a;37(2):244–276. doi: 10.1083/jcb.37.2.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns RR, Palade GE. Studies on blood capillaries. II. Transport of ferritin molecules across the wall of muscle capillaries. J Cell Biol. 1968b;37(2):277–299. doi: 10.1083/jcb.37.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DB, Li SM, Qian XX, Moon C, Zheng J. Tyrosine phosphorylation of caveolin 1 by oxidative stress is reversible and dependent on the c-src tyrosine kinase but not mitogen-activated protein kinase pathways in placental artery endothelial cells. Biol Reprod. 2005;73(4):761–772. doi: 10.1095/biolreprod.105.040881. [DOI] [PubMed] [Google Scholar]
- Cooke FN, Pennington KA, Yang Q, Ealy AD. Several fibroblast growth factors are expressed during pre-attachment bovine conceptus development and regulate interferon-tau expression from trophectoderm. Reproduction. 2009;137(2):259–269. doi: 10.1530/REP-08-0396. [DOI] [PubMed] [Google Scholar]
- Couet J, Li S, Okamoto T, Ikezu T, Lisanti MP. Identification of peptide and protein ligands for the caveolin-scaffolding domain. Implications for the interaction of caveolin with caveolae-associated proteins. J Biol Chem. 1997;272(10):6525–6533. doi: 10.1074/jbc.272.10.6525. [DOI] [PubMed] [Google Scholar]
- Drab M, Verkade P, Elger M, Kasper M, Lohn M, Lauterbach B, Menne J, Lindschau C, Mende F, Luft FC, Schedl A, Haller H, Kurzchalia TV. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science. 2001;293(5539):2449–2452. doi: 10.1126/science.1062688. [DOI] [PubMed] [Google Scholar]
- Duan DS, Werner S, Williams LT. A naturally occurring secreted form of fibroblast growth factor (FGF) receptor 1 binds basic FGF in preference over acidic FGF. J Biol Chem. 1992;267(23):16076–16080. [PubMed] [Google Scholar]
- Fang K, Fu W, Beardsley AR, Sun X, Lisanti MP, Liu J. Overexpression of caveolin-1 inhibits endothelial cell proliferation by arresting the cell cycle at G0/G1 phase. Cell Cycle. 2007;6(2):199–204. doi: 10.4161/cc.6.2.3740. [DOI] [PubMed] [Google Scholar]
- Ferrell JE., Jr. What do scaffold proteins really do? Sci STKE. 2000;2000(52):PE1. doi: 10.1126/stke.2000.52.pe1. [DOI] [PubMed] [Google Scholar]
- Frank PG, Woodman SE, Park DS, Lisanti MP. Caveolin, caveolae, and endothelial cell function. Arterioscler Thromb Vasc Biol. 2003;23(7):1161–1168. doi: 10.1161/01.ATV.0000070546.16946.3A. [DOI] [PubMed] [Google Scholar]
- Furdui CM, Lew ED, Schlessinger J, Anderson KS. Autophosphorylation of FGFR1 kinase is mediated by a sequential and precisely ordered reaction. Mol Cell. 2006;21(5):711–717. doi: 10.1016/j.molcel.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Garcia-Cardena G, Martasek P, Masters BS, Skidd PM, Couet J, Li S, Lisanti MP, Sessa WC. Dissecting the interaction between nitric oxide synthase (NOS) and caveolin. Functional significance of the nos caveolin binding domain in vivo. J Biol Chem. 1997;272(41):25437–25440. doi: 10.1074/jbc.272.41.25437. [DOI] [PubMed] [Google Scholar]
- Glenney JR, Jr., Soppet D. Sequence and expression of caveolin, a protein component of caveolae plasma membrane domains phosphorylated on tyrosine in Rous sarcoma virus-transformed fibroblasts. ProcNatl Acad Sci U S A. 1992;89(21):10517–10521. doi: 10.1073/pnas.89.21.10517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi H, Tsuchiya Y, Nakayama K, Satoh T, Nishida E. Down-regulation of the PI3-kinase/Akt pathway by ERK MAP kinase in growth factor signaling. Genes Cells. 2008;13(9):941–947. doi: 10.1111/j.1365-2443.2008.01218.x. [DOI] [PubMed] [Google Scholar]
- Isshiki M, Ando J, Yamamoto K, Fujita T, Ying Y, Anderson RG. Sites of Ca(2+) wave initiation move with caveolae to the trailing edge of migrating cells. J Cell Sci. 2002a;115(Pt 3):475–484. doi: 10.1242/jcs.115.3.475. [DOI] [PubMed] [Google Scholar]
- Isshiki M, Ying YS, Fujita T, Anderson RG. A molecular sensor detects signal transduction from caveolae in living cells. J Biol Chem. 2002b;277(45):43389–43398. doi: 10.1074/jbc.M205411200. [DOI] [PubMed] [Google Scholar]
- Johnson DE, Lee PL, Lu J, Williams LT. Diverse forms of a receptor for acidic and basic fibroblast growth factors. Mol Cell Biol. 1990;10(9):4728–4736. doi: 10.1128/mcb.10.9.4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrecque L, Royal I, Surprenant DS, Patterson C, Gingras D, Beliveau R. Regulation of vascular endothelial growth factor receptor-2 activity by caveolin-1 and plasma membrane cholesterol. Mol Biol Cell. 2003;14(1):334–347. doi: 10.1091/mbc.E02-07-0379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SM, Zeng LW, Feng L, Chen DB. Rac1-dependent intracellular superoxide formation mediates vascular endothelial growth factor-induced placental angiogenesis in vitro. Endocrinology. 2010;151(11):5315–5325. doi: 10.1210/en.2010-0178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao WX, Feng L, Zhang H, Zheng J, Moore TR, Chen DB. Compartmentalizing VEGF-induced ERK2/1 signaling in placental artery endothelial cell caveolae: a paradoxical role of caveolin-1 in placental angiogenesis in vitro. Mol Endocrinol. 2009;23(9):1428–1444. doi: 10.1210/me.2008-0475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao WX, Feng L, Zheng J, Chen DB. Deciphering mechanisms controlling placental artery endothelial cell migration stimulated by vascular endothelial growth factor. Endocrinology. 2010;151(7):3432–3444. doi: 10.1210/en.2009-1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao WX, Magness RR, Chen DB. Expression of estrogen receptors-alpha and -beta in the pregnant ovine uterine artery endothelial cells in vivo and in vitro. Biol Reprod. 2005;72(3):530–537. doi: 10.1095/biolreprod.104.035949. [DOI] [PubMed] [Google Scholar]
- Liu J, Wang XB, Park DS, Lisanti MP. Caveolin-1 expression enhances endothelial capillary tubule formation. J Biol Chem. 2002;277(12):10661–10668. doi: 10.1074/jbc.M110354200. [DOI] [PubMed] [Google Scholar]
- Liu L, Hausladen A, Zeng M, Que L, Heitman J, Stamler JS. A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature. 2001;410(6827):490–494. doi: 10.1038/35068596. [DOI] [PubMed] [Google Scholar]
- Magness RR. Maternal cardiovascular and other physiologic responses to the endocrinology of pregnancy. In: Bazer FW, editor. The Endocrinology of Pregnancy. Human Press Inc; Totowa NJ: 1999. pp. 507–539. (Chapter 18) [Google Scholar]
- Makarenkova HP, Hoffman MP, Beenken A, Eliseenkova AV, Meech R, Tsau C, Patel VN, Lang RA, Mohammadi M. Differential interactions of FGFs with heparan sulfate control gradient formation and branching morphogenesis. Sci Signal. 2009;2(88):ra55. doi: 10.1126/scisignal.2000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata-Greenwood E, Liao WX, Wang W, Zheng J, Chen DB. Activation of AP-1 transcription factors differentiates FGF2 and vascular endothelial growth factor regulation of endothelial nitric-oxide synthase expression in placental artery endothelial cells. J Biol Chem. 2010;285(23):17348–17358. doi: 10.1074/jbc.M109.092791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata-Greenwood E, Liao WX, Zheng J, Chen DB. Differential activation of multiple signalling pathways dictates eNOS upregulation by FGF2 but not VEGF in placental artery endothelial cells. Placenta. 2008;29(8):708–717. doi: 10.1016/j.placenta.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minshall RD, Tiruppathi C, Vogel SM, Malik AB. Vesicle formation and trafficking in endothelial cells and regulation of endothelial barrier function. Histochem Cell Biol. 2002;117(2):105–112. doi: 10.1007/s00418-001-0367-x. [DOI] [PubMed] [Google Scholar]
- Murakami M, Nguyen LT, Zhuang ZW, Moodie KL, Carmeliet P, Stan RV, Simons M. The FGF system has a key role in regulating vascular integrity. J Clin Invest. 2008;118(10):3355–3366. doi: 10.1172/JCI35298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto T, Schlegel A, Scherer PE, Lisanti MP. Caveolins, a family of scaffolding proteins for organizing “preassembled signaling complexes” at the plasma membrane. J Biol Chem. 1998;273(10):5419–5422. doi: 10.1074/jbc.273.10.5419. [DOI] [PubMed] [Google Scholar]
- Osaki M, Oshimura M, Ito H. PI3K-Akt pathway: its functions and alterations in human cancer. Apoptosis. 2004;9(6):667–676. doi: 10.1023/B:APPT.0000045801.15585.dd. [DOI] [PubMed] [Google Scholar]
- Palade GE, Bruns RR. Structural modulations of plasmalemmal vesicles. J Cell Biol. 1968;37(3):633–649. doi: 10.1083/jcb.37.3.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parat MO, Anand-Apte B, Fox PL. Differential caveolin-1 polarization in endothelial cells during migration in two and three dimensions. Mol Biol Cell. 2003;14(8):3156–3168. doi: 10.1091/mbc.E02-11-0761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Han HJ. Caveolin-1 plays important role in EGF-induced migration and proliferation of mouse embryonic stem cells: involvement of PI3K/Akt and ERK. Am J Physiol Cell Physiol. 2009;297(4):C935–944. doi: 10.1152/ajpcell.00121.2009. [DOI] [PubMed] [Google Scholar]
- Pfarrer C, Weise S, Berisha B, Schams D, Leiser R, Hoffmann B, Schuler G. Fibroblast growth factor (FGF)-1, FGF2, FGF7 and FGF receptors are uniformly expressed in trophoblast giant cells during restricted trophoblast invasion in cows. Placenta. 2006;27(6-7):758–770. doi: 10.1016/j.placenta.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Pike LJ. Growth factor receptors, lipid rafts and caveolae: an evolving story. Biochimica et Biophysica Acta. 2005;1746(3):260–273. doi: 10.1016/j.bbamcr.2005.05.005. [DOI] [PubMed] [Google Scholar]
- Predescu D, Palade GE. Plasmalemmal vesicles represent the large pore system of continuous microvascular endothelium. Am J Physiol. 1993;265(2 Pt 2):H725–733. doi: 10.1152/ajpheart.1993.265.2.H725. [DOI] [PubMed] [Google Scholar]
- Presta M, Dell’Era P, Mitola S, Moroni E, Ronca R, Rusnati M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005;16(2):159–178. doi: 10.1016/j.cytogfr.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, Macaluso F, Russell RG, Li M, Pestell RG, Di Vizio D, Hou H, Jr., Kneitz B, Lagaud G, Christ GJ, Edelmann W, Lisanti MP. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem. 2001;276(41):38121–38138. doi: 10.1074/jbc.M105408200. [DOI] [PubMed] [Google Scholar]
- Reynolds LP, Redmer DA. Angiogenesis in the placenta. Biol Reprod. 2001;64(4):1033–1040. doi: 10.1095/biolreprod64.4.1033. [DOI] [PubMed] [Google Scholar]
- Root LL, Shipley GD. Normal human fibroblasts produce membrane-bound and soluble isoforms of FGFR-1. Mol Cell Biol Res Commun. 2000;3(2):87–97. doi: 10.1006/mcbr.2000.0199. [DOI] [PubMed] [Google Scholar]
- Rothberg KG, Heuser JE, Donzell WC, Ying YS, Glenney JR, Anderson RG. Caveolin, a protein component of caveolae membrane coats. Cell. 1992;68(4):673–682. doi: 10.1016/0092-8674(92)90143-z. [DOI] [PubMed] [Google Scholar]
- Santilman V, Baran J, Anand-Apte B, Fox PL, Parat MO. Caveolin-1 polarization in migrating endothelial cells is directed by substrate topology not chemoattractant gradient. Cell Motil Cytoskeleton. 2006;63(11):673–680. doi: 10.1002/cm.20153. [DOI] [PubMed] [Google Scholar]
- Sloan EK, Stanley KL, Anderson RL. Caveolin-1 inhibits breast cancer growth and metastasis. Oncogene. 2004;23(47):7893–7897. doi: 10.1038/sj.onc.1208062. [DOI] [PubMed] [Google Scholar]
- Song KS, Li S, Okamoto T, Quilliam LA, Sargiacomo M, Lisanti MP. Co-purification and direct interaction of Ras with caveolin, an integral membrane protein of caveolae microdomains: Detergent-free purification of caveolae microdomains. J Biol Chem. 1996;271(16):9690–9697. doi: 10.1074/jbc.271.16.9690. [DOI] [PubMed] [Google Scholar]
- Vihanto MM, Vindis C, Djonov V, Cerretti DP, Huynh-Do U. Caveolin-1 is required for signaling and membrane targeting of EphB1 receptor tyrosine kinase. J Cell Sci. 2006;119(Pt 11):2299–2309. doi: 10.1242/jcs.02946. [DOI] [PubMed] [Google Scholar]
- Xu J, Nakahara M, Crabb JW, Shi E, Matuo Y, Fraser M, Kan M, Hou J, McKeehan WL. Expression and immunochemical analysis of rat and human fibroblast growth factor receptor (flg) isoforms. J Biol Chem. 1992;267(25):17792–17803. [PubMed] [Google Scholar]
- Zheng J, Bird IM, Melsaether AN, Magness RR. Activation of the mitogen-activated protein kinase cascade is necessary but not sufficient for basic fibroblast growth factor- and epidermal growth factor-stimulated expression of endothelial nitric oxide synthase in ovine fetoplacental artery endothelial cells. Endocrinology. 1999;140(3):1399–1407. doi: 10.1210/endo.140.3.6542. [DOI] [PubMed] [Google Scholar]
- Zheng J, Vagnoni KE, Bird IM, Magness RR. Expression of basic fibroblast growth factor, endothelial mitogenic activity, and angiotensin II type-1 receptors in the ovine placenta during the third trimester of pregnancy. Biol Reprod. 1997;56(5):1189–1197. doi: 10.1095/biolreprod56.5.1189. [DOI] [PubMed] [Google Scholar]
- Zheng J, Wen Y, Austin JL, Chen DB. Exogenous nitric oxide stimulates cell proliferation via activation of a mitogen-activated protein kinase pathway in ovine fetoplacental artery endothelial cells. Biol Reprod. 2006;74(2):375–382. doi: 10.1095/biolreprod.105.043190. [DOI] [PubMed] [Google Scholar]
- Zheng J, Wen Y, Song Y, Wang K, Chen DB, Magness RR. Activation of multiple signaling pathways is critical for fibroblast growth factor 2- and vascular endothelial growth factor-stimulated ovine fetoplacental endothelial cell proliferation. Biol Reprod. 2008;78(1):143–150. doi: 10.1095/biolreprod.107.064477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann S, Moelling K. Phosphorylation and regulation of Raf by Akt (protein kinase B) Science. 1999;286(5445):1741–1744. doi: 10.1126/science.286.5445.1741. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.