

Abstract
Hypertrophic scarring and poor intrinsic axon growth capacity constitute major obstacles for spinal cord repair. These processes are tightly regulated by microtubule dynamics. We found that moderate microtubule stabilization decreased scar formation after spinal cord injury (SCI) in rodents via various cellular mechanisms, including dampening of TFG-β signalling. It prevented the accumulation of chondroitin sulfate proteoglycans (CSPGs) and rendered the lesion site permissive for axon regeneration of growth competent sensory neurons. Additionally, microtubule stabilization promoted growth of CNS axons of the Raphe-spinal tract and led to functional improvement. Thus, microtubule stabilization reduces fibrotic scarring and enhances the capacity of axons to grow. Manipulation of microtubules may offer the basis for a multi-targeted therapy after SCI.
Microtubule dynamics regulate key processes during scarring, including cell proliferation, migration and differentiation as well as intracellular trafficking and secretion of extracellular matrix (ECM) molecules (1-2). Moreover, moderate microtubule stabilization prevents axonal retraction and swelling of the axon tip after CNS injury (3), and stimulates axon growth of cultured neurons (4) enabling them to overcome the growth inhibitory effect of CNS myelin (3). We therefore hypothesized that moderate microtubule stabilization with Taxol would facilitate axonal regeneration after SCI by decreasing scar formation and enhancing intrinsic axonal growth.
To test this, we first examined whether Taxol treatment reduces scarring after SCI. Adult rats underwent a dorsal hemisection at the 8th thoracic spinal cord level. Taxol was continuously delivered at the injury site using an intrathecal catheter connected to an osmotic minipump. Seven days post-injury, the lesions of vehicle-treated animals were filled with laminin, fibronectin and collagen IV, hallmarks of a fibrotic scar (n=10; Fig. 1B and fig. S1), which is a major impediment for axon regeneration (5-7). In contrast, in the lesions of animals treated with Taxol at 256 ng/day, a much lower dose than used for chemotherapy (8), fibronectin, laminin, and collagen IV were strongly reduced (percentage of control: 51.9% ± 6.1% (fibronectin) and 41.2 ± 11.4% (laminin); n= 12; p= 0.002; Fig. 1C,D and fig. S1). At this Taxol concentration, astrocytes surrounded the injury as in vehicle controls (Fig. 1B,F); the injury size was equivalent in both groups (1.18 ± 0.09 versus 1.19 ± 0.12 mm2 for the control, n= 7-10 animals/group p= 0.951; Fig. 1E) which suggest that astrocytes isolated the lesion from undamaged CNS tissue (9). Phosphohistone-H3 immunostaining and TUNEL labelling showed that the numbers of proliferating and apoptotic cells were similar between Taxol and vehicle-treated animals (n= 7 animals/group; fig. S2, S3). Hence, Taxol at low doses reduced fibrotic scarring by mechanisms independent of cell proliferation or apoptosis.
Figure 1. Taxol decreases scarring induced by spinal cord injury.

(A) Representation of lesioned spinal cord (box). (B-C) Mid-saggital sections of lesion site from rats treated with (B) vehicle or (C) 256 ng/day 7 days post-injury. Scale bars, 300 μm. (D) Taxol significantly decreases fibrotic scarring (expressed as % of vehicle control; ** p= 0.002) without affecting wound size (E). Taxol does not affect glial compaction at 28 dpi (F). Data expressed as mean ± SEM.
A key event in fibrotic scarring after CNS injury is the activation of transforming growth factor (TGF)-ß signalling. Following SCI, the expression of TGF-ß dramatically increases, which favours fibrosis (10-12). Integrity of the microtubule network is crucial for the transduction of this signal (13). Smad2, the downstream effector of the TGF- ß receptor pathway, binds to microtubules via conventional kinesin-1 (14). We therefore asked whether stabilizing the microtubule network hinders TGF-ß signalling and attenuates fibrogenesis. In extracts from the injury site, Taxol treatment increased the level of detyrosinated microtubules (Fig. 2C), which enables kinesin-1 to bind tightly to microtubules (15). Indeed in Taxol-treated animals, kinesin-1 was enriched in the microtubule fraction of the injury site extracts (Fig. 2D). Far western blot analysis showed that Smad2 directly bound to kinesin-1 from brain and spinal cord extracts (Fig. 2E). This interaction was corroborated by coimmunoprecipitation experiments, which showed that Smad2 and kinesin-1 formed a complex in spinal cord extracts (Fig. 2F). Interestingly, Taxol strongly reduced cargo transport by KIF-5, the motor domain of kinesin-1, as well as by the dynein motor domain BICDN (fig. S4A Fig. 2G,H), suggesting that Taxol would interfere with Smad2 trafficking. In fact, in cultured astrocytes, TGF-ß1 induced nuclear translocation of Smad 2/3 (Fig. 2A); Taxol inhibited 70% of this TGF-ß1-induced nuclear translocation (p= 0.04; Fig. 2A,B) and Smad2/3 colocalized with microtubules (fig. S4B). Time-lapse microscopy showed that overexpressed Smad2-PAGFP moves into the nucleus within minutes after TGF-ß1 stimulation, while Taxol treatment abolished this movement (movie S1, S2). In vivo, 7 days after spinal cord injury, phosphorylated Smad2/3 was located inside the nucleus in 95% of vehicle-treated animals while this translocation only occurred in 30% of the Taxol-treated animals (n= 13 animals/group; Fig 2I). This suggests that Taxol could impair TGF-ß signalling-dependent processes. In fact, in cultured meningeal cells, Taxol reduced the TGF-ß1-stimulated production of fibronectin (Fig. 3C) and markedly impaired TGF-ß1-stimulated migration (90 ± 29% and 103 ± 15% of cell free area in TGF-ß1 plus Taxol 1 and 10 nM, respectively, versus 38 ± 14% in the TGF-ß1 alone; p= 0.003; Fig. 3A,B). Thus, low doses of Taxol interfere with Smad-dependent TGF-ß signalling, reduce extracellular matrix secretion and cell migration, and prevent fibrotic scarring after SCI.
Figure 2. Taxol dampens TGF-ß signalling.

(A-B) In cultured astrocytes, Taxol counteracts the TGF-ß1 induced-nuclear translocation of Smad2/3 (arrowheads) causing cytoplasmic Smad2/3 accumulation (arrow). Results in (B) are mean ± SD (3 independent experiments, * p= 0.041). (C-F) Representative immunoblots. (C) Taxol treament increases total tubulin and decreases tyrosinated tubulin in the lesion site. (D) Kinesin-1 enrichment in microtubule fraction of Taxol-treated lesion site. (E) His-Smad2 binds to kinesin-1 of brain and spinal cord extracts. (F) Smad2 co-immunoprecipitates with kinesin-1. (G) Overlay of sequential binarized peroxisome images, color-coded by time as indicated, for a time series in which KIF5-FRB was recruited to peroxisomes upon addition of rapalog with (right) or without (left) 100 nM Taxol. Blue marks the initial distribution, whereas red marks regions targeted. Scale bars, 10 μm. (H) Time traces of the R90% (see online methods) KIF5-FRB and BICDN-FRB with or without 100 nM Taxol (D). Rapalog is added at 0:00 minutes. Time traces show average ± SEM of 5 (KIF5), 6 (KIF+Taxol), 3 (BICDN), and 8 (BICDN+Taxol) cells. (I) At 7 dpi, Taxol treatment interferes with the nuclear translocation of phospho-Smad 2/3 induced by SCI. Scale bar, 20 μm.
Figure 3. Taxol decreases meningeal cell migration and glycosaminoglycan release in vitro and in vivo.
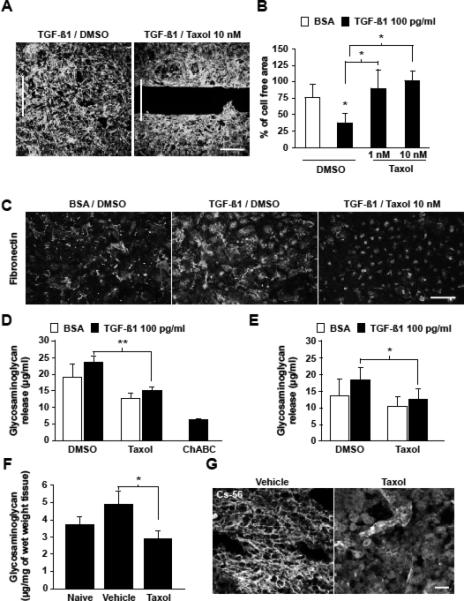
(A-B) 3 DIV, Taxol (1 and 10 nM) decreases meningeal cell migration induced by TGF-ß1. Scale bar, 300 μm; results in (B) are means ± SD from 3 independent experiments; * p= 0.003. (C) Taxol decreases fibronectin deposits induced by TGF-ß1 in meningeal cells. Scale bar, 300 μm. Taxol decreases GAGs released from control and TGF-ß1 stimulated meningeal cells (D) and astrocytes (E) . Results are means ± SD from 3 independent experiments; ** p= 0.008: * p=0.03). (F) Taxol treatment decreases the amount of GAGs induced by SCI. Results are means ± SEM, * p= 0.02. (G) Carbohydrate epitopes of CSPGs (CS-56) are abundant within the ECM in the control group. After Taxol treatment, CSPGs remain cytoplasmic. Scale bar, 75 μm.
Given the reduced expression of ECM proteins at lesion sites in Taxol-treated animals, we asked whether Taxol also affects the production of CSPGs, which are known inhibitors of axon growth. At 7 days post-injury, Taxol decreased the amount of NG2, one of the most abundant CSPGs (16) (fig. S1). Axon growth inhibitory activities of CSPGs mainly reside in their Glycosaminoglycan (GAG) side chains (17-19). Lesion site extracts from Taxol-treated animals showed a significant reduction of GAGs compared to controls (2.4 ± 0.35 versus 4 ± 0.70 μg/mg of tissue; p= 0.026; n= 9 animals/group; Fig. 3F). Consistently, the conditioned medium of cultured meningeal cells and astrocytes treated with 10 nM Taxol showed a 35% and 32% decrease of GAG levels, respectively (p= 0.008 and p= 0.02 respectively; Fig. 3D,E). Moreover, the CSPGs expressed in the Taxol-treated animals localized to the intracellular space instead of scaffolding the cells as observed in the vehicle-treated animals (Fig. 3G). Together, these results demonstrate that administration of low doses of Taxol decreases CSPG expression at the lesion site after SCI.
We next asked whether the Taxol-treated lesion site becomes permissive for regenerating axons in vivo by evaluating the regenerative response of dorsal root ganglion (DRG) neurons. These neurons are set into a growth competent state by injury of their peripheral axons (conditioning), which allows them to regenerate their CNS axons, but only in a scar-free environment (20). We therefore assessed whether the reduction of the scar induced by Taxol is permissive for conditioned axons to grow. Taxol was applied at the lesion for 4 weeks as aforementioned; two weeks after central injury, we conditioned the lumbar L4-6 DRG neurons by transecting the sciatic nerve. Consistent with previous reports (20), 76% of the vehicle-treated animals had no regenerative response but rather showed fiber retraction (Fig. 4A,B). In contrast in Taxol-treated animals, regenerative fibers grew along the edge of the lesion cavity into the lesion site and beyond (Fig. 4A). The longest axons per animal grew an average of 1199 ± 250 μm in the Taxol-treated group versus 176 ± 225 μm in the vehicle-treated animals (Fig. 4B; n= 13 animals/group; p= 0.002). Thus, taxol treatment of the developing scar provides a favourable environment in the CNS for axonal regeneration of growth competent neurons.
Figure 4. Taxol promotes axonal regeneration and functional recovery.

(A) Spinal cord horizontal sections of L4-L6 DRG axons labelled with CTB, 6 weeks after injury. Taxol treatment promotes regeneration of growth competent neurons (A, arrowheads). Scale bars, 50 μm. (B) Longest regenerating axon per animal ± SEM (** p= 0.002). (C) Spinal cord saggital sections stained with anti-5HT antibody 4 weeks after injury. After Taxol treatment, the caudal part of the cord is enriched in serotonergic fibers (C, arrowheads). Scale bars, 75 μm. (D-E) Quantification of 5HT-positive fibers caudal to the lesion ± SEM (*** p= 0.0001 (D) and ** p= 0.002 (E)). (F) Taxol treatment improves locomotor performance over the time.
Since Taxol also enhanced intrinsic axon growth (4) and the elongation of cultured neurons plated on CSPG or CNS myelin components (fig. S5)(3), we assessed whether Taxol treatment alone is sufficient to promote growth of injured CNS axons. To this end, we examined the effect of 4-week continuous Taxol treatment on axonal growth of the Raphe-spinal tract after dorsal hemi-section (21-22). Immunostaining revealed a five-fold increase in the number of serotonin (5HT)-positive fibers in the spinal cord caudal to the lesion in the Taxol-treated animals compared to vehicle (69 ± 12 fibers versus 13± 3 fibers respectively; p= 0.0001; n= 16/group; Fig. 4C-D). The caudal edge of the lesion was enriched in 5HT-positive axons (Fig. 4C,D). Axonal tips showed an increase in number of growth cones compared to retraction bulbs (data not shown) and frequently, out of the retraction bulbs a new process emanated (fig. S6). Additionally, we observed an ectopic distribution of 5HT-fibers coursing along the dorsal part of the cord in Taxol-treated animals. Thus, Taxol induces growth of 5HT-axons after SCI.
We next examined whether Taxol treatment leads to functional recovery after moderate spinal cord contusion (23). Similarly to the hemisection injury, Taxol treatment increased the number of 5HT-positive fibers caudal to the lesion 8 weeks after injury (167 ± 19 fibers versus 88 ± 8 fibers respectively; p= 0.002; n= 10/group; Fig. 4E). We tested locomotor performance by analysing voluntary paw placement using a gridwalk (24). After 2 and 4 weeks, Taxol and vehicle treated animals performed equally on the gridwalk (Fig. 4F). Importantly, however, while vehicle treated animals did not show any additional recovery, Taxol treated animals improved further with only a 5% misstep frequency after 6 and 8 weeks resulting in a 3.4-fold improvement in performance (p= 0.001; Fig. 4F; movies S3, S4). Thus, Taxol-induced functional recovery correlates with its axon growth inducing effect.
Current attempts to elicit axonal regeneration in the injured spinal cord include interference with extrinsic growth inhibitory factors present in CNS myelin and scar tissue (17-18, 25), or interference with their receptors or their signalling pathways (26-29). While these approaches aim to interfere with single inhibitory factors, here, we targeted the cytoskeleton, onto which growth inhibitory signalling pathways converge (27). Our data show that moderate stabilization of microtubules after CNS injury interferes with multiple intracellular processes that prevent axon regeneration. We conclude that Taxol has the potential to offer a multi-targeted therapy for spinal cord injury.
Supplementary Material
Acknowledgments
This work was supported by the Max-Planck-Society, DFG, IFP, the NIH, and the HFSP. We would like to thank Drs. Caroline Hill for the SMAD2-PAGFP plasmid, Axel Ullrich for the HaCaT cells, Klaus Dornmair, Mark Hübener, Rüdiger Klein, Dorothee Neukirchen, Michael Stiess, Sabina Tahirovic, Hartmut Wekerle for critically reading the manuscript, Mr. Van Duc Ha, Mrs. Tanja Irl and the Animal Injury and Repair Core facility from the International Center for Spinal Cord Injury for technical assistance.
Footnotes
Competing interests statement
The authors declare that they have no competing interest.
References
- 1.Liu X, et al. PLoS Med. 2005 Dec;2:e354. doi: 10.1371/journal.pmed.0020354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Westermann S, Weber K. Nat Rev Mol Cell Biol. 2003 Dec;4:938. doi: 10.1038/nrm1260. [DOI] [PubMed] [Google Scholar]
- 3.Erturk A, Hellal F, Enes J, Bradke F. J Neurosci. 2007 Aug 22;27:9169. doi: 10.1523/JNEUROSCI.0612-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Witte H, Neukirchen D, Bradke F. J Cell Biol. 2008 Feb 11;180:619. doi: 10.1083/jcb.200707042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klapka N, Muller HW. J Neurotrauma. 2006 Mar-Apr;23:422. doi: 10.1089/neu.2006.23.422. [DOI] [PubMed] [Google Scholar]
- 6.Shearer MC, et al. Mol Cell Neurosci. 2003 Dec;24:913. doi: 10.1016/j.mcn.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 7.Silver J, Miller JH. Nat Rev Neurosci. 2004 Feb;5:146. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- 8.Rowinsky EK, Cazenave LA, Donehower RC. J Natl Cancer Inst. 1990 Aug 1;82:1247. doi: 10.1093/jnci/82.15.1247. [DOI] [PubMed] [Google Scholar]
- 9.Okada S, et al. Nat Med. 2006 Jul;12:829. doi: 10.1038/nm1425. [DOI] [PubMed] [Google Scholar]
- 10.Schachtrup C, et al. J Neurosci. 2010 Apr 28;30:5843. doi: 10.1523/JNEUROSCI.0137-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moses HL, Coffey RJ, Jr., Leof EB, Lyons RM, Keski-Oja J. J Cell Physiol Suppl Suppl. 1987;5:1. doi: 10.1002/jcp.1041330403. [DOI] [PubMed] [Google Scholar]
- 12.Lindholm D, Castren E, Kiefer R, Zafra F, Thoenen H. J Cell Biol. 1992 Apr;117:395. doi: 10.1083/jcb.117.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Batut J, Howell M, Hill CS. Dev Cell. 2007 Feb;12:261. doi: 10.1016/j.devcel.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 14.Dong C, Li Z, Alvarez R, Jr., Feng XH, Goldschmidt-Clermont PJ. Mol Cell. 2000 Jan;5:27. doi: 10.1016/s1097-2765(00)80400-1. [DOI] [PubMed] [Google Scholar]
- 15.Dunn S, et al. J Cell Sci. 2008 Apr 1;121:1085. doi: 10.1242/jcs.026492. [DOI] [PubMed] [Google Scholar]
- 16.Jones LL, Yamaguchi Y, Stallcup WB, Tuszynski MH. J Neurosci. 2002 Apr 1;22:2792. doi: 10.1523/JNEUROSCI.22-07-02792.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bradbury EJ, et al. Nature. 2002 Apr 11;416:636. doi: 10.1038/416636a. [DOI] [PubMed] [Google Scholar]
- 18.Hurtado A, Podinin H, Oudega M, Grimpe B. Brain. 2008 Oct;131:2596. doi: 10.1093/brain/awn206. [DOI] [PubMed] [Google Scholar]
- 19.Laabs TL, et al. J Neurosci. 2007 Dec 26;27:14494. doi: 10.1523/JNEUROSCI.2807-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ylera B, et al. Curr Biol. 2009 Jun 9;19:930. doi: 10.1016/j.cub.2009.04.017. [DOI] [PubMed] [Google Scholar]
- 21.Dill J, Wang H, Zhou F, Li S. J Neurosci. 2008 Sep 3;28:8914. doi: 10.1523/JNEUROSCI.1178-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pearse DD, et al. Nat Med. 2004 Jun;10:610. doi: 10.1038/nm1056. [DOI] [PubMed] [Google Scholar]
- 23.Scheff SW, Rabchevsky AG, Fugaccia I, Main JA, Lumpp JE., Jr. J Neurotrauma. 2003 Feb;20:179. doi: 10.1089/08977150360547099. [DOI] [PubMed] [Google Scholar]
- 24.Behrmann DL, Bresnahan JC, Beattie MS, Shah BR. J Neurotrauma. 1992;9:197. doi: 10.1089/neu.1992.9.197. Fall. [DOI] [PubMed] [Google Scholar]
- 25.Freund P, et al. Nat Med. 2006 Jul;12:790. doi: 10.1038/nm1436. [DOI] [PubMed] [Google Scholar]
- 26.Dergham P, et al. J Neurosci. 2002 Aug 1;22:6570. doi: 10.1523/JNEUROSCI.22-15-06570.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yiu G, He Z. Nat Rev Neurosci. 2006 Aug;7:617. doi: 10.1038/nrn1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neumann S, Bradke F, Tessier-Lavigne M, Basbaum AI. Neuron. 2002 Jun 13;34:885. doi: 10.1016/s0896-6273(02)00702-x. [DOI] [PubMed] [Google Scholar]
- 29.Qiu J, et al. Neuron. 2002 Jun 13;34:895. doi: 10.1016/s0896-6273(02)00730-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.