

Abstract
Niemann-Pick disease type C (NPC disease) is an incurable cellular lipid trafficking disorder characterized by neurodegeneration and intralysosomal accumulation of cholesterol and glycosphingolipids. Treatment with miglustat, a small imino sugar that reversibly inhibits glucosylceramide synthase, which is necessary for glycosphingolipid synthesis, has been shown to benefit patients with NPC disease. The mechanism(s) and extent of brain cellular changes underlying this benefit are not understood. To investigate the basis of the efficacy of miglustat, cats with disease homologous to the juvenile-onset form of human NPC disease received daily miglustat orally beginning at 3 weeks of age. The plasma half-life of miglustat was 6.6 ± 1.1 hours, with a tmax, Cmax, and area under the plasma concentration-time curve of 1.7 ± 0.6 hours, 20.3 ± 4.6 μg/ml, and 104.1 ± 16.6 μg hours/ml, respectively. Miglustat delayed the onset of neurological signs and increased the lifespan of treated cats, and was associated with decreased GM2 ganglioside accumulation in the cerebellum and improved Purkinje cell survival. Ex vivo examination of microglia from the brains of treated cats revealed normalization of CD1c and class II major histocompatibility complex expression, as well as generation of reactive oxygen species. Together, these results suggest that prolonged Purkinje cell survival, reduced glycosphingolipid accumulation, and/or the modulation of microglial immunophenotype and function contribute to miglustat-induced neurological improvement in treated cats.
Keywords: Animal model, Cholesterol, Feline model, Glucosylceramide synthase, Glycosphingolipid, Miglustat, Niemann Pick
INTRODUCTION
Niemann-Pick disease type C (NPC disease) is an incurable cellular lipid trafficking disorder caused by mutations in the NPC1 or NPC2 genes and resulting in the accumulation of unesterified cholesterol and glycosphingolipids in the endosomal/lysosomal compartments of cells in the CNS and in visceral organs (1). Clinical features include progressive neurological dysfunction, hepatosplenomegaly, and early death. Brain histology and biochemistry reveal widespread neuronal cytoplasmic vacuolization and intracellular cholesterol and ganglioside storage, neuroaxonal dystrophy and neuronal loss most severely affecting Purkinje cells, cortical pyramidal neuron meganeurite formation and ectopic dendritogenesis, gliosis, and inflammation (2–4). The mechanisms by which deficient activity of NPC1 and NPC2 proteins result in clinical disease and neuropathologic alterations are not understood.
The only drug presently known to have measurable positive influence on NPC disease progression in human NPC disease patients is miglustat, a small imino sugar that inhibits glucosylceramide synthase and the synthesis of all glucosylceramide-based glycosphingolipids, including lactosylceramide and the gangliosides (5–14). A randomized controlled study in patients of various ages was performed with therapeutic efficacy assessed primarily by quantification of horizontal saccadic eye movements, and, secondarily by assessments of swallowing, auditory ability, ambulatory ability, and cognition (5, 15). Based on outcome data from this and subsequent studies (5, 8), miglustat was approved for use in the treatment of NPC disease in several countries but remains unapproved in the US pending further proof of efficacy. In these clinical trials there was no histological or biochemical analysis of brain tissue.
The ability of miglustat to ameliorate NPC disease was initially discovered in studies involving murine and feline disease models in which daily administration was found to delay the onset of neurological dysfunction and to increase lifespan (16). The purpose of the present study was to evaluate the pharmacokinetics and efficacy of miglustat in a cohort of cats with NPC disease and to evaluate the means by which miglustat exerts its positive clinical effects.
Naturally occurring feline NPC disease is due to a missense mutation in NPC1 (p.C955S; c.2864G<C) with clinical, neuropathological, and biochemical abnormalities similar to those present in juvenile-onset patients, the most common form of the disease in humans (2, 3, 17–21). The feline model has been critical for identifying the late endosomal/lysosomal accumulation of gangliosides (GM2 and GM3) and unesterified cholesterol (4), for evaluating the association of ganglioside storage with meganeurite formation and ectopic dendritogenesis (16), for correlating neuroaxonal dystrophy with neurological dysfunction (2), and for evaluating efficacy of experimental therapies (16, 22, 23). The current study provides additional strong support for the efficacy of miglustat in delaying neurological signs, increasing lifespan, and improving Purkinje cell survival in NPC disease. We also provide quantitative biochemical data showing moderate reductions in GM2 and GM3 gangliosides and sphingoid bases in brain following miglustat treatment and identify specific effects on microglial phenotype and function that may, in concert, explain its mechanism of action.
MATERIALS AND METHODS
Animals
Cats were raised in the animal colony of the School of Veterinary Medicine, University of Pennsylvania under the guidelines of the National Institutes of Health and USDA for care and use of animals in research. The study was approved by the University of Pennsylvania Institutional Animal Care and Use Committee. Cats were housed in an environment with a temperature of 21°C, 12-hour light cycles, and 12 to 15 air changes per hour. Food and water were offered ad libitum. A PCR-based DNA test was used for diagnosis of the NPC1 missense mutation in peripheral blood leukocytes in cats on the first day after birth (20). Cats were classified as homozygous (NPC), heterozygous for the mutant allele (HET) or lacking the mutant allele (wild type [WT]). Previous studies showed no significant differences in neurological function or pathology between WT and HET cats; therefore, HET cats were included in clinical studies where indicated (21). Neurological examination, post-rotatory nystagmus (PRN), serum biochemical testing, and brainstem auditory evoked response testing were performed as previously described (21, 23).
Miglustat was administered at a dose of 25 mg/kg of powder every 12 hours dissolved in 0.5 ml of water and administered orally via syringe. Miglustat-treated cats (n = 6) received miglustat beginning at 3 weeks of age (the first age at which cats begin to take food other than the dam’s milk), and continued to receive miglustat until death. Both male (n = 2) and female (n = 4) cats were treated with miglustat.
Pharmacokinetics
Pharmacokinetic data on miglustat were obtained in 4 25-week-old WT cats and in 2 8-week-old WT cats following oral administration of 25 mg/kg miglustat. One ml of blood was collected in EDTA at 0 (pre-dose), 0.5, 1, 2, 4, 6, 8, 12, 16, 20, 24, 28, 32, and 36 hours following administration. Blood was centrifuged immediately at 4°C and plasma was collected and stored at −80°C. The concentration of miglustat in plasma was quantified using a validated LC-MS/MS assay with miglitol as internal standard. The limit of quantification was 10 ng/mL and the inter-day coefficient of variation was <12% and the inaccuracy <8%.
Postmortem Examination
Cats were killed using an overdose of barbiturates in accordance with the American Veterinary Medical Association guidelines. All untreated NPC disease cats (NPCuntreated) and 3 miglustat-treated cats (NPC-miglustat) were killed when they were no longer able to maintain sternal recumbency without assistance. Additionally, 3 NPC-miglustat cats were killed at 24 weeks of age (the mean age of death in untreated cats) (21) to compare tissue between age-matched untreated and treated cats. WT and HET cats were killed between 20 and 29 weeks of age for histological and biochemical comparisons. Immediately before death, each cat was given 0.5 mL of heparin (1000 units/mL) i.v. After death the cats were perfused with 500 mL of 0.9% cold saline and samples of brain, liver, spleen, and lung were acquired and frozen. Next, 750 mL of cold 4% paraformaldehyde was perfused into the left ventricle of the heart. After perfusion, further samples of brain, liver, spleen, and lung were collected and fixed in paraformaldehyde for 48 hours. Fixed samples were paraffin-embedded, sectioned, and stained with hematoxylin and eosin (H&E). Immunohistochemistry for calbindin (anti-rat CALB; Swant, Marly, Switzerland), CD18 (Leukocyte Antigen Biology Laboratory, University of California, Davis, CA), GM2 ganglioside (mab10-11; Progenics Pharmaceuticals, Tarrytown, NY) and GM3 ganglioside (DH2, GlycoTech, Gaithersburg, MD) gangliosides were performed. Filipin (Sigma-Aldrich, St. Louis, MO) histochemistry was utilized for identification of unesterified cholesterol sequestration in cells. Tissues were cut on a cryostat or sectioned by either vibratome or microtome for the immunohistochemical and histochemical procedures. Histological procedures were carried out according to published methods (3, 21, 24).
Biochemical Analysis of Lipids
Lipid studies were conducted on frozen tissues essentially as in previous studies (25–27). Water homogenates (20%) were obtained from dissected cerebral gray matter and cerebellum or from liver and total lipids were extracted in chloroform:methanol 1:2 (v/v), as described (25). Protein content was measured on the total homogenates by a modified Lowry procedure (28). Part of the extract was desalted and separated into 2 fractions using 100 mg reverse-phase Bondelut C18 columns (Agilent Technologies, Englewood, CO) (29, 30). The acid fraction eluted with methanol-water 12:1 (v/v) contained all gangliosides and was used without further purification. Total sialic acid was measured by the Svennerholm resorcinol-HCl method and aliquots of this fraction spotted on high performance silica gel G thin layer chromatography (HPTLC) plates (Merck, Darmstadt, Germany) using a Linomat 5 device (Camag, Muttenz, Switzerland).
The plates developed in chloroform-methanol-0.2% CaCl2 55:45:10 (by volume) were sprayed with resorcinol-HCl to visualize the sialic acid moiety of individual gangliosides, and densitometric quantification at 580 nm was performed using a Camag TLCII scanner/Cats software system(25). After normalization to the total sialic acid content and eventual correction of water loss using the protein content, individual concentrations were calculated taking into account the number of sialic acids for each ganglioside. The study of neutral glycolipids (glucosylceramide, lactosylceramide, gangliotriaosylceramide) was done on the second column eluate, obtained with chloroform-methanol 1:2 (v/v). In the brain tissue, separation of glucosylceramide from galactosylceramide was achieved using borate-impregnated HPTLC plates. Densitometry of plates sprayed with the orcinol-sulfuric acid reagent was done at 650 nm (25, 30). Chromatographic systems have been described (25). Analysis of the free sphingoid bases sphingosine and sphinganine was carried out on total lipid extracts by high-performance liquid chromatography using eicosasphinganine (Matreya, Pleasant Gap, PA) as an internal standard. Separation of orthophtalaldehyde derivatives was achieved on a 200 × 5 mm Spherisorb ODS2 column (Waters) and monitored by fluorometry (31).
Biochemical Protein Studies
Western blot analysis of LC3 was carried out according to the following method: Frozen brain tissue was homogenized in ice cold lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% Igepal CA-630, 1% deoxycholic acid, 0.1% SDS supplemented with protease inhibitor cocktail), centrifuged (15000 rpm) for 30 minutes at 4°C and the supernatants (soluble fraction) were collected. Protein concentrations were determined using a BCA protein assay kit. For immunoblotting, samples were analyzed by SDS-PAGE (16% gels) under reducing conditions and transferred to Immuno-blot PVDF membranes. Membranes were blocked in 1x TBS, 0.1% Tween-20, 5% non-fat dry milk, 1% BSA, followed by incubation with an antibody to microtubule-associated protein light chain 3 (LC3, 2 mg/ml) and subsequent incubation with Peroxidase Labeled anti-Rabbit IgG secondary antibody (1:5000). SuperSignal West Pico Chemiluminescent Substrate was used for protein detection on a KODAK 2000R imaging station. Protein quantification for each sample was performed by densitometric analysis using KODAK imaging software and normalized to actin in the same sample. This analysis was represented as LC3-II/Actin normalized to the WT control.
Microglia Isolation and Characterization
Feline microglial cells were isolated via density gradient centrifugation using previously described methods with slight modifications (32). Briefly, samples of brain were collected immediately following saline perfusion and transported in Hankś solution (Gibco Invitrogen, Carlsbad, CA) with 3% fetal calf serum adjusted to pH 7.36, on ice. The meninges were carefully removed and 12 g of brain tissue were mechanically dissociated by mincing through a stainless-steel sieve and enzymatically digested with type II collagenase (Roche Diagnostics, Mannheim, Germany) and DNAse I (Sigma-Aldrich) for 60 minutes at 37°C. The digested CNS preparation was washed twice, resuspended in 45 ml of isotonic Percoll (Amersham Biosciences, Piscataway, NJ) and diluted in Hankś buffer resulting in a density of 1.030 g/ml. For the initial gradient this suspension was underlayered with 5 ml of isotonic Percoll of the density 1.124 g/ml. After centrifugation (1,250 × g for 25 minutes at 20°C) cells from the 1.124 g/ml-density surface were collected, washed, and for further purification transferred to a major gradient consisting of 5 consecutive Percoll-densities (5 ml of the density 1.124 g/ml, 12 ml of 1.072 and 1.060 g/ml each, 8 ml of 1.050, and 1.030 g/ml Percoll) in a 50-ml tube. After centrifugation, feline microglia accumulated specifically on the surfaces of the densities 1.072 and 1.060 g/ml Percoll (33). The cells were collected, washed and subsequently used for the ex vivo examinations. Cell viability was assessed by Trypan blue exclusion.
Phenotypic-expression of feline microglial cells was evaluated by flow cytometry with several monoclonal antibodies either specific for, or cross-reactive with, feline cells. Staining of microglial cells was performed as previously described for the canine species (32). Briefly, after Fc-receptor blockage with normal goat serum IgG (dilution 1:11; Jackson ImmunoResearch Laboratories, Inc., West Grove, PA), primary monoclonal antibodies against the surface molecules CD18, CD11b, CD11c, CD45, CD1c, intercellular adhesion molecule-1 (CD54), B7-1 (CD80), major histocompatibility complex (MHC) class I, MHC class II, CD44, CD14 (directly labeled), CD4, CD8α, CD21, and CD3 (Table 1) were incubated for 30 minutes at 4°C and washed twice with Hankś buffered saline. Secondary antibodies (Table 2) were diluted 1:100 and incubation continued for 30 minutes. Staining against CD3 required fixation and permeabilization of the cells. After washing, cells were resuspended in FACSFlow (BD Biosciences, San Jose, CA) and analyzed immediately ex vivo after staining without fixation with a FACSCalibur (BD Biosciences) using the Cell-Quest software. Negative control samples were stained with the secondary antibodies only. Analysis gates were adjusted to not exceed 2% positive staining with negative controls. For each sample 10,000 live-gated events were analyzed.
Table 1.
Primary Antibodies Used for Immunohistochemistry
| Antibody | Clone | Isotype | Dilution used | Company |
|---|---|---|---|---|
| CD3ε | CD3-12-rat | IgG1 | 1 : 5 | Leukocyte Antigen Biology Laboratory, (LABL), P. F. Moore, University of California, Davis, CA |
| CD4 | FE1.7B12 | IgG1 | 1 : 5 | LABL |
| CD8α | FE1.10E9 | IgG1 | 1 : 5 | LABL |
| CD21 | CA2.1D6 | IgG1 | 1 : 5 | LABL |
| CD18 | Fe3.9F2 | IgG1 | 1 : 5 | LABL |
| CD11b | CA16.3E10 | IgG1 | 1 : 5 | LABL |
| CD1c | Fe5.5C1 | IgG1 | 1 : 5 | LABL |
| MHC II | 42.3 | IgG1 | 1 : 5 | LABL |
| ICAM-1 | RR1-1 | IgG1 | 1 : 50 | Bender MedSystems GmbH, Vienna, Austria |
| CD44 | BAG40A | IgG3 | 1 : 5 | VMRD, Pullman, WA |
| CD14 | TUK4 | IgG2ακ | 1: 7 | Dako, Carpinteria, CA |
| CD11c | CA11.6A1 | IgG1 | 1 : 5 | Serotec, Raleigh, NC |
| CD45 | 25-2C | IgM | 1 : 16 | VMRD |
| MHC I | CF298A | IgG1 | 1 : 16 | VMRD |
| B7-1* | B7.1.66 | IgG1 | 2 : 1 | M. Tompkins, North Carolina State University, Raleigh, NC |
The antibody was prepared as tissue culture supernatant with 0.1% sodium azide (62).
CD = cluster of differentiation, Ig = Immunoglobulin, MHC I = major histocompatibility Complex type I, ICAM = intercellular adhesion molecule.
Table 2.
Secondary Antibodies
| Antibody name | Fragment | Isotype | Dilution used | Company |
|---|---|---|---|---|
| gαm-PE | R-Phycoerythrin F(ab’)2 fragment goat anti-mouse FITC |
IgG | 1 : 100 | Jackson Immuno Research Laboratories, West Grove, PA |
| FITC | conjugated F(ab’)2 fragment goat anti-mouse |
IgM, μ chain-specific | 1 : 100 | Serotec, Raleigh, NC |
PE = phycoerythrin, FITC = fluorescein isothiocyanate, Ig = immunoglobulin.
Microglial generation of reactive oxygen species (ROS) was analyzed as described with slight modifications (34). Briefly, the cell suspension was adjusted to a concentration of 2 × 105 cells in 100 μl and pre-incubated for 15 minutes at 37°C and 5 % CO2. This incubation step was repeated after adding either phorbol myristate acetate (PMA, Sigma-Aldrich) to result in a final concentration of 100 nM or phosphate buffered saline (PBS), which served as negative control. An additional tube with microglial cells only was used as negative reference to evaluate morphology and background fluorescence of the cells. Dihydrorhodamine 123 (Marker Gene Technologies Inc., Eugene, OR) was diluted in dimethyl sulfoxide to a concentration of 15 μg/ml. After adding 20 μl of the diluted dihydrorhodamine 123, incubation was again repeated. Cells were then put on ice in dark surroundings to stop ROS generation, minimize the amount of tube surface adhering cells, and to protect the fluorochromic particles. After addition of FACSFlow samples were measured immediately by flow cytometry in duplicates for each approach and mean values were used for evaluation. The results were evaluated on the basis of percentage of positive (ROS-producing) cells and mean fluorescence intensity (measured by means of fluorescent channel numbers, log values) as a relative means of the intensity of ROS-generation seen as a shift in FL1. Results were evaluated using the multiple histogram analysis of the FACS Cell-Quest Software provided.
Microglial phagocytosis was assessed with heat-killed and lyophilized FITC-labeled Staphylococcus aureus (Invitrogen, Molecular Probes, Eugene, OR) that were resuspended in PBS, sonicated, and adjusted to a concentration of 8 × 108 bacteria/ml. Bacteria were incubated with 20 μl pooled cat serum diluted with PBS (1:5) for 60 minutes at 37°C and 5% CO2 in a humid atmosphere for opsonization. Microglia were mixed with either opsonized, non-opsonized bacteria suspension resulting in a ratio of 1:100, or with PBS (negative control). Incubation was performed as described before with gentle resuspension after 30 minutes to ensure optimal cell-to-cell-contacts. To stop the phagocytosis reaction and minimize adhesion of cells on the tube surface after incubation, cells were put on ice for 15 minutes. After addition of 100 μl FACSFlow into each tube, the percentage of cells performing phagocytosis and phagocytosis intensity (measured as relative means by mean fluorescent channel numbers, values shown in log) was determined by immediate flow cytometric measurement with the FACSCalibur and Cell-Quest software. Phagocytosis measurements were performed in duplicates and mean values were used for evaluation.
Statistical Analysis
Mean values and standard deviations were calculated to describe the findings. The unpaired 2-tailed t test was used to compare data of untreated cats with NP-C with miglustat-treated and normal cats. Values were considered statistically significant at the p < 0.05 (* or †) or p < 0.001 (**) level.
RESULTS
Pharmacokinetics of Miglustat in WT Cats
The plasma half-life of miglustat was 6.6 ± 1.1 hours in 3 WT 25 wk old cats in which it was orally administered. tmax, Cmax, and area under the plasma concentration-time curve in these cats were 1.7 ± 0.6 h, 20.3 ± 4.6 μg/mL, and 104.1 ± 16.6 μg*h/mL respectively. Mean half-life, Tmax, Cmax, and area under the plasma concentration-time curve (AUC) in 2 WT 8-week-old cats were 7.5 hours, 1.29 hours, and 24.7 μg/mL, and 104.1 μg*h/mL, respectively.
Clinical Outcome Data in NPC Disease Cats Treated with Miglustat
The mean time to euthanasia due to disease progression was significantly longer (35.7 ± 0.6 weeks) for NPC-miglustat cats than for the NPC-untreated cats (20.5 ± 4.8 weeks). Increased survival time was due to a delay in onset of the more severe signs of neurological dysfunction typically observed late in disease (Fig. 1A). Specifically, a significant difference in onset of clinical signs of disease was identified in treated cats when compared to untreated 17- and 19-week-old cats, while no significant difference was found in clinical signs between untreated and treated cats at 6 and 12 weeks of age. Remarkably, treated cats maintained the ability to walk for 11 weeks longer than untreated cats but all treated cats eventually developed the full spectrum of clinical signs that were of the same severity as those in untreated cats.
Figure 1.

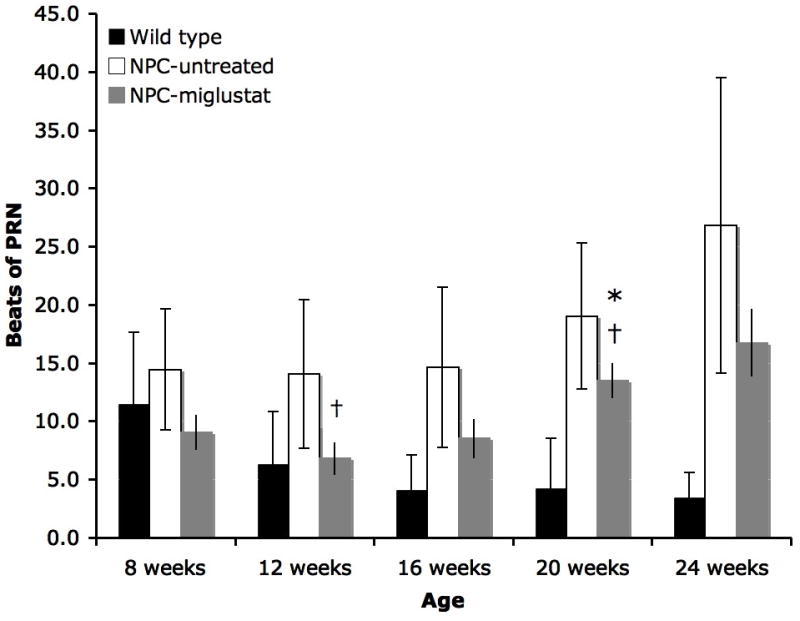
Miglustat delayed onset of neurological dysfunction in Niemann-Pick disease type C (NPC disease) cats. (A) There was a significant delay in the inability to walk and stand in miglustat-treated cats vs. untreated 17- and 19-week-old cats (*p < 0.05). (B) Miglustat treatment also decreased the number of beats of post-rotatory nystagmus (PRN). Significantly, less PRN was identified in NPC-miglustat cats at 12 and 20 weeks of age vs. NPC-untreated cats (†, p < 0.05). NPC-miglustat cats were not significantly different from WT cats in PRN until 20 weeks of age (*), whereas NPC-untreated cats were significantly different from WT cats at 12, 16, 20, and 24 weeks of age.
Untreated NPC-affected cats showed a significantly greater number of beats of PRN vs. WT cats beginning at 12 weeks of age and continued to show greater beats of PRN until death at 24 weeks of age (Fig. 1B). In contrast, treated cats were not significantly different from WT cats until 20 weeks of age. When treated animals were compared to untreated animals, significantly fewer PRN beats were found at 12 and 20 weeks of age (Fig. 1B). Quantitative testing of the auditory system showed no significant differences between NPC-miglustat cats and untreated cats in measures of central conduction time, wave V/I amplitude, or in hearing threshold (data not shown).
NPC-untreated cats showed significant elevations in serum ALT, AST, cholesterol, and chitotriosidase compared to WT cats whereas no differences were identified between NPC-miglustat and NPC-untreated cats in these measures. Finally, no significant differences in body weight were identified between NPC-untreated cats and NPC-miglustat cats (data not shown), which were each significantly lower than control cats beginning at 5 weeks of age and continuing until the time of death.
Histological Studies
Three NPC-miglustat cats were euthanized at 24–25 weeks of age to compare brain histology and biochemistry to age-matched NPC-untreated cats and to 3 other NPC-miglustat cats at terminal stage (35–36 weeks of age). There was widespread accumulation of GM2 and GM3 gangliosides and cholesterol in the cerebral cortex was in the treated and non-treated 24- to 25-week-old NPC-affected cats (Fig. 2). Blinded analysis comparing the two groups for storage of GM2 and GM3 gangliosides revealed qualitatively less accumulation of both gangliosides in NPC-miglustat cats (Fig. 2A, B vs. D, E). As anticipated, WT brain exhibited no detectable GM2 or GM3 ganglioside accumulation (Fig. 2G, H). Unexpectedly, in addition to the reduced ganglioside storage, cholesterol storage, as revealed by levels of filipin staining also appeared to be qualitatively reduced in the treated vs. NPC-untreated cat (Fig. 2C, F). As anticipated, WT cerebral cortex showed no significant filipin labeling (Fig. 2I). NPC disease cats that survived to 35 weeks following treatment also showed GM2 and GM3 gangliosides and cholesterol at detectably lower levels than those observed in the 24-week-old untreated NPC disease cat at end-stage disease (not shown).
Figure 2.

Chronic miglustat treatment leads to detectable reductions in GM2 (A, D, G) and GM3 (B, E, H) ganglioside immunohistochemical staining and in filipin labeling (C, F, I). (A–I) Representative sections of cerebral cortex stained for GM2 (A), GM3 (B) and filipin (C) in an untreated 24-week-old Niemann-Pick disease type C (NPC disease) cat compared to GM2 (D), GM3 (E) and filipin (F) staining in a miglustat-treated NPC disease cat at 25 weeks of age, and GM2 (G), GM3 (H) and filipin (I) staining in an untreated normal young adult cat. Immunostaining for GM2 ganglioside in the untreated animal reveals abundant staining of cell bodies and in smaller processes throughout all layers of the cerebral cortex. Staining in the miglustat-treated cat appears less dense in cell bodies with fewer punctae (assumed to be small dendritic profiles) evident in the neuropil. Similarly, GM3 labeling appears less in the treated cat (E vs. B). Surprisingly, filipin labeling also appears reduced in the treated animal compared to untreated animal (F vs. C). The WT animal, as expected, showed no detectable accumulation of either ganglioside (G, H); neurons with filipin labeling were seen as dark silhouettes lacking evidence of cholesterol accumulation. Bar = 60 μm; the cerebral cortex layer marker in H apply to all panels.
In the cerebellum, NPC-miglustat cats exhibited a remarkable preservation of Purkinje cells as compared to age-matched NPC-untreated cats (Fig. 3). Nonetheless, storage of GM2 ganglioside and cholesterol was prominent in all of the surviving Purkinje cells in both treated and untreated animals (Fig. 3B, C, E, F). Again, however, in terms of the relative levels of GM2 staining observed in the granule cell layer of the cerebellum, this appeared qualitatively less in the treated cat (Fig. 3E vs. 3B). The cerebellum of NPC-miglustat cats that survived to 35 weeks showed loss of Purkinje cells, whereas surviving cells containing GM2 and cholesterol storage similar to the untreated cat at 24 weeks of age (not shown). Finally, no differences were identified between NPC-miglustat and NPC-untreated cats on light microscopic evaluation of the liver.
Figure 3.

Chronic miglustat treatment prolongs Purkinje cell survival in Niemann-Pick disease type C (NPC disease) cerebellum as evidenced by calbindin (A, D, G) and GM2 ganglioside immunohistochemistry (B, E, H) and filipin histochemistry (C, F, I). (A–C) Panels show representative sections of cerebellar cortex from an untreated NPC disease cat at 24 weeks of age compared to a miglustat-treated NPC disease cat at 25 weeks of age (D–F) and an untreated WT young adult cat (G–I). Purkinje cell death in the untreated cat far exceeds that found in treated and WT animals (A vs. D and G). The few surviving Purkinje cells (A, arrows) in the untreated affected animal also exhibit axonal spheroids (A, arrowheads). Staining for GM2 ganglioside reveal abundant staining of surviving Purkinje cells and of granule cells in the granule cell layer (GCL) in both treated and untreated NPC disease cats (B vs. E, respectively), with GM2 staining appearing more intense in granule cells of the non-treated cat (B vs. E). Neurons in normal cat cerebellum do not express detectable levels of GM2 ganglioside (H). Filipin labeling also reveals Purkinje cell loss in the untreated NPC disease cat compared to the treated animal (C vs. F, arrows). The WT cat (I) has an intact row of Purkinje cells as dark silhouettes (arrows). The calibration bar (130 μm) and the cerebellar layer marker in H apply to all panels. Pia = Pial lining on cerebellar surface; ML = molecular layer; PC = Purkinje cell layer; GCL= granule cell layer; WM = cerebellar white matter). Panels A, D and G are also stained with Nissl.
Biochemical Lipid Studies in Brain
Biochemical studies of human patients have shown that sphingolipid abnormalities essentially affect the cerebral gray matter and much less the cerebral white matter (35). This is also true for the cat model (Vanier and Vite, unpublished data) and is well in line with the neuropathological studies. Analyses in cerebrum were, therefore, conducted in carefully dissected gray matter. Because of their limited sizes, cerebellar samples included some white matter. Cholesterol concentrations were not specifically studied because it has previously been shown that the distribution changes observed with filipin staining are quantitatively too small to affect the global levels of cholesterol in brain, even in gray matter (35).
In cerebral gray matter of NPC-untreated cats (aged 21–29 weeks; n = 8), GM2 and GM3 gangliosides, which are normally minor components, showed a 10- to 12-fold increase compared to WT cats (Fig. 4A, B). Glucosylceramide and lactosylceramide, only present in minute amounts in gray matter of unaffected humans (35) or cats (data not shown), were also markedly increased in NPC-untreated cats (Fig. 4C, D), as previously reported for affected patients and mice (30, 35). The rate of migration of the single band observed for each of these lipids is in line with a C18:0 fatty acid moiety, as documented in human NPC disease (35). In cerebral gray matter of miglustat-treated cats (n = 6) there was a minor reduction in the concentrations of GM2 (414 ± 62 vs. 483 ± 46 nmol/g; p = 0.034), and a mean 27% reduction in the concentrations of GM3 gangliosides, with large inter-individual variations (406 ± 129 vs. 563 ± 86 nmol/g; p = 0.017). By biochemical measurement, comparison between 24-week-old and 35- to 36- week-old treated cats did not disclose clear differences. In normal human subjects, cerebellar tissue is known to show a ganglioside pattern strikingly different from that observed in cerebrum, with predominantly polysialogangliosides of the b-series. This was also the case in cats (Fig. 4A). The WT concentrations of GM3 and GM2 gangliosides in cerebellum appeared slightly lower than those in cerebrum (Fig. 4B). In NPC-untreated cats (n = 4), the increase of GM2 was 10- to 12-fold that of WT (i.e. its concentration was about half of that in cerebral cortex), but GM3 was only 3- to 4-fold that of WT (Fig. 4B). In the cerebellum of NPC-miglustat cats (n = 5) there was a mean 35% decrease in storage of GM2 (185 ± 24 vs. 284 ± 63 nmol/g; p = 0.014), and there was no significant change in the concentration of GM3 (Fig. 4B). Again, no clear trend was found comparing 24-week-old and 35- to 36-week-old treated animals. Concentrations of minor neutral glycosphingolipids were only studied in cerebral cortex. There were differences in treated vs. untreated NPC disease cats for glucosylceramide and lactosylceramide (Fig. 4C, D). Miglustat-treated cats showed an approximately 3- fold increase in their concentrations of glucosylceramide and an approximately 2-fold decrease in their concentrations of lactosylceramide. No noticeable change was observed regarding the concentrations of gangliotriaosylceramide (asialo-GM2).
Figure 4.

Gangliosides and minor neutral glycosphingolipids in cerebral and cerebellar gray matter of wild type (WT), untreated and miglustat-treated Niemann-Pick disease type C (NPC disease) cats. (A) Total ganglioside profiles obtained by high performance thin layer chromatography (HPTLC) with sialic acid-specific (resorcinol) staining, showing increases of GM2 and GM3 gangliosides in NPC disease cats. Each lane corresponds to 3 mg wet weight tissue. (B) Quantitative data (expressed as nmol of ganglioside/g wet weight tissue) for monosialogangliosides GM3, GM2 and GM1. Note that the y scales differ for cerebral and cerebellar tissues. Comparison of GM2 levels in treated and untreated cats: p = 0.03 in cerebrum, p = 0.01 in cerebellum. Comparison of GM3 levels in treated and untreated cats: p = 0.02 in cerebrum. (C) Glucosylceramide concentrations in pooled cerebral gray matter tissue from 3 miglustat-treated vs. 2 untreated NPC disease cats, separated from galactosylceramide on borate-impregnated HPTLC plates, visualized by orcinol-sulfuric reagent. An equivalent of 15 mg wet weight tissue was spotted (less for quantification). The reference glucosylceramide was from spleen and therefore had 2 bands of different fatty acids). (D) Lactosylceramide in cerebral gray matter tissue from miglustat-treated and untreated NPC disease cats separated on HPTLC plates and visualized by an orcinol-sulfuric acid reagent. The amounts spotted are indicated. Abbreviations: WT = wild type; mig = miglustat; untr’d = untreated; Glc-cer = glucosylceramide; Gal-cer = galactosylceramide; Lac-cer = lactosylceramide; GA2 = gangliotriaosylceramide.
Free sphingoid bases, sphingosine (Fig. 5), and sphinganine were also studied. In untreated affected cats, a 3-fold increase was observed both in cerebral gray matter and in cerebellum, compared to WT age-matched cats. A proportional or higher increase in sphinganine (not shown) was also observed. In cerebrum of miglustat-treated cats (n = 6) there was a small but significant reduction in sphingosine concentrations compared to those in untreated cats (n = 8) (113 ± 19 vs. 149 ± 14 pmol/mg protein; p = 0.001). A similar, although not significant trend (possibly due to the smaller number of samples studied) was observed in cerebellum.
Figure 5.

Free sphingosine concentrations in cerebral and cerebellar gray matter of wild type (WT), untreated and miglustat-treated Niemann-Pick disease type C (NPC disease) cats. Concentrations are expressed as pmol/mg protein. Comparison between miglustat-treated and untreated cats in cerebral gray matter: p = 0.001.
Lipid Studies in Liver Tissue
As expected, miglustat has no effect on cholesterol, sphingomyelin or bis(monoacylglycero)phosphate (BMP) (Fig. 6A). This also applied to the levels of free sphingosine and sphinganine. Sphingosine showed an approximately 50-fold increase in affected untreated cats (1675 ± 155 pmol/mg protein (n = 4) vs. a normal value of 37 ± 15 (n = 7). Values of 1375, 1940 and 2250 pmol/mg protein were found in 3 miglustat-treated cats. With regard to glycosphingolipids, liver tissue from miglustat-treated cats showed no significant reduction vs. untreated cats in the very abnormal levels of glucosylceramide, lactosylceramide and globotriaosylceramide (Fig. 6B). GM3 ganglioside was increased approximately 5 times in affected untreated cats (1.1–1.5 μmol/g tissue) vs. normal cats (0.2–0.3 μmol/g) and remained unchanged in miglustat-treated cats (0.9–1.4 μmol/g). We were thus unable to demonstrate any effect of miglustat (histologically or biochemically) on liver lipid storage in cats.
Figure 6.

Total lipids and neutral glycosphingolipids in liver tissue of wild type (WT), untreated and miglustat-treated Niemann-Pick disease type C (NPC disease) cats. (A) Total lipid profiles in non-acidic and acidic fractions. An equivalent of 2 mg wet weight tissue was applied to the high performance thin layer chromatography (HPTLC) plate, revealed by the anisaldehyde spray. (B) Neutral glycosphingolipids separated on HPTLC plates (equivalent spotted of 12 mg wet weight tissue) and visualized by orcinolsulfuric acid reagent. BMP = bis(monoacylglycero)phosphate; Glc-cer = glucosylceramide; Gb3 = globotriaosylceramide; Lac-cer = lactosylceramide; mig = miglustat; untr’d = untreated.
Microglia Immunophenotype
Recent studies suggest that inflammation in CNS neuronal storage disorders might play an important role in the pathogenesis of neurodegeneration and its therapy (36, 37). Therefore, we characterized microglial immunophenotype and function ex vivo to evaluate a possible impact on the pathogenesis of NPC disease. Feline microglia cells were identified on the basis of size (FSC = forward scatter), complexity (SSC = side scatter), and their characteristic CD18-positive and CD45low immunophenotype. For most parameters examined 10,000 live-gated events could be evaluated in flow cytometry. Microglial purities of 86.1 to 99.0% (× = 95.3) were used for the ex vivo examinations. The low percentage of CD4-positive (× = 2.9%), CD8α-positive (× = 2.6%), CD21-positive (× = 2.4%), and CD3-positive (× = 1.9%) cells indicative of T and B lymphocytes revealed an insignificant contamination with these cells.
In healthy age-matched WT cats, the isolated microglial cells appeared as a homogenous population of relatively small cells (Fig. 7A). In contrast, the microglia cell population of NPC-untreated cats displayed a distinctly increased diversity in size and complexity (Fig. 7B, C); less diversity was apparent in miglustat-treated cats (Fig. 7C).
Figure 7.

(A–C) Microglia populations in a FACS dot plot preparation shown as size (FSC, abscissa) vs. complexity (SSC, ordinate) of a healthy age-matched control cat (A), a Niemann-Pick disease type C (NPC disease)-affected untreated cat (B), and an NPC-affected miglustat-treated cat (C). Microglia in healthy cats are a homogenous population of relatively small cells (A), whereas the microglia population in NPC-affected cats show increased diversity in size and complexity (B, C), which may reflect different states of activation. FSC = forward scatter, SSC = side scatter. MIF = mean fluorescence intensity.
Eleven antibodies were used to characterize microglial immunophenotype. Two parameters were assessed for each surface antigen examined: the percentage of positive microglia cells and the mean fluorescence intensity (measured by mean fluorescent channel numbers) as a relative mean for level of expression. The percentage of CD1c-positive cells and the expression intensity of CD1c were inversely affected by disease. NPC-untreated cats showed a decrease in CD1c-positive cells but an increase in expression compared to WT cats (p = 0.0164, Fig. 8A, B). In contrast, 35 wk old NPC-miglustat cats, with clinical signs as severe as those found in untreated 24 wk old cats, showed percentage and expression of CD1c that were more similar to those of WT than to those of NPC-untreated cats. Despite the variation within the group of 4 NPC miglustat-treated cats at the 2 ages examined, significantly lower expression intensity was found in the miglustat-treated cats compared to NPC-untreated cats (p = 0.04, Fig. 8B). In contrast, the expression intensity of CD45 was significantly upregulated in miglustat-treated cats (p = 0.02) compared to WT cats, although the percentage of CD45-positive microglia was unaltered (data not shown).
Figure 8.

Microglial expression of CD1c and intensity of CD1c expression and major histocompatibility complex type I (MHC I) in healthy, Niemann-Pick disease type C (NPC disease)-affected untreated and NPC-affected miglustat-treated cats. (A) The percentage of CD1c-expressing microglia showed a tendency for downregulation in NPC-affected untreated and 24-week-old miglustat-treated cats (A). Cats affected by NPC display upregulated expression intensity of CD1c and MHC I (B, C). w = weeks, CD = cluster of differentiation. (*p < 0.05)
Upregulation of the expression intensity was also seen for the surface antigens B7-1 (CD80) (data not shown), CD14 (data not shown), and MHC I (Fig. 8C) in NPC– untreated cats vs. WT cats. The expression intensity of MHC I was also significantly less in NPC-miglustat cats at both 24 and 35 weeks of age vs. NPC-untreated cats (Fig. 8C; p = 0.02). Intercellular adhesion molecule-1 and CD44 expression intensity were significant upregulated in miglustat-treated cats vs. WT cats (p = 0.004 and 0.04, respectively). For CD44 there was a tendency for upregulation in NPC-untreated cats compared to the controls (p = 0.08). No difference in expression or expression intensity between NPC-untreated and WT cats could be demonstrated for the integrins CD18, CD11b, and CD11b, or for MHC II.
Generation of ROS
To evaluate microglial generation of oxygen radicals on the pathogenesis of NPC and their modulation by miglustat we performed an ROS generation test. Feline microglia could be triggered to produce ROS, which is reflected by the high percentage of positive cells (PBS and PMA) when compared to the negative control (Fig. 9). The percentage of ROS-generating microglia was significantly lower in NPC-untreated cats compared to WT (p = 0.02 for PBS both and PMA) and NPC-miglustat cats (p = 0.02 for both PBS and PMA; Fig. 9). There was no difference in the enhancement of ROS generation by stimulation with PMA. The intensity of ROS generation was significantly enhanced in NPC-untreated cats compared to WT cats (p = 0.04 for PBS and PMA; Fig. 9B). The difference between the groups of cats was already very distinct in comparison to the negative control preparation, which was 4.3-fold higher in untreated and 5.4-fold higher in NPC–miglustat cats (p = 0.0001 and p = 0.04, respectively), and compared to age-matched WT cats. The 2 35-week-old miglustat-treated cats demonstrated a distinctly higher ROS generation intensity than the 2 24-week-old cats (Fig. 9B), leading to a high standard deviation within this group of treated cats. Therefore, only a tendency for an upregulated ROS generation intensity could be recognized compared to healthy control cats (p = 0.1).
Figure 9.

Microglial generation of reactive oxygen species (ROS) in healthy agematched controls (WT), Niemann Pick disease type C (NPC)-affected untreated, and miglustat-treated cats. (A) A significantly lower percentage of microglia could be triggered to generate ROS in NPC-affected untreated cats compared to the miglustat-treated and healthy control cats (p = 0.02). (B) In contrast, ROS generation intensity in NPC-affected untreated cats was significantly greater than that in WT cats (p = 0.04) whereas in miglustat-treated cats in comparable age groups there was only a tendency for enhanced ROS generation intensity (p = 0.0973). (C, D) Percentage of microglia phagocytosing of Staphylococcus aureus (C) and phagocytosis intensity (D) in WT, NPC-affected untreated and miglustat-treated cats. Microglia from all groups could be triggered to ingest bacteria (C). There was greater phagocytosis intensity in NPC-affected untreated vs. WT cats (D) (p = 0.006). w = weeks, nops = non-opsonized bacteria, ops = opsonized bacteria, * = p < 0.05 w = weeks, PBS = phosphate-buffered saline, PMA= Phorbol-12-Myristate-13-Acetate, (* p < 0.05).
Phagocytosis
Feline microglia showed a distinct phagocytosis of FITC-labeled Staphylococci, reflected by the high percentage of positive cells (non-opsonized and opsonized bacteria) compared to the negative control (Fig. 9C). No differences were noted between age-matched WT and cats affected with NPC either treated or untreated (Fig. 9C).
A significantly enhanced phagocytosis of opsonized Staphylococci could be demonstrated in the NPC-untreated cats compared to the age-matched WT cats (p = 0.006, Fig. 9D). This significant difference could also be detected for the negative control preparation, which showed a 3.5-fold higher intensity in the NPC-untreated cats compared to the age-matched WT cats (p = 0.0001).
DISCUSSION
Miglustat is approved by the FDA for treatment of adult patients with mild to moderate type 1 Gaucher disease. Although it is not currently approved for the treatment of NPC disease in the US, it is approved for this purpose in the European Union by the European Medicines Agency and in other countries (Switzerland, Liechtenstein, Iceland, Norway, Brazil, South Korea, Australia, Russia, New Zealand, Turkey, Colombia, Mexico, Chile, Argentina, and Canada). We have evaluated the use of miglustat in the feline model, which recapitulates both the neuropathological and biochemical abnormalities observed in juvenile-onset patients, the most common subset of human NPC patients (21). The feline model, along with the murine model, was the first in which miglustat administration was shown to ameliorate clinical neurological disease (16). Two affected cats treated at 7 and 13 weeks of age with 50 mg/kg/day miglustat for 3 weeks and 8 weeks, respectively, showed a delay in the onset of intention tremor and ataxia, increased survival of Purkinje cells, and an apparent reduction in brain ganglioside accumulation (16). The dose used for our study was chosen based on efficacious results from this initial study in the absence of any signs of untoward effects (16). The current work extends the results of the previous investigation while adding new quantitative brain and liver biochemistry as well as evaluating microglial activation. In the current study both dose and age at which treatment was instituted were strictly regulated. The pharmacokinetic study in WT cats shows that oral administration of a single dose of 25 mg/kg miglustat results in similar tmax and half-life as found in humans in whom a single dose of 100 mg given orally resulted in a maximum concentration at 2 to 2.5 hours and a half-life of 7.3 hours. In contrast, both Cmax (862 ng/ml) and AUC (9502 ng*h/ml) measures in humans were lower than those found in cats reflecting differences in dose between species. Clinically, the onset of specific neurological dysfunction was compared in treated and untreated cohorts in order to evaluate the effect of treatment on these measures. Miglustat given at 25 mg/kg every 12 hours beginning at 3 weeks of age, prior to the onset of any neurological deficits, and continued throughout life resulted in significant delay in time to recumbency and time to abnormalities in PRN. The amelioration of cerebellar pathological alterations, particularly involving the flocculonodular lobes, is proposed to be responsible for the improvement in the nystagmus seen in treated cats. The delay in onset of these neurological abnormalities, along with the lack of treatment effect on serum transaminase activity and the lack of effect on hepatic histology or biochemistry suggest that the remarkable increase in lifespan by 15 weeks in treated cats was due to amelioration of the neurological disease alone.
The principal mechanism of action of miglustat is believed to be inhibition of glucosylceramide synthase and subsequent reduction in the biosynthesis of glucosylceramide and complex glycosphingolipids (GSLs) (38, 39). We evaluated the histological and biochemical effects of miglustat in the brain of the feline model of NPC disease. Cerebella from 24-week-old NPC-miglustat cats showed remarkable preservation of Purkinje cells when compared to aged-matched NPC-untreated cats that were at end-stage disease. This finding is fully consistent with evidence of preserved motor system function. Despite the presence of many GM2-storing Purkinje cells, there was a 35% reduction in storage of GM2 ganglioside. Immunocytochemical studies suggested that the GM2 reduction identified biochemically likely occurred through the presence of less GM2 storage in neurons of the granule cell layer. Overall, these findings suggest that the mechanism of action of miglustat in treating NPC disease may indeed have been through reduction in GSL synthesis. While small decreases in GM2 and GM3 gangliosides were evident in cerebral cortex when examined both by immunohistochemistry and biochemically, the extent of these decreases in the latter analysis did not reach significance. Of note is the fact that in untreated affected cats, the load of GM2 and above all GM3 storage is less in cerebellum compared to cerebrum (Fig. 4B). The cerebral cortex of NPC-miglustat cats also showed a clear reduction in lactosylceramide, but, paradoxically, there was an approximately 2-fold increase in glucosylceramide. In normal brain, glucosylceramide is present only in trace amounts (10 nmol/g in human brain [35]), but considerable increases in glucosylceramide occur in all species affected with NPC disease; humans (305 ± 135 nmol/g, n = 6 [35]), mice (30), and cats (Fig. 4C). The administration of miglustat to normal mice also resulted in marked increases in brain glucosylceramide (40); similarly, treating Sandhoff disease mice (which do not accumulate glucosylceramide) with miglustat resulted in >10-fold increases in this GSL (41). The mechanism for this observed increase in brain glucosylceramide concentration in miglustat-treated animals has been ascribed to a secondary inhibitory effect of miglustat on the non-lysosomal enzyme beta-glucosidase 2 (40, 42–44). The survival benefit of miglustat therapy found in Sandhoff mice (41) was postulated to be due in part to the elevation in brain glucosylceramide concentrations, which has been shown to stimulate neuronal growth (45), and/or to decreases in sphingosine-1-phosphate levels, which can reduce astroglial proliferation (46). Whether the increase in glucosylceramide seen in treated cats could be beneficial is probably questionable since in human NPC disease the observed levels are often comparable to those reported in Gaucher disease type 2 brains. The concentration of sphingosine-1-phosphate was not measured.
The critical question of whether the efficacy of miglustat is due primarily to its ability to inhibit GSL synthesis remains controversial. It is clear from the studies here that miglustat has at best a weak ability to repress ganglioside storage in cerebellum in NPC disease. While it is possible that this mechanism of action is sufficient to result in Purkinje cell survival, there are other hypotheses. In addition to GSL storage, cholesterol and sphingosine accumulation also occurs in NPC brain. One of the miglustat-treated cats in the current study showed a qualitative decrease in neuronal cholesterol storage in the cerebral cortex, as assessed by filipin staining; however, a similar decrease was not observed in Purkinje cells. Unfortunately, because cholesterol storage in neurons corresponds more to an abnormal cell distribution than to an overall mass increase (47), biochemical cholesterol measurements on brain gray matter lipid extracts (with thus minimal myelin content) are unable to detect differences even between normal and untreated, affected NPC subjects (35); therefore, that analysis was not performed. While reductions in sequestered cholesterol would not be anticipated based on the known pharmacological effects of miglustat, genetic studies in mice in which complex ganglioside production was limited did show significantly reduced filipin-labeling in most neurons (49). Sphingosine accumulation, resulting in deleterious effects on cellular cholesterol metabolism and calcium homeostasis, has also been postulated to result in neurodegeneration in NPC disease (48). The present study is the first to document a 3-fold, small but significant, increase in free sphingosine concentrations in feline NPC disease brain to levels quite similar to those observed in murine NPC (27), and also to reveal only a small decrease in overall brain sphingosine concentrations following effective miglustat therapy. These data suggest that miglustat is unlikely to have a major effect through modulating sphingosine concentrations.
Finally, recent studies suggest that a critical component of NPC disease pathogenesis may be linked to an increase in GSL synthesis following a block in salvage of GM2 and GM3 gangliosides from the late endosomal/lysosomal system (49). A similar egress block and increase in synthesis is well established for cholesterol in NPC disease (50, 51). If correct, such an increase in GSL production would be expected to produce abnormally heightened levels of complex gangliosides in the plasmalemma, which could in turn cause signaling abnormalities in neurons and other cells by altering the makeup of GSL-cholesterol microdomains (52). The impact of miglustat could therefore be limited simply to reducing GSL synthesis sufficiently to ameliorate this phenomenon. Changes in patterns of ganglioside expression on the surface of neurons or glia could also lead to alterations in neuron-microglial interactions. A direct effect of miglustat on limiting the inflammatory response has recently been proposed (41). Microglia, the resident macrophages of the brain, can be activated to release pro-inflammatory mediators resulting in amplification of the inflammatory response (53) and neuronal cell death (54). Indeed, bone marrow transplantation reduced neurodegeneration and improved neurological function in Sandhoff disease and metachromatic leukodystrophy (55, 56) presumably due to normalization of microglial function. Additionally, because microglia also carry the NPC1 mutation, their inflammatory response may be more aggressive and inappropriate (57). Microglial characterization of cats affected by NPC revealed an altered phenotype in terms of size and complexity, and an upregulated immunophenotype and function reflecting distinct signs of their activation (58, 59). However, some surface antigens were only upregulated in cats treated with miglustat. In contrast, ROS generation was significantly lower in NPC disease cats whereas miglustat-treated cats, at both 24 and 35 weeks of age, displayed higher levels that were comparable to those of healthy WT cats. The results suggest that miglustat does not prevent microglial activation but may have a modulatory effect on certain aspects of microglial immunophenotype and function that correlates with delayed progression of neurological signs. An upregulation of various surface molecules such as CD18, CD11b/c, CD1c, MHC I and MHC II, and CD45 was also seen in canine distemper virus (CDV) CNS infection and other inflammatory CNS diseases (60, 61). This immunophenotype enables them to present antigen, incite leukocyte adhesion and aggregation leading to recruitment into the CNS, and mediation of the immune response. Moreover, the functional tests revealed enhanced phagocytosis and generation of ROS in CDV and other inflammatory CNS diseases (60, 61). The enhanced ROS generation correlated with severity of demyelination in CDV infection (32). While in NPC the percentage of microglia generating ROS was reduced in untreated NPC disease cats vs. WT and miglustat-treated cats, the generation intensity was higher in both NPC groups vs. the WT. The net effect of microglial ROS generation and its modulation by miglustat is therefore not known. Miglustat-treated NPC disease cats showed a percentage of ROS generating microglia comparable to the WT. ROS generation intensity was enhanced in microglia of NPC diseased cats compared to the WT; due to the heterogeneity in miglustat treated cats of different ages this did not reach significance. Because miglustat-treated cats did show enhanced ROS generation it is not known whether an enhanced ROS generation might have a positive impact on disease progression/pathogenesis in NPC disease.
Recent studies in Npc1−/− mice show that correction of the NPC defect in Purkinje cells alone is sufficient to prevent neuronal degeneration, and that microgliosis is ameliorated in areas where Purkinje cells did not degenerate (62). That reactive microglia occur through the CNS of Npc1−/− mice prior to neuronal degeneration has also been reported (63). Because miglustat slowed Purkinje cell loss but did not prevent it, and because microglia were not evaluated from cats at preclinical stages, we were unable to evaluate whether microglial activation occurs prior to neurodegeneration. However, the microglial data suggest that miglustat does modulate microglial function.
In conclusion, miglustat has been shown to be efficacious in delaying clinical disease, increasing longevity, and ameliorating Purkinje cell death in the feline model of NPC disease. In addition to the effect miglustat has on decreasing cerebellar GM2 accumulation, increases in cerebral glucosylceramide concentration, decreases in sphingosine concentration, and altered microglia phenotype were also identified. It is likely that each of these may contribute to the mechanistic basis of miglustat-induced neurological improvement in treated cats.
Acknowledgments
Sources of support: Ara Parseghian Medical Research Foundation (CHV), Dana’s Angels Research Trust (CHV), NCRR02512 (Mark E. Haskins), NIH NS053677 (SUW). Miglustat provided by Actelion Pharmaceuticals Ltd. V.M. Stein was supported by the German Research Foundation (DFG-STE 1069/2-1).
References
- 1.Vanier MT. Niemann-Pick disease type C. Orphanet J Rare Dis. 2010;5:16. doi: 10.1186/1750-1172-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.March PA, Thrall MA, Brown DE, et al. GABAergic neuroaxonal dystrophy and other cytopathological alterations in feline Niemann-Pick disease type C. Acta Neuropathol. 1997;94:164–72. doi: 10.1007/s004010050689. [DOI] [PubMed] [Google Scholar]
- 3.Zervas M, Dobrenis K, Walkley SU. Neurons in Niemann-Pick disease type C accumulate gangliosides as well as unesterified cholesterol and undergo dendritic and axonal alterations. J Neuropathol Exp Neurol. 2001;60:49–64. doi: 10.1093/jnen/60.1.49. [DOI] [PubMed] [Google Scholar]
- 4.Walkley SU, Suzuki K. Consequences of NPC1 and NPC2 loss of function in mammalian neurons. Biochim Biophys Acta. 2004;1685:48–62. doi: 10.1016/j.bbalip.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 5.Patterson MC, Vecchio D, Jacklin E, et al. Long-term miglustat therapy in children with Niemann-Pick disease type C. J Child Neurol. 2010e;25:300–5. doi: 10.1177/0883073809344222. [DOI] [PubMed] [Google Scholar]
- 6.Fecarotta S, Amitrano M, Romano A, et al. The videofluoroscopic swallowing study shows a sustained improvement of dysphagia in children with Niemann-Pick disease type C after therapy with miglustat. Am J Med Genet A. 2011;155A:540–7. doi: 10.1002/ajmg.a.33847. [DOI] [PubMed] [Google Scholar]
- 7.Pineda M, Perez-Poyato MS, O’Callaghan M, et al. Clinical experience with miglustat therapy in pediatric patients with Niemann-Pick disease type C: a case series. Mol Genet Metab. 2010;99:358–66. doi: 10.1016/j.ymgme.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 8.Wraith JE, Vecchio D, Jacklin E, et al. Miglustat in adult and juvenile patients with Niemann-Pick disease type C: long-term data from a clinical trial. Mol Genet Metab. 2010;99:351–7. doi: 10.1016/j.ymgme.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 9.Zarowski M, Steinborn B, Gurda B, et al. Treatment of cataplexy in Niemann-Pick disease type C with the use of miglustat. Eur J Paediatr Neurol. 2011;15:84–7. doi: 10.1016/j.ejpn.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 10.Chien YH, Lee NC, Tsai LK, et al. Treatment of Niemann-Pick disease type C in two children with miglustat: initial responses and maintenance of effects over 1 year. J Inherit Metab Dis. 2007;30:826. doi: 10.1007/s10545-007-0630-y. [DOI] [PubMed] [Google Scholar]
- 11.Patterson MC, Vecchio D, Prady H, et al. Miglustat for treatment of Niemann-Pick C disease: a randomised controlled study. Lancet Neurol. 2007;6:765–72. doi: 10.1016/S1474-4422(07)70194-1. [DOI] [PubMed] [Google Scholar]
- 12.Santos ML, Raskin S, Telles DS, et al. Treatment of a child diagnosed with Niemann-Pick disease type C with miglustat: A case report in Brazil. J Inherit Metab Dis. 2008;31 (Suppl 2):S357–61. doi: 10.1007/s10545-008-0923-9. [DOI] [PubMed] [Google Scholar]
- 13.Galanaud D, Tourbah A, Lehericy S, et al. 24 month-treatment with miglustat of three patients with Niemann-Pick disease type C: follow up using brain spectroscopy. Mol Genet Metab. 2009;96:55–8. doi: 10.1016/j.ymgme.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 14.Pineda M, Wraith JE, Mengel E, et al. Miglustat in patients with Niemann-Pick disease Type C (NP-C): a multicenter observational retrospective cohort study. Mol Genet Metab. 2009;98:243–9. doi: 10.1016/j.ymgme.2009.07.003. [DOI] [PubMed] [Google Scholar]
- 15.Patterson MC, Vecchio D, Prady H, et al. Miglustat for treatment of Niemann-Pick C disease: a randomised controlled study. [see comment] Lancet Neurol. 2007;6:765–72. doi: 10.1016/S1474-4422(07)70194-1. [DOI] [PubMed] [Google Scholar]
- 16.Zervas M, Somers KL, Thrall MA, Walkley SU. Critical role for glycosphingolipids in Niemann-Pick disease type C. Curr Biol. 2001;11:1283–7. doi: 10.1016/s0960-9822(01)00396-7. [DOI] [PubMed] [Google Scholar]
- 17.Lowenthal AC, Cummings JF, Wenger DA, et al. Feline sphingolipidosis resembling Niemann-Pick disease type C. Acta Neuropathol. 1990;81:189–97. doi: 10.1007/BF00334507. [DOI] [PubMed] [Google Scholar]
- 18.Brown DE, Thrall MA, Walkley SU, et al. Feline Niemann-Pick disease type C. Am J Pathol. 1994;144:1412–5. [PMC free article] [PubMed] [Google Scholar]
- 19.Munana KR, Luttgen PJ, Thrall MA, et al. Neurological manifestations of Niemann-Pick disease type C in cats. J Vet Intern Med. 1994;8:117–21. doi: 10.1111/j.1939-1676.1994.tb03208.x. [DOI] [PubMed] [Google Scholar]
- 20.Somers KL, Royals MA, Carstea ED, et al. Mutation analysis of feline Niemann-Pick C1 disease. Mol Genet Metab. 2003;79:99–103. doi: 10.1016/s1096-7192(03)00074-x. [DOI] [PubMed] [Google Scholar]
- 21.Vite CH, Ding W, Bryan C, et al. Clinical, electrophysiological, and serum biochemical measures of progressive neurological and hepatic dysfunction in feline Niemann-Pick type C disease. Pediatr Res. 2008;64:544–9. doi: 10.1203/PDR.0b013e318184d2ce. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Somers KL, Brown DE, Fulton R, et al. Effects of dietary cholesterol restriction in a feline model of Niemann-Pick type C disease. J Inherit Metab Dis. 2001;24:427–36. doi: 10.1023/a:1010588112003. [DOI] [PubMed] [Google Scholar]
- 23.Ward S, O’Donnell P, Fernandez S, Vite CH. 2-hydroxypropyl-beta-cyclodextrin raises hearing threshold in normal cats and in cats with Niemann-Pick type C disease. Pediatr Res. 2010;68:52–6. doi: 10.1203/PDR.0b013e3181df4623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Micsenyi MC, Dobrenis K, Stephney G, et al. Neuropathology of the Mcoln1(−/−) knockout mouse model of mucolipidosis type IV. J Neuropathol Exp Neurol. 2009;68:125–35. doi: 10.1097/NEN.0b013e3181942cf0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fujita N, Suzuki K, Vanier MT, et al. Targeted disruption of the mouse sphingolipid activator protein gene: a complex phenotype, including severe leukodystrophy and wide-spread storage of multiple sphingolipids. Hum Mol Genet. 1996;5:711–25. doi: 10.1093/hmg/5.6.711. [DOI] [PubMed] [Google Scholar]
- 26.Sleat DE, Wiseman JA, El-Banna M, et al. Genetic evidence for nonredundant functional cooperativity between NPC1 and NPC2 in lipid transport. Proc Natl Acad Sci U S A. 2004;101:5886–91. doi: 10.1073/pnas.0308456101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davidson CD, Ali NF, Micsenyi MC, et al. Chronic cyclodextrin treatment of murine Niemann-Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression. PLoS One. 2009;4:e6951. doi: 10.1371/journal.pone.0006951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hartree EF. Determination of protein: a modification of the Lowry method that gives a linear photometric response. Anal Biochem. 1972;48:422–7. doi: 10.1016/0003-2697(72)90094-2. [DOI] [PubMed] [Google Scholar]
- 29.Kyrklund T. Two procedures to remove polar contaminants from a crude brain lipid extract by using prepacked reversed-phase columns. Lipids. 1987;22:274–7. doi: 10.1007/BF02533991. [DOI] [PubMed] [Google Scholar]
- 30.Liu Y, Wu YP, Wada R, et al. Alleviation of neuronal ganglioside storage does not improve the clinical course of the Niemann-Pick C disease mouse. Hum Mol Genet. 2000;9:1087–92. doi: 10.1093/hmg/9.7.1087. [DOI] [PubMed] [Google Scholar]
- 31.Rodriguez-Lafrasse C, Rousson R, Pentchev PG, et al. Free sphingoid bases in tissues from patients with type C Niemann-Pick disease and other lysosomal storage disorders. Biochim Biophys Acta. 1994;1226:138–44. doi: 10.1016/0925-4439(94)90021-3. [DOI] [PubMed] [Google Scholar]
- 32.Stein VM, Czub M, Hansen R, et al. Characterization of canine microglial cells isolated ex vivo. Vet Immunol Immunopathol. 2004;99:73–85. doi: 10.1016/j.vetimm.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 33.Hein A, Martin JP, Koehren F, et al. In vivo infection of ramified microglia from adult cat central nervous system by feline immunodeficiency virus. Virology. 2000;268:420–9. doi: 10.1006/viro.1999.0152. [DOI] [PubMed] [Google Scholar]
- 34.Emmendorffer A, Hecht M, Lohmann-Matthes ML, et al. A fast and easy method to determine the production of reactive oxygen intermediates by human and murine phagocytes using dihydrorhodamine 123. J Immunol Methods. 1990;131:269–75. doi: 10.1016/0022-1759(90)90198-5. [DOI] [PubMed] [Google Scholar]
- 35.Vanier MT. Lipid changes in Niemann-Pick disease type C brain: personal experience and review of the literature. Neurochem Res. 1999;24:481–9. doi: 10.1023/a:1022575511354. [DOI] [PubMed] [Google Scholar]
- 36.Jeyakumar M, Thomas R, Elliot-Smith E, et al. Central nervous system inflammation is a hallmark of pathogenesis in mouse models of GM1 and GM2 gangliosidosis. Brain. 2003;126:974–87. doi: 10.1093/brain/awg089. [DOI] [PubMed] [Google Scholar]
- 37.Liu B, Li H, Repa JJ, et al. Genetic variations and treatments that affect the lifespan of the NPC1 mouse. J Lipid Res. 2008;49:663–9. doi: 10.1194/jlr.M700525-JLR200. [DOI] [PubMed] [Google Scholar]
- 38.Platt FM, Neises GR, Dwek RA, Butters TD. N-butyldeoxynojirimycin is a novel inhibitor of glycolipid biosynthesis. J Biol Chem. 1994;269:8362–5. [PubMed] [Google Scholar]
- 39.Platt FM, Jeyakumar M. Substrate reduction therapy. Acta Paediatr Suppl. 2008;97:88–93. doi: 10.1111/j.1651-2227.2008.00656.x. [DOI] [PubMed] [Google Scholar]
- 40.Walden CM, Sandhoff R, Chuang CC, et al. Accumulation of glucosylceramide in murine testis, caused by inhibition of beta-glucosidase 2: implications for spermatogenesis. The Journal of biological chemistry. 2007;282:32655–64. doi: 10.1074/jbc.M702387200. [DOI] [PubMed] [Google Scholar]
- 41.Ashe KM, Bangari D, Li L, et al. Iminosugar-based inhibitors of glucosylceramide synthase increase brain glycosphingolipids and survival in a mouse model of Sandhoff disease. PLoS One. 2011;6:e21758. doi: 10.1371/journal.pone.0021758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Overkleeft HS, Renkema GH, Neele J, et al. Generation of specific deoxynojirimycin-type inhibitors of the non-lysosomal glucosylceramidase. J Biol Chem. 1998;273:26522–7. doi: 10.1074/jbc.273.41.26522. [DOI] [PubMed] [Google Scholar]
- 43.Yildiz Y, Matern H, Thompson B, et al. Mutation of beta-glucosidase 2 causes glycolipid storage disease and impaired male fertility. J Clin Invest. 2006;116:2985–94. doi: 10.1172/JCI29224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boot RG, Verhoek M, Donker-Koopman W, et al. Identification of the non-lysosomal glucosylceramidase as beta-glucosidase 2. J Biol Chem. 2007;282:1305–12. doi: 10.1074/jbc.M610544200. [DOI] [PubMed] [Google Scholar]
- 45.Bodennec J, Pelled D, Riebeling C, et al. Phosphatidylcholine synthesis is elevated in neuronal models of Gaucher disease due to direct activation of CTP:phosphocholine cytidylyltransferase by glucosylceramide. FASEB J. 2002;16:1814–6. doi: 10.1096/fj.02-0149fje. [DOI] [PubMed] [Google Scholar]
- 46.Wu YP, Mizugishi K, Bektas M, Sandhoff R, Proia RL. Sphingosine kinase 1/S1P receptor signaling axis controls glial proliferation in mice with Sandhoff disease. Hum Mol Genet. 2008;17:2257–64. doi: 10.1093/hmg/ddn126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Karten B, Vance DE, Campenot RB, Vance JE. Cholesterol accumulates in cell bodies, but is decreased in distal axons, of Niemann-Pick C1-deficient neurons. J Neurochem. 2002;83:1154–63. doi: 10.1046/j.1471-4159.2002.01220.x. [DOI] [PubMed] [Google Scholar]
- 48.Lloyd-Evans E, Morgan AJ, He X, et al. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat Med. 2008;14:1247–55. doi: 10.1038/nm.1876. [DOI] [PubMed] [Google Scholar]
- 49.Zhou S, Davidson C, McGlynn R, et al. Endosomal/lysosomal processing of gangliosides affects neuronal cholesterol sequestration in Niemann-Pick disease type C. Am J Pathol. 2011;179:890–902. doi: 10.1016/j.ajpath.2011.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xie C, Turley SD, Pentchev PG, et al. Cholesterol balance and metabolism in mice with loss of function of Niemann-Pick C protein. Am J Physiol. 1999;276:E336–44. doi: 10.1152/ajpendo.1999.276.2.E336. [DOI] [PubMed] [Google Scholar]
- 51.Pentchev PG, Blanchette-Mackie EJ, Dawidowicz EA. The NP-C gene: a key to pathways of intracellular cholesterol transport. Trends Cell Biol. 1994;4:365–9. doi: 10.1016/0962-8924(94)90086-8. [DOI] [PubMed] [Google Scholar]
- 52.Lopez PH, Schnaar RL. Gangliosides in cell recognition and membrane protein regulation. Curr Opin Struct Biol. 2009;19:549–57. doi: 10.1016/j.sbi.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Culbert AA, Skaper SD, Howlett DR, et al. MAPK-activated protein kinase 2 deficiency in microglia inhibits pro-inflammatory mediator release and resultant neurotoxicity. Relevance to neuroinflammation in a transgenic mouse model of Alzheimer disease. J Biol Chem. 2006;281:23658–67. doi: 10.1074/jbc.M513646200. [DOI] [PubMed] [Google Scholar]
- 54.Skaper SD, Facci L, Culbert AA, et al. P2X(7) receptors on microglial cells mediate injury to cortical neurons in vitro. Glia. 2006;54:234–42. doi: 10.1002/glia.20379. [DOI] [PubMed] [Google Scholar]
- 55.Wada R, Tifft CJ, Proia RL. Microglial activation precedes acute neurodegeneration in Sandhoff disease and is suppressed by bone marrow transplantation. Proc Natl Acad Sci U S A. 2000;97:10954–9. doi: 10.1073/pnas.97.20.10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Biffi A, De Palma M, Quattrini A, et al. Correction of metachromatic leukodystrophy in the mouse model by transplantation of genetically modified hematopoietic stem cells. J Clin Invest. 2004;113:1118–29. doi: 10.1172/JCI19205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Repa JJ, Li H, Frank-Cannon TC, et al. Liver X receptor activation enhances cholesterol loss from the brain, decreases neuroinflammation, and increases survival of the NPC1 mouse. J Neurosci. 2007;27:14470–80. doi: 10.1523/JNEUROSCI.4823-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–8. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- 59.Streit WJ. Microglial cells. In: Kettenmann H, Ransom BR, editors. Neuroglia. New York: Oxford University Press; 1995. pp. 85–97. [Google Scholar]
- 60.Stein VM, Czub M, Schreiner N, et al. Microglial cell activation in demyelinating canine distemper lesions. J Neuroimmunol. 2004;153:122–31. doi: 10.1016/j.jneuroim.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 61.Stein VM, Baumgartner W, Kreienbrock L, et al. Canine microglial cells: stereotypy in immunophenotype and specificity in function? Vet Immunol Immunopathol. 2006;113:277–87. doi: 10.1016/j.vetimm.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 62.Lopez ME, Klein AD, Dimbil UJ, Scott MP. Anatomically defined neuron-based rescue of neurodegenerative Niemann-Pick type C disorder. J Neurosci. 2011;31:4367–78. doi: 10.1523/JNEUROSCI.5981-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Baudry M, Yao Y, Simmons D, Liu J, Bi X. Postnatal development of inflammation in a murine model of Niemann-Pick type C disease: immunohistochemical observations of microglia and astroglia. Exp Neurol. 2003;184:887–903. doi: 10.1016/S0014-4886(03)00345-5. [DOI] [PubMed] [Google Scholar]