

Abstract
Parkinson’s disease (PD) is caused by the progressive degeneration of dopaminergic neurons in the substantia nigra. Although the etiology for most PD remains elusive, the identification of specific genetic defects in familial cases of PD and the signaling pathways governed by these genes has provided tremendous insight into PD pathogenesis. Mutations in the leucine-rich repeat kinase 2 (LRRK2) gene are frequently found in familial and sporadic PD. Although current knowledge regarding the regulatory mechanisms of LRRK2 activation is limited, it is becoming increasingly evident that aberrant kinase activity of the pathologic mutants of LRRK2 is associated with neurodegeneration, suggesting that the kinase activity of LRRK2 is a potential therapeutic target. In addition, LRRK2 inhibitors might provide valuable tools to understand the pathophysiological and physiological roles of LRRK2 as well as the etiology of PD. We discuss here the potential and feasibility of targeting LRRK2 as a therapeutic strategy for PD.
Pathology and treatment of PD
Parkinson’s disease (PD) is the second most prevalent chronic neurodegenerative disease. It is age-related, affecting approximately 2% of the population over 60 years of age, and rising to 5% over 80 years of age [1, 2]. PD patients experience major motor symptoms, including rigidity, tremor, postural instability, and bradykinesia. Although a variety of neurons are affected to some degree in PD, the abnormalities in movement are primarily induced by loss and or dysfunction of dopaminergic (DA) neurons in the substantia nigra (SN) pars compacta (SNpc).
Currently, there is no cure for PD, but medications can alleviate some of the symptoms of PD. The mainstay of treatment for PD patients suffering from motor symptoms is dopamine replacement therapy including (i) levopopa (L-DOPA, the precursor of dopamine, which is converted to dopamine by dopa-decarboxylase); (ii) dopamine receptor agonists; (iii) inhibitors of monoamine oxidase (MAO-B), which metabolizes dopamine in brain; and (iv) inhibitors of catechol O-methyltransferase (COMT), which break downs L-DOPA. Surgical therapies such as pallidotomy and deep brain stimulation offer patients adjunctive therapy to assist in the management of the motoric symptoms of PD including tremor, motor fluctuations, dykinesia and dystonia [3]. Various therapies are also used to treat many of the non-motor manifestations of PD, such as autonomic dysfunction, somnolence and other sleep disorders, cognitive issues, anxiety, depression, psychosis and others [4]. The primary goals of the aforementioned therapies are to relieve the motoric and non-motoric symptoms of PD, rather than to cure or modify the progression of PD. Therefore, the development of new drugs to reverse, prevent, or slow down the progress of PD is one of the major goals, if not the primary goal, for PD research.
The discovery of neuroprotective or disease modifying therapy for PD has spectacularly failed, in that there are no proven or approved neuroprotective or disease modifying therapies for PD [5]. Some reasons for failure may include the use of acute intoxication models such as the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and 6-hydroxy-dopamine (6-OHDA) models of PD to discover and test new neuroprotective therapies. Although these models elicit selective loss of DA neurons, they may not accurately recapitulate the degenerative process of PD. Other causes of failure may include poor clinical trial design, starting therapy too late in the course of disease, the lack of sufficient target engagement in the brain, as well as engaging targets that may not be critical in the pathogenesis of PD.
All these issues remain crucial barriers to the ultimate development of neuroprotective or disease modifying therapy for PD. Although the etiology of sporadic PD remains unclear, several genes associated with hereditary PD provide definable targets that can be utilized for rationale drug discovery [6]. For example, mutations in α-synuclein, leucine rich repeat (LRR) kinase 2 (LRRK2), parkin, PTEN-induced kinase-1 (PINK-1), and DJ-1 have been suggested to cause familial PD in various ethnic groups [7]. In particular, mutations in LRRK2 are the most common mutations in sporadic and familial late-onset PD [8]. Patients with mutations in LRRK2 display typical features of PD and for the most part show similar pathology to that of sporadic PD. While familial PD may be less common than sporadic PD, the identification of specific disease-causing mutations and aberrant signaling that is the consequence of these mutations are likely to enhance our understanding of the etiology of PD and more importantly, provide important insights into formulating therapeutic strategies for PD.
LRRK2 biology and pathobiology
From the standpoint of drug development, LRRK2 is an attractive PD therapeutic target. Mutations in LRRK2 are a common cause of PD. LRRK2 mutations were first described in 2004 in families with dominantly inherited PD [9, 10]. LRRK2 shows broad expression in various regions of the brain, including the olfactory bulb, striatum, cortex, hippocampus, midbrain, brain stem and cerebellum [11]. PD caused by LRRK2 mutations is for the most part indistinguishable from sporadic PD [12]. Patients with LRRK2 mutations have loss of DA neurons in SNpc neurons, and the majority of cases have α-synuclein positive Lewy body pathology [13]. Since the original description of disease-segregating mutations in LRRK2, over 40 mutations have been reported in LRRK2 and at least 7 are pathogenic [7, 8]. The most common mutation of LRRK2, G2019S, is found in a wide range of ethnic groups and in 1–3% of sporadic and 4–8% of familial cases [7, 8]. Among North Africa Arabs, 39% of PD patients have the G2019S mutation, with familial cases accounting for 40% and sporadic cases contributing 33%. In the United States Jewish population, 13% of PD patients have the G2019S mutation with familial cases accounting for 23% and sporadic cases contributing 10%. Moreover, two independent genome-wide association studies indicate that variants within the LRRK2 locus are major risk factors for sporadic PD, consistent with the idea that perturbations in LRRK2 are a major cause of PD [14, 15].
LRRK2 is a large protein (280 KDa). Sequence homology analysis and functional characterization reveal it has the highest similarity to mixed-lineage kinases (MLK) that typically have both serine/threonine and tyrosine kinase activities, although LRRK2 does not seem to have tyrosine kinase activity [16, 17]. MLKs are part of the mitogen-activated protein kinase (MAPK) family and act as MAPK kinase kinases (MAPKKKs) to initiate and transduce a wide range of cellular responses [18]. How and whether LRRK2 functions as a MAPKKK is not known because the mechanism underlying its activation and its downstream kinase effectors are not well characterized. Several proteomic and random peptide analyses suggest that LRRK2 is a serine/threonine kinase and prefers threonine residues as the phosphorylation site [17, 19, 20]. Consistent with the possibility that LRRK2 may function as an MLK/MAPKKK are the extent and the number of physiologic process that may be regulated by LRRK2. These include a role in neurite outgrowth and guidance [21, 22], protein translation through regulation of microRNA processing [23] and vesicle storage and mobilization within the recycling pool [24]. However, its physiological and pathological functions remain to be fully characterized.
LRRK2 has multiple protein domains (Figure 1), including protein-protein binding domains, such as the LRR domain and the WD40 domain [25]. An interesting feature of LRRK2 is that it also has two distinct but functionally linked enzymatic domains, a Ras of complex (Roc) GTPase domain and a protein kinase domain that are linked by a carboxy-terminal of Roc (COR) sequence [26].
Figure 1. Schematic diagram of LRRK2 domains and pathogenic mutations.

LRRK2 has multiple protein domains including ANK (ankyrin-like repeat), LRR (leucine rich repeat), ROC (Ras of complex protein) GTPase, COR (C-terminal of ROC), kinase, and WD40. ANK, LRR, and WD40 are protein-protein interaction domains. The ROC and kinase domains have enzymatic activity. Multiple mutations in LRRK2 are found in PD patients. Seven pathogenic mutations are located on ROC, COR, and kinase domains.
Notably, multiple pathogenic mutations (I1371V, R1441C, R1441G, R1441H, Y1699C, Y1699G, G2019S, and I2020T) are located within the GTPase and the kinase domains or within the COR domain (Figure 1). The majority of these mutants have abnormally high kinase activity when compared to wild-type LRRK2 (for review see [27]). Although the effects of some mutants still remain controversial (presumably due to different protein sources, assay methods and substrates used in the kinase assay), several lines of evidence strongly suggest that the kinase activity of LRRK2 is associated with neuronal toxicity and degeneration of dopaminergic neurons [16, 17, 28–30]. Initial studies showed that neuronal toxicity is induced by transient expression of pathological LRRK2 mutants, but not by LRRK2 variants that harbor a kinase-inactivating point mutation [28, 30]. Among the LRKK2 mutants, the G2019S mutant consistently displays robust kinase activity that is substantially higher than wild-type LRRK2 [27]. The G2019S mutant is of particular interest because it is the most common mutation found in familial and sporadic PD patients, and patients with this mutation are clinically indistinguishable from idiopathic PD [12]. Although there is relatively strong evidence that pathologic LRRK2 kinase activity is responsible for the neurodegeneration in LRRK2-associated PD, recent studies suggest that the GTPase activity of LRRK2 may also contribute to the toxicity of the pathogenic LRRK2 mutants [31, 32].
As noted above, one of the consistent pathological features of patients with LRRK2 mutations is α-synuclein positive Lewy body pathology [13]. Thus, it is possible that inhibitors of LRRK2 neurodegeneration may not only provide disease modification for patients with LRRK2 mutations, but also for sporadic PD. Whether aberrant LRRK2 kinase and/or GTPase plays a role in sporadic PD is not known. Consistent with the idea that LRRK2 may contribute to the pathology of sporadic PD is the observation that overexpression of LRRK2 G2109S markedly enhanced the neuropathological abnormalities in A53T α-synuclein transgenic mice (a model of sporadic PD), whereas knockout of LRRK2 reduced the abnormalities [33]. These results suggest that inhibition of LRRK2 might be an option for treating α-synuclein-induced neurodegeneration. Thus, inhibitors of LRRK2 kinase and/or GTPase activity might tackle a common pathogenic mechanism in PD (i.e. pathologic LRRK2 kinase and/or GTPase activity), providing the possibility of a general therapeutic disease modifying approach in PD. Although recent studies suggest that the neurodegenerative phenotypes in another A53T α-synuclein transgenic mouse model are independent of LRRK2 [34]. Future studies will be required to sort out the relative contributions of LRRK2 kinase and/or GTPase activity in sporadic PD pathogenesis.
Animal models of LRRK2-induced disease
Several LRRK2 transgenic animals show Parkinsonian-like phenotypes. Over-expression of LRRK2 in Drosophila induces a progressive adult onset loss of dopaminergic neurons, impaired mobility and a shortened lifespan [35–37]. LRRK2-transgenic C. elegans also show age-dependent dopaminergic neurodegeneration, behavioral deficits, and locomotor dysfunction [38, 39]. Notably, over-expression of LRRK2 mutants, which have higher kinase activities as compared to wild-type LRRK2, induces more severe phenotypes. So far, multiple lines of LRRK2 mouse models for several different pathogenic mutants have been developed, but most of the LRRK2 transgenic mice do not show specific degeneration of dopaminergic neurons [33, 40–42]. One exception is a transgenic model that shows modest late-age degeneration of DA neurons [43]. Despite the limited pathology in many of the LRRK2 transgenic mouse models, they display abnormalities in the nigrostriatal system, such as impaired dopamine release and behavioral deficits [44]. These phenotypes might reflect some of the earliest neuronal dysfunction induced by pathogenic LRRK2 mutations. It is not clear why LRRK2 transgenic mice do not show substantial pathology characteristic of PD. It might be related to partial penetration of the LRRK2 mutations in humans. For example, penetrance of the G2019S mutation is age-related, reaching approximately 80% in carriers by 80 years of age [12]. Because penetrance is incomplete even at advanced ages in humans, additional genetic and/or environmental factors might be required for dopaminergic degeneration in both humans and mice.
Unlike transgenic mice, transient expression of the pathogenic LRRK2 mutant, G2019S using herpes simplex virus (HSV) amplicons or adenoviral vectors to deliver LRRK2 genes into the striatum and the SN of mice induces degeneration of TH positive neurons in the SN [29, 45]. In the absence of LRRK2-linked PD mouse models with substantial degeneration of DA neurons, the C. elegans and Drosophila model systems, as well as the viral-mediated gene delivery systems in rodents are currently the models of choice to test the neuroprotecitve efficacy of potential LRRK2 inhibitors. Induced pluripotent stem cells (iPSCs) derived from a patients carrying LRRK2 mutations have the potential to offer an additional platform for development of drugs specifically targeted at pathophysiologic mechanisms caused by LRRK2 mutations in human neurons [46, 47].
Kinases as therapeutic targets in PD
Protein kinases are appealing pharmaceutical drug targets because of their prominent roles in various kinds of diseases such as cancer, inflammation, arthritis, diabetes, cardiovascular disease, and neurodegenerative diseases. Multiple small molecule kinase inhibitors have been approved by USA FDA and are available in the market, including imatinib (Gleevec), sorafenib (Nexavar), sunitinib (Sutent), rapamycin (Sirolimus) to name a few [48]. Potential druggable kinase-related signaling pathways in PD include protein kinase Cδ, the MLK-c-jun N-terminal kinase (JNK) signaling cascade, and AKT/protein kinase B (PKB) signaling cascade, all of which are kinases implicated in programmed cell death [49, 50]. CEP1347, a MLK inhibitor was particularly promising as it showed neuroprotective effects in a variety of PD and neurodegenerative models [51]. Despite several lines of evidence both in vitro and in vivo that suggested a high therapeutic potential, the clinical trial of CEP1347 has been discontinued because of the lack of significant therapeutic efficacy [52]. The failure of CEP1347 illustrates the challenge and the potential pitfalls of evaluating kinase inhibitors as neuroprotective agents in PD, including the necessity to demonstrate that any future kinase inhibitor reaches therapeutic levels in the CNS and that it does not interfere with other signal pathways [53].
Screening considerations for LRRK2 inhibitors
Several points need to be considered wh en developing suitable screening systems to identify specific LRRK2 kinase inhibitors. Compound screening should be performed with proteins of high purity and physiologically relevant substrates. The low efficiency and purity of full-length recombinant LRRK2 proteins and the lack of clear physiological LRRK2 kinase substrates has hampered the development of LRRK2 kinase inhibitors. Mammalian protein expression systems such as HEK-293 cells are widely used for the purification of full-length recombinant LRRK2 proteins [29]. These mammalian expression systems are efficient for small-scale expression and for biochemical analysis, but the cost of large-scale expression limits the suitability for high throughput screening (HTS). There is also the possibility of contamination by co-purifying kinases. Baculovirus purification of N-terminal-truncated wild type and pathologic mutants of LRRK2 (970–2527) which display kinase activity comparable to full-length LRRK2 is capable of providing sufficient purity and quantity for HTS [54]. Ideally, the availability of pure full-length LRRK2 in sufficient quantities would greatly facilitate the development of specific LRRK2 inhibitors, as the N-terminal-truncated LRRK2 may remove steric hindrance and allow the accommodation of more promiscuous inhibitors to reach the active site.
Similarities in the active site of kinases often make it difficult to develop highly specific kinase inhibitors. Often even highly selective kinase inhibitors inhibit other structurally similar kinases. Typically, the DFG and APE motifs in the activation loop of kinases are conserved. However, LRRK2 differs from other Raf and MLK kinases in that it has DYG (instead of DFG) and it is modified as DYS in the LRRK2 G2019S mutant. This different and modified activation loop in LRRK2 could provide enough structural differences to develop highly specific LRRK2 kinase inhibitors with little activity against other kinases. In addition, it will be essential to have specific physiologic substrates and cellular assays for the rational design, screening and confirmation of highly specific kinase inhibitors. Physiological substrates of LRKK2 substrates have yet to be determined and/or confirmed. However, there have been efforts to design substrates that can be readily and specifically phosphorylated by LRRK2 in vitro. Moesin [55], 4E-BP1 [56], MKK3/6, MKK4/7 [57, 58], MBP [16, 17] and LRRK2 itself have been shown to be phosphorylated by LRRK2 in vitro, although LRRK2-dependent phosphorylation of these potential substrates cannot be detected in cells. Recent studies suggest that ArfGAP1 is a physiologic substrate whose phosphorylation and activity is regulated in vivo by LRRK2 [32]. It will be critical to develop validated cell-based assays for LRRK2 kinase screening and evaluation.
Through KESTREL (kinase substrate tracking and elucidation) screening and subsequent biochemical analysis, moesin, a member of the ERM (ezrine/radixin/moesin), was shown to be phosphorylated by LRRK2 at Thr558, and a 15 amino acid peptide encompassing Thr558 (LRRKtide, RLGRDKYKTLRQIRQ) was shown to be efficiently phosphorylated by LRRK2 [55]. Through a positional scanning peptide library approach, an optimal phosphorylation motif for LRRK2 was evaluated. Combining the motif sequence (WWRFYTLRRA) with the LRRKtide sequence gave rise to the Nictide (RLGWWRFYTLRRARQGNTKQP) sequence [19]. The Nictide sequence was more efficiently phosphorylated by LRRK2 as compared to either the moesin protein or LRRKtide sequence. Another report provided a sequence, F/Y-x-T-R/K, as a consensus phosphorylation motif of LRRK2 [20]. The −2 and +2 position of this sequence has similarity with LRRKtide and Nictide. Together, these analyses provide valuable information for identification of potential LRRK2 substrates and phosphorylation sites. In the absence of physiological substrates, these peptides can be used as alternative substrates in LRRK2 kinase enzyme assays.
Conventional screening systems to identify kinase inhibitors such as measuring of incorporation of 32P or immunoassay of phosphorylated residues are usually too costly and/or labor-intensive for HTS. However, these assays are routinely used for confirmation and produce high quality signals for monitoring kinase activity. In addition to these conventional methods, HTS platforms have been applied to discover potent LRRK2-specific inhibitors. To date, reported cases of compound screening for LRRK2 kinase inhibitors have employed TR-FRET (time-resolved fluorescence resonance energy transfer) [59, 60] and AlphaScreen (amplified luminescent proximity homogenous assay) technology [61] using moesin proteins or LRRKtide peptide as kinase substrates as well as chemical proteomics [62] and high throughput kinase profiling [63]. These approaches to develop LRRK2-specific assay systems, although still in their infancy, reflect the continuous and increasing efforts in LRRK2 kinase inhibitor discovery and development.
LRRK2 kinase inhibitors
Many of the LRRK2 kinase inhibitors identified to date were discovered by using libraries of defined kinase inhibitors (Table 1). In the first screenings for inhibitors of LRRK2 kinase, studies used chemical libraries that consisted of specific inhibitors against different kinase families, including protein kinase A, protein kinase C, MAPK, MAPKK, casein kinase (CK) I/II, Ca2+/calmodulin-dependent (CAM) kinase II, glycogen synthase kinase-3 (GSK-3), Rho kinase (ROCK), Raf kinase, myosin light chain kinase (MLCK), cyclin-dependent kinases, and MLKs [64]. Four compounds potently inhibited LRRK2 kinase activity and shared a basic indolocarbazole structure: Go6976, staurosporine, K-252a, and K-252b., Additional inhibitors were evaluated through a similar strategy, and H-1152, a ROCK inhibitor, and sunitinib, a receptor tyrosine kinase (RTK) inhibitor, were found to efficiently inhibit LRRK2 [19]. Nichols et al. [19] also defined a pharmacological approach to validate whether substrates are phosphorylated by LRRK2 by identifying a LRRK2 A2016T mutant that is normally active, but resistant to H-1152 or sunitinib. Kinase specificity profiling against a panel of 85 protein kinases revealed that H-1152 potently inhibited four kinases [LRRK2 G2019S, ROCK2, aurora B kinase, and brain selective kinase 2 (BRSK2)], and sunitinib inhibited two kinases [LRRK2 G2019S and pleckstrin homology kinase (PHK)] [19]. Sunitinib is clinically used for the treatment of renal cell carcinoma and imatinib-resistant gastrointestinal stromal tumors. Even though these reports showed promising results in vitro, there were no in vivo studies determining whether these compounds would have therapeutic efficacy in inhibiting LRRK2.
Table 1.
Inhibitors of LRRK2 kinase
| Compound | Structure | IC50 (µM) | Remarks | Ref |
|---|---|---|---|---|
| Staurosporine |  |
0.001 (WT) | Inhibits LRRK2 autophosphorylation and LRRK2-induced MBP phosphorylation. Disrupts LRRK2 dimers. Non-specific kinase inhibitor. | [29, 64] |
| K252a |  |
0.025 (WT) | Inhibits LRRK2 autophosphorylation and LRRK2-induced MBP phosphorylation Serine/threonine and tyrosine kinase inhibitor. | [64] |
| K252b |  |
0.05 (WT) | Inhibits LRRK2 autophosphorylation and LRRK2-induced MBP phosphorylation. Ectokinase inhibitor. | [64] |
| Gö6976 |  |
0.25 (WT) | Inhibits LRRK2 autophosphorylation and LRRK2-induced MBP phosphorylation. Protein kinase C Inhibitor. | [64] |
| Sunitinib |  |
0.079 (WT) 0.019 (G2019S) | Inhibits LRRK2-mediated phosphorylation of LRRKtide and Nictide. Resistant to LRRK2 A2016T mutant. Receptor tyrosine kinase Inhibitor. | [19] |
| H-1152 | 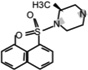 |
0.244 (WT) 0.15 (G2019S) | Inhibits LRRK2-mediated phosphorylation of LRRKtide and Nictide. Resistant to LRRK2 A2016T mutant. Rho kinase inhibitor | [19] |
| GW5074 |  |
0.88 (WT) 0.22 (G2019S) | Inhibits LRRK2 autophosphorylation and MBP and 4E-BP phosphorylation. Inhibits mutant LRRK2-mediated toxicity in primary neurons and loss of dopaminergic neuron in the substantia nigra in the HSV-LRRK2 G2019S model of neurodegeneration. It is neuroprotective in LRRK2 G2019S-transgenic C. elegans and Drosophila models. | [29, 65] |
| Indirubin-3’-monooxime |  |
1.750 (WT) 0.38 (G2019S) | Inhibits LRRK2 autophosphorylation and MBP and 4E-BP phosphorylation. Inhibits mutant LRRK2-mediated toxicity in primary neurons and loss of dopaminergic neuron in the substantia nigra in the HSV-LRRK2 G2019S model of neurodegeneration.. | [29] |
| Sorafenib | 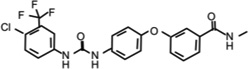 |
14.56 (WT) 1.17 (G2019S) | Inhibits LRRK2 autophosphosrylation and LRRK2-mediated MBP phosphorylation. Inhibits mutant LRRK2-mediated toxicity in primary neurons. It is neuroprotective in LRRK2 G2019S-transgenic C. elegans and Drosophila models. | [29, 65] |
| CZC-25146 | 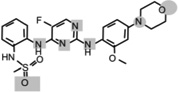 |
0.0048 (WT) 0.0069 (G2019S) | Potent and selective LRRK2 inhibitor. Inhibits mutant LRRK2-mediated toxicity in primary rodent and human neurons. Displays poor brain penentration. | [62] |
| CZC-54252 | 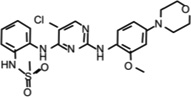 |
0.0013 (WT) 0.0019 (G2019S) | Potent and selective LRRK2 inhibitor. Inhibits mutant LRRK2-mediated toxicity in human neurons. Displays poor brain penentration. | [62] |
| LRRK2-IN-1 |  |
0.013 (WT) 0.006 (G2019S) | Potent and selective LRRK2 inhibitor. Induces loss of 14-3-3 binding to LRRK2. Promotes dephosphorylation of Ser910 and Ser935 on LRRK2. Promotes relocaliztion of LRRK2 to aggregate and fibrillar-like structures that resemble inclusion bodies. Unable to efficiently cross the blood-brain barrier. Resistant to LRRK2 A2016T mutant. | [63] |
Eight inhibitors of LRRK2 kinase were identified by screening a commercially available chemical library (Biomol) that is composed of 84 kinase and phosphatase inhibitors, including staurosporine, GF109203X, Ro 31–8220, 5-iodotubercidin, GW5074, indirubin-3′-monooxime, SP 600125 and damnacanthal [29]. For in vivo validation, a herpes simplex virus (HSV) amplicon-based mouse model was developed to examine LRRK2-mediated dopaminergic neuronal toxicity. After stereostatic injection of HSV- LRRK2 virus in the brain, HSV-LRRK2 G2019S induced loss of dopaminergic neurons in the SN. Intraperitoneal administration of the two brain-penetrable inhibitors of LRRK2, GW5074 or indirubin-3-monooxime, potently reduced the loss of dopaminergic neurons [29]. Sorafenib another Raf kinase inhibitor like GW5074 inhibited LRRK2 and displayed neuroprotection in a LRRK2 G2019S-transgenic C. elegans model and prevented behavioral deficits in a LRRK2 G2019S-transgenic Drosophila model. ZM336372, another Raf kinase inhibitor, which has no effect on LRRK2 activity was not protective [65]. Thus, Raf kinase inhibition is not responsible for the protective effects of GW5074. These results provided in vivo proof-of-principle that small molecule inhibitors of LRRK2 kinase activity can protect against LRRK2-induced neurodegeneration. Because GW5074 and indirubin-3-monooxime have different kinase inhibition profiles [66, 67], and because inhibitors of the same class that do not inhibit LRRK2 kinase activity fail to protect against LRRK2 G2019S-induced neurodegeneration, it is likely that inhibition of LRRK2 kinase activity accounts for the neuroprotection. A variant of LRRK2 that harbors a kinase-inactivating point mutation HSV-LRRK2-G2019S/D1994A, also failed to cause loss of dopaminergic neurons, which is consistent with the theory that LRRK2 kinase activity is required for cell death in vivo [29].
Novel strategies, such as chemical proteomics [62, 68] and high-throughput kinase-profiling [63, 69] have been applied to identify selective and potent LRRK2 kinase inhibitors. The pharmacochemical research group of Cellzome AG identified potent LRRK2-selective inhibitors, CZC-25146 and CZC-54252, by using a chemoproteomic strategy [62]. Both compounds strongly inhibited LRRK2 WT (IC50 ≅ 1 – 5 nM) and LRRK2 G2019S (IC50 ≅ 2 – 7 nM) and attenuated LRRK2 G2019S- and R1441C-mediated toxicity in primary human and rodent neurons. Furthermore, the specificity of the compounds was assessed against 184 different protein kinases and one lipid kinase by applying Kinobeads™ technology [68]. CZC-25146 showed a very clean profile because it only inhibited an additional five kinases [62]. Despite the potency and specificity of CZC-25146, it is unlikely to be applied clinically due to its poor pharmacokinetic properties that indicate it will have limited brain penetration [62].
Another LRRK2 kinase inhibitor identified through HTS is LRRK2-IN-1 [63]. When binding affinity of LRRK2-IN-1 was analyzed for more than 470 kinases using three different complementary approaches (KINOMEscan, Dundee profiling, and KiNativ technology), 12 kinases showed comparable binding affinity with LRRK2. Although it potently inhibited LRRK2 activity, its blood-brain barrier (BBB) penetration was poor, and it is unclear whether it can block LRRK2-mediated neurotoxicity. All the LRRK2 inhibitors identified so far are unlikely to be applied clinically due to either poor brain penetrance or limitations in specificity. However, the structural information gleaned from these studies provides valuable information for the development of more specific, potent and brain penetrable inhibitors.
As with any kinase inhibitor development for human use, issues related to safety will need to be carefully evaluated. This is particularly important for a chronic disease like PD. Potential problems have already been identified in LRRK2 knockout mice (See Text Box 1) that may complicated the development and ultimate clinical use of inhibitors of LRRK2 kinase activity.
Text Box 1. Concerns in developing LRRK2 kinase inhibitors.
One of the major considerations in developing pharmaceutical drugs, is to limit adverse side effects. Since inhibition of LRRK2 for the treatment and prevention of progression of PD will necessitate long-term inhibition of LRRK2, knockout mice provide an important model system to understand the potential long-term consequences of LRRK2 inhibition. A few reports suggest that knocking out LRRK2 does have potential deleterious consequences. Two independent studies of mice lacking endogenous LRRK2 indicate that the absence of LRRK2 leads to pathophysiological abnormalities in kidney including kidney darkening and age-dependent atrophy and accumulation of lipofuscin granules. One knockout line targeting the promoter and exon 1 and another targeting exons 29 and 30, which encodes the first half of the ROC domain, show accumulation and aggregation of α-synuclein and impaired autophagy-lysosomal pathways [83]. In a separate study, knocking out the kinase domain (exon 41) in mice led to early-onset (age 6 weeks) in the number and size of secondary lysosomes in kidney proximal tubule cells and lamellar bodies in lung type II cells, while mice harboring a kinase-inactivating point mutation in LRRK2 only exhibited abnormalities in the kidney [84]. The genetic effect in the kidney was mimicked in wild type mice treated with a LRRK2-selective inhibitor suggesting that the observed abnormalities were not due to developmental effects of knocking out LRRK2 embryonically [84]. Another set of investigators found that LRRK2 deficiency conferred enhanced susceptibility to experimental colitis in mice [85]. LRRK2 has been shown to be a potent inhibitor of the transcription factor NFAT that plays a crucial role in regulating immune responses. Moreover, LRRK2 negatively regulated NFAT-driven innate immune responses, implying a role for LRRK2 in inflammatory bowel disease [85]. All these unexpected functions of LRRK2 outside of the nervous system need to be critically evaluated as LRRK2 therapeutics move forward for the treatment of PD.
Beyond the kinase domain
To date, the catalytic core of the LRRK2 kinase domain has been targeted for screening LRRK2 kinase inhibitors by using ATP-competitive compounds. Little is known about the regulatory mechanisms of LRRK2 activation due to the lack of information about the physiological and pathological regulators and/or downstream effectors of LRRK2. However, multiple lines of evidence suggest self-regulation is a mechanism for controlling its kinase activity, which is mediated, in part, by its GTPase domain and homodimerization (Figure 2). Given the homology of LRRK2 with the MLK subfamily and other MAPKKKs, which are regulated by small GTPases, LRRK2 kinase activity might be regulated by its own GTPase domain in an intrinsic manner. Interestingly, GTP binding significantly enhances the kinase activity of LRRK2 and pathogenic mutants of LRRK2 that show higher GTP binding or reduced GTP hydrolysis have enhanced kinase activity [16, 30, 70–72]. Neurotoxicity induced by some pathogenic mutants of LRRK2 requires GTP binding [17, 30, 31]. Using truncated constructs of LRRK2, Xiong et al. [31] showed that reducing LRRK2 GTP hydrolysis enhanced LRRK2 toxicity, whereas enhancing LRRK2 GTP hydrolysis was protective against LRRK2 toxicity. Studies also indicate that the GTPase activity of full-length LRRK2 plays an important role in LRRK2 toxicity both in vitro and in vivo. The familial LRRK2 mutations, which have decreased GTP hydrolysis, such as R1441C, R1441G are toxic [72, 73]. In addition, LRRK2 G2019S also has decreased GTP hydrolysis [32]. ArfGAP1 was recently shown to be the first GTPase-activating protein (GAP) for LRRK2 [32] and ARHGEF7 may be a guanine nucleotide exchange factor (GEF) for LRRK2 [74]. As a GAP for LRKK2, ArfGAP1 enhances both wild-type and mutant LRRK2 GTP hydrolysis and protects against LRRK2 toxicity in vitro and in vivo, so it is likely that agents that enhance LRRK2 GTP hydrolysis such as LRRK2 GAP activators or GEF inhibitors might be attractive therapeutic targets.
Figure 2. Drug targets for regulation of LRRK2 activity.

A. Kinase Activity. Drugs targeting the kinase activity of LRRK2 have tremendous promise as disease modifying agents for the treatment of PD. Current inhibitors of LRRK2 kinase activity are targeted toward the ATP binding site and are based on inhibition of LRRK2 autophosphorylation and pseudosubstrate phosphorylation. It is conceivable that inhibition of a key pathogenic LRRK2 phosphorylation substrate may also be an attractive therapeutic target.
B. GTPase Activity. Another prominent candidate target is the GTPase activity. Because reducing LRRK2 GTP hydrolysis enhances LRRK2 toxicity and increasing LRRK2 GTP hydrolysis is protective against LRRK2 toxicity, strategies that increase LRRK2’s GAP (ArfGAP1) activity or decrease LRRK2’s GEF activity (unknown) might be attractive therapeutic targets.
C. Dimerization. Drugs targeting the regions of LRRK2 that are critical for its dimerization, such as the WD40 domain or compounds that disrupt dimerization are potential drug candidates for inhibition of LRRK2’s activity.
Another possible intervention point for regulating LRRK2 kinase activity is dimerization. Indeed, dimerization is a common phenomenon among protein kinases for which dimerization often mediates auto-regulation or modulates downstream signaling molecules. Dimerization of LRRK2 was first suggested through co-immunoprecipitation experiments by using differentially tagged full-length and fragments of LRRK2 [57]. LRRK2 self associates, and the kinase domain was dispensable for this the self-association. The dimerization of LRRK2 may be required for the kinase-active state of LRRK2 [75]. The crystal structure of the ROC domain of LRRK2 revealed that LRRK2 exists as a dimer and that the COR domain following the ROC domain may function as a hinge between the ROC domain and the kinase domain [76]. When the ROC domain was used as a bait in a yeast two-hybrid system, it bound to the N-terminus (18–186) of LRRK2, the C-terminus of the LRR domain, the N-terminus of the ROC domain (1123–1200), and the C-terminus of the WD40 domain (2084–2217) [77]. Although the ROC domain could bind to multiple domains within LRRK2, its deletion was not sufficient to distort LRRK2 dimerization, suggesting an additional interaction region(s) within LRRK2 mediating the dimerization. LRRK2 has multiple protein binding domains, including ankyrin repeat region (ANK), leucine-rich repeat (LRR), and the WD40 domain (Figure 1). An N-terminal deletion (including the ANK and LRR domains) did not diminish dimerization, but a WD40 truncated-LRRK2 could not be dimerized. These studies suggest that the ROC and the WD40 domains might mediate LRRK2 dimerization, but the WD40 domain appears to be more critical. Structural analysis comparing LRRK2 and LRRK1, a LRRK2 homolog, which is non-pathogenic in PD, has revealed that the WD40 domain is important in LRRK2 PD pathology, and WD40-truncated LRRK2 fails to exhibit autophosphorylation and neuronal toxicity [77, 78]. Thus, interfering with the WD40 domain-mediated LRRK2 dimerization process might be another potential therapeutic approach for treating LRRK2-linked PD. Despite the evidence suggesting that LRRK2 exists and functions as a dimer, recent studies indicate that the monomeric form has kinase activity and it is the predominant species in cells [79, 80].
The interaction regions between a kinase and its regulators/effectors can be targeted for selective inhibition, but these interactions often involve a relatively broad, tight binding region, and they show strong associations that are mediated by hydrophobic contacts, hydrogen and ionic bonds [50]. These characteristics present a barrier for small molecules to access the interface and block the interactions [81]. Instead, peptides or nucleotide aptamers might offer a strategy for disrupting protein-protein interactions [82], which could modulate or interfere with the LRRK2 dimerization process. Thus, a better understanding regarding the binding properties of LRRK2 and the regulatory mechanisms and the importance of the dimerization process is warranted to potentially develop specific and selective LRRK2 inhibitors.
Concluding remarks
Since the discovery of mutations in LRRK2 as a major cause of PD, there has been rapid progress in the development of LRRK2 inhibitors. These include the first-in-class relatively selective and specific LRRK2 inhibitors, and proof-of-concept experiments that show inhibition of LRRK2 has tremendous potential to be a disease modifying therapy in the treatment of PD. Despite these advances, the development of specific brain penetrable LRRK2 kinase inhibitors are yet to be reported. Additional in-depth information about LRRK2 enzyme kinetics, structure of the active kinase core, identification of specific physiological regulators and effectors and identification of regulatory mechanisms of activation and inhibition will be required to develop safe and effective LRRK2 inhibitors for the treatment of PD.
Acknowledgements
This work was supported by grants from the NIH NS38377. T.M.D. is the Leonard and Madlyn Abramson Professor in Neurodegenerative Diseases. The authors acknowledge the joint participation by the Adrienne Helis Malvin Medical Research Foundation through its direct engagement in the continuous active conduct of medical research in conjunction with The Johns Hopkins Hospital and the Johns Hopkins University School of Medicine and the Foundation’s Parkinson’s Disease Program No. M-1.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lees AJ, et al. Parkinson's disease. Lancet. 2009;373:2055–2066. doi: 10.1016/S0140-6736(09)60492-X. [DOI] [PubMed] [Google Scholar]
- 2.Savitt JM, et al. Diagnosis and treatment of Parkinson disease: molecules to medicine. J Clin Invest. 2006;116:1744–1754. doi: 10.1172/JCI29178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bronstein JM, et al. Deep brain stimulation for Parkinson disease: an expert consensus and review of key issues. Arch Neurol. 2011;68:165. doi: 10.1001/archneurol.2010.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chaudhuri KR, Odin P. The challenge of non-motor symptoms in Parkinson's disease. Prog Brain Res. 2010;184:325–341. doi: 10.1016/S0079-6123(10)84017-8. [DOI] [PubMed] [Google Scholar]
- 5.Smith Y, et al. Parkinson's Disease Therapeutics: New Developments and Challenges Since the Introduction of Levodopa. Neuropsychopharmacology. 2011 doi: 10.1038/npp.2011.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thomas B, Beal MF. Parkinson's disease. Hum Mol Genet. 2007;16(Spec No. 2):R183–R194. doi: 10.1093/hmg/ddm159. [DOI] [PubMed] [Google Scholar]
- 7.Martin I, et al. Recent advances in the genetics of Parkinson's disease. Annu Rev Genomics Hum Genet. 2011;12:301–325. doi: 10.1146/annurev-genom-082410-101440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dachsel JC, Farrer MJ. LRRK2 and Parkinson disease. Arch Neurol. 2010;67:542–547. doi: 10.1001/archneurol.2010.79. [DOI] [PubMed] [Google Scholar]
- 9.Paisan-Ruiz C, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 10.Zimprich A, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 11.Biskup S, et al. Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Annals of Neurology. 2006;60:557–569. doi: 10.1002/ana.21019. [DOI] [PubMed] [Google Scholar]
- 12.Healy DG, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson's disease: a case-control study. Lancet neurology. 2008;7:583–590. doi: 10.1016/S1474-4422(08)70117-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ross OA, et al. Lrrk2 and Lewy body disease. Annals of Neurology. 2006;59:388–393. doi: 10.1002/ana.20731. [DOI] [PubMed] [Google Scholar]
- 14.Satake W, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson's disease. Nature Genetics. 2009;41:1303–1307. doi: 10.1038/ng.485. [DOI] [PubMed] [Google Scholar]
- 15.Simon-Sanchez J, et al. Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nature Genetics. 2009;41:1308–1312. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.West AB, et al. Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A. 2005;102:16842–16847. doi: 10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.West AB, et al. Parkinson's disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum Mol Genet. 2007;16:223–232. doi: 10.1093/hmg/ddl471. [DOI] [PubMed] [Google Scholar]
- 18.Gallo KA, Johnson GL. Mixed-lineage kinase control of JNK and p38 MAPK pathways. Nat Rev Mol Cell Biol. 2002;3:663–672. doi: 10.1038/nrm906. [DOI] [PubMed] [Google Scholar]
- 19.Nichols RJ, et al. Substrate specificity and inhibitors of LRRK2, a protein kinase mutated in Parkinson's disease. Biochem J. 2009;424:47–60. doi: 10.1042/BJ20091035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pungaliya PP, et al. Identification and characterization of a leucine-rich repeat kinase 2 (LRRK2) consensus phosphorylation motif. PLoS One. 2010;5:e13672. doi: 10.1371/journal.pone.0013672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.MacLeod D, et al. The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron. 2006;52:587–593. doi: 10.1016/j.neuron.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 22.Sakaguchi-Nakashima A, et al. LRK-1, a C. elegans PARK8-related kinase, regulates axonal-dendritic polarity of SV proteins. Curr Biol. 2007;17:592–598. doi: 10.1016/j.cub.2007.01.074. [DOI] [PubMed] [Google Scholar]
- 23.Gehrke S, et al. Pathogenic LRRK2 negatively regulates microRNA-mediated translational repression. Nature. 2010;466:637–641. doi: 10.1038/nature09191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Piccoli G, et al. LRRK2 controls synaptic vesicle storage and mobilization within the recycling pool. The Journal of Neuroscience. 2011;31:2225–2237. doi: 10.1523/JNEUROSCI.3730-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mata IF, et al. LRRK2 in Parkinson's disease: protein domains and functional insights. Trends Neurosci. 2006;29:286–293. doi: 10.1016/j.tins.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 26.Cookson MR. The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson's disease. Nature reviews. Neuroscience. 2010;11:791–797. doi: 10.1038/nrn2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Greggio E, Cookson MR. Leucine-rich repeat kinase 2 mutations and Parkinson's disease: three questions. ASN Neuro. 2009;1 doi: 10.1042/AN20090007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Greggio E, et al. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis. 2006;23:329–341. doi: 10.1016/j.nbd.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 29.Lee BD, et al. Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson's disease. Nat Med. 2010;16:998–1000. doi: 10.1038/nm.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith WW, et al. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci. 2006;9:1231–1233. doi: 10.1038/nn1776. [DOI] [PubMed] [Google Scholar]
- 31.Xiong Y, et al. GTPase activity plays a key role in the pathobiology of LRRK2. PLoS Genet. 2010;6 doi: 10.1371/journal.pgen.1000902. e1000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xiong Y, et al. ArfGAP1 Is a GTPase Activating Protein for LRRK2: Reciprocal Regulation of ArfGAP1 by LRRK2. The Journal of Neuroscience. 2012;32:3877–3886. doi: 10.1523/JNEUROSCI.4566-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin X, et al. Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson's-disease-related mutant alpha-synuclein. Neuron. 2009;64:807–827. doi: 10.1016/j.neuron.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Daher JP, et al. Neurodegenerative phenotypes in an A53T alpha-synuclein transgenic mouse model are independent of LRRK2. Hum Mol Genet. 2012 doi: 10.1093/hmg/dds057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Z, et al. A Drosophila model for LRRK2-linked parkinsonism. Proc Natl Acad Sci U S A. 2008;105:2693–2698. doi: 10.1073/pnas.0708452105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ng CH, et al. Parkin protects against LRRK2 G2019S mutant-induced dopaminergic neurodegeneration in Drosophila. The Journal of Neuroscience. 2009;29:11257–11262. doi: 10.1523/JNEUROSCI.2375-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Venderova K, et al. Leucine-Rich Repeat Kinase 2 interacts with Parkin, DJ-1 and PINK-1 in a Drosophila melanogaster model of Parkinson's disease. Hum Mol Genet. 2009;18:4390–4404. doi: 10.1093/hmg/ddp394. [DOI] [PubMed] [Google Scholar]
- 38.Saha S, et al. LRRK2 modulates vulnerability to mitochondrial dysfunction in Caenorhabditis elegans. The Journal of Neuroscience. 2009;29:9210–9218. doi: 10.1523/JNEUROSCI.2281-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yao C, et al. LRRK2-mediated neurodegeneration and dysfunction of dopaminergic neurons in a Caenorhabditis elegans model of Parkinson's disease. Neurobiol Dis. 2010;40:73–81. doi: 10.1016/j.nbd.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li X, et al. Enhanced striatal dopamine transmission and motor performance with LRRK2 overexpression in mice is eliminated by familial Parkinson's disease mutation G2019S. The Journal of Neuroscience. 2010;30:1788–1797. doi: 10.1523/JNEUROSCI.5604-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li Y, et al. Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of Parkinson's disease. Nat Neurosci. 2009;12:826–828. doi: 10.1038/nn.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang L, et al. The chaperone activity of heat shock protein 90 is critical for maintaining the stability of leucine-rich repeat kinase 2. The Journal of Neuroscience. 2008;28:3384–3391. doi: 10.1523/JNEUROSCI.0185-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ramonet D, et al. Dopaminergicn euronal loss, reduced neurite complexity and autophagic abnormalities in transgenic mice expressing G2019S mutant LRRK2. PLoS One. 2011;6:e18568. doi: 10.1371/journal.pone.0018568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dawson TM, et al. Genetic animal models of Parkinson's disease. Neuron. 2010;66:646–661. doi: 10.1016/j.neuron.2010.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dusonchet J, et al. A rat model of progressive nigral neurodegeneration induced by the Parkinson's disease-associated G2019S mutation in LRRK2. The Journal of Neuroscience. 2011;31:907–912. doi: 10.1523/JNEUROSCI.5092-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nguyen HN, et al. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell. 2011;8:267–280. doi: 10.1016/j.stem.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sanchez-Danes A, et al. Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson's disease. EMBO Molecular Medicine, in press. 2012 doi: 10.1002/emmm.201200215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Noble ME, et al. Protein kinase inhibitors: insights into drug design from structure. Science. 2004;303:1800–1805. doi: 10.1126/science.1095920. [DOI] [PubMed] [Google Scholar]
- 49.Burke RE. Inhibition of mitogen-activated protein kinase and stimulation of Akt kinase signaling pathways: Two approaches with therapeutic potential in the treatment of neurodegenerative disease. Pharmacol Ther. 2007;114:261–277. doi: 10.1016/j.pharmthera.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cuny GD. Kinase inhibitors as potential therapeutics for acute and chronic neurodegenerative conditions. Curr Pharm Des. 2009;15:3919–3939. doi: 10.2174/138161209789649330. [DOI] [PubMed] [Google Scholar]
- 51.Wang LH, et al. Mixed-lineage kinases: a target for the prevention of neurodegeneration. Annual review of Pharmacology and Toxicology. 2004;44:451–474. doi: 10.1146/annurev.pharmtox.44.101802.121840. [DOI] [PubMed] [Google Scholar]
- 52.Mixed lineage kinase inhibitor CEP-1347 fails to delay disability in early Parkinson disease. Neurology. 2007;69:1480–1490. doi: 10.1212/01.wnl.0000277648.63931.c0. [DOI] [PubMed] [Google Scholar]
- 53.Wang LH, Johnson EM., Jr Mixed lineage kinase inhibitor CEP-1347 fails to delay disability in early Parkinson disease. Neurology. 2008;71:462. doi: 10.1212/01.wnl.0000324506.93877.5e. author reply 462–463. [DOI] [PubMed] [Google Scholar]
- 54.Anand VS, et al. Investigation of leucine-rich repeat kinase 2: enzymological properties and novel assays. FEBS J. 2009;276:466–478. doi: 10.1111/j.1742-4658.2008.06789.x. [DOI] [PubMed] [Google Scholar]
- 55.Jaleel M, et al. LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson's disease mutants affect kinase activity. Biochem J. 2007;405:307–317. doi: 10.1042/BJ20070209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Imai Y, et al. Phosphorylation of 4E-BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. EMBO J. 2008;27:2432–2443. doi: 10.1038/emboj.2008.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gloeckner CJ, et al. The Parkinson disease-associated protein kinase LRRK2 exhibits MAPKKK activity and phosphorylates MKK3/6 and MKK4/7, in vitro. J Neurochem. 2009;109:959–968. doi: 10.1111/j.1471-4159.2009.06024.x. [DOI] [PubMed] [Google Scholar]
- 58.Hsu CH, et al. MKK6 binds and regulates expression of Parkinson's disease-related protein LRRK2. J Neurochem. 2010;112:1593–1604. doi: 10.1111/j.1471-4159.2010.06568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu M, et al. Development of a mechanism-based high-throughput screen assay for leucine-rich repeat kinase 2--discovery of LRRK2 inhibitors. Anal Biochem. 2010;404:186–192. doi: 10.1016/j.ab.2010.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Reichling LJ, Riddle SM. Leucine-rich repeat kinase 2 mutants I2020T and G2019S exhibit altered kinase inhibitor sensitivity. Biochem Biophys Res Commun. 2009;384:255–258. doi: 10.1016/j.bbrc.2009.04.098. [DOI] [PubMed] [Google Scholar]
- 61.Pedro L, et al. Development of a high-throughput AlphaScreen assay measuring full-length LRRK2(G2019S) kinase activity using moesin protein substrate. Anal Biochem. 2010;404:45–51. doi: 10.1016/j.ab.2010.04.028. [DOI] [PubMed] [Google Scholar]
- 62.Ramsden N, et al. Chemoproteomics-Based Design of Potent LRRK2-Selective Lead Compounds That Attenuate Parkinson's Disease-Related Toxicity in Human Neurons. ACS Chem Biol. 2011;6:1021–1028. doi: 10.1021/cb2002413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Deng X, et al. Characterization of a selective inhibitor of the Parkinson's disease kinase LRRK2. Nat Chem Biol. 2011;7:203–205. doi: 10.1038/nchembio.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Covy JP, Giasson BI. Identification of compounds that inhibit the kinase activity of leucine-rich repeat kinase 2. Biochem Biophys Res Commun. 2009;378:473–477. doi: 10.1016/j.bbrc.2008.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu Z, et al. Inhibitors of LRRK2 kinase attenuate neurodegeneration and Parkinson-like phenotypes in Caenorhabditis elegans and Drosophila Parkinson's disease models. Hum Mol Genet. 2011;20:3933–3942. doi: 10.1093/hmg/ddr312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bain J, et al. The specificities of protein kinase inhibitors: an update. Biochem J. 2003;371:199–204. doi: 10.1042/BJ20021535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bain J, et al. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bantscheff M, et al. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat Biotechnol. 2007;25:1035–1044. doi: 10.1038/nbt1328. [DOI] [PubMed] [Google Scholar]
- 69.Goldstein DM, et al. High-throughput kinase profiling as a platform for drug discovery. Nat Rev Drug Discov. 2008;7:391–397. doi: 10.1038/nrd2541. [DOI] [PubMed] [Google Scholar]
- 70.Ito G, et al. GTP binding is essential to the protein kinase activity of LRRK2, a causative gene product for familial Parkinson's disease. Biochemistry. 2007;46:1380–1388. doi: 10.1021/bi061960m. [DOI] [PubMed] [Google Scholar]
- 71.Korr D, et al. LRRK1 protein kinase activity is stimulated upon binding of GTP to its Roc domain. Cell Signal. 2006;18:910–920. doi: 10.1016/j.cellsig.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 72.Lewis PA, et al. The R1441C mutation of LRRK2 disrupts GTP hydrolysis. Biochem Biophys Res Commun. 2007;357:668–671. doi: 10.1016/j.bbrc.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li X, et al. Leucine-rich repeat kinase 2 (LRRK2)/PARK8 possesses GTPase activity that is altered in familial Parkinson's disease R1441C/G mutants. J Neurochem. 2007;103:238–247. doi: 10.1111/j.1471-4159.2007.04743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Haebig K, et al. ARHGEF7 (Beta-PIX) acts as guanine nucleotide exchange factor for leucine-rich repeat kinase 2. PLoS One. 2010;5:e13762. doi: 10.1371/journal.pone.0013762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sen S, et al. Dependence of leucine-rich repeat kinase 2 (LRRK2) kinase activity on dimerization. J Biol Chem. 2009;284:36346–36356. doi: 10.1074/jbc.M109.025437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Deng J, et al. Structure of the ROC domain from the Parkinson's disease-associated leucine-rich repeat kinase 2 reveals a dimeric GTPase. Proc Natl Acad Sci U S A. 2008;105:1499–1504. doi: 10.1073/pnas.0709098105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Greggio E, et al. The Parkinson disease-associated leucine-rich repeat kinase 2 (LRRK2) is a dimer that undergoes intramolecular autophosphorylation. J Biol Chem. 2008;283:16906–16914. doi: 10.1074/jbc.M708718200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jorgensen ND, et al. The WD40 domain is required for LRRK2 neurotoxicity. PLoS One. 2009;4:e8463. doi: 10.1371/journal.pone.0008463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Berger Z, et al. Membrane localization of LRRK2 is associated with increased formation of the highly active LRRK2 dimer and changes in its phosphorylation. Biochemistry. 2010;49:5511–5523. doi: 10.1021/bi100157u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ito G, Iwatsubo T. Re-examination of the dimerization state of leucine-rich repeat kinase 2: predominance of the monomeric form. Biochem J. 2011 doi: 10.1042/BJ20111215. [DOI] [PubMed] [Google Scholar]
- 81.Whitty A, Kumaravel G. Between a rock and a hard place? Nat Chem Biol. 2006;2:112–118. doi: 10.1038/nchembio0306-112. [DOI] [PubMed] [Google Scholar]
- 82.Bogoyevitch MA, et al. Peptide inhibitors of protein kinases-discovery, characterisation and use. Biochim Biophys Acta. 2005;1754:79–99. doi: 10.1016/j.bbapap.2005.07.025. [DOI] [PubMed] [Google Scholar]
- 83.Tong Y, et al. Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of alpha-synuclein, and apoptotic cell death in aged mice. Proc Natl Acad Sci U S A. 2010;107:9879–9884. doi: 10.1073/pnas.1004676107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Herzig MC, et al. LRRK2 protein levels are determined by kinase function and are crucial for kidney and lung homeostasis in mice. Hum Mol Genet. 2011;20:4209–4223. doi: 10.1093/hmg/ddr348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liu Z, et al. The kinase LRRK2 is a regulator of the transcription factor NFAT that modulates the severity of inflammatory bowel disease. Nature immunology. 2011;12:1063–1070. doi: 10.1038/ni.2113. [DOI] [PMC free article] [PubMed] [Google Scholar]