

Abstract
The differentiation of naïve T cells into distinct subsets of effector T cells is critical for effective immunity against a wide variety of infectious agents in the environment. Activation of innate immune responses by Candida through pattern recognition receptors directs the subsequent development of naïve T cells into Th17 cells, which are essential for effective mucosal immunity against fungi. Thorough analyses of cohorts of patients with unusual susceptibility to chronic mucocutaneous candidiasis (CMC) resulting from Th17 deficiency has confirmed the role of Th17 cells and Th17 cytokines in human host defense against Candida and has provided valuable insight into the complex process of Th17 cell development.
Keywords: T helper cells, Candida, fungi, Th17 cell, pathogen recognition receptors, cytokines, transcription factors, differentiation, chronic mucocutaneous candidiasis
INTRODUCTION
Effective host defense against a wide variety of pathogens in our environment requires well-coordinated innate and adaptive immune responses. The innate immune system detects the presence of invading pathogens through widely expressed pathogen recognition receptors (i.e. Toll-like receptors (TLRs), NOD-like receptors, RIG-I-like receptors, C-type lectins), leading to the subsequent activation of an adaptive immune response, which destroys pathogens incompletely eliminated by the innate immune system (reviewed in1). The innate immune system is germline encoded and is characterized by limited diversity against invariant pathogen associated molecular patterns, whereas adaptive immune responses are characterized by much greater diversity and specificity resulting from somatic recombination. The development and regulation of distinct subsets of T helper cells is essential for protection against a wide range of pathogens (i.e. extracellular versus intracellular).
T helper cells are T cells that provide help or activating signals to other immune cells through the production of cytokines. The first characterization of Th1 and Th2 T helper cell subsets occurred over two decades ago and was based upon the profile of cytokines each subset secreted2. Th1 cells are characterized by production of IFNγ and are required for defense against intracellular pathogens and mediate delayed type hypersensitivity. IFNγ enhances the bactericidal activities of macrophages, upregulates MHC class I and class II expression, and promotes production of opsonizing antibodies by B cells. Th2 cells are characterized by production of IL-4, IL-5, and IL-13 and are involved in defense against parasitic infections, mediate production of IgE, and promote eosinophilic inflammation3. The production of IL-4 supports the production of neutralizing antibodies against extracellular bacteria that characterize humoral immunity.
Subsequent analysis of T cell differentiation has revealed far greater complexity in the T helper cell subsets that can be generated. These T helper cells are defined by their expression of specific transcription factors and functions. For instance, T regulatory cells (Treg) are characterized by expression of forkhead box P3 (FOXP3) and are critical for the maintenance of self tolerance. However, there is plasticity in T cell differentiation, since Treg cells can be induced to lose FOXP3 expression and adopt a Th1 cell phenotype capable of IFNγ production and autoimmune destruction4. More recently, an additional subset of effector T cell, the Th17 subset, has been identified 5. The development of Th17 cells is dependent upon exposure of naïve T cells to a complex milieu of cytokines that leads to expression of a unique set of transcription factors required for Th17 differentiation (described below). Similar to Tregs, plasticity of Th17 cells has been observed in lymphopenic environments in mouse models in which Th17 cells can convert into IFNγ secreting Th1 cells6. Thus, characterization of the cytokines, signaling pathways, and transcription factors that direct T cell differentiation is critical for understanding T cell differentiation and function. Identification of novel mutations in genes that impair T cell differentiation and function in immune deficient patients will expand our knowledge of the role of specific T cell subsets in host defense against pathogens.
Both mouse models and analysis of Th17 cells in humans have demonstrated that Th17 cells play a critical role in mucosal immunity7, 8. The recent descriptions of infectious susceptibility to CMC in patients with defects in Th17 development and function resulting from single gene mutations or as part of a syndrome have served to clearly define the role of Th17 T cells in human host defense. The complex processes of Th17 cell development have been previously described in this issue and elsewhere7, 9, and it will only be mentioned below in relation to other immunodeficiency diseases which impact Th17 cell development and function. Knowledge of the mechanisms of Th17 cell differentiation and inborn errors of Th17 cell function in patients has expanded rapidly over the past several years. This review will summarize the clinical aspects of patients with genetic defects that result in Th17 deficiency.
The IL-17 Cytokine Family
The IL-17 cytokine family consists of six members (IL-17A-F). Among IL-17 family members, IL-17A and IL-17F share the highest amino acid sequence homology (50%)10. The receptors for IL-17 include a family of five receptors (IL-17RA-E). Heterodimers of IL-17RA and IL-17RC constitute the receptor for IL-17A and IL-17F. IL-17A and IL-17F are preferentially produced by Th17 cells (along with IL-21 and IL-22). IL-17A and IL-17F form homo-and heterodimers and induce expression of pro-inflammatory cytokines (IL-1, IL-6, TNFα, G-CSF, GM-CSF), chemokines (CXCL1, CXCL5, IL-8, CCL2, CCL7), antimicrobial peptides(AMPs) (defensins, S100), and matrix metalloproteinases (MMP1, MMP3, MP13) from the endothelium, epithelia, and fibroblasts9. As a result, Th17 cells can promote granulopoesis and recruitment of neutrophils to sites of infection and provide host defense against extracellular bacteria and fungi. Interestingly, a variety of other T cell types, including CD8+ T cells (Tc17), γδ T cells (γδ-17), and NKT cells (NKT-17) can produce IL-17A and IL-17F (reviewed in11). Further, innate immune cells including neutrophils, monocytes, NK cells, and lymphoid tissue inducer (LTi)-like cells can produce IL-17A and IL-17F. Finally, IL-17A has been shown to be produced by intestinal Paneth cells12, whereas colonic epithelial cells have been shown to express IL-17F mRNA, suggesting that a variety of cell types may play roles in host defense against fungi and extracellular bacteria8. Less is known about the regulation of IL-17 from these cell types.
DEVELOPMENT OF TH17 CELLS
The differentiation of naïve T cells into Th17 cells is a highly complex process and is incompletely understood. Th17 cell development is directed by multiple cytokines, including IL-1β, IL-6, TGFβ, IL-21, and IL-23 (Fig. 1), which lead to activation of the transcription factors STAT3 and IRF4 and subsequent expression of retinoic acid receptor-related orphan receptor γt (RORγt)7, 13–17. These cytokines are produced by activated T cells and antigen presenting cells stimulated through pathogen recognition receptors (i.e. Dectin-1/2, TLR 2, 4, Mincle) in response to pathogen associated molecular patterns (β-glucans, lipoproteins, peptidoglycan, zymosan) found within the cell walls of fungi and bacteria18. Several other transcription factors, including RORα, basic leucine zipper transcription factor (Batf), Runx1, and IκBζ can cooperate with RORγt to regulate Th17 differentiation (reviewed in9). Additionally, the transcription factors SMAD-3 and ATF-2 are required for expression of IL-23 p19, which promotes Th17 differentiation19.
Figure 1. Summary of Th17 development and human diseases that impair Th17 development or function.
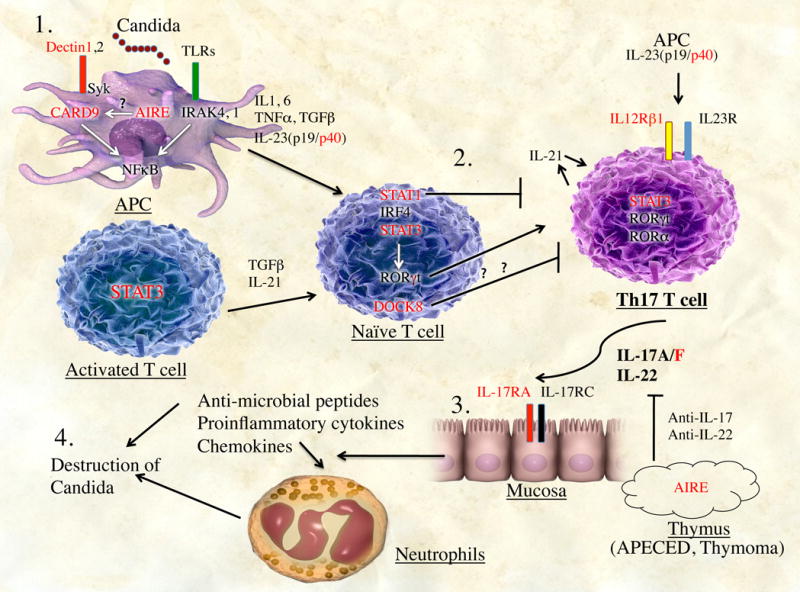
1, IL-1β, IL-6, IL-23, TNF-α, and TGF-β are secreted after activation of antigen-presenting cells (APC) by fungal cell-wall components. Additionally, activated T cells produce TGF-β and IL-21. 2, Exposure of naive T cells to this cytokine milieu induces their differentiation into TH17 cells. Gain-of-function mutations in patients with STAT1 as well as STAT3 functional deficiency and DOCK8 deficiency block TH17 differentiation. 3, TH17 cells produce IL-17A/F and IL-22, which induces production of proinflammatory cytokines, chemokines, and antimicrobial peptides by mucosal cells, leading to recruitment of neutrophils to sites of infection and destruction of Candida species (4). Genes in red represent currently known mutations that impair TH17 cell development or TH17 effector functions, as described in the text.
Insights into the cytokine milieu required for Th17 development in humans has been gained from the study of patients with defects in these cytokine pathways. Myeloid differentiation primary response gene-88 (MyD88) and interleukin-1 receptor associated kinase-4 (IRAK-4) are essential effectors of the IL-1R and TLR signaling pathways. Studies by de Beaucoudray et al20 demonstrated that Th17 T cell populations are intact in patients with IRAK-4 and MyD88 deficiency, who are unable to respond to IL-1β. This observation suggests that while IL-1β may promote Th17 differentiation, it is not absolutely required. Since all TLR signaling (with the exception of TLR3) is also blocked in these patients, TLR-induced IL-6 production is absent21. The lack of IL-1 responsiveness and impaired IL-6 production in MyD88- and IRAK-4-deficient patients is manifested clinically by reduced fever and reduced acute phase reactant production in these patients during invasive bacterial infections. Given that IL-6 plays a critical role in Th17 development, it is clear that IL-6 induced by the stimulation of other pathogen recognition receptors (PRRs) is adequate for Th17 cell development. Other functional sources of IL-6 in MyD88- and IRAK-4-deficient patients include activation of TLR3, NOD-like receptors, RIG-I-like receptors, and C-type lectins.
In contrast, IL12p40 and IL12Rβ1-deficient patients, both of whom have impaired IL-23 signaling, have reduced populations of Th17 cells20. IL12p40 and IL23p19 form heterodimers to make active IL-23, while IL12Rβ1 and IL23R heterodimerize to form the active IL-23 receptor. The reduction in Th17 populations in IL12p40 and IL12Rβ1-deficient patients is not as severe as in STAT3 deficient patients, suggesting that IL-23 is important for Th17 development and/or maintenance, but some redundancy may exist to allow reduced Th17 development. Since candidal infections in IL12p40 and IL12Rβ1-deficient patients are not common, it is likely that the majority of these patients retain adequate Th17 function to prevent susceptibility to CMC20. Additionally, IL-12 has been shown to play a role in anti-candidal immunity. Since the IL12p40 and IL12Rβ1-deficient patients are obviously deficient in IL-12 responses, which accounts for their susceptibility to mycobacterial infections, identification of IL-23R or IL23p19 deficient patients would likely provide more specific insight into the importance of IL-23 in human Th17 differentiation and host defense against Candida.
CMC DISEASE AS PART OF A SYNDROME
Candida albicans is a commensal organism that can cause transient mucocutaneous infections of skin, nails, and oral and genital mucosae in healthy individuals. In immunocompromised individuals, however, C albicans can cause chronic mucocutaneous and sometimes fatal invasive infections. Neutrophil defects are associated with systemic candidiasis and susceptibility to a wide variety of bacteria. CMC disease and invasive candidiasis has also been described in a large Iranian family with a homozygous nonsense mutation in CARD922, a signaling protein that along with Syk tyrosine kinase is downstream of the anti-fungal receptors Dectin-1, Dectin-2, and Mincle (Fig. 1). Severe T cell deficiencies, such as severe combined immunodeficiencies and HIV, are associated with CMC disease, but they also are associated with other bacterial, viral, and opportunistic infections. Infectious susceptibility that is primarily limited to CMC represents a unique subset of immune deficiency.
To date, several complex human diseases have been identified that impair Th17 cell differentiation and can result in susceptibility to CMC disease as one manifestation of a more complex syndrome (Table 1). These include autosomal dominant hyper IgE syndrome (AD-HIES) with mutations in signal transducer and activator of transcription-3 (STAT3)23 and autosomal recessive (AR)-HIES with mutations in dedicator of cytokinesis gene-8 (DOCK8)24. Furthermore, autoimmune polyendocrine syndrome type 1 (APS-1), also known as autoimmune polyendocrinopathy, candidiasis, and ectodermal dystrophy (APECED) and some cases of thymoma also result in susceptibility to CMC through impairment of Th17 functions25. These secondary causes of Th17 function have provided useful insights into the mechanisms that regulate human Th17 effector development and function and serve to confirm the essential role of Th17 cells in mucosal immunity against Candida.
Table 1.
Conditions that impair TH17 development
| CONDITION | PHENOTYPE | GENOTYPE |
|---|---|---|
| TH17 deficiency within a syndrome | ||
| AD-HIES | Elevated IgE, S.aureus abscesses, pneunmonia, pneumatocele formation, candidiasis | STAT3 mutations |
| AR-HIES | Elevated IgE, eosinophilia, atopy, recurrent sinopulmonary infections, Herpes virus, candidiasis | DOCK8 mutations |
| GOF-STAT1 | Pneumonia, P. jirovecii pneumonia, CMV, autoimmunity, cancers, candidiasis | STAT1 mutations within exon 10 |
| APS1/APECED | Autoimmune polyendocrinopathy, ectodermal dystrophy, candidiasis | AIRE mutations |
| Thymoma | Autoimmunity, myasthenia gravis, rarely candidiasis | Absent AIRE expression in tumor |
| Pure Th17 deficiency | ||
| IL-17 receptor IL-17 ligand |
Candidal dermatitis, S aureus dermatitis | IL-17RA, IL-17F mutations |
AD-HIES due to mutations in STAT3
AD-HIES due to mutations in STAT3 is a multisystem immune deficiency characterized by highly elevated serum levels of IgE, local and invasive infections with S. aureus that result in cold abscesses and recurrent pneumonia with pneumatocele formation, skeletal abnormalities, and coarse facial features26–28. Additionally, development of CMC is a common complication in these patients. The cytokines IL-6 and IL-21, which activate STAT3, play significant roles in the development of human Th17 T cells29, as discussed above. As previously mentioned, STAT3 activation induces the expression of RORγt, a transcription factor required for the development of Th17 T cells and the subsequent expression of IL-17, IL-21, IL-22, and IL23R, which play essential roles in Th17 function. Since patients with AD-HIES due to STAT3 mutations are susceptible to CMC disease and STAT3 is essential for Th17 cell development and Th17 cells play an important role in mucosal immunity against Candida, Ma et al23 evaluated these patients for potential defects in Th17 development. Analysis of T cell blasts generated from AD-HIES total CD4+ T cells revealed significant reductions in IL-17+ T cells and subsequent stimulation of these CD4+ T cells resulted in no production of IL-17. Furthermore, production of IL-22, a Th17-derived cytokine involved in epithelial and mucosal immunity, was absent from AD-HIES CD4+ T cells. Consistent with the essential role of STAT3 in RORγt expression, upregulation of RORγt expression was reduced in stimulated AD-HIES CD4+ T cells compared to healthy controls. Furthermore, sorted naïve CD4+ T cells from AD-HIES patients failed to differentiate into Th17 T cells when stimulated with anti-CD2 and anti-CD3 plus anti-CD28 in the presence of IL-1β plus IL-6 or IL-23, confirming the requirement for STAT3 in Th17 differentiation. Consistent with these observations, Milner et al30 found that blood cells obtained from AD-HIES patients that were stimulated overnight with S. aureus or Candida failed to generate memory CD4+IL-17+ T cells. Thus, analysis of T cells from patients with STAT3 mutations has clearly demonstrated an essential role for STAT3 in Th17 differentiation and mucosal and epithelial immunity against Candida and S aureus 23, 30, 31(Fig. 1). As mentioned above, Tc17, γδ-17, and NKT-17 are also sources of Th17 cytokines11; however, they either do not produce Th17 cytokines or they produce inadequate quantities of Th17 cytokines in AD-HIES patients to prevent CMC disease. This suggests that STAT3 is likely to be involved in Th17 cytokine production in Tc17 cells, γδ-17 cells, and NKT-17 cells.
DOCK8
AR-HIES due to mutations in DOCK8 is characterized by highly elevated serum IgE levels, hypereosinophilia, recurrent sinopulmonary infections, unusual susceptibility to herpesvirus infections, candidal dermatitis, and atopy32–34. In addition, DOCK8 deficient patients tend to be T cell lymphopenic with poor T cell proliferation after activation with anti-CD3 and anti-CD28. The molecular basis of cutaneous candidal infections in these patients is incompletely understood at present. Previous analysis of a cohort of AR-HIES patient, some of whom were later confirmed to have DOCK8 deficiency, revealed that these patients have defects in Th17 differentiation24. RORγt expression, which is critical for Th17 differentiation, was markedly reduced in peripheral T cells from patients with AD-HIES due to STAT3 mutations, as well as in AR-HIES patients. Interestingly, in vitro induction of RORγt expression by naïve T cells was intact in the AR-HIES, but not AD-HIES patients. These data suggest that the initial steps of Th17 differentiation are intact in the AR-HIES patients, but subsequent steps of differentiation are impaired (Fig. 1). IL-17 production was impaired in both groups of HIES patients, although the impairment was more severe in the patients with STAT3 mutations. Thus, defective Th17 differentiation occurs by a different mechanism in AR-HIES versus AD-HIES due to STAT3 mutations. It is likely that impaired Th17 differentiation and IL-17 production contributes to the susceptibility of AR-HIES patients with DOCK8 mutations to candidal dermatitis. It is also possible that a more global impairment in T cell function in DOCK8 deficient patients, as demonstrated clinically by their susceptibility to viral infections, also contributes to candidal infections. A clearer understanding of the cause(s) of impaired Th17 differentiation in DOCK8 patients may help to further elucidate the complex process of Th17 differentiation.
Gain of function mutations in STAT1
In a search for genetic causes of AD-CMC disease, two groups of investigators independently identified heterozygous mutations in STAT1 as a novel cause of deficiency of Th17 differentiation and AD-CMC. In one study, van de Veerdonk et al35 analyzed five families that had severe CMC disease. Other documented infections included pneumonia and one case of Pneumocystis jirovecii pneumonia with CMV infection. Additionally, several patients suffered from autoimmune disease (autoimmune hepatitis, autoimmune hemolysis, hypothyroidism) and a few developed cancers (esophageal cancer, squamous cell carcinoma). Functional analysis of blood cells revealed defects in Candida-stimulated production of interferon-γ, IL-17, and IL-22. Further functional analyses demonstrated defective functioning of IL-12 and IL-23 signaling pathways. These studies were followed by array-based sequence capture followed by next generation sequencing of 100 candidates genes relevant to IL-12 and IL-23 signaling and Th1 and Th17 responses. This analysis identified heterozygous mutations within highly conserved residues in exon 10 of STAT1, which lies within coiled-coil domain.
Simultaneously, Liu et al36 analyzed additional genetic causes of CMC disease by applying a genome-wide strategy based upon whole-exome sequencing. All patients suffered CMC disease and several of these patients suffered autoimmune disorders (thyroid autoimmunity, systemic lupus erythematosis), cerebral aneurysms, or squamous cell carcinoma. Candidate variations were selected by filtering out known polymorphisms obtained from several databases. This analysis identified heterozygous variations in the STAT1 gene all within the coiled-coil domain. Functional analysis of one of the STAT1 mutant alleles (STAT1R274Q) revealed enhanced activation of a gamma activated sequence reporter construct in response to IFNα, IFNγ, and IL-27. The increased transcriptional activation by the STAT1R274Q allele was associated with impaired nuclear dephosphorylation. Thus, STAT1 mutations within the coiled-coil domain appeared to be gain of function mutations. Stimulation of EBV-immortalized B cells and fibroblasts from patients with gain of function mutations in STAT1 resulted in augmented responses to IFNα, IFNγ, and IL-27, cytokines known to be antagonistic to Th17 development37, 38. Interestingly, cellular responses to predominantly STAT3 activating cytokines that promote Th17 differentiation, like IL-6 and IL-21, also resulted in increased STAT1 phosphorylation in EBV B cells from patients with gain of function STAT1 mutations compared to healthy controls. The enhanced STAT1 activation may be significant since STAT1 activation has been shown to be antagonistic to Th17 development13(Fig. 1). Finally, percentages of IL17A+, IL17F+, and IL-22+ T cells, as well as production of IL-17A and IL-22 in patients with gain of function STAT1 mutations were significantly lower than healthy controls and patients with loss of function STAT1 mutations, providing the molecular basis for CMC disease in these patients. Interestingly, enhanced cellular responses to type 1 interferon in these patients may underlie a predisposition to autoimmune disorders observed in several of these patients, since autoimmune disorders have been associated with an increased type 1 interferon signature39, 40. A fascinating aspect of the role of STAT1 in human immune deficiencies is that mutations in one gene can lead to three very different phenotypes: CMC disease36, mycobacterial disease41, or susceptibility to viral disease42.
APS1/APECED and Thymoma
The syndrome of autoimmune polyendocrine syndrome type 1 (APS-1), or autoimmune polyendocrinopathy, candidiasis, and ectodermal dystrophy (APECED) is the result of autosomal recessive mutations in autoimmune regulator (AIRE) gene43. AIRE is a transcription factor expressed by medullary thymic epithelial cells, which is responsible for the expression of a wide variety of tissue specific antigens within the thymus. Expression of these tissue specific antigens in the thymus leads to deletional self tolerance in T cells maturing within the thymus. However, the lack of functional AIRE in APECED patients results in impaired central T cell tolerance, leading to the generation of autoantibodies. The clinical presentation of APECED is variable, but the majority of patients develop CMC disease, hypoparathyroidism, and Addison’s disease25. CMC is the sole immune deficiency in APECED patients and is one of the earliest manifestations of the syndrome. Neutralizing antibodies against cytokines, in particular type 1 interferons and Th17 cytokines, are the most prevalent autoantibodies in APECED patients (reviewed in25). This high prevalence of autoantibodies against type 1 interferons and IL-22 is virtually diagnostic for APECED. Autoantibodies against IL-17A, IL-17F, and IL-22 appear to be more prevalent in APECED patients with CMC than those without CMC, suggesting a causal role in CMC (Fig. 1). Kisand et al44 also observed that IL-17 and IL-22 production was markedly decreased from Candida-stimulated blood cells derived from APECED patients with CMC disease, perhaps due to autoimmune destruction of IL-17+ and IL-22+ T cells. Interestingly, Pedroza et al45 observed that the AIRE protein can also function as a signaling effector downstream of Dectin-1 in blood cells and a monocytic cell line (Fig. 1). These observations suggest that AIRE mutations in APECED may contribute to impaired innate responses to Candida, which could subsequently impair IL-17 and IL-22 production, although this remains to be formally evaluated.
Thymomas are tumors that originate from thymic epithelial cells. The majority of these tumors fail to express AIRE (Fig. 1). Thymomas are associated with autoimmune manifestations, most notably with antibodies against muscle acetylcholine receptors that cause myasthenia gravis. Autoantibodies against type 1 interferons are also common in thymoma patients; however, autoantibodies against Th17 cytokines are rare44. As a result, CMC is only rarely associated with thymoma. For unknown reasons, APECED and thymoma patients, despite having high titers of neutralizing antibodies against IFNα and IFNω, do not have increased susceptibility to viral infections. Possible explanations are that IFNβ and IFNλ may compensate or local concentrations of type 1 interferons may be adequate to activate biological responses. The rare occurrence of CMC disease in thymoma patients with anti-IL-17 cytokine antibodies provides additional circumstantial evidence of the importance of IL-17 cytokines in CMC disease.
INBORN ERRORS OF IMMUNITY RESULTING IN PURE CMC DISEASE
Inborn errors of IL-17RA and IL-17F
Recently, two genetic causes of pure CMC disease have been identified in humans (Table 1). Puel et al46 studied individuals and families with infectious susceptibility limited to CMC for inborn errors of IL-17, IL-17R, and IL-22. Other known causes of CMC disease were excluded clinically and genetically. Evaluation of an infant who was the product of a consanguineous union that presented with candidal dermatitis and subsequent Staphylococcus aureus dermatitis resulted in the first description of autosomal recessive CMC disease due to a homozygous nonsense mutation in the IL-17RA gene (Q248X/Q248X). The Q248X/Q248X mutation resulted in absent expression of IL-17RA on the patient’s blood cells and fibroblasts and a complete absence of responsiveness to IL-17A/F. Responsiveness to IL-17 cytokines was restored to the patient’s fibroblasts by transfection with wild type IL-17RA, confirming the lack of IL-17RA expression as the cause the patient’s CMC disease.
Additionally, Puel46 et al evaluated a multiplex family with autosomal dominant inheritance of CMC disease, identifying a heterozygous missense mutation in the IL-17F gene. The mutation resulted in the substitution of the serine residue at position 65 of the protein with a leucine residue (S65L). The S65 residue is conserved across all mammalian species and is believed to be involved in ligand-receptor interaction. The production of inflammatory cytokines by normal fibroblasts and blood cells elicited by mutant IL-17F(S65L) homodimers was severely impaired. In addition, stimulation of normal fibroblasts and keratinocytes with heterodimers consisting of IL-17F(S65L)/IL-17FWT or IL-17F(S65L)/IL-17A resulted in severely impaired production of inflammatory cytokines, demonstrating a dominant negative effect of IL-17F(S65L). Interestingly, the IL-17F(S65L) mutation was also found in two apparently healthy members of the family members, suggesting incomplete clinical penetrance. At present, although experience is very limited with patients having genetic defects in IL-17F or IL-17RA, these observations support a role for Th17 cells and Th17 cytokines that is limited to mucosal immunity against Candida, and possibly S aureus (Fig. 1).
CONCLUSION
The study of multiple cohorts of patients with Th17 deficiency that result from unique genetic defects has been invaluable in defining the role Th17 cells and Th17 cytokines in human mucosal host defense against Candida, and possibly to S. aureus. Many of these disorders cause susceptibility to more than one pathogen, including Candida. In the cases of STAT1, STAT3, IL12Rβ1, DOCK8, IL-17RA, and IL-17F mutations, thorough analyses of the molecular mechanisms that underlie these disorders has revealed defective Th17 cell development or effector function that results in susceptibility to CMC disease. Some of these disorders have associated autoimmunity. Interestingly, CMC disease is a sequelae of APS-1 and, rarely, of thymoma, as a result of neutralizing autoantibodies against IL-17 and IL-22. The common link among all these disorders, of course, is a deficiency in Th17 effector function. CMC disease in Th17 deficient patients is likely secondary to the lack of IL-17-induced production of anti-microbial peptides by mucosal cells and impaired recruitment of neutrophils that clear the Candida. Analysis of Th17 cell populations in MyD88 and IRAK-4 deficient patients was worthwhile because although they do not suffer CMC disease, the finding of normal Th17 cell populations in these patients demonstrated that IL-1β is not absolutely required for Th17 cell development in humans. In rare cases, oral candidiasis has been observed in patients with mutations in NEMO/IKKγ, who have reduced NFκB activation47. Th17 cell development is severely impaired in c-Rel−/− mice, suggesting a role for NFκB in Th17 development48. Further analysis of patients with unusual susceptibility to CMC disease is likely to provide additional insights into the regulation of Th17 cell development and function.
What is known?
Th17 T cells and Th17 cytokines mediate mucosal immunity against fungi, and, perhaps, S. aureus in humans.
Th17 cells appear to be redundant for most other forms of immunity.
Although IL-1β promotes Th17 development in mouse models, it is not absolutely required for Th17 development in humans.
Analysis of STAT1 mutations reveals great phenotypic variation: CMC disease, mycobacterial disease, or viral disease.
STAT3 is a critical mediator of Th17 T cell development and resistance to CMC disease.
What is still unknown?
Will Th17 cytokines have therapeutic use in select cases of severe CMC disease?
Could use of Th17 cytokines clinically result in autoimmunity?
What is the contribution of Tc17 cells, γδ-17 cells, NKT-17 cells, neutrophils, monocytes, and LTi-like cells to mucosal immunity?
What is the mechanism of impaired Th17 development and candidiasis in DOCK8 patients?
Will analysis of additional patients and families with CMC disease identify novel genes important for Th17 development?
Will analysis of additional patients and families with CMC disease help to clarify the process of Th17 development in humans?
Acknowledgments
DRM is supported by NIH grant 5K08AI76625.
Abbreviations used
- AD
Autosomal dominant
- AIRE
Autoimmune regulator
- AMP
Antimicrobial peptide
- APC
Antigen presenting cell
- APECED
Autoimmune polyendocrineopathy-candidiasis ectodermal dystrophy
- APS-1
Autoimmune polyendocrine syndrome 1
- ATF-2
Activating transcription factor-2
- AR
Autosomal recessive
- BATF
Basic leucine zipper transcription factor, ATF-like
- CARD9
Caspase recruitment domain-containg protein 9
- CMC
Chronic mucocutaneous cadidiasis
- CMV
Cytomegalovirus
- DOCK8
Dedicator of cytokinesis 8
- EBV
Epstein Barr virus
- FOXP3
Forkhead box P3
- HIES
Hyper IgE syndrome
- IκBζ
Inhibitor kappa B zeta
- IRAK-4
Interleukin-1 receptor-associated kinase-4
- NFκB
Nuclear factor kappa B
- MINCLE
Macrophage-inducible C-type lectin
- MyD88
Myeloid differentiation factor-88
- NEMO
NFκB essential modifier
- NKT cell
Natural killer T cell
- NOD
Nucleotide oligomerization domain
- PRR
Pattern recognition receptor
- RIG-I
Retinoic acid inducible gene-I
- ROR
Retinoic acid-related orphan receptor
- RUNX1
Runt-related transcription factor 1
- STAT
Signal transducer and activator of transcription
- Th cell
T helper cell
- Treg
T regulatory cell
- TLR
Toll-like receptor
Footnotes
The author has no conflicts of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 34:637–50. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 2.Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. 1989;7:145–73. doi: 10.1146/annurev.iy.07.040189.001045. [DOI] [PubMed] [Google Scholar]
- 3.Abbas AK, Murphy KM, Sher A. Functional diversity of helper T lymphocytes. Nature. 1996;383:787–93. doi: 10.1038/383787a0. [DOI] [PubMed] [Google Scholar]
- 4.Zhou X, Bailey-Bucktrout S, Jeker LT, Bluestone JA. Plasticity of CD4(+) FoxP3(+) T cells. Curr Opin Immunol. 2009;21:281–5. doi: 10.1016/j.coi.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bettelli E, Korn T, Kuchroo VK. Th17: the third member of the effector T cell trilogy. Curr Opin Immunol. 2007;19:652–7. doi: 10.1016/j.coi.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martin-Orozco N, Chung Y, Chang SH, Wang YH, Dong C. Th17 cells promote pancreatic inflammation but only induce diabetes efficiently in lymphopenic hosts after conversion into Th1 cells. Eur J Immunol. 2009;39:216–24. doi: 10.1002/eji.200838475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 8.Ishigame H, Kakuta S, Nagai T, Kadoki M, Nambu A, Komiyama Y, et al. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity. 2009;30:108–19. doi: 10.1016/j.immuni.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 9.Iwakura Y, Ishigame H, Saijo S, Nakae S. Functional specialization of interleukin-17 family members. Immunity. 34:149–62. doi: 10.1016/j.immuni.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 10.Kolls JK, Linden A. Interleukin-17 family members and inflammation. Immunity. 2004;21:467–76. doi: 10.1016/j.immuni.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 11.Cua DJ, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol. 10:479–89. doi: 10.1038/nri2800. [DOI] [PubMed] [Google Scholar]
- 12.Takahashi N, Vanlaere I, de Rycke R, Cauwels A, Joosten LA, Lubberts E, et al. IL-17 produced by Paneth cells drives TNF-induced shock. J Exp Med. 2008;205:1755–61. doi: 10.1084/jem.20080588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hirahara K, Ghoreschi K, Laurence A, Yang XP, Kanno Y, O’Shea JJ. Signal transduction pathways and transcriptional regulation in Th17 cell differentiation. Cytokine Growth Factor Rev. 21:425–34. doi: 10.1016/j.cytogfr.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou L, Littman DR. Transcriptional regulatory networks in Th17 cell differentiation. Curr Opin Immunol. 2009;21:146–52. doi: 10.1016/j.coi.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8:942–9. doi: 10.1038/ni1496. [DOI] [PubMed] [Google Scholar]
- 16.Manel N, Unutmaz D, Littman DR. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat Immunol. 2008;9:641–9. doi: 10.1038/ni.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang L, Anderson DE, Baecher-Allan C, Hastings WD, Bettelli E, Oukka M, et al. IL-21 and TGF-beta are required for differentiation of human T(H)17 cells. Nature. 2008;454:350–2. doi: 10.1038/nature07021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vautier S, da Sousa MG, Brown GD. C-type lectins, fungi and Th17 responses. Cytokine Growth Factor Rev. 21:405–12. doi: 10.1016/j.cytogfr.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Al-Salleeh F, Petro TM. Promoter analysis reveals critical roles for SMAD-3 and ATF-2 in expression of IL-23 p19 in macrophages. J Immunol. 2008;181:4523–33. doi: 10.4049/jimmunol.181.7.4523. [DOI] [PubMed] [Google Scholar]
- 20.de Beaucoudrey L, Puel A, Filipe-Santos O, Cobat A, Ghandil P, Chrabieh M, et al. Mutations in STAT3 and IL12RB1 impair the development of human IL-17-producing T cells. J Exp Med. 2008;205:1543–50. doi: 10.1084/jem.20080321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Picard C, von Bernuth H, Ghandil P, Chrabieh M, Levy O, Arkwright PD, et al. Clinical features and outcome of patients with IRAK-4 and MyD88 deficiency. Medicine (Baltimore) 89:403–25. doi: 10.1097/MD.0b013e3181fd8ec3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Glocker EO, Hennigs A, Nabavi M, Schaffer AA, Woellner C, Salzer U, et al. A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N Engl J Med. 2009;361:1727–35. doi: 10.1056/NEJMoa0810719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma CS, Chew GY, Simpson N, Priyadarshi A, Wong M, Grimbacher B, et al. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J Exp Med. 2008;205:1551–7. doi: 10.1084/jem.20080218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Al Khatib S, Keles S, Garcia-Lloret M, Karakoc-Aydiner E, Reisli I, Artac H, et al. Defects along the T(H)17 differentiation pathway underlie genetically distinct forms of the hyper IgE syndrome. J Allergy Clin Immunol. 2009;124:342–8. 8 e1–5. doi: 10.1016/j.jaci.2009.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kisand K, Lilic D, Casanova JL, Peterson P, Meager A, Willcox N. Mucocutaneous candidiasis and autoimmunity against cytokines in APECED and thymoma patients: clinical and pathogenetic implications. Eur J Immunol. 41:1517–27. doi: 10.1002/eji.201041253. [DOI] [PubMed] [Google Scholar]
- 26.Buckley RH. Primary immunodeficiency diseases: dissectors of the immune system. Immunol Rev. 2002;185:206–19. doi: 10.1034/j.1600-065x.2002.18517.x. [DOI] [PubMed] [Google Scholar]
- 27.Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, et al. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med. 2007;357:1608–19. doi: 10.1056/NEJMoa073687. [DOI] [PubMed] [Google Scholar]
- 28.Grimbacher B, Holland SM, Puck JM. Hyper-IgE syndromes. Immunol Rev. 2005;203:244–50. doi: 10.1111/j.0105-2896.2005.00228.x. [DOI] [PubMed] [Google Scholar]
- 29.McGeachy MJ, Cua DJ. Th17 cell differentiation: the long and winding road. Immunity. 2008;28:445–53. doi: 10.1016/j.immuni.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 30.Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, et al. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature. 2008;452:773–6. doi: 10.1038/nature06764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Minegishi Y, Saito M, Nagasawa M, Takada H, Hara T, Tsuchiya S, et al. Molecular explanation for the contradiction between systemic Th17 defect and localized bacterial infection in hyper-IgE syndrome. J Exp Med. 2009;206:1291–301. doi: 10.1084/jem.20082767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Q, Davis JC, Lamborn IT, Freeman AF, Jing H, Favreau AJ, et al. Combined immunodeficiency associated with DOCK8 mutations. N Engl J Med. 2009;361:2046–55. doi: 10.1056/NEJMoa0905506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Engelhardt KR, McGhee S, Winkler S, Sassi A, Woellner C, Lopez-Herrera G, et al. Large deletions and point mutations involving the dedicator of cytokinesis 8 (DOCK8) in the autosomal-recessive form of hyper-IgE syndrome. J Allergy Clin Immunol. 2009;124:1289–302. e4. doi: 10.1016/j.jaci.2009.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chu EY, Freeman AF, Jing H, Cowen EW, Davis J, Su HC, et al. Cutaneous Manifestations of DOCK8 Deficiency Syndrome. Arch Dermatol. doi: 10.1001/archdermatol.2011.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van de Veerdonk FL, Plantinga TS, Hoischen A, Smeekens SP, Joosten LA, Gilissen C, et al. STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N Engl J Med. 365:54–61. doi: 10.1056/NEJMoa1100102. [DOI] [PubMed] [Google Scholar]
- 36.Liu L, Okada S, Kong XF, Kreins AY, Cypowyj S, Abhyankar A, et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med. 208:1635–48. doi: 10.1084/jem.20110958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Diveu C, McGeachy MJ, Boniface K, Stumhofer JS, Sathe M, Joyce-Shaikh B, et al. IL-27 blocks RORc expression to inhibit lineage commitment of Th17 cells. J Immunol. 2009;182:5748–56. doi: 10.4049/jimmunol.0801162. [DOI] [PubMed] [Google Scholar]
- 38.Ramgolam VS, Sha Y, Jin J, Zhang X, Markovic-Plese S. IFN-beta inhibits human Th17 cell differentiation. J Immunol. 2009;183:5418–27. doi: 10.4049/jimmunol.0803227. [DOI] [PubMed] [Google Scholar]
- 39.Oppenheim Y, Ban Y, Tomer Y. Interferon induced Autoimmune Thyroid Disease (AITD): a model for human autoimmunity. Autoimmun Rev. 2004;3:388–93. doi: 10.1016/j.autrev.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 40.Crow MK, Kirou KA, Wohlgemuth J. Microarray analysis of interferon-regulated genes in SLE. Autoimmunity. 2003;36:481–90. doi: 10.1080/08916930310001625952. [DOI] [PubMed] [Google Scholar]
- 41.Dupuis S, Dargemont C, Fieschi C, Thomassin N, Rosenzweig S, Harris J, et al. Impairment of mycobacterial but not viral immunity by a germline human STAT1 mutation. Science. 2001;293:300–3. doi: 10.1126/science.1061154. [DOI] [PubMed] [Google Scholar]
- 42.Dupuis S, Jouanguy E, Al-Hajjar S, Fieschi C, Al-Mohsen IZ, Al-Jumaah S, et al. Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat Genet. 2003;33:388–91. doi: 10.1038/ng1097. [DOI] [PubMed] [Google Scholar]
- 43.Bjorses P, Aaltonen J, Horelli-Kuitunen N, Yaspo ML, Peltonen L. Gene defect behind APECED: a new clue to autoimmunity. Hum Mol Genet. 1998;7:1547–53. doi: 10.1093/hmg/7.10.1547. [DOI] [PubMed] [Google Scholar]
- 44.Kisand K, Boe Wolff AS, Podkrajsek KT, Tserel L, Link M, Kisand KV, et al. Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. J Exp Med. 207:299–308. doi: 10.1084/jem.20091669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pedroza LA, Kumar V, Sanborn KB, Mace EM, Niinikoski H, Nadeau K, et al. Autoimmune regulator (AIRE) contributes to Dectin-1-induced TNF-alpha production and complexes with caspase recruitment domain-containing protein 9 (CARD9), spleen tyrosine kinase (Syk), and Dectin-1. J Allergy Clin Immunol. doi: 10.1016/j.jaci.2011.08.027. [DOI] [PubMed] [Google Scholar]
- 46.Puel A, Cypowyj S, Bustamante J, Wright JF, Liu L, Lim HK, et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science. 332:65–8. doi: 10.1126/science.1200439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hanson EP, Monaco-Shawver L, Solt LA, Madge LA, Banerjee PP, May MJ, et al. Hypomorphic nuclear factor-kappaB essential modulator mutation database and reconstitution system identifies phenotypic and immunologic diversity. J Allergy Clin Immunol. 2008;122:1169–77. e16. doi: 10.1016/j.jaci.2008.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen G, Hardy K, Pagler E, Ma L, Lee S, Gerondakis S, et al. The NF-kappaB transcription factor c-Rel is required for Th17 effector cell development in experimental autoimmune encephalomyelitis. J Immunol. 187:4483–91. doi: 10.4049/jimmunol.1101757. [DOI] [PubMed] [Google Scholar]