

Abstract
Acute lung injury (ALI) is characterized by inflammatory disruption of the alveolar–vascular barrier, resulting in severe respiratory compromise. Inhibition of the intercellular messenger protein, Group V phospholipase A2 (gVPLA2), blocks vascular permeability caused by LPS both in vivo and in vitro. In this investigation we studied the mechanism by which recombinant gVPLA2 increases permeability of cultured human pulmonary endothelial cells (EC). Exogenous gVPLA2 (500 nM), a highly hydrolytic enzyme, caused a significant increase in EC permeability that began within minutes and persisted for >10 hours. However, the major hydrolysis products of gVPLA2 (Lyso-PC, Lyso-PG, LPA, arachidonic acid) did not cause EC structural rearrangement or loss of barrier function at concentrations <10 μM. Higher concentrations (≥ 30 μM) of these membrane hydrolysis products caused some increased permeability but were associated with EC toxicity (measured by propidium iodide incorporation) that did not occur with barrier disruption by gVPLA2 (500 nM). Pharmacologic inhibition of multiple intracellular signaling pathways induced by gVPLA2 activity (ERK, p38, PI3K, cytosolic gIVPLA2) also did not prevent EC barrier disruption by gVPLA2. Finally, pretreatment with heparinase to prevent internalization of gVPLA2 did not inhibit EC barrier disruption by gVPLA2. Our data thus indicate that gVPLA2 increases pulmonary EC permeability directly through action as a membrane hydrolytic agent. Disruption of EC barrier function does not depend upon membrane hydrolysis products, gVPLA2 internalization, or upregulation of downstream intracellular signaling.
Keywords: phospholipase A2, vascular permeability, cytoskeleton, actin, acute lung injury, barrier function
Despite advances in supportive care and ventilator management, the most severe cases of acute lung injury/acute respiratory distress syndrome (ALI/ARDS) continue to cause unacceptably high mortality rates in afflicted patients.[1,2] Because effective pharmacologic intervention for ALI/ARDS is not available,[3,4] improved understanding of the underlying pathophysiology is needed to develop targeted therapies. A critical early step in the pathogenesis of ALI/ARDS is the disruption of the lung vascular endothelial cell (EC) barrier by inflammatory stimuli, leading to pulmonary edema and subsequent respiratory compromise.[5] Endothelial barrier function is primarily regulated by the structural arrangement of the EC actin cytoskeleton linkages to the cell membrane and underlying junctional complexes.[6] Investigations into the mechanisms by which inflammatory signals disrupt EC barrier function therefore provide insights into pathways that potentially may be exploited therapeutically.
Secretory phospholipase A2 (sPLA2) lipolytic enzymes catalyze the cleavage of fatty acids from the sn-2 position of phospholipids[7,8] and have been implicated in the pathogenesis of ALI in both animals[9] and patients.[10,11] At least 10 different sPLA2 enzymes with varying tissue distributions and phospholipase activities have been identified in mammals.[7] Recent data implicate a functional role for the 14 kDa secretory group V PLA2 (gVPLA2) enzyme in ALI pathophysiology. Inhibition of gVPLA2 by specific blocking antibody[12] or pharmacologic inhibition[13] significantly attenuates vascular permeability caused by LPS in mice. In addition, deletion of the gene encoding gVPLA2 in mice (pla2g5-/- knockout) blocks increases in multiple indices of lung injury after LPS.[12] Studies performed in vitro using cultured human pulmonary EC have demonstrated that disruption of the endothelial barrier by LPS can be blocked by inhibition of gVPLA2.[14] Moreover, the extracellular application of recombinant gVPLA2 directly increases permeability of cultured human pulmonary EC.[14] Accordingly, prior studies strongly support an important mechanistic role for gVPLA2 in the development of ALI-associated permeability both in vivo and in vitro. However, the mechanism by which gVPLA2 increases EC permeability remains unclear.
The objective of this present study was to further characterize in vitro the potential pathway(s) responsible for disruption of pulmonary EC barrier function by gVPLA2. We hypothesize that one of three putative mechanisms might account for the development of EC barrier dysfunction caused by gVPLA2: (1) direct outer membrane hydrolysis; (2) secondary effects induced by products of gVPLA2 membrane hydrolysis; and (3) induction of intracellular signaling pathways. It is likely that direct hydrolysis of the EC outer membrane by gVPLA2 physically disrupts its integrity to increase permeability. The second possibility is that the products of membrane hydrolysis generated by gVPLA2 are the primary agents that initiate downstream signaling events that result in EC barrier dysfunction. gVPLA2 activity generates multiple products with potential biologic effects, including free fatty acid, arachidonic acid (AA), lysophosphatidylcholine (lyso-PC), lysophosphatidylglycerol (lyso-PG), lysophosphatidic acid (LPA), and others.[7,8] A final possibility is that gVPLA2 activity at the EC membrane induces intracellular signaling pathways to produce downstream effects (e.g., junctional complex disruption) resulting in barrier dysfunction.
In this study, we now demonstrate that the primary membrane hydrolysis products generated by gVPLA2 do not duplicate the increased permeability caused by gVPLA2 itself in cultured pulmonary EC. In addition, multiple intracellular signaling pathways induced by gVPLA2 in pulmonary EC do not participate in barrier disruption. Thus, our data indicate that gVPLA 2increases pulmonary EC permeability through direct hydrolytic action at the EC membrane and provide mechanistic insights into an important inflammatory signal that participates in the generation of vascular leak during ALI syndromes.
MATERIALS AND METHODS
Reagents
Recombinant human gVPLA2 was purchased from Cayman Chemical (Ann Arbor, Mich.). Arachidonic acid, lyso-PC, lyso-PG, LPA were obtained from Avanti Polar Lipids (Alabaster, Ala.). Pharmacologic inhibitors UO126, SB203580, TFMK, and LY294002 were obtained from EMD Chemicals (Gibbstown, N.J.). Heparinase I was obtained from Sigma-Aldrich Chemical (St. Louis, Mo.). Antibodies were obtained as follows: pan-ERK, phospho-ERK, pan-p38, phospho-p38, pan-AKT, phospho-AKT, pan-gIVaPLA2, phospho-gIVaPLA2 from Cell Signaling (Beverly, Mass.), mouse anti-VE-cadherin antibody (Santa Cruz Biotechnology, Santa Cruz, Calif.). Texas-Red phalloidin was obtained from Invitrogen (Carlsbad, Calif.). All other reagents were obtained from Sigma unless otherwise noted.
Cell culture
Human pulmonary artery endothelial cells (HPAEC) and human lung microvascular endothelial cells (HLMVEC) were obtained from Lonza (Walkersville, Md.) and cultured according to the manufacturer's instructions as previously described.[15] EC (Passages 6–9) were grown in Endothelial Growth Medium-2 (EGM-2) at 37°C in a 5% CO2 incubator. The medium was changed 1 day prior to experimentation.
Transendothelial monolayer electrical resistance
EC were grown to confluency in polycarbonate wells containing evaporated gold microelectrodes, and Transendothelial monolayer electrical resistance (TER) measurements were performed using an electrical cell-substrate impedance sensing system (ECIS; Applied Biophysics, Troy, N.Y.) as previously described in detail.[16] TER values from each microelectrode were expressed as normalized resistance and pooled as discrete time points and plotted versus time as the mean±SEM.
Dextran transwell permeability assay
A transendothelial permeability assay was performed as per the manufacturer's instructions utilizing labeled tracer flux across confluent EC grown on confluent polycarbonate filters (Vascular Permeability Assay Kit, ML) as previously described.[14] Briefly, EC on transwell inserts were exposed to gVPLA2 (500 nM) or lyso-PC (1–30 μM) for two hours. FITC-labeled dextran (~40 kDa) was added to the luminal compartment for an additional two hours, and FITC-dextran clearance across the filter to the abluminal compartment was measured by relative fluorescence excitation at 485 nm and emission at 530 nm. Data were expressed as arbitrary fluorescence units.
Immunofluorescence
EC were grown on gelatinized cover slips before exposure to various conditions as described for individual experiments. EC were then fixed in 3.7% formaldehyde for 10 minutes, permeabilized with 0.25% Triton-X100 for five minutes, washed in PBS, blocked with 2% BSA in TBS-T for one hour, and then incubated for one hour at room temperature with the primary antibody of interest. After washing, EC were incubated with the appropriate secondary antibody conjugated to immunofluorescent dyes (or Texas-Red conjugated phalloidin for actin staining) for 1 hour at room temperature. Final washing was performed with TBS-T, and coverslips were mounted using Prolong Anti-Fade Reagent (Invitrogen) and analyzed using a Nikon Eclipse TE2000-s inverted microscope and Adobe Photoshop 7.0.
Immunoblotting analysis
Cultured EC were stimulated with either vehicle control or 500 nM gVPLA2 for 1-15 min at 37°C. Treated EC were subsequently washed with cold Ca2+/Mg-free PBS and lysed with 0.3% SDS lysis buffer containing protease inhibitors (1 mM EDTA, 1 mM PMSF, 1 mM sodium orthovanadate, 1 mM sodium fluoride, 0.2 TIU/ml aprotinin, 10 μM leupeptin, 5 μM pepstatin A). Sample proteins were separated with 4–15% SDS-PAGE gels (Bio-Rad, Hercules, Calif.) and transferred onto Immobilion-P PVDF membranes (Millipore). Membranes were then immunoblotted with primary antibodies (1:500–1000, 4°C, overnight) followed by secondary antibodies conjugated to HRP (1:5000, room temperature, 30 minutes). Protein expression was detected with enhanced chemiluminescence (Pierce ECL or SuperSignal West Dura, Pierce Biotechnology, Rockford, Ill.) on Biomax MR film (Kodak, Rochester, NY). Multiple blots were scanned and quantitatively analyzed using ImageQuant software (v5.2; Molecular Dynamics, Piscataway, N.J.).
Determination of toxicity on human pulmonary artery endothelial cells
To determine the cytotoxic effect of membrane hydrolysis products on EC, propidium iodide staining was assessed as previously described for eosinophils.[17] Aliquots of 0.5 × 106 EC were incubated for 30 minutes at 37°C with 10–50 μM lyso-PC, lyso-PG, LPA, or arachidonic acid in a total volume of 250 μl. Propidium iodide at 5 μg/ml was added to the medium of drug-treated cells, and the cell suspension was immediately analyzed by flow cytometry, or plated for fluorescent imaging. Red fluorescence intensity was determined by flow cytometry on at least 10,000 cells from each sample, and the percentage of stained cells was analyzed using the Cellquest software.
Statistical analysis
Data were expressed as mean ± SEM. Statistical analyses among groups were performed using standard Student's t-test. Statistical significance in all cases was defined at P<0.05.
RESULTS
gVPLA2 membrane hydrolysis products do not induce EC barrier disruption
Prior studies have demonstrated that 0–500 nM recombinant gVPLA2 increases the permeability of cultured microvascular and macrovascular human pulmonary EC in a concentration-dependent fashion.[14] Differential barrier properties of these two classes of EC have been described in some models.[18,19] Our data indicate that gVPLA2 disrupts human lung microvascular EC (HLMVEC) permeability in a qualitative manner similar to macrovascular human pulmonary artery EC (HPAEC). In this study, we chose to use HPAEC for most analyses because the magnitude of disruption induced by gVPLA2 in general is greater than that observed in microvascular cells. To determine the effects of gVPLA2 and individual gVPLA2 hydrolysis products on human pulmonary EC barrier function, we first measured the transendothelial monolayer resistance (TER), a highly sensitive method for obtaining real-time permeability data.[15,16]
As previously observed,[14] 500 nM of gVPLA2 causes a rapid decrease in TER (which correlates with increased permeability and disruption of barrier function) in both HPAEC and HLMVEC by ~30–50% for>10 hours (Fig. 1A-C). In contrast, multiple hydrolysis products known to be produced by gVPLA2 activity at the cell membrane (arachidonic acid [AA], lysophosphatidylcholine [lyso-PC], lysophosphatidylglycerol [lyso-PG], lysophosphatidic acid [LPA])[7,8] did not decrease TER when added individually to HPAEC at concentrations<1 μM (Fig. 1A). Further TER studies revealed that AA, Lyso-PG, and LPA all failed to significantly alter HPAEC barrier function until their concentration was>10 μM (data not shown).
Figure 1.

Effects of gVPLA2 hydrolysis products on EC permeability. Human lung EC were stimulated at time 0 (arrow) and TER measurements taken as follows: (A) HPAEC: vehicle (black), gVPLA2 500 nM (red), 1 μM each of LPA (purple), AA (aqua), lyso-PC (yellow), lyso-PG (green). Mean±S.E. shown for each timepoint. N=3–5 independent experiments. P<0.05 for gVPLA2 vs. all other conditions. (B) HPAEC: vehicle (black), gVPLA2 (red), or lyso-PC 5 μM (yellow), 10 μM (aqua), 30 μM (purple). N=4–5. (C) HLMVEC: vehicle (black), gVPLA2 (red), or lyso-PC 5 μM (yellow), 10 μM (aqua), 30 μM (purple). N=3–8.
Lyso-PC is expected to be the major lysophospholipid produced by gVPLA2 at the cell surface because mammalian cell membranes are enriched in its precursor, PC, and gVPLA2 demonstrates greater hydrolytic activity for PC than other glycerophospholipids.[20] In additional TER studies, at least 30 μM lyso-PC was required to increase permeability in both HPAEC and HLMVEC (Fig. 1B and C). To determine the effects of lyso-PC on EC permeability to larger particles, a transwell assay utilizing labeled dextran (~40 kD) was employed. As previously demonstrated for HPAEC,[14] gVPLA2 (500 nM) significantly increased HLMVEC permeability to dextran as a cumulative measurement after two hours of incubation (Fig. 2). However, lyso-PC did not increase HLMVEC permeability to larger particles at concentrations less than 30 μM (Fig. 2). Interestingly, 30 μM lyso-PC induced dramatically more permeability in HLMVEC as measured by labeled dextran than by TER (Figures 1C and 2). The cause of this quantitative discrepancy is unclear, but it likely relates to the differential cell properties assessed by each technique. The TER assay uses the passage of electrical current across the cell monolayer to estimate permeability. This current can pass via paracellular pathways as in the dextran assay, but it also can transit directly through the cells, or pass between the cells and the underlying matrix.[21] In certain situations, the resistance to current under the cells can dominate the effects of the paracellular compartment so that the overall TER reading reflects this property to a greater extent than the flow of current between the cells,[22] thus potentially leading to a discrepancy with labeled macromolecule assessments of permeability. However, regardless of the assay used, all the data reported here are consistent with the primary observation that lyso-PC produced by gVPLA2 activity is unlikely to be a primary mediator of EC permeability induced by gVPLA2.
Figure 2.
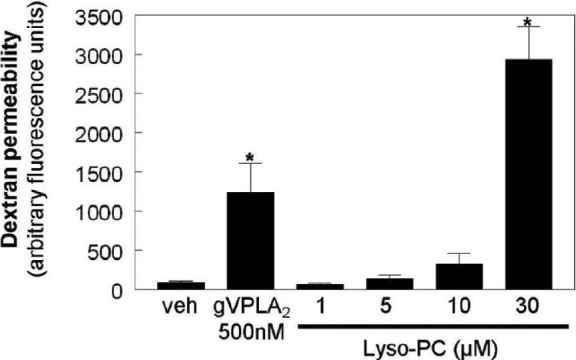
The effects of Lyso-PC on endothelial permeability to labeled dextran. HLMVEC were cultured on polycarbonate filters as described in the Materials and Methods section and then stimulated with vehicle, gVPLA2 (500 nM), or Lyso-PC (1–30 μM) for 2 hours. FITC-labeled dextran was added to the luminal compartment, and clearance across the EC monolayer was assayed after 2 hours by relative fluorescence excitation. Data are expressed as arbitrary fluorescence units. N=3 independent experiments. (*P<0.05 vs. all other conditions).
Immunofluorescent analysis was performed to assess the effects of lyso-PC on EC cytoskeletal structure. Consistent with prior observations,[14] incubation of HPAEC with gVPLA2 (500 nM, 30 minutes) produced increased actin stress fibers, intercellular gap formation, and disruption of peripheral VE-cadherin staining (the major cell-cell junction protein in EC;[23] Fig. 3). These structural changes are known to cause increased permeability in cultured EC.[6] In contrast, HPAEC incubated with lyso-PC at concentrations<30 μM exhibited stable or increased cortical actin staining and intact intercellular VE-cadherin distribution (Fig. 3), a pattern indicative of intact barrier function. Although 30 μM of lyso-PC resulted in some stress fibers and large gap formation between EC, this high concentration produced cell toxicity not seen with gVPLA2 alone. Propidium iodide staining demonstrated significantly increased toxicity in HPAEC after incubation with 30 μM lyso-PC (50% of cells with positive uptake) compared with vehicle controls (20.5%, P<0.05) or those incubated with 500 nM gVPLA2 (24%, P<0.05; Fig. 4). In addition, three other major products of gVPLA2 hydrolysis (Lyso-PG, LPA, and arachidonic acid) induced significant toxicity at concentrations at which they increase HPAEC permeability (30–50 μM; Fig. 5). These findings do not support a role for lyso-PC, lyso-PG, LPA, or arachidonic acid generation by gVPLA2 as a mechanistic contributor to pulmonary EC permeability induced by gVPLA2 in vitro.
Figure 3.

The effects of Lyso-PC on endothelial structure. HPAEC were incubated for 30 minutes with vehicle, recombinant gVPLA2 (500 nM), or Lyso-PC (1–30 μM as indicated) and then fixed and stained for F-actin (red) or VE-cadherin (green) as described in the Materials and Methods section. The F-actin and VE-cadherin images represent the same cell field for each condition. Arrows indicate intercellular gaps or disruption of intercellular VE-cadherin bands. Images are representative of three to four independent experiments.
Figure 4.

High concentrations of Lyso-PC induce EC toxicity. HPAEC were incubated with vehicle, gVPLA2 500 nM, or Lyso-PC 10–50 μM for 30 minutes and then incubated with propidium iodide as per the Materials and Methods section. (A) Representative bright field (top) and fluorescent (bottom) images are shown. The same cell field is depicted for each image pairing (20× magnification). Red indicates propidium iodide incorporation. (B) Propidium iodide incorporation was quantified by flow cytometry as described in the Materials and Methods section. Mean±S.E. shown. N=3 independent experiments. *P<0.05 for Lyso-PC 30–50 μM vs. all other conditions.
Figure 5.

High concentrations of membrane hydrolysis products induce endothelial toxicity. HPAEC were incubated with vehicle, gVPLA2 (500 nM), Lyso-PG, LPA, or arachidonic acid (30–50 μM) for 30 minutes. Propidium iodide incorporation was quantified by flow cytometry as described in the Materials and Methods section. Mean±S.E. shown for each condition. N=3–9 per condition. *P<0.01 vs. vehicle control.
Intracellular signaling pathways are not required for gVPLA2-induced EC barrier disruption
Previous studies in various cell types have demonstrated that gVPLA2 stimulates upregulation of multiple downstream signaling cascades, including the ERK, p38, Akt, and cytoplasmic gIVaPLA2 pathways.[24,25] To determine the effects of gVPLA2 on these pathways in HPAEC, Western blot analyses were performed at various timepoints following gVPLA2 stimulation. Phosphorylated ERK (indicative of ERK activation) was significantly increased within 3 minutes in HPAEC following gVPLA2 stimulation (Fig. 6A), while no activation of p38 or Akt occurs (Fig. 6B and C). Downstream cytoplasmic gIVaPLA2 also was rapidly activated following gVPLA2 (Fig. 6D). Thus, the ERK and gIVaPLA2 pathways are induced in HPAEC by gVPLA2 during the timeframe in which the initiation of permeability occurs (Fig. 1A). Because lyso-PC is the major lysophospholipid produced by gVPLA2, its effects on ERK activation in pulmonary EC were assessed. Lyso-PC (10 μM) rapidly activated ERK in HPAEC within 3–5 minutes, which declined thereafter (Fig. 7). Thus, it is possible that some portion of the ERK phosphorylation induced by gVPLA2 is a result of lyso-PC generation.
Figure 6.

The effects of gVPLA2 on HPAEC intracellular signaling. HPAEC were stimulated with vehicle or gVPLA2 (500 nM) for 1–15 minutes, and then cell lysates were collected and analyzed by Western blotting as described in the Materials and Methods section for expression of total (pan) and phosphorylated ERK (A), p38 (B), Akt (C), and gIVaPLA2 (D). Representative blots for each condition are shown. Bar graphs represent results of densitometric quantification of multiple independent experiments. N=3–7 per condition. *P<0.05 vs. vehicle control.
Figure 7.

The effects of lyso-PC on ERK phosphorylation. HPAEC were stimulated with vehicle or lyso-PC (10 μM) for 1–15 minutes, and then cell lysates were collected and analyzed by Western blotting as described in the Materials and Methods section for expression of total (pan) and phosphorylated ERK. Representative blots are shown. Bar graphs represent results of densitometric quantification of multiple independent experiments. *P<0.05 vs. vehicle control. N=3 independent experiments per condition.
We next determined if pharmacologic inhibition of any of these signaling pathways affected pulmonary EC barrier disruption by gVPLA2. HPAEC were preincubated with UO126 (ERK inhibitor), SB203580 (p38 inhibitor), LY294002 (PI3 kinase inhibitor), or TFMK (gIVPLA2 inhibitor) for 30 minutes prior to stimulation with gVPLA2 . Inhibitor concentrations were selected based upon preliminary experiments demonstrating their effectiveness in blocking agonist-induced activation (Figure 8 demonstrates that 10 μM UO126 prevents ERK phosphorylation by gVPLA2). Pooled data from multiple TER experiments revealed that HPAEC barrier disruption by gVPLA2 is not dependent on activation of any of these pathways (Fig. 8). In addition, immunofluorescent analysis of EC structure demonstrated that ERK inhibition (UO126, 10 μM) did not block the increased actin stress fibers, intercellular gap formation, and disruption of peripheral VE-cadherin produced by gVPLA2 (500 nM, five minutes; Fig. 9). In fact, HPAEC incubated with both UO126 and gVPLA2 exhibited more pronounced disruption of VE-cadherin staining than that caused by gVPLA2 alone (Fig. 9). These results indicate that pulmonary EC permeability induced by gVPLA2 in vitro does not require ERK, p38, Akt, or gIVaPLA2 signaling.
Figure 8.

Effects of intracellular signaling pathways on permeability induced by gVPLA2. Top: HPAEC were incubated with UO126 (0–10 μM) for 30 minutes before stimulation with gVPLA2 (500 nM) for 10 minutes. ERK phosphorylation was determined by Western. Bottom: HPAEC were incubated for 30 minutes with vehicle, UO126 10 μM (ERK inhibitor), SB203580 10 μM (p38), LY294002 25 μM (PI3 kinase), or TFMK 30 μM (gIVPLA2) and then stimulated with gVPLA2. Pooled TER data from multiple independent experiments are expressed as maximal % change in TER from baseline in 5 hours. *P<0.05 for all conditions vs. vehicle. N=4–9
Figure 9.
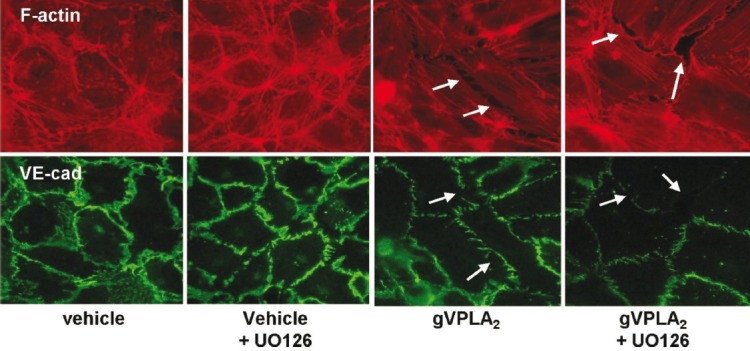
ERK inhibition fails to block the effects of gVPLA2 on endothelial structure. HPAEC were preincubated for 30 minutes with vehicle or UO126 (10 μM). The cells were then stimulated for 5 minutes with either vehicle control or gVPLA2 (500 nM) and then fixed and stained for F-actin (red) or VE-cadherin (green) as described in the Materials and Methods section. The F-actin and VE-cadherin images represent the same cell field for each condition. Arrows indicate intercellular gaps or disruption of intercellular VE-cadherin bands. Images are representative of three to four independent experiments.
gVPLA2 internalization is not required for EC barrier disruption
gVPLA2 also enters mammalian cells through heparin proteoglycan binding at the outer surface to act intracellularly on the perinuclear membrane. This activation generates bioactive lipid mediators both by cytosolic gIVaPLA2-dependent[26,27] and gIVaPLA2-independent mechanisms.[28,29] However, internalization of gVPLA2 into EC did not cause increased membrane permeability. HPAEC were preincubated for several hours with heparinase I to remove cell surface heparin sulfate glycosaminoglycans as previously described.[30] To ensure adequate removal of the cell surface heparin sulfate moieties, we used significantly higher concentrations of heparinase I in this study (1–10 units/ml) compared with prior reports (15 mU/ml).[30] Baseline HPAEC TER was not affected by incubation with heparinase I (data not shown). Despite pretreatment with these high concentrations of heparinase I, HPAEC barrier disruption by gVPLA2 was not inhibited (Fig. 10). Rather, there was a trend toward increased barrier dysfunction after gVPLA2 in EC pretreated with the higher concentration of heparinase I (10 units/ml, Fig. 10), suggesting that inhibition of gVPLA2 internalization increases permeability.
Figure 10.
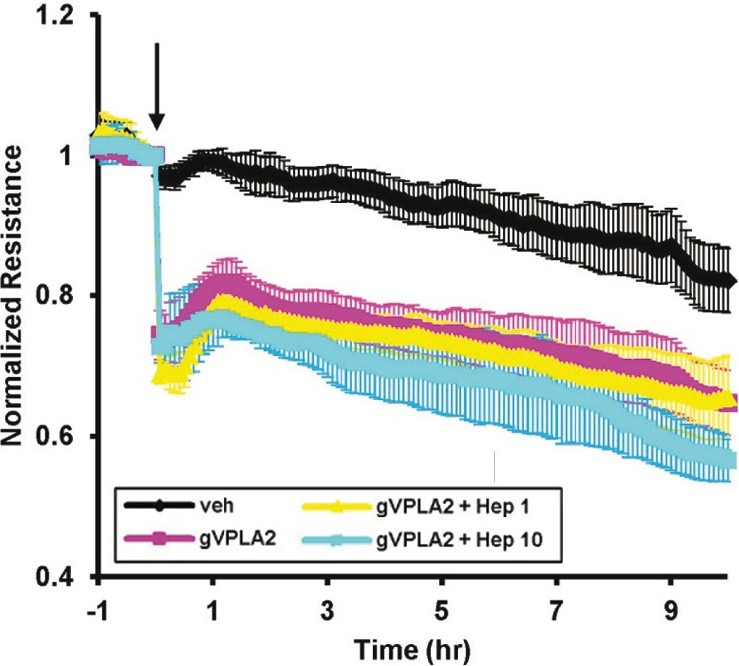
Heparinase fails to attenuate permeability induced by gVPLA2. HPAEC were cultured on gold microelectrodes, and real-time TER measurements were taken as per the Materials and Methods section. EC were preincubated for 3–4 hours with vehicle or heparinase I at the indicated concentrations (1–10 unit/ml). At time 0 (arrow), EC were then stimulated with vehicle or gVPLA2 500 nM as follows: vehicle only (black line), gVPLA2 only (purple line), gVPLA2 + heparinase I 1 unit/ml (yellow line), gVPLA2 + heparinase I 10 units/ml (aqua line). Mean±S.E. shown for each timepoint. N=3–5 independent experiments per condition.
DISCUSSION
The objective of this investigation was to characterize in vitro the pathway responsible for disruption of pulmonary EC barrier function caused by gVPLA2. We have reported previously that inhibition of gVPLA2 by specific monoclonal antibody or genetic deletion of gVPLA2 blocks ALI caused by LPS in mice,[12] while gVPLA2 blocking antibody prevents pulmonary EC barrier disruption induced by LPS in vitro.[14] Animal studies by other groups further support an important role for gVPLA2 in the pathophysiology of ALI. Transgenic mice overexpressing gVPLA2 develop fatal respiratory failure shortly after birth that pathophysiologically resembles ALI.[31] Recent reports using the general sPLA2 inhibitor LY374388 to attenuate LPS-induced ALI in mice[13] or acute cardiogenic pulmonary edema in a mouse model[32] suggest that this protective effect is largely due to gVPLA2 inhibition. Thus, gVPLA2 may be a critical target for therapeutic modulation of pathways responsible for the development of pulmonary edema, and further exploration of its mechanistic effects is warranted.
We have recently reported that the addition of recombinant gVPLA2 to the culture media rapidly induces sustained disruption of EC barrier function in both macrovascular and microvascular pulmonary EC;[14] however, the mechanism by which gVPLA2 produced this effect was not defined. Although the structurally related enzyme, gIIaPLA2, has been suggested to participate in induction of ALI,[33] it did not increase pulmonary EC permeability in vitro.[14] gVPLA2 has much higher affinity than gIIaPLA2 for zwitterionic phosphatidylcholine-rich outer plasma membranes and therefore greater ability to generate free fatty acids and lysophospholipids at these surfaces.[34] gVPLA2 is unique among sPLA2 enzymes in having a dual ability binding to bind cell membranes via two mechanisms, through cationic residues at the C-terminus which bind to cell surface heparan sulfate proteoglycan (like gIIaPLA2), and via direct binding with membrane phosphatidylcholine (like gXPLA2).[34] These properties make gVPLA2 a more effective paracrine agent during inflammation than gIIaPLA2 because the former enzyme exhibits greater transcellular lipolytic activity[35] These differences may explain why attempts to modulate the type gIIaPLA2 form with the relatively specific inhibitor LY315920NA/S-5920 failed to improve mortality in a study of 250 patients with severe sepsis[36] and suggest that gVPLA2 may be a better target for ALI therapy.
Our results now further elucidate and simplify the mechanism by which gVPLA2 causes pulmonary EC permeability in vitro. We investigated the hypothesis that the products of membrane hydrolysis generated by gVPLA2 are the primary agents that initiate subsequent events leading to EC barrier dysfunction. Major products produced by gVPLA2 activity include arachidonic acid (AA), lysophosphatidylcholine (lyso-PC), lysophosphatidylglycerol (lyso-PG), and lysophosphatidic acid (LPA)[7,8] Lyso-PC is expected to be the major lysophospholipid produced by gVPLA2 at the cell surface because mammalian cell membranes are enriched in its precursor, phosphatidylcholine, and gVPLA2 demonstrates greater hydrolytic activity for PC than other glycerophospholipids.[20] Prior work by others has demonstrated increased permeability caused by lyso-PC in human dermal microvascular EC and bovine pulmonary microvessel EC,[37] human coronary artery EC,[38] and human gastric epithelial cells.[39] However, in all these studies,>10 μM lyso-PC was needed to increase permeability.
In our study, neither lyso-PC nor any of the other products studied (AA, lyso-PG, LPA) disrupt pulmonary EC barrier function at concentrations similar to gVPLA2 alone (Fig. 1A). They are able to increase permeability in both macro- and microvascular pulmonary EC only at relatively high concentrations (≥ 30 μM) (Figs. 1 and 2) that are associated with cell toxicity (Figs. 4 and 5). Lyso-PC concentrations in this range previously have been shown to be toxic to eosinophils.[17] Propidium iodide staining demonstrates significantly increased toxicity in HPAEC after incubation with 30 μM lyso-PC (50% of cells with positive uptake) compared with vehicle controls (20.5%, P<0.05) and 500 nM gVPLA2 (24%, P<0.05) (Fig. 4). Moreover, three of the other major products of gVPLA2 hydrolysis (Lyso-PG, LPA, and AA) induce significant toxicity within the concentration range at which they begin to increase HPAEC permeability (30–50 μM) (Fig. 5). Because gVPLA2 alone does not induce cell toxicity at the range of concentrations associated with permeability (Fig. 4), it is unlikely that it generates membrane hydrolysis products sufficient to produce barrier dysfunction.
It previously has been reported that albumin and other serum proteins can bind to Lyso-PC to inhibit its bioactivity in assays of EC permeability.[37] In this study, lyso-PC concentrations as low as 2 μM significantly decreased TER in human dermal microvascular EC when the concentration of albumin in the media was lowered.[37] However, our TER experiments were performed in serum free media without albumin, which eliminates any potential inhibitory effect on lyso-PC bioavailability by these proteins. Therefore, human pulmonary EC appear to be less sensitive to barrier disruption by lyso-PC than human dermal EC. Consistent with this observation, lyso-PC infusion failed to increase pulmonary microvascular permeability (assessed by fluid filtration coefficient (Kf)) in isolated perfused dog lungs.[40]
These data do not support a role for lyso-PC or other gVPLA2 membrane hydrolysis products as important mechanistic contributors to pulmonary EC permeability induced by gVPLA2. However, it is important to consider the limitations of our current findings. Given that membrane hydrolysis by gVPLA2 produces dozens of products,[7,8] the possibility exists that some of these other compounds may participate in barrier disruption. It is not feasible to test every possible hydrolysis product. Accordingly, we chose to focus on those likely generated in the highest concentrations (e.g., lyso-PC).
Previous studies in various cell types have demonstrated that gVPLA2 stimulates upregulation of multiple intracellular signaling cascades, including the ERK, p38, Akt, and cytoplasmic gIVaPLA2 pathways.[24,25] Although our results demonstrate rapid activation of ERK and gIVaPLA2 in HPAEC following gVPLA2 stimulation (Fig. 6), pharmacologic inhibition of these pathways fails to block barrier disruption by gVPLA2 (Fig. 8) or structural cytoskeletal changes (Fig. 9). Although ERK activation occurs in association with multiple barrier-regulatory agonists in EC, this activity is not always necessary for changes in permeability. For example, pharmacologic ERK inhibition attenuates barrier disruption induced by ADAM-15 in HUVEC,[41] but not permeability increases induced by nocodazole in bovine PAEC,[42] despite both agonists producing significant ERK phosphorylation. Differences in cell type and/or agonist-specific activation by other complementary pathways likely account for this variation in ERK effects. Our data do not support a functional role for ERK signaling in pulmonary EC permeability induced by gVPLA2 (Fig. 8). Activation of cytosolic gIVaPLA2 increases intracellular AA levels leading to the production of eicosanoids and other inflammatory signaling molecules,[43] and pharmacologic inhibition of gIVaPLA2 or its genetic deletion partially protects against ALI in mice.[44,45] However, these protective effects in vivo could be due to inhibition of gIVaPLA2 signaling in PMNs and other immune cells, as little data exist that demonstrate a direct role of gIVaPLA2 in EC permeability. Data presented here argue against a functional role for gIVaPLA2 signaling in pulmonary EC permeability induced by gVPLA2 in vitro (Fig. 8).
Although gVPLA2 can enter mammalian cells in a heparin proteoglycan-dependent manner and act intracellularly on the perinuclear membrane to generate additional bioactive lipid mediators through both cytosolic gIVaPLA2-dependent[26,27] and gIVaPLA2-independent mechanisms,[28,29] this cellular uptake does not appear to be necessary for gVPLA2 to cause increased permeability. Removal of EC surface heparin sulfate glycosaminoglycans with high concentrations of heparinase I fails to block HPAEC TER decrease by gVPLA2 (Fig. 10).
Our results suggest that gVPLA2 increases pulmonary EC permeability through direct action at the EC membrane that does not require membrane hydrolysis products, gVPLA2 internalization, or downstream intracellular signaling. We previously have noted increased actin stress fiber formation and adherens junction disruption induced by gVPLA2 in human pulmonary EC.[14] One possibility is that direct membrane action of gVPLA2 results in pore formation and subsequent calcium influx which can result in MLCK and/or Rho activation, both of which can produce stress fiber formation and junctional disassembly.[6] Another potential explanation is that gVPLA2 activity disrupts the cell surface caveolin-enriched microdomains that are necessary for optimal EC barrier function.[46,47] Detailed studies are ongoing to more precisely determine the mechanisms responsible for mediating increased EC permeability caused by gVPLA2 action at the cell membrane.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Erickson SE, Martin GS, Davis JL, Matthay MA, Eisner MD. Recent trends in acute lung injury mortality: 1996-2005. Crit Care Med. 2009;37:1574–9. doi: 10.1097/CCM.0b013e31819fefdf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, et al. Incidence and outcomes of acute lung injury. N Engl J Med. 2005;353:1685–93. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- 3.Bosma KJ, Taneja R, Lewis JF. Pharmacotherapy for prevention and treatment of acute respiratory distress syndrome: Current and experimental approaches. Drugs. 2010;70:1255–82. doi: 10.2165/10898570-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wheeler AP, Bernard GR. Acute lung injury and the acute respiratory distress syndrome: A clinical review. Lancet. 2007;369:1553–64. doi: 10.1016/S0140-6736(07)60604-7. [DOI] [PubMed] [Google Scholar]
- 5.Maniatis NA, Kotanidou A, Catravas JD, Orfanos SE. Endothelial pathomechanisms in acute lung injury. Vascul Pharmacol. 2008;49:119–33. doi: 10.1016/j.vph.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol. 2001;91:1487–500. doi: 10.1152/jappl.2001.91.4.1487. [DOI] [PubMed] [Google Scholar]
- 7.Boyanovsky BB, Webb NR. Biology of secretory phospholipase A2. Cardiovasc Drugs Ther. 2009;23:61–72. doi: 10.1007/s10557-008-6134-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lambeau G, Gelb MH. Biochemistry and physiology of mammalian secreted phospholipases A2. Annu Rev Biochem. 2008;77:495–520. doi: 10.1146/annurev.biochem.76.062405.154007. [DOI] [PubMed] [Google Scholar]
- 9.Edelson JD, Vadas P, Villar J, Mullen JB, Pruzanski W. Acute lung injury induced by phospholipase A 2 .Structural and functional changes. Am Rev Respir Dis. 1991;143(5 Pt 1):1102–9. doi: 10.1164/ajrccm/143.5_Pt_1.1102. [DOI] [PubMed] [Google Scholar]
- 10.Kim DK, Fukuda T, Thompson BT, Cockrill B, Hales C, Bonventre JV. Bronchoalveolar lavage fluid phospholipase A 2 activities are increased in human adult respiratory distress syndrome. Am J Physiol. 1995;269(1 Pt 1):L109–18. doi: 10.1152/ajplung.1995.269.1.L109. [DOI] [PubMed] [Google Scholar]
- 11.Nakos G, Kitsiouli E, Hatzidaki E, Koulouras V, Touqui L, Lekka ME. Phospholipases A2 and platelet-activating-factor acetylhydrolase in patients with acute respiratory distress syndrome. Crit Care Med. 2005;33:772–9. doi: 10.1097/01.ccm.0000158519.80090.74. [DOI] [PubMed] [Google Scholar]
- 12.Muñoz NM, Meliton AY, Meliton LN, Dudek SM, Leff AR. Secretory group V phospholipase A 2 regulates acute lung injury and neutrophilic inflammation caused by LPS in mice. Am J Physiol Lung Cell Mol Physiol. 2009;296:L879–87. doi: 10.1152/ajplung.90580.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sato R, Yamaga S, Watanabe K, Hishiyama S, Kawabata K, Kobayashi T, et al. Inhibition of secretory phospholipase A 2 activity attenuates lipopolysaccharide-induced acute lung injury in a mouse model. Exp Lung Res. 2010;36:191–200. doi: 10.3109/01902140903288026. [DOI] [PubMed] [Google Scholar]
- 14.Dudek SM, Munoz NM, Desai A, Osan CM, Meliton AY, Leff AR. Group V phospholipase A2 mediates barrier disruption of human pulmonary endothelial cells caused by LPS in vitro. Am J Respir Cell Mol Biol. 2011;44:361–8. doi: 10.1165/rcmb.2009-0446OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dudek SM, Jacobson JR, Chiang ET, Birukov KG, Wang P, Zhan X, et al. Pulmonary endothelial cell barrier enhancement by sphingosine 1-phosphate: roles for cortactin and myosin light chain kinase. J Biol Chem. 2004;279:24692–700. doi: 10.1074/jbc.M313969200. [DOI] [PubMed] [Google Scholar]
- 16.Garcia JG, Liu F, Verin AD, Birukova A, Dechert MA, Gerthoffer WT, et al. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Invest. 2001;108:689–701. doi: 10.1172/JCI12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu X, Learoyd J, Butt S, Zhu L, Usatyuk PV, Natarajan V, et al. Regulation of eosinophil adhesion by lysophosphatidylcholine via a non-store-operated Ca2+ channel. Am J Respir Cell Mol Biol. 2007;36:585–93. doi: 10.1165/rcmb.2006-0391OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Troyanovsky B, Alvarez DF, King JA, Schaphorst KL. Thrombin enhances the barrier function of rat microvascular endothelium in a PAR-1-dependent manner. Am J Physiol Lung Cell Mol Physiol. 2008;294:L266–75. doi: 10.1152/ajplung.00107.2007. [DOI] [PubMed] [Google Scholar]
- 19.Wu S, Cioffi EA, Alvarez D, Sayner SL, Chen H, Cioffi DL, et al. Essential role of a Ca2+-selective, store-operated current (ISOC) in endothelial cell permeability: determinants of the vascular leak site. Circ Res. 2005;96:856–63. doi: 10.1161/01.RES.0000163632.67282.1f. [DOI] [PubMed] [Google Scholar]
- 20.Pruzanski W, Lambeau G, Lazdunski M, Cho W, Kopilov J, Kuksis A. Hydrolysis of minor glycerophospholipids of plasma lipoproteins by human group IIA, V and X secretory phospholipases A2. Biochim Biophys Acta. 2007;1771:5–19. doi: 10.1016/j.bbalip.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 21.Lo CM, Keese CR, Giaever I. Impedance analysis of MDCK cells measured by electric cell-substrate impedance sensing. Biophys J. 1995;69:2800–7. doi: 10.1016/S0006-3495(95)80153-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lo CM, Keese CR, Giaever I. Cell-substrate contact: another factor may influence transepithelial electrical resistance of cell layers cultured on permeable filters. Exp Cell Res. 1999;250:576–80. doi: 10.1006/excr.1999.4538. [DOI] [PubMed] [Google Scholar]
- 23.Vestweber D, Winderlich M, Cagna G, Nottebaum AF. Cell adhesion dynamics at endothelial junctions: VE-cadherin as a major player. Trends Cell Biol. 2009;19:8–15. doi: 10.1016/j.tcb.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 24.Kikawada E, Bonventre JV, Arm JP. Group V secretory PLA2 regulates TLR2-dependent eicosanoid generation in mouse mast cells through amplification of ERK and cPLA2alpha activation. Blood. 2007;110:561–7. doi: 10.1182/blood-2006-10-052258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim YJ, Kim KP, Han SK, Munoz NM, Zhu X, Sano H, et al. Group V phospholipase A2 induces leukotriene biosynthesis in human neutrophils through the activation of group IVA phospholipase A2. J Biol Chem. 2002;277:36479–88. doi: 10.1074/jbc.M205399200. [DOI] [PubMed] [Google Scholar]
- 26.Han SK, Kim KP, Koduri R, Bittova L, Munoz NM, Leff AR, et al. Roles of Trp31 in high membrane binding and proinflammatory activity of human group V phospholipase A2. J Biol Chem. 1999;274:11881–8. doi: 10.1074/jbc.274.17.11881. [DOI] [PubMed] [Google Scholar]
- 27.Kim KP, Rafter JD, Bittova L, Han SK, Snitko Y, Munoz NM, et al. Mechanism of human group V phospholipase A 2 (PLA2)-induced leukotriene biosynthesis in human neutrophils.A potential role of heparan sulfate binding in PLA2 internalization and degradation. J Biol Chem. 2001;276:11126–34. doi: 10.1074/jbc.M004604200. [DOI] [PubMed] [Google Scholar]
- 28.Munoz NM, Kim YJ, Meliton AY, Kim KP, Han SK, Boetticher E, et al. Human group V phospholipase A2 induces group IVA phospholipase A2-independent cysteinyl leukotriene synthesis in human eosinophils. J Biol Chem. 2003;278:38813–20. doi: 10.1074/jbc.M302476200. [DOI] [PubMed] [Google Scholar]
- 29.Munoz NM, Meliton AY, Lambertino A, Boetticher E, Learoyd J, Sultan F, et al. Transcellular secretion of group V phospholipase A 2 from epithelium induces beta 2-integrin-mediated adhesion and synthesis of leukotriene C4 in eosinophils. J Immunol. 2006;177:574–82. doi: 10.4049/jimmunol.177.1.574. [DOI] [PubMed] [Google Scholar]
- 30.Dull RO, Dinavahi R, Schwartz L, Humphries DE, Berry D, Sasisekharan R, et al. Lung endothelial heparan sulfates mediate cationic peptide-induced barrier dysfunction: A new role for the glycocalyx. Am J Physiol Lung Cell Mol Physiol. 2003;285:L986–95. doi: 10.1152/ajplung.00022.2003. [DOI] [PubMed] [Google Scholar]
- 31.Ohtsuki M, Taketomi Y, Arata S, Masuda S, Ishikawa Y, Ishii T, et al. Transgenic expression of group V, but not group X, secreted phospholipase A 2 in mice leads to neonatal lethality because of lung dysfunction. J Biol Chem. 2006;281:36420–33. doi: 10.1074/jbc.M607975200. [DOI] [PubMed] [Google Scholar]
- 32.Kawabata K, Fujioka D, Kobayashi T, Saito Y, Obata JE, Nakamura T, et al. Inhibition of secretory phospholipase A 2 activity attenuates acute cardiogenic pulmonary edema induced by isoproterenol infusion in mice after myocardial infarction. J Cardiovasc Pharmacol. 2010;56:369–78. doi: 10.1097/FJC.0b013e3181ef1aab. [DOI] [PubMed] [Google Scholar]
- 33.Kitsiouli E, Nakos G, Lekka ME. Phospholipase A 2 subclasses in acute respiratory distress syndrome. Biochim Biophys Acta. 2009;1792:941–53. doi: 10.1016/j.bbadis.2009.06.007. [DOI] [PubMed] [Google Scholar]
- 34.Balestrieri B, Arm JP. Group V sPLA2: Classical and novel functions. Biochim Biophys Acta. 2006;1761:1280–8. doi: 10.1016/j.bbalip.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 35.Wijewickrama GT, Kim JH, Kim YJ, Abraham A, Oh Y, Ananthanarayanan B, et al. Systematic evaluation of transcellular activities of secretory phospholipases A2. High activity of group V phospholipases A2 to induce eicosanoid biosynthesis in neighboring inflammatory cells. J Biol Chem. 2006;281:10935–44. doi: 10.1074/jbc.M512657200. [DOI] [PubMed] [Google Scholar]
- 36.Zeiher BG, Steingrub J, Laterre PF, Dmitrienko A, Fukiishi Y, Abraham E. LY315920NA/S-5920, a selective inhibitor of group IIA secretory phospholipase A 2 , fails to improve clinical outcome for patients with severe sepsis. Crit Care Med. 2005;33:1741–8. doi: 10.1097/01.ccm.0000171540.54520.69. [DOI] [PubMed] [Google Scholar]
- 37.Huang F, Subbaiah PV, Holian O, Zhang J, Johnson A, Gertzberg N, et al. Lysophosphatidylcholine increases endothelial permeability: Role of PKCalpha and RhoA cross talk. Am J Physiol Lung Cell Mol Physiol. 2005;289:L176–85. doi: 10.1152/ajplung.00003.2005. [DOI] [PubMed] [Google Scholar]
- 38.Yan S, Chai H, Wang H, Yang H, Nan B, Yao Q, et al. Effects of lysophosphatidylcholine on monolayer cell permeability of human coronary artery endothelial cells. Surgery. 2005;138:464–73. doi: 10.1016/j.surg.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 39.Dial EJ, Tran DM, Romero JJ, Zayat M, Lichtenberger LM. A direct role for secretory phospholipase A 2 and lysophosphatidylcholine in the mediation of LPS-induced gastric injury. Shock. 2010;33:634–8. doi: 10.1097/SHK.0b013e3181cb9266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Butler BD, Davies I, Drake RE. Effect of lysophosphatidylcholine on the filtration coefficient in intact dog lungs. Am J Physiol. 1989;257(5 Pt 2):H1466–70. doi: 10.1152/ajpheart.1989.257.5.H1466. [DOI] [PubMed] [Google Scholar]
- 41.Sun C, Wu MH, Guo M, Day ML, Lee ES, Yuan SY. ADAM15 regulates endothelial permeability and neutrophil migration via Src/ERK1/2 signalling. Cardiovasc Res. 2010;87:348–55. doi: 10.1093/cvr/cvq060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Birukova AA, Birukov KG, Gorshkov B, Liu F, Garcia JG, Verin AD. MAP kinases in lung endothelial permeability induced by microtubule disassembly. Am J Physiol Lung Cell Mol Physiol. 2005;289:L75–84. doi: 10.1152/ajplung.00447.2004. [DOI] [PubMed] [Google Scholar]
- 43.Niknami M, Patel M, Witting PK, Dong Q. Molecules in focus: Cytosolic phospholipase A2-alpha. Int J Biochem Cell Biol. 2009;41:994–7. doi: 10.1016/j.biocel.2008.07.017. [DOI] [PubMed] [Google Scholar]
- 44.Nagase T, Uozumi N, Aoki-Nagase T, Terawaki K, Ishii S, Tomita T, et al. A potent inhibitor of cytosolic phospholipase A 2 , arachidonyl trifluoromethyl ketone, attenuates LPS-induced lung injury in mice. Am J Physiol Lung Cell Mol Physiol. 2003;284:L720–6. doi: 10.1152/ajplung.00396.2002. [DOI] [PubMed] [Google Scholar]
- 45.Nagase T, Uozumi N, Ishii S, Kume K, Izumi T, Ouchi Y, et al. Acute lung injury by sepsis and acid aspiration: a key role for cytosolic phospholipase A2. Nat Immunol. 2000;1:42–6. doi: 10.1038/76897. [DOI] [PubMed] [Google Scholar]
- 46.Singleton PA, Chatchavalvanich S, Fu P, Xing J, Birukova AA, Fortune JA, et al. Akt-mediated transactivation of the S1P1 receptor in caveolin-enriched microdomains regulates endothelial barrier enhancement by oxidized phospholipids. Circ Res. 2009;104:978–86. doi: 10.1161/CIRCRESAHA.108.193367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Singleton PA, Dudek SM, Ma SF, Garcia JG. Transactivation of sphingosine 1-phosphate receptors is essential for vascular barrier regulation.Novel role for hyaluronan and CD44 receptor family. J Biol Chem. 2006;281:34381–93. doi: 10.1074/jbc.M603680200. [DOI] [PubMed] [Google Scholar]