

Abstract
Aging is characterized by a progressive loss of muscle mass and muscle strength. Declines in skeletal muscle mitochondria are thought to play a primary role in this process. Mitochondria are the major producers of reactive oxygen species, which damage DNA, proteins, and lipids if not rapidly quenched. Animal and human studies typically show that skeletal muscle mitochondria are altered with aging, including increased mutations in mitochondrial DNA, decreased activity of some mitochondrial enzymes, altered respiration with reduced maximal capacity at least in sedentary individuals, and reduced total mitochondrial content with increased morphological changes. However, there has been much controversy over measurements of mitochondrial energy production, which may largely be explained by differences in approach and by whether physical activity is controlled for. These changes may in turn alter mitochondrial dynamics, such as fusion and fission rates, and mitochondrially induced apoptosis, which may also lead to net muscle fiber loss and age-related sarcopenia. Fortunately, strategies such as exercise and caloric restriction that reduce oxidative damage also improve mitochondrial function. While these strategies may not completely prevent the primary effects of aging, they may help to attenuate the rate of decline.
1. Introduction
Around the fourth decade of life, both muscle mass and strength begin to decline [1], and these declines accelerate with advancing age [2]. The loss of muscle mass occurs at a rate of just under 1% per year [3] and appears to be an unavoidable consequence of aging, although it can be slowed by exercise, especially resistance training [4–6]. A significant concern is that as one ages, changes in muscle mass and strength tend to be dissociated. Data from the Baltimore Longitudinal Study of Aging [7] and the Health ABC study [3] showed using DXA and CT that muscle strength declined three times faster than muscle mass, suggesting a decrease in muscle “quality.” This posits that along with an overall reduction in tissue mass, changes are occurring within the skeletal muscle to affect strength. Changes such as accumulation of intra- and extra-myocellular lipids, improper folding of structural and contractile proteins, and mitochondrial dysfunction are thought to occur with age and are the topic of intense scrutiny [8–10].
Dysfunctional mitochondria in particular are thought to play a key role in muscle function decline, as the mitochondria are the main producers of both cellular energy and free radicals. Alterations in mitochondria have been noted in aging, including decreased total volume, increased oxidative damage, and reduced oxidative capacity. These biochemical and bioenergetic changes are accompanied by perturbations in cellular dynamics, such as a decrease in mitochondrial biogenesis and an increase in mitochondrially mediated apoptosis (Figure 1). These changes may underlie not only a loss of muscle quality with age, but also other common age-associated pathologies such as ectopic lipid infiltration, systemic inflammation, and insulin resistance [10]. In this paper, we examine the evidence for age-related changes in skeletal muscle mitochondria, with a focus on studies conducted in humans.
Figure 1.
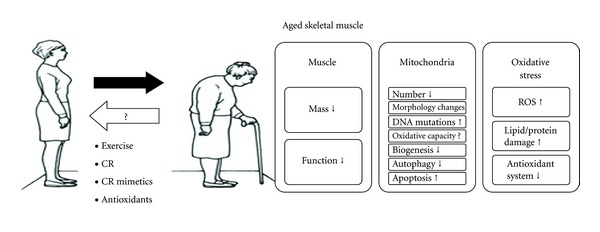
This cartoon describes the changes in skeletal muscle with aging on the right side of the figure. Both the mass and function of skeletal muscle are decreased in elderly people. Furthermore, at the mitochondrial level, the number of mitochondria is decreased in parallel with changes in mitochondrial morphology. Mitochondrial DNA, oxidative capacity, biogenesis, and autophagy are all decreased in conjunction with an increased number of DNA mutations and increased levels of apoptosis. Finally, oxidative stress is increased in the muscles of elderly people in association with cellular lipid, protein, and DNA damage. The bottom left of the cartoon shows that exercise, caloric restriction, caloric restriction mimetics, and antioxidants can all delay the aging of skeletal muscle.
1.1. Overview of Mitochondria
Mitochondria are double-membrane encoded organelles with their own genome that consume oxygen and substrates to generate the vast majority of ATP while producing reactive oxygen species in the process. They also participate in a wide range of other cellular processes, including signal transduction, cell cycle regulation, oxidative stress, thermogenesis, and apoptosis. In doing so, they are highly dynamic organelles that are continuously remodeling through biogenesis, fission, fusion, and autophagy, thus responding to and modulating cellular dynamics. For instance, they undergo biogenesis to meet increased energy demands in response to exercise, and they ensure cellular quality by initiating an apoptotic program to remove defective cells.
Mitochondria transduce energy from substrates through the tricarboxylic acid (TCA) cycle and the electron transport system (ETS) to generate ATP. The ETS consists of multipolypeptide complexes (I–V) embedded in the inner mitochondrial membrane (IMM) that receive electrons from reducing equivalents NADH and FADH2, generated by dehydrogenase activity in the TCA cycle. The electrons are transferred along the complexes with O2 serving as the final acceptor at complex IV [11] (Figure 2). The reduction potential (propensity to accept an electron) increases along the chain of complexes, and the energy generated is sufficient to drive the translocation of hydrogen ions across the IMM. This creates a proton gradient and membrane potential (collectively termed the proton motive force) that drives the synthesis of ATP as protons flow back to the matrix via complex V (ATP synthase). This process is also called oxidative phosphorylation (OXPHOS). However, the ETS is not a perfectly efficient system, and significant (and highly variable) proton “leak” occurs by the movement of hydrogen ions back into the matrix space that is not mediated through complex V. In this manner, proton transfer can be uncoupled from ATP phosphorylation, and this inefficiency contributes to the demand for reducing equivalents. Mitochondria are therefore thought to have a much greater capacity to generate ATP than what is usually required [12].
Figure 2.

Mitochondrial processes are both static (a) and dynamic (b). (a) depicts the classical movement of electrons along complexes I–IV embedded in the inner mitochondrial membrane with the generation of a proton gradient (membrane potential). The proton gradient causes hydrogen ions to flow back into the mitochondrial matrix via complex V (ATP synthase), producing ATP in the process. (b) depicts the processes of mitochondrial fusion and fission. Mitochondria can undergo constriction and division (1), mediated by Drp1, which bonds and localizes to the constriction site via an interaction with the receptor-like protein Fis1 (2). During fusion, a tether of the Mfn1/2 to collateral mitochondrial Mfn1/2 conjoins the outer membranes. Opa1, an inner membrane GTPase protein, facilitates the fusion of the inner membrane, cristae formation, and unifying of compartments.
Because of this metabolic latitude, many assume that mild impairment in mitochondrial function per se does not cause cellular disturbances associated with aging and with chronic diseases such as insulin resistance and type 2 diabetes [12]. Indeed, whether mitochondrial dysfunction is a cause or a consequence of cellular impairment and the aging process is a subject of intense debate. Moreover, the definition of mitochondrial dysfunction itself has been the subject of controversy. For example, alterations in mitochondrial mRNA transcripts may not result in changes in protein levels, so it is not wholly clear whether this state—whether compensatory or not—can be counted as a disruption in normal function [13].
In this paper, we focus on the aging of skeletal muscle (SM) mitochondria, with an eye towards the putative role of mitochondria in SM aging. We look at several different aspects of mitochondrial function—which we define to mean any process that involves the mitochondria—and explore the evidence for dysfunction, which we define to mean any deviation from normal function. Section 2 examines the impact of aging on the biochemical and bioenergetic pathways in SM mitochondria. Section 3 explores the changes in mitochondrial and cellular dynamics, which may further exacerbate the age-related decline in muscular function. The review concludes in Section 4 with an overview of strategies to attenuate the aging of SM mitochondria.
2. Biochemical and Bioenergetic Aging of Mitochondria
During the aging process, mitochondria are characterized by changes in oxidative stress, a decay in mitochondrial DNA, a reduction in some enzyme activities, and alterations in mitochondrial respiration. These biochemical changes are accompanied by phenotypic changes in the mitochondria themselves. In old age, a significant proportion of the mitochondrial organelles are abnormally enlarged and more rounded in shape (reviewed in [14, 15]). An increasing proportion of them are also depolarized or nonfunctional, perhaps indicative of defects in mitochondrial turnover [15–17]. Within the mitochondria themselves, shortened mitochondrial cristae and vacuolization of the matrix are apparent, which lead to homogenization of the materials within the mitochondrial compartments [14, 18]. Coupled with these morphological changes, the density of mitochondria in SM substantially drops [13, 19–21], as shown for example, by electron microscopy in the vastus lateralis muscle of people over 60 years of age [20].
2.1. Increased Oxidative Damage: ROS Production and Scavenging
Within the cell, the mitochondria constitute the major source of reactive oxygen species (ROS). Mitochondrial complexes I and III are the main sites of superoxide generation and contribute the most to ROS production [22]. The reactive oxygen species, including O2 ∙− and H2O2, can cause oxidative damage to surrounding structures and the particularly vulnerable mtDNA, which is in close proximity to the primary site of ROS production. Oxidation by ROS results in the synthesis of faulty proteins, oxidized lipids, and mtDNA mutations, which may lead to cellular and mitochondrial dysfunction. These processes are implicated in the mitochondrial theory of aging [23, 24], which holds that the accumulation of ROS damage over time leads to age-associated mitochondrial impairment. In general, ROS production is found to be increased in aged muscle in both the subsarcolemmal and intermyofibrillar pools of mitochondria [25] (though some report a decrease in ROS production with age [26]). This increase in ROS production is associated with oxidation of ETS complex V, leading to decreased ATP production [27, 28], increased levels of 8-oxodeoxyguanosine (8-oxoG) from DNA oxidation [25, 29], increased levels of protein carbonyls, and increased nitration [30]. In particular, proteomic studies have found increased nitration of complex II and altered carbonylation of complex I, complex V, and isocitrate dehydrogenase (reviewed in [31]).
Cumulative oxidative damage may in part be attributed to a reduction in ETS activity that would extend the length of time that electrons remain at complexes I and III, increasing the potential for donation of electrons to oxygen [32]. In theory, it could also be attributed to reduced activity of antioxidant defenses, including manganese superoxide dismutase (MnSOD), catalase (CAT), and glutathione peroxidase (GPx). These enzymes work together to convert O2 ∙− to H2O2, which is then further reduced to H2O. Data on antioxidant enzyme activity and aging is mixed, with some studies showing increased activity [33–35] while others show reduced enzyme activity [36–38]. We recently found in a group of elderly adults aged 70–84 years that the expression of total superoxide dismutase (SOD) was unchanged compared to that of a group of young (20–34 years old) BMI-matched individuals; however, urinary isoprostanes (a marker of whole-body oxidative damage) was increased by 38% [10]. Furthermore, TCA cycle activity measured as citrate synthase (CS) activity was 19% lower, and mitochondrial capacity was 17% lower in the elderly, although mitochondrial content (OXPHOS protein) was unchanged. Safdar et al. recently showed that the protein content of MnSOD (mitochondria-specific) was significantly reduced in active and inactive older adults compared to their younger counterparts; however, MnSOD activity in the older subjects who were recreationally active was similar to that of young subjects [39]. Together, these data suggest that oxidative stress is increased with age without an accompanying increase in antioxidant activity and may be linked with mitochondrial dysfunction. Exercise and caloric restriction may alleviate the age-associated oxidative stress by upregulating antioxidant enzyme activity even in the face of reduced antioxidant protein content (Figure 3).
Figure 3.

The electron transport system (ETS) of the inner mitochondrial membrane is the primary site of reactive oxygen species (ROS) production and therefore the main source of oxidative stress (damage to proteins, lipids, and DNA) in the mitochondria and in the cell. Free radical superoxide anions (O2 ∙−) are generated when electrons are donated from complexes I and III of the ETS to O2 instead of the appropriate ETS subunit. 2–4% of total oxygen consumption may go toward the production of ROS instead of energy as ATP. Scavenging enzymes represent an important mitochondrial defense mechanism against oxidative stress by neutralizing O2 ∙− within the mitochondrial matrix (superoxide dismutase; MnSOD = SOD2) and catalyzing the reduction of mitochondrial SOD2-generated H2O2 to nontoxic H2O in the mitochondria and the cell (glutathione peroxidase and catalase). Mitochondria in young muscle (a) are numerous and efficient. With age (b), muscle mitochondria become less numerous and seem to develop impaired function associated with reduced oxidative capacity. Through lifestyle changes such as exercise, and caloric restriction, and caloric restriction mimetics, we hypothesize that antioxidative enzymes are upregulated, and that most of the above impairments in aged muscle may be improved (c).
If oxidative stress contributes to mitochondrial dysfunction, can mitochondrial dysfunction be reversed by changing the cellular oxidative status? A recent study attempted to answer this question using a transgenic mouse model to express human catalase targeted to the mitochondria (MCAT) [40]. Mitochondrial function was assessed both in vitro and in vivo in wildtype young (3–6 months old) and older (15–18 months old) lean healthy mice versus age-matched MCAT transgenic littermates. Whereas the older wildtype mice exhibited all the “usual” deleterious metabolic impairments including increased oxidative damage, reduced mitochondrial content (~30%) and function, increased intramuscular lipid (~70%), and marked muscle insulin resistance (~35%), the older MCAT mice resembled the young mice and demonstrated none of the age-associated impairments. These data suggest that by increasing ROS scavenging and reducing the oxidized state of the cell, many age-related deficits can be prevented.
2.2. Increased Uncoupling?
Another mechanism to reduce oxidative stress may be mediated by attenuation of the proton motive force, thereby decreasing the potential for electrons to form oxygen radicals. This could be accomplished by increasing respiration or by increasing proton leak. Rodent studies on age-associated changes in proton leak are mixed. Iossa et al. [41] showed that proton leak was decreased with age, resulting in increased mitochondrial efficiency. Conversely, reduced coupling was found in aged mice using an in vivo approach to measure the P/O ratio [42]. Data in aged humans are scarce. We recently found that the in vivo P/O ratio determined by ATP turnover (demand) and oxygen uptake of the vastus lateralis was 21% lower in elderly compared to young adults [10], suggesting that less ATP was produced per oxygen uptake in the elderly adults. This lends support to earlier work by Amara et al. [43] showing that mild uncoupling protects against age-related declines in mitochondrial function and cellular ATP concentration. Furthermore, we found that a lower P/O ratio was associated with lower SOD activity, indicating a possible link between cellular antioxidant activity and mitochondrial coupling. In particular, increased oxidative stress with nonupregulated antioxidant defense may induce mitochondrial uncoupling in order to reduce the oxidative potential. This potential adaptive mechanism should be further explored.
2.3. Decay of Mitochondrial DNA
Age appears to affect both mitochondrial DNA (mtDNA) content and integrity. Mammalian cells typically contain on the order of 1,000–10,000 copies of mtDNA, which code for less than 10% of all mitochondrial proteins; the rest is encoded by nuclear DNA [44, 45]. At 16.6 kbp long, each double-stranded circular mtDNA molecule encodes 13 genes involved in the ETS (subunits of complexes I, III, IV, and V), 22 tRNAs, and 2 rRNAs [46]. A majority of studies [47–50], though not all [51], report that mtDNA copy number decreases with age in human SM and may be caused at least in part by oxidative damage, as the age-associated decline in mtDNA copy number tends to be greater in more oxidative fibers [52].
The decline in mtDNA is also accompanied by an increase in mtDNA damage, particularly deletions and oxidative lesions, but also point mutations, tandem duplications, and rearrangements (reviewed in [53, 54]; in particular [36–38, 48, 55–64]). mtDNA is particularly susceptible to oxidative damage because it is near the major site of ROS production, lacks protective histones, and has weaker DNA repair mechanisms [65, 66]. One study found that deletions affect up to 70% of mtDNA molecules in the SM of individuals aged 80 years and older [56]. However, there are disagreements over mtDNA mutation frequencies (reviewed in [54]) and to what extent ROS production is responsible; there is increasing evidence that perhaps a majority of mutations are due to the inherent error rate of mtDNA polymerase gamma (Pol gamma) [67].
The functional impact of mtDNA damage is still a matter of debate both in general (reviewed in [54, 67, 68]) and in the context of sarcopenia (reviewed in [69]). Is mtDNA damage a consequence or a cause of the aging process? On one hand, the onset of a mitochondrial decline in energy production occurs before mtDNA mutations are often detectable [70]. On the other hand, several studies in humans reveal strong correlations between mtDNA mutation rates and bioenergetic deficiency (typically complex IV) or muscle fiber atrophy [43, 55, 62, 63, 71–74]. In addition, specific point mutations cluster at mtDNA replication control sites, which may reduce gene transcription and be at least partially responsible for declining levels of protein synthesis with age [60, 75]. Other evidence comes from mtDNA mutator mice, which are genetically engineered to have defects in the proofreading function of Pol gamma. They accumulate mtDNA mutations at an accelerated rate, have more abnormal mitochondria, and exhibit premature aging, sarcopenia, and reduced lifespan [76–78].
2.4. Alterations in Mitochondrial mRNA and Protein Levels
The expression of most mitochondrial genes, including cytochrome c [79], does not change with age. However, declines in gene transcripts are observed for several of the polypeptide components of complexes I, IV, and V, whereas for complexes II and III, declines are evident for only a couple components with most being unchanged [73, 80–84]. In addition, the transcripts of some nuclear genes encoding mitochondrial regulatory proteins, a few TCA proteins (including one transcript variant of citrate synthase), and some glycolysis enzymes appear to be altered in aged human SM [48, 52, 80, 83, 84]. Many similar changes in gene expression (or lack thereof) have been observed in monkeys and rats [52, 85–87]. However, there are discrepancies among reports, which are at least in part explained by muscle-specific differences in gene expression [52, 88]. Whether these alterations in gene transcripts of energy-producing pathways affect muscle aging is unclear. While much evidence points to a functional role, Giresi et al. found that the key genes associated with sarcopenia are involved in inflammation, apoptosis, and regulating mRNA splicing, not in OXPHOS [89].
Of course, age-related alterations in mRNA levels may not induce similar changes in protein abundances, which are controlled by the balance between synthesis and degradation [13, 90]. Both mitochondrial protein synthesis [75] and proteolysis via ubiquitin-proteasome and lysosomal degradation systems (reviewed in [91, 92]) are known to decline with age. The majority of mitochondrial proteins, including polypeptide components of the ETS complexes, have not been found to change with age [10, 93]. Notable exceptions include complex II, which tends to increase with age at least in rodents [86, 94, 95], and complex IV, which tends to decrease with age in both humans [50, 96, 97] and rodents [86, 98]. This is supported by observations of increased complex IV-deficient and complex II-hyperactive fibers with age [99–101]. For complexes I, III, and V, proteomic analysis shows that the occurrence of abnormal polypeptides tends to increase with age in both human [102] and rodent SM [86, 94, 98, 103], but is decreased in human studies using other approaches [48, 50, 96]. Proteins involved in glycolysis tend to be unchanged or reduced with age, which is consistent with the observed shift from glycolytic to more oxidative metabolism, while the data on TCA proteins (in particular, citrate synthase and isocitrate dehydrogenase) is mixed [48, 50, 86, 94–96, 98, 102, 103].
2.5. A Decline in Mitochondrial Energy Production?
It is unclear whether these age-related changes in turn affect mitochondrial energy production. When physical activity is not a criterion in subject selection, substantial declines in enzyme activity are often found to occur with age. Most such studies in human SM report that complex I and complex IV activities decrease substantially, perhaps because these two complexes have more of their subunits encoded by the more vuinerable mtDNA than the other complexes [1, 21, 56, 58, 75, 79, 90, 97, 104, 105]. Similar results have been reported in rodents and dogs [2, 5, 52, 106–112], though there have been exceptions both in humans [93, 113, 114] and rodents [86, 94, 109]. The activity of complex II, which is encoded entirely by the nuclear genome, appears not to change with age in either human [1, 56, 64, 113, 114] or animal [2, 86, 108] SM, with a few exceptions [39, 79, 94, 105]. Data regarding complex III activity is both less available and more mixed, with some studies reporting no change in human [113, 114] or animal [2, 108] SM, but others reporting a decrease in activity [5, 56]. Interestingly, one study found evidence of an age-related decline in complex III activity in females but not males [90]. Finally, the activities of enzymes involved in the TCA cycle and glycolytic pathways tend to be unchanged or decline with age, and in some cases (e.g., citrate synthase), the trend is not clear [10, 21, 26, 39, 48, 56, 75, 93, 96, 97, 104, 105, 111–113, 115–120]. Some of these discrepancies may be explained by the differential impact of aging on different muscles. For example, one study in rats found a decrease in complex IV activity in the lateral but not medial gastrocnemius [52], and a study in humans found that citrate synthase activity was negatively correlated with age in the gastrocnemius but not the vastus lateralis [121]. Other inconsistencies may be explained by differences in isolation or techniques, or in the normalization of enzymatic activity. For example, Picard et al. found that measurements on isolated mitochondria tend to exaggerate the declines in mitochondrial function in comparison to measurements in intact mitochondria in permeabilized myofibers [86].
Enzymatic changes may in turn affect mitochondrial respiration and ATP flux. Short et al. found that the maximal capacity for ATP synthesis drops by 8% per decade or 5% when normalizing to mitochondrial protein content [48]. Similar results concerning mitochondrial respiration were reported by Trounce et al. [122]. But other human studies have found no evidence of a decline in mitochondrial respiration [26, 93, 115, 117], and there is no consistent pattern regarding how the respiratory control ratio—the ratio of states III and IV respiration activities—changes with age. Similarly, there are conflicting reports on fatty acid-linked mitochondrial respiration including reduced carnitine palmitoyltransferase-dependent and -independent pathways with age (particularly nonphosphorylating respiration [48, 86, 108]), while others see no age-related changes [26, 115, 117]. In vivo techniques have also been used to measure oxidative capacity. In vivo measurements of SM oxidative activity are typically performed using phosphorous magnetic resonance spectroscopy (31P-MRS) to probe the kinetics of phosphocreatine and its recovery time following muscle contraction. Some in vivo studies in older humans [10, 20, 118, 120], but not all [119, 123, 124], and at least one in mice [125] have found evidence of reductions in maximal ATP flux. In particular, Conley et al. found that elderly adults have a 50% reduction in oxidative capacity per muscle volume and a 30% reduction per mitochondrial volume [20]. An MRS study on basal ATP flux in humans, however, found no decline in flux with aging [43].
However, it has become increasingly clear that most of the declines in mitochondrial function attributed to chronological age are instead a result of physical inactivity. When physical activity levels are matched between young and old subjects or physical activity is otherwise taken into account, most studies find no age-related changes in mitochondrial enzyme activities, mitochondrial respiration, or ATP flux [13, 39, 50, 101, 126–129]. Interestingly, those studies that do report age-related declines even after matching for activity levels tend to involve sedentary young and old subjects [104, 114, 130], indicating that declines in function with aging may occur predominantly in sedentary individuals. In particular, in ex vivo studies, the activities of ETS enzymes and citrate synthase and mitochondrial respiration show strong dependences on physical activity level that are independent of age [101, 131, 132]. Similar results are also reported in mice [98, 133]. In vivo MRS studies on activity-matched subjects also tend to find no evidence of a change in mitochondrial oxidative capacity—in this case, maximal ATP flux—between young and old subjects [134–136]. However, one in vivo MRS study in activity-matched sedentary individuals did report a reduction in basal oxidation and phosphorylation [130]. Also, glycolytic flux is lower in older activity-matched people [135, 136], and a recent MRS study showed that oxidative capacity does indeed change in some muscles with age but also demonstrated that physical activity is intimately linked with oxidative capacity [128]. Interestingly, there is evidence that physical activity can reverse the age-related declines in most but not all mitochondrial markers of energy production [50, 132], which while encouraging, nonetheless indicates that there are residual declines in a small subset of markers that cannot be completely prevented.
3. Age-Related Changes in Mitochondrial Dynamics
3.1. Decreased Mitochondrial Biogenesis
Once thought of as relatively static round organelles, mitochondria are now recognized as highly dynamic, existing in networks that are constantly being remodeled by biogenesis, fusion and fission, and degradative processes such as autophagy. Through these dynamics, they both respond to and drive cellular processes, including apoptosis, whose dysregulation is thought to be a key factor in sarcopenia.
Mitochondrial biogenesis is the expansion of existing mitochondrial content—whether through growth of the mitochondrial network (increase in mitochondrial mass) or division of preexisting mitochondria (increase in mitochondrial number; also discussed later in this paper). It is triggered when the energy demand exceeds respiratory capacity—in particular, in response to exercise, stress, hypoxia, nutrient availability, hormones (including insulin), ROS production, and temperature (reviewed in [137, 138]). Once biogenesis is triggered, the nuclear genome produces mitochondrial regulatory factors, which are then imported into the mitochondria to initiate replication and transcription of mtDNA, ultimately resulting in expansion of the mitochondrial network. The initial triggers are thought to arise from signaling cascades involving the energy sensor AMP kinase (AMPK) and/or alterations in Ca2+ flux and protein kinases such as calcium/calmodulin-dependent kinases (CAMKs), protein kinase C (PKC), and p38 MAPK (particularly in response to muscle contraction) (reviewed in [138]). These signals in turn induce the expression of the peroxisome proliferator-activated receptor (PPAR) gamma coactivator (PGC-1) family of cotranscription factors, particularly PGC-1α, which is also directly activated by AMPK [139] and p38 MAPK [140–142], as well as many key signaling molecules like SIRT1 [143, 144] and transducers of regulated CREB (cAMP response element-binding protein)-binding proteins (TORCs) [145].
As the master regulator of biogenesis, PGC-1α coordinates and cooperates with multiple cotranscription factors—including PPARs, myocyte enhancing factors (MEFs), and CREB—to induce the transcription of nuclear genes encoding mitochondrial proteins [146, 147]. Most importantly, PGC-1α activates the nuclear respiratory factors 1 and 2 (Nrf-1, Nrf-2) on the promoters [148, 149], thereby driving the transcription of an even greater assortment of nuclear-encoded mitochondrial proteins (reviewed in [137, 138, 150]), including mitochondrial transcription factor A (Tfam). Tfam controls mtDNA transcription and content and organizes mtDNA into nucleoid-like structures, which are thought to maintain mtDNA integrity [151]. The Tfam precursor protein, along with numerous other mitochondrial precursor proteins, are targeted to the mitochondria by chaperones and then imported via the mitochondrial protein import machinery (reviewed in [152–155]). Import is accomplished through a set of aqueous pores formed from translocases of the outer membrane (TOMs; e.g., Tom20) and translocases of the inner membrane (TIMs) through pathways that depend on whether the polypeptide is destined for the outer mitochondrial membrane (OMM), IMM, or mitochondrial matrix. Once imported, the precursor proteins are modified, folded, and assembled into their final form. In particular, Tfam can then act on mtDNA driving its replication through Pol gamma and its transcription through mitochondrial RNA polymerase [156]. Finally, the translated ETS polypeptides from the nuclear and mitochondrial genomes are assembled into multisubunit enzymes.
With increasing age, the density of mitochondria in SM drops substantially [13, 19–21, 25], suggesting an overall decline in mitochondrial biogenesis. Moreover, there is an impairment in AMPK-stimulated biogenesis in old age [157]; however, the reasons why are largely unknown. Declining PGC-1α levels could explain the reduction in mitochondrial biogenesis, as overexpression of PGC-1α in the SM of aged mice improved oxidative capacity, suppressed mitochondrial degradation, and prevented muscle atrophy [158]. These improvements were accompanied by attenuation of the age-related increase in inflammatory cytokines and by prevention of insulin resistance [158]. However, measurements of PGC-1α in aged SM are not definitive: one study found a decline in protein abundance in rats [25], and some studies of gene expression have reported reduced expression [96, 112, 157], while others found no change [10, 159]. Similarly, reports on the gene and protein expressions of both Nrf-1 and Tfam are conflicting [25, 50, 80, 96, 159, 160]; however, preliminary evidence suggests that Nrf-1 binding to the Tfam promoter appears to increase in old age [160].
3.2. Changes in Fission and Fusion?
Mitochondrial dynamics are also influenced by the balance between fission and fusion (Figure 2(b)). Fission, or division, of mitochondria is required to transmit mitochondria among dividing cells and to meet increased ATP demands. Fission also plays a key role in maintaining mitochondrial quality and mtDNA integrity, as it allows dysfunctional mitochondria to be severed from the network and to be removed by autophagy [161, 162]; indeed, mitochondria excised by fission often have a lower membrane potential, a target for autophagy [16]. In mammals, fission is known to be orchestrated by two proteins: Fis1 and the GTPase dynamin-related protein Drp1. Fis1, which localizes in the OMM, recruits the cytosolic protein Drp1 [162, 163]. Once recruited to the OMM, Drp1 wraps around and constricts the membrane, initiating fission (reviewed in [164]). One study found that activation of the fission machinery was sufficient to induce muscular atrophy [165], while another study found that in yeast, increased fission leads to a shortened lifespan and a greater sensitivity to ROS-induced apoptosis [166]. Because Fis1 protein levels are elevated in aging rat SM [94] and Drp1 transcripts tend to be lower in older humans [21], age-associated increases in fission may indeed contribute to sarcopenia. On the other hand, suppression of Fis1 or Drp1 produces elongated mitochondrial networks and a senescent phenotype but increases ROS production and mtDNA damage [163, 167, 168].
The counterpart of fission is fusion, which unifies mitochondria. Fusion allows mixing of mitochondrial compartments, facilitating equilibration of OXPHOS proteins, energy exchange, and complementation of the mitochondrial genome [169, 170]. In aging, this mixing is thought to prevent mutations in mtDNA from resulting in respiratory dysfunction; however, it also permits the accumulation of mutated mtDNA that might otherwise be removed via fission and autophagy [170, 171]. For these reasons, fusion plays a role in regulating mtDNA integrity and respiratory function [172–174]. In humans, fusion is known to be controlled by optic atrophy 1 (Opa1) and the two GTPases mitofusin 1 and 2 (the isoforms Mfn1 and Mfn2). Mfn1 and Mfn2 are located in the OMM, where they promote tethering and fusion [162], while Opa1 facilitates fusion from its localization on the IMM (reviewed in [164]). Opa1 also assists in regulating degradative processes: it regulates apoptosis by keeping the inner mitochondrial cristae junctions tight to prevent cytochrome c release, which triggers apoptosis [175, 176], and its cleavage may be involved in flagging dysfunctional mitochondria for autophagic removal [176]. One study reported that Mfn2 gene expression was lower in the SM of older humans [21]. Interestingly, muscle-specific Mfn1- and Mfn2-knockout mice experience enhanced mitochondrial proliferation and increased mutations in and depletion of mtDNA; these changes occur in parallel with accelerated muscle loss [177]. A mutation in the other key fusion protein, Opa1, is associated with reduced oxidative phosphorylation and ATP production in human SM [178]. Thus, age-associated changes in the dynamical remodeling processes of fission and fusion likely affect mtDNA integrity, respiratory function, ROS production, and cellular senescence.
3.3. Alterations in Mitochondrial Turnover
Mitochondrial turnover is executed predominantly by the autophagy-lysosome system, a cellular housekeeping system that degrades mitochondria as well as other cellular components. Removal of mitochondria through the autophagy-lysosome system is known as “mitophagy.” In mitophagy, dysfunctional mitochondria are recognized and engulfed in a double-membrane structure called a phagophore or preautophagosome. Once the engulfing process is complete, vesicles called autophagosomes form. The autophagosomes then fuse with the lysosome, producing autolysosomes, and the contents are hydrolytically degraded and recycled (reviewed in [16, 179, 180]). The process is mediated through a collection of several autophagy gene (Atg) products. In yeast, the recognition process is enacted through Atg32, a mitophagy-specific receptor on the OMM [181], and through other key players, such as Atg8 and its activator Atg7. Mitophagy in mammals has been less well characterized, but the mammalian homologues to Atg32 and Atg8 are believed to be Nix and LC3, respectively [180, 182]. In addition, mitophagy in mammalian cells may be carried out through ubiquitination of OMM proteins, followed by recognition via an LC3 complex (reviewed in [180]).
Relative to other mitochondrial dynamics, there is less known about the role of mitophagy in the aging of SM. Evidence suggests that mitophagy selectively removes defective mitochondria that are depolarized or produce excessive ROS (reviewed in [16, 17, 179, 183]). In corroboration, suppression of autophagy results in increased ROS production, reduced oxygen consumption, and higher mtDNA mutation rates [184, 185]. With age, autophagy has been found to decline both in general (reviewed in [186]) and in the SM of aged rats [187]. While the repercussions are still unknown, mitophagy has been negatively correlated with oxidative damage and apoptosis [187], suggesting that reduced autophagy rates may contribute to muscular dysfunction. This is confirmed by studies on muscle-specific Atg7 knockout mice, who accumulate abnormal mitochondria, have lower resting oxygen consumption, and experience increased oxidative stress and higher rates of apoptosis; these mice also suffer from muscle atrophy, weakness, and myofibril degeneration [188, 189]. Furthermore, studies in a few species indicate that enhanced autophagy may increase lifespan (reviewed in [190]).
Mitochondrial quality control and degradation are also modulated by the ubiquitin-proteasome system, which removes oxidized proteins and short-lived proteins (reviewed in [191]). Evidence from studies in mammals has suggested that ubiquitin-proteasome activity declines with age in SM and may contribute to muscular atrophy (reviewed in [91, 192]). However, aging may differentially affect the components of the ubiquitin-proteasome system [193], as some genes involved—including some proteasome subunits and ubiquitin-specific proteases—are expressed at higher levels in SM from older humans, yet others are unchanged or decreased with age [49, 84, 94, 194–196]. Finally, changes in proteasomal activity may be fiber type-specific [197]: in particular, one study in humans reported that ubiquitin protein levels increase in fast-twitch muscle fibers, which may explain why type II fibers atrophy faster with age [198].
3.4. Increased Mitochondria-Mediated Apoptosis
Mitochondria also respond to and modulate cellular dynamics through apoptotic signaling, which is activated when either OXPHOS or their redox potential is disrupted, or in response to proapoptotic signals (reviewed in [199]). Mitochondria can induce apoptosis through either caspase-dependent or caspase-independent signaling mechanisms (reviewed in [199–201]). Caspase-dependent signaling is dependent on the release of cytochrome c from within the mitochondria, which triggers a cascade of actions by cysteine proteases called caspases that results in apoptosis. In brief, cytochrome c and other proapoptotic factors are released from the mitochondria. Once released, cytochrome c joins with apoptotic protease activating factor-1 (Apaf-1) and procaspase-9 to form a complex called the apoptosome. The apoptosome then cleaves and activates procaspase-9, which acts on caspase-3. Caspase-3 in turn activates a caspase-activated DNase (CAD) to degrade DNA and initiates cellular degradation. In the caspase-independent pathway, which is also known as “mitoptosis,” the mitochondria release apoptosis-inducing factor (AIF) and endonuclease G (EndoG), which then induce chromatin condensation and DNA fragmentation. There are multiple points in these pathways that are regulated by pro- and anti-apoptotic proteins. In particular, the release of apoptotic triggers appears to be modulated through two mechanisms: (1) the balance of proapoptotic (e.g., Bax) and anti-apoptotic proteins (e.g., Bcl-2), particularly from the Bcl-2 family, which control OMM stability and form the mitochondrial apoptosis-induced channel (MAC), and (2) the mitochondrial permeability transition pore (mPTP). One example of the direct connection between mitochondrial and cellular dynamics is that mitochondrial fragmentation occurs around the time that proapoptotic factors are released from the mitochondria, and this step is contingent upon increased fission through Drp1 as well as a block in mitochondrial fusion (reviewed in [202]).
Apoptosis increases significantly with age and likely contributes to sarcopenia and other age-associated declines (reviewed in [201, 203, 204]). Though it is difficult to prove this conclusively, many studies have correlated rates of apoptosis with markers of sarcopenia or SM function (reviewed in [201, 203, 204]). In humans, the percent of apoptotic cells tends to increase with age, though generally more so in type II fibers [194, 205, 206]. The weight of evidence from both human and animal studies suggests that the caspase-independent pathway is upregulated with age, while the caspase-dependent pathway is not (reviewed in [201]). In particular, one study found an increase in AIF gene transcripts in SM from older people, but no change in Bax, Bcl-2, or caspase-3 expression [207], and another study in humans reported no change in caspase-3 or -7 activity [194]. This is also supported by animal studies, which have found that the mPTP becomes more susceptible towards being opened [25, 208, 209] and that the levels and activities of caspase-independent apoptotic proteins AIF and EndoG increase [25, 112, 210–213] with age. Recent evidence in rats suggests that apoptotic susceptibilities and markers are age- and fiber type-specific, which may explain some of the mixed results, particularly in regard to the caspase-dependent pathway [25, 214–216]. Interestingly, a study showed that disuse atrophy increased caspase-3 activity in young rats but not old and dramatically increased EndoG levels in old rats but not young, indicating that older SM likely responds to apoptotic stimuli through different signaling pathways than younger SM [212].
4. Strategies to Attenuate Mitochondrial Aging
4.1. Exercise
Exercise training has long been known to induce mitochondrial biogenesis, upregulate SM gene expression and protein synthesis, and increase SM oxidative capacity [217, 218]. It is apparent from previous research that physical activity decreases during aging [219]. Therefore, it remains unclear whether the abnormalities in mitochondrial function are a primary effect of aging or are due to the associated decline in physical activity. This issue is compounded by the cross-sectional nature of some studies without objective control for activity level [130, 220–222]. For example, we recently found that mitochondrial capacity was reduced in elderly subjects compared to their young BMI-matched counterparts [10]. In this study, we included only sedentary individuals, defined as less than 2 hours of intentional physical activity per week. Despite careful screening for activity levels using questionnaires, we found that daily physical activity was significantly lower than reported in the elderly group. This suggests that independent of exercise training, simply living an active lifestyle may have a significant impact on mitochondrial function; however, we cannot determine the cause and effect due to the cross-sectional nature of the analysis.
There is strong evidence that exercise training can improve SM mitochondrial function in elderly adults [84, 96, 97, 223–225] and may also protect against age-associated apoptosis [226]. Short et al. found that 4 months of aerobic exercise in older adults increased protein synthesis, mitochondrial enzyme activity (citrate synthase and cytochrome c oxidase), and expression of genes involved in mitochondrial content and biogenesis to levels similar to those in younger adults [97, 227]. In a separate study, 12 weeks of aerobic exercise training increased mitochondrial content and activity, particularly in the subsarcolemmal fraction [224]. Exercise has also been shown to increase the activity of antioxidant enzymes and heat shock proteins [228], potentially reducing ROS production and decreasing the potential for mitochondrial oxidative damage thought to occur during aging. Indeed, Parise et al. showed that resistance exercise in older adults increased antioxidant content (but not activity), decreased oxidative damage (8-OHdG), and increased complex IV activity [229, 230].
Despite these significant differences in mitochondrial parameters, most data show that some impairment remains. For example, the usual age-associated decline in mitochondrial oxidative capacity was absent in older adults who were chronically endurance trained. However, chronic exercise did not completely restore the expression of mitochondrial proteins, mtDNA content, and mitochondrial transcription factors to the levels of younger subjects, suggesting a persisting, independent effect of age [50]. Supporting this, Melov et al. used an “omics” approach to show that after 6 months of exercise training, the transcriptional signature of aging was substantially but incompletely reversed back to the transcriptome of younger adults [84]. In all, the results to date indicate that exercise can help to attenuate age-associated changes in SM mitochondria. However, it does not completely prevent these changes. The data available are rather limited and apply mostly to short-term exercise interventions (i.e., 12–24 weeks) rather than chronic exercise. In addition, the impact of active lifestyle changes—for example, decreasing sedentary time and increasing “nonexercise” activity—on mitochondrial function in elderly adults may be significant [231] and needs further investigation, as this may be a more logical approach rather than prescribing an exercise regimen.
4.2. Caloric Restriction
Caloric restriction, which typically involves consuming 20–40% fewer calories than normal, also preserves mitochondrial health and attenuates SM decline with age. Caloric restriction (CR) is recognized as the most robust intervention that retards both primary aging (natural age-related deterioration) and secondary aging (accelerated aging due to disease and negative lifestyle behaviors), thereby increasing both median and maximal lifespan in many species. While CR studies in primates and humans are largely ongoing, studies in rodents consistently show that CR extends maximum life span by up to 50% and reduces the incidence of many age-associated diseases, including cancer and metabolic diseases (reviewed in [232]). Preliminary evidence in rhesus monkeys indicates similar effects can be expected in primates [231].
The benefits ascribed to CR are believed to be due in large part to reductions in oxidative stress (reviewed in [233]). In primates, a decade-long CR intervention resulted in marked decreases in oxidative damage to lipids and proteins [234]; in aged rats, CR also reduces ROS production [159, 235, 236]. As a result, aged CR animals exhibit fewer mtDNA and nuclear DNA mutations and less oxidative damage to SM mitochondria than their ad libitum-fed counterparts [99, 236–240]. Some of these findings have been replicated in the still ongoing CR trial in humans, dubbed CALERIE, which reported that CR subjects had less mtDNA damage and more mtDNA content than controls [241]. Microarray and RT-PCR experiments confirm that CR increases transcripts of genes involved in ROS scavenging, including SOD and GPx, and decreases transcripts from stress response genes [196, 242].
Calorie restriction appears to modulate mitochondrial efficiency, content, and function. CR lowers energy expenditure in animals and humans by producing mitochondria that consume less oxygen yet are able to maintain normal levels of ATP production (reviewed in [232]; in particular, [159, 237, 241–243]). It is generally believed that this energetic adaptation is mediated via decreased proton leak, which has been confirmed in rodent studies, and that decreased proton leak is in turn enabled by the shift to a less oxidative milieu [159, 237, 244]. In terms of mitochondrial content and function, CR does not affect the gene expression, protein level, or activity of citrate synthase, nor the activities of other TCA proteins [111, 112, 241, 242, 245]. However, CR may affect some ETS enzymes. CR reduced the age-associated accumulation of complex IV-negative and complex II-hyperactive fibers in rats [99] and in rhesus monkeys [100]. Only complex IV activity, however, is consistently responsive to CR, showing increased activity in comparison to that in aged rodents and usually similar activity to their younger counterparts [110–112, 246]. Nonetheless, CR may prevent the decline in the activities of complexes I–III in some muscles [111]. However, a study in rhesus monkeys found that about 9 years of CR only attenuated the decline in gene expression of the complex II iron-protein subunit [85], and a 14-week CR intervention where the rats were fed at 70% of ad libitum levels found no increases in any mitochondrial gene transcripts or proteins [245]. CR rodents did have fewer ETS abnormalities, but the abnormalities were otherwise similar to controls, suggesting that CR only affects the onset of fiber atrophy [240, 247]. However, in primate studies, CR did not alter the number of fibers with ETS abnormalities, but nonetheless preserved muscle fiber [100, 248].
CR also affects mitochondrial dynamics. In rodent SM, CR also increases mitochondrial biogenesis relative to controls by slowing the decline in PGC-1α gene expression with age, which may be at least partially responsible for maintaining oxidative capacity in aged CR animals [110, 112]. The CALERIE study in humans also reported increased transcripts from several genes involved in mitochondrial biogenesis, including PGC-1α, Tfam, and SIRT1 [241]. It is not clear, however, whether CR affects the mitochondrial metabolic regulator AMPK, as there have been mixed results [249, 250]. In addition, CR attenuated the decline in mitophagy in the SM of old rodents [187] and the decline in ubiquitin-proteasome activity in monkeys [251]. It also reduced apoptosis susceptibility by promoting a remodeling of caspases and caspase-related proteins to favor decreased likelihood of cytochrome c release from the mitochondria [187, 252–254]. These findings are particularly important as mitochondrial dysfunction and apoptosis have been proposed as key mediators of sarcopenia.
Combined, these results suggest that CR reduces oxidative stress and remodels mitochondrial dynamics to promote the production of more fuel-efficient mitochondria that produce less ROS and to favor the removal of dysfunctional mitochondria [187, 243, 255]. Regardless of the underlying mechanism, CR has repeatedly been shown in rodents to either partially or fully oppose the hallmarks of sarcopenia, including the age-related declines in muscle force, fiber cross-sectional area, fiber number, and fiber type (reviewed in [99, 247, 254, 256–258]). Similar findings have been reported in CR studies in rhesus monkeys [100, 248].
4.3. Mimetics
Currently, there is strong interest in using a CR- or exercise-mimetic to attenuate age-related mitochondrial dysfunction. The best known of these is resveratrol (3,5,4,9-trihydroxystilbene), a phytoalexin that is abundant in red wine. In the context of secondary aging, resveratrol's effects on mitochondria and SM have been well studied. In rodent models involving metabolic pathology, resveratrol improves mtDNA copy number and function, increases mitochondrial biogenesis, improves exercise capacity and motor function, and mitigates metabolic dysfunction [259–261]. On a molecular level, resveratrol induces an increase in the expression of PGC-1α, Tfam, and UCP3, and increases SIRT1, AMPK, and PGC-1α activation [259, 260]. In humans, a recent 30-day study of resveratrol supplementation in 11 obese but metabolically healthy men reported improved mitochondrial respiration in the presence of fatty acid-derived substrate, increased AMPK and citrate synthase activity, and higher SIRT1 and PGC-1α protein levels; however, no changes in mitochondrial content were observed [262]. Interestingly, while resveratrol was originally identified as a potent activator of SIRT1, there is controversy surrounding its mode of action, and it may exert much of its effects through AMPK [260, 263, 264].
Rodent studies consistently show improved mitochondrial health with resveratrol (reviewed in [265]). For example, in senescence-accelerated prone mice, those given resveratrol had higher physical endurance, maximal force contraction, and oxygen consumption, and higher levels of transcripts from PGC-1α and ETS genes [266]. Aged rats given resveratrol exhibited similar responses; they also displayed reduced levels of oxidative stress but no changes in most apoptotic markers, indicating the improvements were mediated through changes in redox status and not apoptotic pathways [267]. Like CR, resveratrol's effects on mitochondrial biogenesis are believed to be mediated in large part via reductions in mitochondrial ROS production and via upregulation of fatty acid catabolism coupled with a downregulation in fatty acid synthesis [137]. However, one recent study of long-term resveratrol supplementation in mice did not find any improvement in the age-related declines in PGC-1α, mitochondrial content, muscle mass, and maximal isometric force production; though resveratrol did preserve type II fiber contractile function and reduce oxidative stress [268].
5. Conclusions
In summary, animal and human data consistently show that skeletal muscle mitochondria are altered in aging, including increased mutations in mitochondrial DNA, decreased expression of some mitochondrial proteins, reduced enzyme activity and altered respiration with reduced maximal capacity in sedentary adults, and reduced total mitochondrial content with increased morphological changes. Since the primary role of mitochondria is to produce ATP to maintain the energy status of the cell, shifts in respiratory activity and capacity can lower the membrane potential, reduce cellular ATP concentration, and signal cellular apoptotic events. Increased apoptosis without correspondingly increased protein synthesis will eventually lead to net muscle fiber loss. All of these factors probably contribute to age-associated sarcopenia, and mounting evidence suggests that most of these age-related changes can be either prevented or attenuated through increased physical activity.
There is some thought that the accumulation of oxidative damage caused by long-term ROS production is responsible for these changes with age. Indeed, blocking ROS formation by the targeted expression of antioxidant enzymes ameliorates age-associated dysfunction and returns mitochondrial parameters to those of young animals (Figure 3). Whether this is also true for humans is unknown; however, strategies that improve mitochondrial function, such as exercise and caloric restriction, also reduce ROS production and increase antioxidant defenses. Exercise is also known to be a powerful stimulant of mitochondrial biogenesis and oxidative capacity, although exercise training in elderly adults probably does not completely reverse the primary effects of aging. However, exercise training—even adopting active lifestyle habits—may clearly reduce the rate of mitochondrial decline and attenuate the aging phenotype. Whether CR- and exercise-mimetics, including resveratrol, work as effectively remains to be determined and is an area of active research.
In conclusion, there is clearly a need for more research in this field and particularly to:
figure out which age-related changes are universal and which depend on physical activity or lifestyle habits or gender;
unravel how cells compensate for oxidative damage and mitochondrial dysfunction;
elucidate how fusion, fission, and autophagy work together to ensure quality control and to remove defective mitochondria; and
determine which mitochondrial declines can be slowed down (or even reversed) by healthier lifestyles.
We expect this to continue be a fruitful and exciting area of study in years to come.
References
- 1.Frontera WR, Hughes VA, Lutz KJ, Evans WJ. A cross-sectional study of muscle strength and mass in 45- to 78-yr-old men and women. Journal of Applied Physiology. 1991;71(2):644–650. doi: 10.1152/jappl.1991.71.2.644. [DOI] [PubMed] [Google Scholar]
- 2.Hughes VA, Frontera WR, Wood M, et al. Longitudinal muscle strength changes in older adults: influence of muscle mass, physical activity, and health. The Journals of Gerontology A. 2001;56(5):B209–B217. doi: 10.1093/gerona/56.5.b209. [DOI] [PubMed] [Google Scholar]
- 3.Goodpaster BH, Park SW, Harris TB, et al. The loss of skeletal muscle strength, mass, and quality in older adults: the Health, Aging and Body Composition Study. Journals of Gerontology A. 2006;61(10):1059–1064. doi: 10.1093/gerona/61.10.1059. [DOI] [PubMed] [Google Scholar]
- 4.Fiatarone MA, O'Neill EF, Ryan ND, Clements KM, Solares GR, Nelson ME. Exercise training and nutritional supplementation for physical frailty in very elderly people. The New England journal of medicine. 1994;330(25):1769–1775. doi: 10.1056/NEJM199406233302501. [DOI] [PubMed] [Google Scholar]
- 5.Goodpaster BH, Chomentowski P, Ward BK, et al. Effects of physical activity on strength and skeletal muscle fat infiltration in older adults: a randomized controlled trial. Journal of Applied Physiology. 2008;105(5):1498–1503. doi: 10.1152/japplphysiol.90425.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nelson ME, Fiatarone MA, Morganti CM, Trice I, Greenberg RA, Evans WJ. Effects of high-intensity strength training on multiple risk factors for osteoporotic fractures: a randomized controlled trial. Journal of the American Medical Association. 1994;272(24):1909–1914. doi: 10.1001/jama.1994.03520240037038. [DOI] [PubMed] [Google Scholar]
- 7.Metter EJ, Lynch N, Conwit R, Lindle R, Tobin J, Hurley B. Muscle quality and age: cross-sectional and longitudinal comparisons. The Journals of Gerontology A. 1999;54(5):B207–B218. doi: 10.1093/gerona/54.5.b207. [DOI] [PubMed] [Google Scholar]
- 8.Cree MG, Newcomer BR, Katsanos CS, et al. Intramuscular and liver triglycerides are increased in the elderly. The Journal of Clinical Endocrinology and Metabolism. 2004;89(8):3864–3871. doi: 10.1210/jc.2003-031986. [DOI] [PubMed] [Google Scholar]
- 9.Hipkiss AR. Mitochondrial dysfunction, proteotoxicity, and aging: causes or effects, and the possible impact of NAD+-controlled protein glycation. Advances in Clinical Chemistry. 2010;50:123–150. [PubMed] [Google Scholar]
- 10.Johannsen DL, Conley KE, Bajpeyi S, et al. Ectopic lipid accumulation and reduced glucose tolerance in elderly adults are accompanied by altered skeletal muscle mitochondrial activity. The Journal of Clinical Endocrinology and Metabolism. 2012;97(1):242–250. doi: 10.1210/jc.2011-1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saraste M. Oxidative phosphorylation at the fin de siecle. Science. 1999;283(5407):1488–1493. doi: 10.1126/science.283.5407.1488. [DOI] [PubMed] [Google Scholar]
- 12.Holloszy JO. Skeletal muscle,“mitochondrial deficiency” does not mediate insulin resistance. The American Journal of Clinical Nutrition. 2009;89(1):463S–466S. doi: 10.3945/ajcn.2008.26717C. [DOI] [PubMed] [Google Scholar]
- 13.Miller BF, Robinson MM, Bruss MD, Hellerstein M, Hamilton KL. A comprehensive assessment of mitochondrial protein synthesis and cellular proliferation with age and caloric restriction. Aging Cell. 2012;11(1):150–161. doi: 10.1111/j.1474-9726.2011.00769.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shigenaga MK, Hagen TM, Ames BN. Oxidative damage and mitochondrial decay in aging. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(23):10771–10778. doi: 10.1073/pnas.91.23.10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Terman A, Kurz T, Navratil M, Arriaga EA, Brunk UT. Mitochondrial turnover and aging of long-lived postmitotic cells: the mitochondrial-lysosomal axis theory of aging. Antioxidants and Redox Signaling. 2010;12(4):503–535. doi: 10.1089/ars.2009.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Twig G, Hyde B, Shirihai OS. Mitochondrial fusion, fission and autophagy as a quality control axis: the bioenergetic view. Biochimica et Biophysica Acta. 2008;1777(9):1092–1097. doi: 10.1016/j.bbabio.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Twig G, Elorza A, Molina AJ, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. The EMBO Journal. 2008;27(2):433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beregi E, Regius O, Huttl T, Gobl Z. Age-related changes in the skeletal muscle cells. Zeitschrift fur Gerontologie. 1988;21(2):83–86. [PubMed] [Google Scholar]
- 19.Poggi P, Marchetti C, Scelsi R. Automatic morphometric analysis of skeletal muscle fibers in the aging man. Anatomical Record. 1987;217(1):30–34. doi: 10.1002/ar.1092170106. [DOI] [PubMed] [Google Scholar]
- 20.Conley KE, Jubrias SA, Esselman PC. Oxidative capacity and ageing in human muscle. The Journal of Physiology. 2000;5261, part 1:203–210. doi: 10.1111/j.1469-7793.2000.t01-1-00203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crane JD, Devries MC, Safdar A, Hamadeh MJ, Tarnopolsky MA. The effect of aging on human skeletal muscle mitochondrial and intramyocellular lipid ultrastructure. The Journals of Gerontology A. 2010;65(2):119–128. doi: 10.1093/gerona/glp179. [DOI] [PubMed] [Google Scholar]
- 22.Nakamura S, Takamura T, Matsuzawa-Nagata N, et al. Palmitate induces insulin resistance in H4IIEC3 hepatocytes through reactive oxygen species produced by mitochondria. The Journal of Biological Chemistry. 2009;284(22):14809–14818. doi: 10.1074/jbc.M901488200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harman D. Aging: a theory based on free radical and radiation chemistry. Journal of Gerontology. 1956;11(3):298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 24.Harman D. Free radical theory of aging: an update: increasing the functional life span. Annals of the New York Academy of Sciences. 2006;1067:10–21. doi: 10.1196/annals.1354.003. [DOI] [PubMed] [Google Scholar]
- 25.Chabi B, Ljubicic V, Menzies KJ, Huang JH, Saleem A, Hood DA. Mitochondrial function and apoptotic susceptibility in aging skeletal muscle. Aging Cell. 2008;7(1):2–12. doi: 10.1111/j.1474-9726.2007.00347.x. [DOI] [PubMed] [Google Scholar]
- 26.Hütter E, Skovbro M, Lener B, et al. Oxidative stress and mitochondrial impairment can be separated from lipofuscin accumulation in aged human skeletal muscle. Aging Cell. 2007;6(2):245–256. doi: 10.1111/j.1474-9726.2007.00282.x. [DOI] [PubMed] [Google Scholar]
- 27.Mansouri A, Muller FL, Liu Y, et al. Alterations in mitochondrial function, hydrogen peroxide release and oxidative damage in mouse hind-limb skeletal muscle during aging. Mechanisms of Ageing and Development. 2006;127(3):298–306. doi: 10.1016/j.mad.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 28.Yarian CS, Rebrin I, Sohal RS. Aconitase and ATP synthase are targets of malondialdehyde modification and undergo an age-related decrease in activity in mouse heart mitochondria. Biochemical and Biophysical Research Communications. 2005;330(1):151–156. doi: 10.1016/j.bbrc.2005.02.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pesce V, Cormio A, Fracasso F, et al. Age-related mitochondrial genotypic and phenotypic alterations in human skeletal muscle. Free Radical Biology and Medicine. 2001;30(11):1223–1233. doi: 10.1016/s0891-5849(01)00517-2. [DOI] [PubMed] [Google Scholar]
- 30.Beal MF. Oxidatively modified proteins in aging and disease. Free Radical Biology & Medicine. 2002;32(9):797–803. doi: 10.1016/s0891-5849(02)00780-3. [DOI] [PubMed] [Google Scholar]
- 31.Staunton L, O'Connell K, Ohlendieck K. Proteomic profiling of mitochondrial enzymes during skeletal muscle aging. Journal of Aging Research. 2011;2011:9 pages. doi: 10.4061/2011/908035.908035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kushnareva Y, Murphy AN, Andreyev A. Complex I-mediated reactive oxygen species generation: modulation by cytochrome c and NAD(P)+ oxidation-reduction state. The Biochemical Journal. 2002;368, part 2:545–553. doi: 10.1042/BJ20021121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barreiro E, Coronell C, Laviña B, Ramírez-Sarmiento A, Orozco-Levi M, Gea J. Aging, sex differences, and oxidative stress in human respiratory and limb muscles. Free Radical Biology and Medicine. 2006;41(5):797–809. doi: 10.1016/j.freeradbiomed.2006.05.027. [DOI] [PubMed] [Google Scholar]
- 34.Ji LL, Dillon D, Wu E. Alteration of antioxidant enzymes with aging in rat skeletal muscle and liver. The American Journal of Physiology. 1990;258(4, part 2):918–923. doi: 10.1152/ajpregu.1990.258.4.R918. [DOI] [PubMed] [Google Scholar]
- 35.Luhtala TA, Roecker EB, Pugh T, Feuers RJ, Weindruch R. Dietary restriction attenuates age-related increases in rat skeletal muscle antioxidant enzyme activities. Journals of Gerontology. 1994;49(5):B231–B238. doi: 10.1093/geronj/49.5.b231. [DOI] [PubMed] [Google Scholar]
- 36.Armeni T, Principato G, Quiles JL, Pieri C, Bompadre S, Battino M. Mitochondrial dysfunctions during aging: vitamin E deficiency or caloric restriction—two different ways of modulating stress. Journal of Bioenergetics and Biomembranes. 2003;35(2):181–191. doi: 10.1023/a:1023754305218. [DOI] [PubMed] [Google Scholar]
- 37.Ren J, Li Q, Wu S, Li SY, Babcock SA. Cardiac overexpression of antioxidant catalase attenuates aging-induced cardiomyocyte relaxation dysfunction. Mechanisms of Ageing and Development. 2007;128(3):276–285. doi: 10.1016/j.mad.2006.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xia E, Rao G, Van Remmen H, Heydari AR, Richardson A. Activities of antioxidant enzymes in various tissues of male Fischer 344 rats are altered by food restriction. Journal of Nutrition. 1995;125(2):195–201. doi: 10.1093/jn/125.2.195. [DOI] [PubMed] [Google Scholar]
- 39.Safdar A, Hamadeh MJ, Kaczor JJ, Raha S, Debeer J, Tarnopolsky MA. Aberrant mitochondrial homeostasis in the skeletal muscle of sedentary older adults. PLoS ONE. 2010;5(5):p. e10778. doi: 10.1371/journal.pone.0010778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee HY, Choi CS, Birkenfeld AL, Alves TC, Jornayvaz FR, Jurczak MJ. Targeted expression of catalase to mitochondria prevents age-associated reductions in mitochondrial function and insulin resistance. Cell Metabolism. 2010;12(6):668–674. doi: 10.1016/j.cmet.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iossa S, Mollica MP, Lionetti L, Crescenzo R, Tasso R, Liverini G. A possible link between skeletal muscle mitochondrial efficiency and age-induced insulin resistance. Diabetes. 2004;53(11):2861–2866. doi: 10.2337/diabetes.53.11.2861. [DOI] [PubMed] [Google Scholar]
- 42.Marcinek DJ, Schenkman KA, Ciesielski WA, Lee D, Conley KE. Reduced mitochondrial coupling in vivo alters cellular energetics in aged mouse skeletal muscle. The Journal of Physiology. 2005;569, part 2:467–473. doi: 10.1113/jphysiol.2005.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Amara CE, Shankland EG, Jubrias SA, Marcinek DJ, Kushmerick MJ, Conley KE. Mild mitochondrial uncoupling impacts cellular aging in human muscles in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(3):1057–1062. doi: 10.1073/pnas.0610131104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lopez MF, Kristal BS, Chernokalskaya E, Lazarev A, Shestopalov AI, Bogdanova A, et al. High-throughput profiling of the mitochondrial proteome using affinity fractionation and automation. Electrophoresis. 2000;21(16):3427–3440. doi: 10.1002/1522-2683(20001001)21:16<3427::AID-ELPS3427>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 45.Calvo S, Jain M, Xie X, Sheth SA, Chang B, Goldberger OA, et al. Systematic identification of human mitochondrial disease genes through integrative genomics. Nature Genetics. 2006;38(5):576–582. doi: 10.1038/ng1776. [DOI] [PubMed] [Google Scholar]
- 46.Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290(5806):457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 47.Menshikova EV, Ritov VB, Fairfull L, Ferrell RE, Kelley DE, Goodpaster BH. Effects of exercise on mitochondrial content and function in aging human skeletal muscle. Journals of Gerontology A. 2006;61(6):534–540. doi: 10.1093/gerona/61.6.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Short KR, Bigelow ML, Kahl J, Singh R, Coenen-Schimke J, Raghavakaimal S, et al. Decline in skeletal muscle mitochondrial function with aging in humans. . Proceedings of the National Academy of Sciences of the United States of America. 2005;102(15):5618–5623. doi: 10.1073/pnas.0501559102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Welle S, Bhatt K, Shah B, Needler N, Delehanty JM, Thornton CA. Reduced amount of mitochondrial DNA in aged human muscle. Journal of Applied Physiology. 2003;94(4):1479–1484. doi: 10.1152/japplphysiol.01061.2002. [DOI] [PubMed] [Google Scholar]
- 50.Lanza IR, Short DK, Short KR, Raghavakaimal S, Basu R, Joyner MJ, et al. Endurance exercise as a countermeasure for aging. Diabetes. 2008;57(11):2933–2942. doi: 10.2337/db08-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barrientos A, Casademont J, Cardellach F, et al. Qualitative and quantitative changes in skeletal muscle mtDNA and expression of mitochondrial-encoded genes in the human aging process. Biochemical and Molecular Medicine. 1997;62(2):165–171. doi: 10.1006/bmme.1997.2647. [DOI] [PubMed] [Google Scholar]
- 52.Barazzoni R, Short KR, Nair KS. Effects of aging on mitochondrial DNA copy number and cytochrome c oxidase gene expression in rat skeletal muscle, liver, and heart. Journal of Biological Chemistry. 2000;275(5):3343–3347. doi: 10.1074/jbc.275.5.3343. [DOI] [PubMed] [Google Scholar]
- 53.McKenzie D, Bua E, McKiernan S, Cao Z, Wanagat J, Aiken JM. Mitochondrial DNA deletion mutations: a causal role in sarcopenia. European Journal of Biochemistry. 2002;269(8):2010–2015. doi: 10.1046/j.1432-1033.2002.02867.x. [DOI] [PubMed] [Google Scholar]
- 54.Khrapko K, Vijg J. Mitochondrial DNA mutations and aging: devils in the details? Trends in Genetics. 2009;25(2):91–98. doi: 10.1016/j.tig.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bua E, Johnson J, Herbst A, et al. Mitochondrial DNA-deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. American Journal of Human Genetics. 2006;79(3):469–480. doi: 10.1086/507132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chabi B, Mousson de Camaret B, Chevrollier A, Boisgard S, Stepien G. Random mtDNA deletions and functional consequence in aged human skeletal muscle. Biochemical and Biophysical Research Communications. 2005;332(2):542–549. doi: 10.1016/j.bbrc.2005.04.153. [DOI] [PubMed] [Google Scholar]
- 57.Melov S, Shoffner JM, Kaufman A, Wallace DC. Marked increase in the number and variety of mitochondrial DNA rearrangements in aging human skeletal muscle. Nucleic Acids Research. 1995;23(20):4122–4126. doi: 10.1093/nar/23.20.4122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cooper JM, Mann VM, Schapira AHV. Analyses of mitochondrial respiratory chain function and mitochondrial DNA deletion in human skeletal muscle: effect of ageing. Journal of the Neurological Sciences. 1992;113(1):91–98. doi: 10.1016/0022-510x(92)90270-u. [DOI] [PubMed] [Google Scholar]
- 59.Pallotti F, Chen X, Bonilla E, Schon EA. Evidence that specific mtDNA point mutations may not accumulate in skeletal muscle during normal human aging. American Journal of Human Genetics. 1996;59(3):591–602. [PMC free article] [PubMed] [Google Scholar]
- 60.Wang Y, Michikawa Y, Mallidis C, Bai Y, Woodhouse L, Yarasheski KE, et al. Muscle-specific mutations accumulate with aging in critical human mtDNA control sites for replication. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(7):4022–4027. doi: 10.1073/pnas.061013598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Michikawa Y, Mazzucchelli F, Bresolin N, Scarlato G, Attardi G. Aging-dependent large accumulation of point mutations in the human mtDNA control region for replication. Science. 1999;286(5440):774–779. doi: 10.1126/science.286.5440.774. [DOI] [PubMed] [Google Scholar]
- 62.Wanagat J, Cao Z, Pathare P, Aiken JM. Mitochondrial DNA deletion mutations colocalize with segmental electron transport system abnormalities, muscle fiber atrophy, fiber splitting, and oxidative damage in sarcopenia. The FASEB Journal. 2001;15(2):322–332. doi: 10.1096/fj.00-0320com. [DOI] [PubMed] [Google Scholar]
- 63.Torii K, Sugiyama S, Tanaka M, Takagi K, Hanaki Y, Iida K, et al. Aging-associated deletions of human diaphragmatic mitochondrial DNA. American Journal of Respiratory Cell and Molecular Biology. 1992;6(5):543–549. doi: 10.1165/ajrcmb/6.5.543. [DOI] [PubMed] [Google Scholar]
- 64.Fattoretti P, Vecchiet J, Felzani G, et al. Succinic dehydrogenase activity in human muscle mitochondria during aging: a quantitative cytochemical investigation. Mechanisms of Ageing and Development. 2001;122(15):1841–1848. doi: 10.1016/s0047-6374(01)00320-7. [DOI] [PubMed] [Google Scholar]
- 65.Bohr VA, Stevnsner T, de Souza-Pinto NC. Mitochondrial DNA repair of oxidative damage in mammalian cells. Gene. 2002;286(1):127–134. doi: 10.1016/s0378-1119(01)00813-7. [DOI] [PubMed] [Google Scholar]
- 66.Yakes FM, Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(2):514–519. doi: 10.1073/pnas.94.2.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Larsson NG. Somatic mitochondrial DNA mutations in mammalian aging. Annual Review of Biochemistry. 2010;79:683–706. doi: 10.1146/annurev-biochem-060408-093701. [DOI] [PubMed] [Google Scholar]
- 68.Meissner C. Mutations of mitochondrial DNA—cause or consequence of the ageing process? Z Gerontol Geriatr. 2007;40(5):325–333. doi: 10.1007/s00391-007-0481-z. [DOI] [PubMed] [Google Scholar]
- 69.Hiona A, Leeuwenburgh C. The role of mitochondrial DNA mutations in aging and sarcopenia: implications for the mitochondrial vicious cycle theory of aging. Experimental Gerontology. 2008;43(1):24–33. doi: 10.1016/j.exger.2007.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Conley KE, Marcinek DJ, Villarin J. Mitochondrial dysfunction and age. Current Opinion in Clinical Nutrition and Metabolic Care. 2007;10(6):688–692. doi: 10.1097/MCO.0b013e3282f0dbfb. [DOI] [PubMed] [Google Scholar]
- 71.Brierley EJ, Johnson MA, Lightowlers RN, James OF, Turnbull DM. Role of mitochondrial DNA mutations in human aging: implications for the central nervous system and muscle. Annals of Neurology. 1998;43(2):217–223. doi: 10.1002/ana.410430212. [DOI] [PubMed] [Google Scholar]
- 72.Pesce V, Cormio A, Fracasso F, et al. Age-related mitochondrial genotypic and phenotypic alterations in human skeletal muscle. Free Radical Biology and Medicine. 2001;30(11):1223–1233. doi: 10.1016/s0891-5849(01)00517-2. [DOI] [PubMed] [Google Scholar]
- 73.Fayet G, Jansson M, Sternberg D, et al. Ageing muscle: clonal expansions of mitochondrial DNA point mutations and deletions cause focal impairment of mitochondrial function. Neuromuscular Disorders. 2002;12(5):484–493. doi: 10.1016/s0960-8966(01)00332-7. [DOI] [PubMed] [Google Scholar]
- 74.Lee HC, Pang CY, Hsu HS, Wei YH. Ageing-associated tandem duplications in the D-loop of mitochondrial DNA of human muscle. FEBS Letters. 1994;354(1):79–83. doi: 10.1016/0014-5793(94)01063-3. [DOI] [PubMed] [Google Scholar]
- 75.Rooyackers OE, Adey DB, Ades PA, Nair KS. Effect of age on in vivo rates of mitochondrial protein synthesis in human skeletal muscle. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(26):15364–15369. doi: 10.1073/pnas.93.26.15364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Trifunovic A, Wredenberg A, Falkenberg M, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429(6990):417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 77.Kujoth GC, Hiona A, Pugh TD, et al. Medicine: mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309(5733):481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 78.Hiona A, Sanz A, Kujoth GC, et al. Mitochondrial DNA mutations induce mitochondrial dysfunction, apoptosis and sarcopenia in skeletal muscle of mitochondrial DNA mutator mice. PLoS ONE. 2010;5(7) doi: 10.1371/journal.pone.0011468.e11468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Boffoli D, Scacco SC, Vergari R, Solarino G, Santacroce G, Papa S. Decline with age of the respiratory chain activity in human skeletal muscle. Biochimica et Biophysica Acta. 1994;1226(1):73–82. doi: 10.1016/0925-4439(94)90061-2. [DOI] [PubMed] [Google Scholar]
- 80.Welle S, Bhatt K, Thornton CA. High-abundance mRNAs in human muscle: comparison between young and old. Journal of Applied Physiology. 2000;89(1):297–304. doi: 10.1152/jappl.2000.89.1.297. [DOI] [PubMed] [Google Scholar]
- 81.Welle S, Brooks AI, Delehanty JM, et al. Skeletal muscle gene expression profiles in 20–29 year old and 65–71 year old women. Experimental Gerontology. 2004;39(3):369–377. doi: 10.1016/j.exger.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 82.Zahn JM, Sonu R, Vogel H, Crane E, Mazan-Mamczarz K, Rabkin R. Transcriptional profiling of aging in human muscle reveals a common aging signature. PLoS Genetics. 2006;2(7):p. e115. doi: 10.1371/journal.pgen.0020115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Welle S, Brooks AI, Delehanty JM, Needler N, Thornton CA. Gene expression profile of aging in human muscle. Physiol Genomics. 2003;14(2):149–159. doi: 10.1152/physiolgenomics.00049.2003. [DOI] [PubMed] [Google Scholar]
- 84.Melov S, Tarnopolsky MA, Beckman K, Felkey K, Hubbard A. Resistance exercise reverses aging in human skeletal muscle. PLoS ONE. 2007;2(5):p. e465. doi: 10.1371/journal.pone.0000465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kayo T, Allison DB, Weindruch R, Prolla TA. Influences of aging and caloric restriction on the transcriptional profile of skeletal muscle from rhesus monkeys. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(9):5093–5098. doi: 10.1073/pnas.081061898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Picard M, Ritchie D, Wright KJ, et al. Mitochondrial functional impairment with aging is exaggerated in isolated mitochondria compared to permeabilized myofibers. Aging Cell. 2010;9(6):1032–1046. doi: 10.1111/j.1474-9726.2010.00628.x. [DOI] [PubMed] [Google Scholar]
- 87.de Magalhaes JP, Curado J, Church GM. Meta-analysis of age-related gene expression profiles identifies common signatures of aging. Bioinformatics. 2009;25(7):875–881. doi: 10.1093/bioinformatics/btp073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lyons CN, Mathieu-Costello O, Moyes CD. Regulation of skeletal muscle mitochondrial content during aging. Journals of Gerontology A. 2006;61(1):3–13. doi: 10.1093/gerona/61.1.3. [DOI] [PubMed] [Google Scholar]
- 89.Giresi PG, Stevenson EJ, Theilhaber J, et al. Identification of a molecular signature of sarcopenia. Physiological Genomics. 2005;21:253–263. doi: 10.1152/physiolgenomics.00249.2004. [DOI] [PubMed] [Google Scholar]
- 90.Mootha VK, Bunkenborg J, Olsen JV, et al. Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell. 2003;115(5):629–640. doi: 10.1016/s0092-8674(03)00926-7. [DOI] [PubMed] [Google Scholar]
- 91.Combaret L, Dardevet D, Béchet D, Taillandier D, Mosoni L, Attaix D. Skeletal muscle proteolysis in aging. Current Opinion in Clinical Nutrition and Metabolic Care. 2009;12(1):37–41. doi: 10.1097/MCO.0b013e32831b9c31. [DOI] [PubMed] [Google Scholar]
- 92.Irving BA, Robinson MM, Nair KS. Age effect on myocellular remodeling: response to exercise and nutrition in humans. Ageing Research Reviews. 2012;11(3):374–389. doi: 10.1016/j.arr.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gianni P, Jan KJ, Douglas MJ, Stuart PM, Tarnopolsky MA. Oxidative stress and the mitochondrial theory of aging in human skeletal muscle. Experimental Gerontology. 2004;39(9):1391–400. doi: 10.1016/j.exger.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 94.O’Connell K, Ohlendieck K. Proteomic DIGE analysis of the mitochondria-enriched fraction from aged rat skeletal muscle. Proteomics. 2009;9(24):5509–5524. doi: 10.1002/pmic.200900472. [DOI] [PubMed] [Google Scholar]
- 95.Piec I, Listrat A, Alliot J, Chambon C, Taylor RG, Bechet D. Differential proteome analysis of aging in rat skeletal muscle. The FASEB Journal. 2005;19(9):1143–1145. doi: 10.1096/fj.04-3084fje. [DOI] [PubMed] [Google Scholar]
- 96.Ghosh S, Lertwattanarak R, Lefort N, Molina-Carrion M, Joya-Galeana J, Bowen BP. Reduction in reactive oxygen species production by mitochondria from elderly subjects with normal and impaired glucose tolerance. Diabetes. 2011;60(8):2051–2060. doi: 10.2337/db11-0121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Short KR, Vittone JL, Bigelow ML, et al. Impact of aerobic exercise training on age-related changes in insulin sensitivity and muscle oxidative capacity. Diabetes. 2003;52(8):1888–1896. doi: 10.2337/diabetes.52.8.1888. [DOI] [PubMed] [Google Scholar]
- 98.Alves RMP, Vitorino R, Figueiredo P, Duarte JA, Ferreira R, Amado F. Lifelong physical activity modulation of the skeletal muscle mitochondrial proteome in mice. Journals of Gerontology A. 2010;65(8):832–842. doi: 10.1093/gerona/glq081. [DOI] [PubMed] [Google Scholar]
- 99.Aspnes LE, Lee CM, Weindruch R, Chung SS, Roecker EB, Aiken JM. Caloric restriction reduces fiber loss and mitochondrial abnormalities in aged rat muscle. The FASEB Journal. 1997;11(7):573–581. doi: 10.1096/fasebj.11.7.9212081. [DOI] [PubMed] [Google Scholar]
- 100.McKiernan SH, Colman RJ, Aiken E, et al. Cellular adaptation contributes to calorie restriction-induced preservation of skeletal muscle in aged rhesus monkeys. Experimental Gerontology. 2012;47(3):229–236. doi: 10.1016/j.exger.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Brierley EJ, Johnson MA, James OF, Turnbull DM. Effects of physical activity and age on mitochondrial function. Monthly Journal of the Association of Physicians. 1996;89(4):251–258. doi: 10.1093/qjmed/89.4.251. [DOI] [PubMed] [Google Scholar]
- 102.Gelfi C, Vigano A, Ripamonti M, et al. The human muscle proteome in aging. Journal of Proteome Research. 2006;5(6):1344–1353. doi: 10.1021/pr050414x. [DOI] [PubMed] [Google Scholar]
- 103.Donoghue P, Staunton L, Mullen E, Manning G, Ohlendieck K. DIGE analysis of rat skeletal muscle proteins using nonionic detergent phase extraction of young adult versus aged gastrocnemius tissue. Journal of Proteomics. 2010;73(8):1441–1453. doi: 10.1016/j.jprot.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 104.Tonkonogi M, Fernström M, Walsh B, et al. Reduced oxidative power but unchanged antioxidative capacity in skeletal muscle from aged humans. Pflugers Archiv European Journal of Physiology. 2003;446(2):261–269. doi: 10.1007/s00424-003-1044-9. [DOI] [PubMed] [Google Scholar]
- 105.Coggan AR, Spina RJ, King DS, et al. Histochemical and enzymatic comparison of the gastrocnemius muscle of young and elderly men and women. Journals of Gerontology. 1992;47(3):B71–B76. doi: 10.1093/geronj/47.3.b71. [DOI] [PubMed] [Google Scholar]
- 106.Lenaz G, Bovina C, Castelluccio C, et al. Mitochondrial complex I defects in aging. Molecular and Cellular Biochemistry. 1997;174(1-2):329–333. [PubMed] [Google Scholar]
- 107.Kumaran S, Subathra M, Balu M, Panneerselvam C. Age-associated decreased activities of mitochondrial electron transport chain complexes in heart and skeletal muscle: role of L-carnitine. Chemico-Biological Interactions. 2004;148(1-2):11–18. doi: 10.1016/j.cbi.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 108.Kerner J, Turkaly PJ, Minkler PE, Hoppel CL. Aging skeletal muscle mitochondria in the rat: decreased uncoupling protein-3 content. American Journal of Physiology. 2001;281(5):E1054–E1062. doi: 10.1152/ajpendo.2001.281.5.E1054. [DOI] [PubMed] [Google Scholar]
- 109.Mansouri A, Muller FL, Liu Y, et al. Alterations in mitochondrial function, hydrogen peroxide release and oxidative damage in mouse hind-limb skeletal muscle during aging. Mechanisms of Ageing and Development. 2006;127(3):298–306. doi: 10.1016/j.mad.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 110.Hepple RT, Baker DJ, McConkey M, Murynka T, Norris R. Caloric restriction protects mitochondrial function with aging in skeletal and cardiac muscles. Rejuvenation Research. 2006;9(2):219–222. doi: 10.1089/rej.2006.9.219. [DOI] [PubMed] [Google Scholar]
- 111.Hepple RT, Baker DJ, Kaczor JJ, Krause DJ. Long-term caloric restriction abrogates the age-related decline in skeletal muscle aerobic function. The FASEB Journal. 2005;19(10):1320–1322. doi: 10.1096/fj.04-3535fje. [DOI] [PubMed] [Google Scholar]
- 112.Baker DJ, Betik AC, Krause DJ, Hepple RT. No decline in skeletal muscle oxidative capacity with aging in long-term calorically restricted rats: effects are independent of mitochondrial DNA integrity. Journals of Gerontology A. 2006;61(7):675–684. doi: 10.1093/gerona/61.7.675. [DOI] [PubMed] [Google Scholar]
- 113.Capel F, Rimbert V, Lioger D, et al. Due to reverse electron transfer, mitochondrial H2O2 release increases with age in human vastus lateralis muscle although oxidative capacity is preserved. Mechanisms of Ageing and Development. 2005;126(4):505–511. doi: 10.1016/j.mad.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 114.Pastoris O, Boschi F, Verri M, et al. The effects of aging on enzyme activities and metabolite concentrations in skeletal muscle from sedentary male and female subjects. Experimental Gerontology. 2000;35(1):95–104. doi: 10.1016/s0531-5565(99)00077-7. [DOI] [PubMed] [Google Scholar]
- 115.Rasmussen UF, Krustrup P, Kjær M, Rasmussen HN. Experimental evidence against the mitochondrial theory of aging A study of isolated human skeletal muscle mitochondria. Experimental Gerontology. 2003;38(8):877–886. doi: 10.1016/s0531-5565(03)00092-5. [DOI] [PubMed] [Google Scholar]
- 116.Larsson L, Sjodin B, Karlsson J. Histochemical and biochemical changes in human skeletal muscle with age in sedentary males, age 22–65 yrs. Acta Physiologica Scandinavica. 1978;103(1):31–39. doi: 10.1111/j.1748-1716.1978.tb06187.x. [DOI] [PubMed] [Google Scholar]
- 117.Rasmussen UF, Krustrup P, Kjaer M, Rasmussen HN. Human skeletal muscle mitochondrial metabolism in youth and senescence: no signs of functional changes in ATP formation and mitochondrial oxidative capacity. Pflugers Archiv European Journal of Physiology. 2003;446(2):270–278. doi: 10.1007/s00424-003-1022-2. [DOI] [PubMed] [Google Scholar]
- 118.McCully KK, Fielding RA, Evans WJ, Leigh JS, Posner JD. Relationships between in vivo and in vitro measurements of metabolism in young and old human calf muscles. Journal of Applied Physiology. 1993;75(2):813–819. doi: 10.1152/jappl.1993.75.2.813. [DOI] [PubMed] [Google Scholar]
- 119.Chilibeck PD, McCreary CR, Marsh GD, et al. Evaluation of muscle oxidative potential by 31P-MRS during incremental exercise in old and young humans. European Journal of Applied Physiology and Occupational Physiology. 1998;78(5):460–465. doi: 10.1007/s004210050446. [DOI] [PubMed] [Google Scholar]
- 120.McCully KK, Forciea MA, Hack LM, et al. Muscle metabolism in older subjects using 31P magnetic resonance spectroscopy. Canadian Journal of Physiology and Pharmacology. 1991;69(5):576–580. doi: 10.1139/y91-084. [DOI] [PubMed] [Google Scholar]
- 121.Houmard JA, Weidner ML, Gavigan KE, Tyndall GL, Hickey MS, Alshami A. Fiber type and citrate synthase activity in the human gastrocnemius and vastus lateralis with aging. Journal of Applied Physiology. 1998;85(4):1337–1341. doi: 10.1152/jappl.1998.85.4.1337. [DOI] [PubMed] [Google Scholar]
- 122.Trounce I, Byrne E, Marzuki S. Decline in skeletal muscle mitochondrial respiratory chain function: possible factor in ageing. The Lancet. 1989;1(8639):637–639. doi: 10.1016/s0140-6736(89)92143-0. [DOI] [PubMed] [Google Scholar]
- 123.Schunk K, Pitton M, Düber C, Kersjes W, Schadmand-Fischer S, Thelen M. Dynamic phosphorus-31 magnetic resonance spectroscopy of the quadriceps muscle: effects of age and sex on spectroscopic results. Investigative Radiology. 1999;34(2):116–125. doi: 10.1097/00004424-199902000-00004. [DOI] [PubMed] [Google Scholar]
- 124.Taylor DJ, Kemp GJ, Thompson CH, Radda GK. Ageing: effects on oxidative function of skeletal muscle in vivo. Molecular and Cellular Biochemistry. 1997;174(1-2):1321–1322. [PubMed] [Google Scholar]
- 125.Marcinek DJ, Schenkman KA, Ciesielski WA, Lee D, Conley KE. Reduced mitochondrial coupling in vivo alters cellular energetics in aged mouse skeletal muscle. The Journal of Physiology. 2005;569, part 2:467–473. doi: 10.1113/jphysiol.2005.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Barrientos A, Casademont J, Rötig A, et al. Absence of relationship between the level of electron transport chain activities and aging in human skeletal muscle. Biochemical and Biophysical Research Communications. 1996;229(2):536–539. doi: 10.1006/bbrc.1996.1839. [DOI] [PubMed] [Google Scholar]
- 127.Brierly EJ, Johnson MA, Bowman A, et al. Mitochondrial function in muscle from elderly athletes. Annals of Neurology. 1997;41(1):114–116. doi: 10.1002/ana.410410120. [DOI] [PubMed] [Google Scholar]
- 128.Larsen RG, Callahan DM, Foulis SA, Kent-Braun JA. Age-related changes in oxidative capacity differ between locomotory muscles and are associated with physical activity behavior. Applied Physiology, Nutrition and Metabolism. 2012;37(1):88–99. doi: 10.1139/h11-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Waters DL, Brooks WM, Qualls CR, Baumgartner RN. Skeletal muscle mitochondrial function and lean body mass in healthy exercising elderly. Mechanisms of Ageing and Development. 2003;124(3):301–309. doi: 10.1016/s0047-6374(02)00197-5. [DOI] [PubMed] [Google Scholar]
- 130.Petersen KF, Befroy D, Dufour S, et al. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. 2003;300(5622):1140–1142. doi: 10.1126/science.1082889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Proctor DN, Sinning WE, Walro JM, Sieck GC, Lemon PWR. Oxidative capacity of human muscle fiber types: effects of age and training status. Journal of Applied Physiology. 1995;78(6):2033–2038. doi: 10.1152/jappl.1995.78.6.2033. [DOI] [PubMed] [Google Scholar]
- 132.Rimbert V, Boirie Y, Bedu M, Hocquette JF, Ritz P, Morio B. Muscle fat oxidative capacity is not impaired by age but by physical inactivity: association with insulin sensitivity. The FASEB Journal. 2004;18(6):737–739. doi: 10.1096/fj.03-1104fje. [DOI] [PubMed] [Google Scholar]
- 133.Figueiredo PA, Powers SK, Ferreira RM, Amado F, Appell HJ, Duarte JA. Impact of lifelong sedentary behavior on mitochondrial function of mice skeletal muscle. The Journals of Gerontology A. 2009;64(9):927–939. doi: 10.1093/gerona/glp066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kent-Braun JA, Ng AV. Skeletal muscle oxidative capacity in young and older women and men. Journal of Applied Physiology. 2000;89(3):1072–1078. doi: 10.1152/jappl.2000.89.3.1072. [DOI] [PubMed] [Google Scholar]
- 135.Lanza IR, Befroy DE, Kent-Braun JA. Age-related changes in ATP-producing pathways in human skeletal muscle in vivo. Journal of Applied Physiology. 2005;99(5):1736–1744. doi: 10.1152/japplphysiol.00566.2005. [DOI] [PubMed] [Google Scholar]
- 136.Lanza IR, Larsen RG, Kent-Braun JA. Effects of old age on human skeletal muscle energetics during fatiguing contractions with and without blood flow. The Journal of Physiology. 2007;583, part 3:1093–1105. doi: 10.1113/jphysiol.2007.138362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lopez-Lluch G, Irusta PM, Navas P, de Cabo R. Mitochondrial biogenesis and healthy aging. Experimental Gerontology. 2008;43(9):813–819. doi: 10.1016/j.exger.2008.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hood DA, Irrcher I, Ljubicic V, Joseph AM. Coordination of metabolic plasticity in skeletal muscle. The Journal of Experimental Biology. 2006;209, part 2:2265–2275. doi: 10.1242/jeb.02182. [DOI] [PubMed] [Google Scholar]
- 139.Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(29):12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Puigserver P, Rhee J, Lin J, et al. Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARgamma coactivator-1. Molecular Cell. 2001;8(5):971–982. doi: 10.1016/s1097-2765(01)00390-2. [DOI] [PubMed] [Google Scholar]
- 141.Knutti D, Kressler D, Kralli A. Regulation of the transcriptional coactivator PGC-1 via MAPK-sensitive interaction with a repressor. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(17):9713–9718. doi: 10.1073/pnas.171184698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Akimoto T, Pohnert SC, Li P, et al. Exercise stimulates Pgc-1alpha transcription in skeletal muscle through activation of the p38 MAPK pathway. The Journal of Biological Chemistry. 2005;280(20):19587–19593. doi: 10.1074/jbc.M408862200. [DOI] [PubMed] [Google Scholar]
- 143.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature. 2005;434(7029):113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 144.Gerhart-Hines Z, Rodgers JT, Bare O, et al. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. The EMBO Journal. 2007;26(7):1913–1923. doi: 10.1038/sj.emboj.7601633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wu Z, Huang X, Feng Y, et al. Transducer of regulated CREB-binding proteins (TORCs) induce PGC-1α transcription and mitochondrial biogenesis in muscle cells. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(39):14379–14384. doi: 10.1073/pnas.0606714103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92(6):829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 147.Puigserver P, Adelmant G, Wu Z, et al. Activation of PPARγ coactivator-1 through transcription factor docking. Science. 1999;286(5443):1368–1371. doi: 10.1126/science.286.5443.1368. [DOI] [PubMed] [Google Scholar]
- 148.Virbasius JV, Virbasius CA, Scarpulla RC. Identity of GABP with NRF-2, a multisubunit activator of cytochrome oxidase expression, reveals a cellular role for and ETS domain activator of viral promoters. Genes and Development. 1993;7(3):380–392. doi: 10.1101/gad.7.3.380. [DOI] [PubMed] [Google Scholar]
- 149.Virbasius CMA, Virbasius JV, Scarpulla RC. NRF-1, an activator involved in nuclear-mitochondrial interactions, utilizes a new DNA-binding domain conserved in a family of developmental regulators. Genes and Development. 1993;7(12 A):2431–2445. doi: 10.1101/gad.7.12a.2431. [DOI] [PubMed] [Google Scholar]
- 150.Scarpulla RC. Nuclear control of respiratory chain expression by nuclear respiratory factors and PGC-1-related coactivator. Annals of the New York Academy of Sciences. 2008;1147:321–334. doi: 10.1196/annals.1427.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Virbasius JV, Scarpulla RC. Activation of the human mitochondrial transcription factor A gene by nuclear respiratory factors: a potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(4):1309–1313. doi: 10.1073/pnas.91.4.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Hood DA, Joseph AM. Mitochondrial assembly: protein import. The Proceedings of the Nutrition Society. 2004;63(2):293–300. doi: 10.1079/PNS2004342. [DOI] [PubMed] [Google Scholar]
- 153.Diaz F, Moraes CT. Mitochondrial biogenesis and turnover. Cell Calcium. 2008;44(1):24–35. doi: 10.1016/j.ceca.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hood DA, Adhihetty PJ, Colavecchia M, et al. Mitochondrial biogenesis and the role of the protein import pathway. Medicine and Science in Sports and Exercise. 2003;35(1):86–94. doi: 10.1097/00005768-200301000-00015. [DOI] [PubMed] [Google Scholar]
- 155.Neupert W, Herrmann JM. Translocation of proteins into mitochondria. Annual Review of Biochemistry. 2007;76:723–749. doi: 10.1146/annurev.biochem.76.052705.163409. [DOI] [PubMed] [Google Scholar]
- 156.Gleyzer N, Vercauteren K, Scarpulla RC. Control of mitochondrial transcription specificity factors (TFB1M and TFB2M) by nuclear respiratory factors (NRF-1 and NRF-2) and PGC-1 family coactivators. Molecular and Cellular Biology. 2005;25(4):1354–1366. doi: 10.1128/MCB.25.4.1354-1366.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Reznick RM, Zong H, Li J, et al. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metabolism. 2007;5(2):151–156. doi: 10.1016/j.cmet.2007.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Wenz T, Rossi SG, Rotundo RL, Spiegelman BM, Moraes CT. Increased muscle PGC-1α expression protects from sarcopenia and metabolic disease during aging. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(48):20405–20410. doi: 10.1073/pnas.0911570106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 159.Bevilacqua L, Ramsey JJ, Hagopian K, Weindruch R, Harper ME. Effects of short- and medium-term calorie restriction on muscle mitochondrial proton leak and reactive oxygen species production. American Journal of Physiology. 2004;286(5):E852–E861. doi: 10.1152/ajpendo.00367.2003. [DOI] [PubMed] [Google Scholar]
- 160.Lezza AMS, Pesce V, Cormio A, et al. Increased expression of mitochondrial transcription factor A and nuclear respiratory factor-1 in skeletal muscle from aged human subjects. FEBS Letters. 2001;501(1–3):74–78. doi: 10.1016/s0014-5793(01)02628-x. [DOI] [PubMed] [Google Scholar]
- 161.Frazier AE, Kiu C, Stojanovski D, Hoogenraad NJ, Ryan MT. Mitochondrial morphology and distribution in mammalian cells. Biological Chemistry. 2006;387(12):1551–1558. doi: 10.1515/BC.2006.193. [DOI] [PubMed] [Google Scholar]
- 162.Benard G, Karbowski M. Mitochondrial fusion and division: regulation and role in cell viability. Seminars in Cell & Developmental Biology. 2009;20(3):365–374. doi: 10.1016/j.semcdb.2008.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Stojanovski D, Koutsopoulos OS, Okamoto K, Ryan MT. Levels of human Fis1 at the mitochondrial outer membrane regulate mitochondrial morphology. Journal of Cell Science. 2004;117, part 7:1201–1210. doi: 10.1242/jcs.01058. [DOI] [PubMed] [Google Scholar]
- 164.Chan DC. Mitochondrial fusion and fission in mammals. Annual Review of Cell and Developmental Biology. 2006;22:79–99. doi: 10.1146/annurev.cellbio.22.010305.104638. [DOI] [PubMed] [Google Scholar]
- 165.Romanello V, Guadagnin E, Gomes L, et al. Mitochondrial fission and remodelling contributes to muscle atrophy. The EMBO Journal. 2010;29(10):1774–1785. doi: 10.1038/emboj.2010.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Scheckhuber CQ, Wanger RA, Mignat CA, Osiewacz HD. Unopposed mitochondrial fission leads to severe lifespan shortening. Cell Cycle. 2011;10(18):3105–3110. doi: 10.4161/cc.10.18.17196. [DOI] [PubMed] [Google Scholar]
- 167.Lee S, Jeong SY, Lim WC, et al. Mitochondrial fission and fusion mediators, hFis1 and OPA1, modulate cellular senescence. Journal of Biological Chemistry. 2007;282(31):22977–22983. doi: 10.1074/jbc.M700679200. [DOI] [PubMed] [Google Scholar]
- 168.Smirnova E, Shurland DL, Ryazantsev SN, van der Bliek AM. A human dynamin-related protein controls the distribution of mitochondria. The Journal of Cell Biology. 1998;143(2):351–358. doi: 10.1083/jcb.143.2.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ono T, Isobe K, Nakada K, Hayashi JI. Human cells are protected from mitochondrial dysfunction by complementation of DNA products in fused mitochondria. Nature Genetics. 2001;28(3):272–275. doi: 10.1038/90116. [DOI] [PubMed] [Google Scholar]
- 170.Sato A, Nakada K, Hayashi JI. Mitochondrial dynamics and aging: mitochondrial interaction preventing individuals from expression of respiratory deficiency caused by mutant mtDNA. Biochimica et Biophysica Acta. 2006;1763(5-6):473–481. doi: 10.1016/j.bbamcr.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 171.Kowald A, Kirkwood TB. Evolution of the mitochondrial fusion-fission cycle and its role in aging. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(25):10237–10242. doi: 10.1073/pnas.1101604108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kirkwood SP, Munn EA, Brooks GA. Mitochondrial reticulum in limb skeletal muscle. The American Journal of Physiology. 1986;3, part 1:C395–C402. doi: 10.1152/ajpcell.1986.251.3.C395. [DOI] [PubMed] [Google Scholar]
- 173.Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. The Journal of Biological Chemistry. 2005;280(28):26185–26192. doi: 10.1074/jbc.M503062200. [DOI] [PubMed] [Google Scholar]
- 174.Bach D, Pich S, Soriano FX, et al. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism: a novel regulatory mechanism altered in obesity. Journal of Biological Chemistry. 2003;278(19):17190–17197. doi: 10.1074/jbc.M212754200. [DOI] [PubMed] [Google Scholar]
- 175.Cipolat S, Rudka T, Hartmann D, et al. Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling. Cell. 2006;126(1):163–175. doi: 10.1016/j.cell.2006.06.021. [DOI] [PubMed] [Google Scholar]
- 176.Duvezin-Caubet S, Jagasia R, Wagener J, et al. Proteolytic processing of OPA1 links mitochondrial dysfunction to alterations in mitochondrial morphology. Journal of Biological Chemistry. 2006;281(49):37972–37979. doi: 10.1074/jbc.M606059200. [DOI] [PubMed] [Google Scholar]
- 177.Chen H, Vermulst M, Wang YE, et al. Mitochondrial fusion is required for mtdna stability in skeletal muscle and tolerance of mtDNA mutations. Cell. 2010;141(2):280–289. doi: 10.1016/j.cell.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Lodi R, Tonon C, Valentino ML, et al. Deficit of in vivo mitochondrial ATP production in OPA1-related dominant optic atrophy. Annals of Neurology. 2004;56(5):719–723. doi: 10.1002/ana.20278. [DOI] [PubMed] [Google Scholar]
- 179.Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Archives of Biochemistry and Biophysics. 2007;462(2):245–253. doi: 10.1016/j.abb.2007.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Wang K, Klionsky DJ. Mitochondria removal by autophagy. Autophagy. 2011;7(3):297–300. doi: 10.4161/auto.7.3.14502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Okamoto K, Kondo-Okamoto N, Ohsumi Y. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Developmental Cell. 2009;17(1):87–97. doi: 10.1016/j.devcel.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 182.Kanki T. Nix, a receptor protein for mitophagy in mammals. Autophagy. 2010;6(3):433–435. doi: 10.4161/auto.6.3.11420. [DOI] [PubMed] [Google Scholar]
- 183.Gu Y, Wang C, Cohen A. Effect of IGF-1 on the balance between autophagy of dysfunctional mitochondria and apoptosis. FEBS Letters. 2004;577(3):357–360. doi: 10.1016/j.febslet.2004.10.040. [DOI] [PubMed] [Google Scholar]
- 184.Zhang Y, Qi H, Taylor R, Xu W, Liu LF, Jin S. The role of autophagy in mitochondria maintenance: characterization of mitochondrial functions in autophagy-deficient S. cerevisiae strains. Autophagy. 2007;3(4):337–346. doi: 10.4161/auto.4127. [DOI] [PubMed] [Google Scholar]
- 185.Cavallini G, Donati A, Taddei M, Bergamini E. Evidence for selective mitochondrial autophagy and failure in aging. Autophagy. 2007;3(1):26–27. doi: 10.4161/auto.3268. [DOI] [PubMed] [Google Scholar]
- 186.Cuervo AM, Bergamini E, Brunk UT, Dröge W, Ffrench M, Terman A. Autophagy and aging: the importance of maintaining “clean” cells. Autophagy. 2005;1(3):131–140. doi: 10.4161/auto.1.3.2017. [DOI] [PubMed] [Google Scholar]
- 187.Wohlgemuth SE, Seo AY, Marzetti E, Lees HA, Leeuwenburgh C. Skeletal muscle autophagy and apoptosis during aging: effects of calorie restriction and life-long exercise. Experimental Gerontology. 2010;45(2):138–148. doi: 10.1016/j.exger.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Masiero E, Sandri M. Autophagy inhibition induces atrophy and myopathy in adult skeletal muscles. Autophagy. 2010;6(2):307–309. doi: 10.4161/auto.6.2.11137. [DOI] [PubMed] [Google Scholar]
- 189.Wu JJ, Quijano C, Chen E, et al. Mitochondrial dysfunction and oxidative stress mediate the physiological impairment induced by the disruption of autophagy. Aging. 2009;1(4):425–437. doi: 10.18632/aging.100038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Vellai T, Takacs-Vellai K, Sass M, Klionsky DJ. The regulation of aging: does autophagy underlie longevity? Trends in Cell Biology. 2009;19(10):487–494. doi: 10.1016/j.tcb.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Taylor EB, Rutter J. Mitochondrial quality control by the ubiquitin-proteasome system. Biochemical Society Transactions. 2011;39(5):1509–1513. doi: 10.1042/BST0391509. [DOI] [PubMed] [Google Scholar]
- 192.Low P. The role of ubiquitin-proteasome system in ageing. General and Comparative Endocrinology. 2011;172(1):39–43. doi: 10.1016/j.ygcen.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 193.Altun M, Besche HC, Overkleeft HS, et al. Muscle wasting in aged, sarcopenic rats is associated with enhanced activity of the ubiquitin proteasome pathway. Journal of Biological Chemistry. 2010;285(51):39597–39608. doi: 10.1074/jbc.M110.129718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Whitman SA, Wacker MJ, Richmond SR, Godard MP. Contributions of the ubiquitin-proteasome pathway and apoptosis to human skeletal muscle wasting with age. Pflugers Archiv European Journal of Physiology. 2005;450(6):437–446. doi: 10.1007/s00424-005-1473-8. [DOI] [PubMed] [Google Scholar]
- 195.Dalbo VJ, Roberts MD, Hassell SE, Brown RD, Kerksick CM. Effects of age on serum hormone concentrations and intramuscular proteolytic signaling before and after a single bout of resistance training. The Journal of Strength & Conditioning Research. 2011;25(1):1–9. doi: 10.1519/JSC.0b013e3181fc5a68. [DOI] [PubMed] [Google Scholar]
- 196.Lee CK, Klopp RG, Weindruch R, Prolla TA. Gene expression profile of aging and its retardation by caloric restriction. Science. 1999;285(5432):1390–1393. doi: 10.1126/science.285.5432.1390. [DOI] [PubMed] [Google Scholar]
- 197.Strucksberg KH, Tangavelou K, Schröder R, Clemen CS. Proteasomal activity in skeletal muscle: a matter of assay design, muscle type, and age. Analytical Biochemistry. 2010;399(2):225–229. doi: 10.1016/j.ab.2009.12.026. [DOI] [PubMed] [Google Scholar]
- 198.Cai D, Lee KK, Li M, Tang MK, Chan KM. Ubiquitin expression is up-regulated in human and rat skeletal muscles during aging. Archives of Biochemistry and Biophysics. 2004;425(1):42–50. doi: 10.1016/j.abb.2004.02.027. [DOI] [PubMed] [Google Scholar]
- 199.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281(5381):1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 200.Jeong SY, Seol DW. The role of mitochondria in apoptosis. BMB Reports. 2008;41(1):11–22. doi: 10.5483/bmbrep.2008.41.1.011. [DOI] [PubMed] [Google Scholar]
- 201.Marzetti E, Hwang JC, Lees HA, et al. Mitochondrial death effectors: relevance to sarcopenia and disuse muscle atrophy. Biochimica et Biophysica. 2010;1800(3):235–244. doi: 10.1016/j.bbagen.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Suen DF, Norris KL, Youle RJ. Mitochondrial dynamics and apoptosis. Genes & Development. 2008;22(12):1577–1590. doi: 10.1101/gad.1658508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Dirks AJ, Leeuwenburgh C. The role of apoptosis in age-related skeletal muscle atrophy. Sports Medicine. 2005;35(6):473–483. doi: 10.2165/00007256-200535060-00002. [DOI] [PubMed] [Google Scholar]
- 204.Marzetti E, Leeuwenburgh C. Skeletal muscle apoptosis, sarcopenia and frailty at old age. Experimental Gerontology. 2006;41(12):1234–1238. doi: 10.1016/j.exger.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 205.Malmgren LT, Jones CE, Bookman LM. Muscle fiber and satellite cell apoptosis in the aging human thyroarytenoid muscle: a stereological study with confocal laser scanning microscopy. Otolaryngology. 2001;125(1):34–39. doi: 10.1067/mhn.2001.116449. [DOI] [PubMed] [Google Scholar]
- 206.Koseki T, Inohara N, Chen S, Nunez G. ARC, an inhibitor of apoptosis expressed in skeletal muscle and heart that interacts selectively with caspases. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(9):5156–5160. doi: 10.1073/pnas.95.9.5156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Park SY, Kim HY, Lee JH, Yoon KH, Chang MS, Park SK. The age-dependent induction of apoptosis-inducing factor (AIF) in the human semitendinosus skeletal muscle. Cellular and Molecular Biology Letters. 2010;15(1):1–12. doi: 10.2478/s11658-009-0030-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Tamilselvan J, Jayaraman G, Sivarajan K, Panneerselvam C. Age-dependent upregulation of p53 and cytochrome c release and susceptibility to apoptosis in skeletal muscle fiber of aged rats: role of carnitine and lipoic acid. Free Radical Biology and Medicine. 2007;43(12):1656–1669. doi: 10.1016/j.freeradbiomed.2007.08.028. [DOI] [PubMed] [Google Scholar]
- 209.Seo AY, Xu J, Servais S, et al. Mitochondrial iron accumulation with age and functional consequences. Aging Cell. 2008;7(5):706–716. doi: 10.1111/j.1474-9726.2008.00418.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Dirks A, Leeuwenburgh C. Apoptosis in skeletal muscle with aging. American Journal of Physiology Regulatory. 2002;282(2):519–527. doi: 10.1152/ajpregu.00458.2001. [DOI] [PubMed] [Google Scholar]
- 211.Marzetti E, Wohlgemuth SE, Lees HA, Chung HY, Giovannini S, Leeuwenburgh C. Age-related activation of mitochondrial caspase-independent apoptotic signaling in rat gastrocnemius muscle. Mechanisms of Ageing and Development. 2008;129(9):542–549. doi: 10.1016/j.mad.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Leeuwenburgh C, Gurley CM, Strotman BA, Dupont-Versteegden EE. Age-related differences in apoptosis with disuse atrophy in soleus muscle. American Journal of Physiology. 2005;288(5):R1288–R1296. doi: 10.1152/ajpregu.00576.2004. [DOI] [PubMed] [Google Scholar]
- 213.Siu PM, Pistilli EE, Alway SE. Apoptotic responses to hindlimb suspension in gastrocnemius muscles from young adult and aged rats. American Journal of Physiology. 2005;289(4):R1015–R1026. doi: 10.1152/ajpregu.00198.2005. [DOI] [PubMed] [Google Scholar]
- 214.Rice KM, Blough ER. Sarcopenia-related apoptosis is regulated differently in fast- and slow-twitch muscles of the aging F344/N × BN rat model. Mechanisms of Ageing and Development. 2006;127(8):670–679. doi: 10.1016/j.mad.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 215.McMillan EM, Quadrilatero J. Differential apoptosis-related protein expression, mitochondrial properties, proteolytic enzyme activity, and DNA fragmentation between skeletal muscles. American Journal of Physiology. 2011;300(3):R531–R543. doi: 10.1152/ajpregu.00488.2010. [DOI] [PubMed] [Google Scholar]
- 216.Adhihetty PJ, Ljubicic V, Menzies KJ, Hood DA. Differential susceptibility of subsarcolemmal and intermyofibrillar mitochondria to apoptotic stimuli. American Journal of Physiology. 2005;289(4):C994–C1001. doi: 10.1152/ajpcell.00031.2005. [DOI] [PubMed] [Google Scholar]
- 217.Holloszy JO. Biochemical adaptations in muscle. Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. Journal of Biological Chemistry. 1967;242(9):2278–2282. [PubMed] [Google Scholar]
- 218.Holloszy JO. Adaptations of muscular tissue to training. Progress in Cardiovascular Diseases. 1976;18(6):445–458. doi: 10.1016/0033-0620(76)90011-6. [DOI] [PubMed] [Google Scholar]
- 219.Johannsen DL, DeLany JP, Frisard MI, et al. Physical activity in aging: comparison among young, aged, and nonagenarian individuals. Journal of Applied Physiology. 2008;105(2):495–501. doi: 10.1152/japplphysiol.90450.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Coggan AR, Spina RJ, King DS, et al. Histochemical and enzymatic comparison of the gastrocnemius muscle of young and elderly men and women. Journals of Gerontology. 1992;47(3):B71–B76. doi: 10.1093/geronj/47.3.b71. [DOI] [PubMed] [Google Scholar]
- 221.Cooper JM, Mann VM, Schapira AH. Analyses of mitochondrial respiratory chain function and mitochondrial DNA deletion in human skeletal muscle: effect of ageing. Journal of the Neurological Sciences. 1992;113(1):91–98. doi: 10.1016/0022-510x(92)90270-u. [DOI] [PubMed] [Google Scholar]
- 222.Short KR, Bigelow ML, Kahl J, et al. Decline in skeletal muscle mitochondrial function with aging in humans. Proceedings of the National Academy of Sciences. 2005;102(15):5618–5623. doi: 10.1073/pnas.0501559102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Jubrias SA, Esselman PC, Price LB, Cress ME, Conley KE. Large energetic adaptations of elderly muscle to resistance and endurance training. Journal of Applied Physiology. 2001;90(5):1663–1670. doi: 10.1152/jappl.2001.90.5.1663. [DOI] [PubMed] [Google Scholar]
- 224.Menshikova EV, Ritov VB, Fairfull L, Ferrell RE, Kelley DE, Goodpaster BH. Effects of exercise on mitochondrial content and function in aging human skeletal muscle. The Journals of Gerontology A. 2006;61(6):534–540. doi: 10.1093/gerona/61.6.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Short KR. Mitochondrial ATP measurements. American Journal of Physiology Regulatory. 2004;287(1):R243–R245. doi: 10.1152/ajpregu.00606.2003. [DOI] [PubMed] [Google Scholar]
- 226.Song W, Kwak HB, Lawler JM. Exercise training attenuates age-induced changes in apoptotic signaling in rat skeletal muscle. Antioxidants and Redox Signaling. 2006;8(3-4):517–528. doi: 10.1089/ars.2006.8.517. [DOI] [PubMed] [Google Scholar]
- 227.Short KR, Vittone JL, Bigelow ML, Proctor DN, Nair KS. Age and aerobic exercise training effects on whole body and muscle protein metabolism. American Journal of Physiology. 2004;286(1):92–101. doi: 10.1152/ajpendo.00366.2003. [DOI] [PubMed] [Google Scholar]
- 228.Leeuwenburgh C, Fiebig R, Chandwaney R, Ji Li Li Aging and exercise training in skeletal muscle: responses of glutathione and antioxidant enzyme systems. American Journal of Physiology. 1994;267(2, part 2):R439–R445. doi: 10.1152/ajpregu.1994.267.2.R439. [DOI] [PubMed] [Google Scholar]
- 229.Parise G, Brose AN, Tarnopolsky MA. Resistance exercise training decreases oxidative damage to DNA and increases cytochrome oxidase activity in older adults. Experimental Gerontology. 2005;40(3):173–180. doi: 10.1016/j.exger.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 230.Parise G, Phillips SM, Kaczor JJ, Tarnopolsky MA. Antioxidant enzyme activity is up-regulated after unilateral resistance exercise training in older adults. Free Radical Biology and Medicine. 2005;39(2):289–295. doi: 10.1016/j.freeradbiomed.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 231.Colman RJ, Anderson RM, Johnson SC, et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009;325(5937):201–204. doi: 10.1126/science.1173635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Speakman JR, Mitchell SE. Caloric restriction. Molecular Aspects of Medicine. 2011;32(3):159–221. doi: 10.1016/j.mam.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 233.Merry BJ. Oxidative stress and mitochondrial function with aging—the effects of calorie restriction. Aging Cell. 2004;3(1):7–12. doi: 10.1046/j.1474-9728.2003.00074.x. [DOI] [PubMed] [Google Scholar]
- 234.Zainal TA, Oberley TD, Allison DB, Szweda LI, Weindruch R. Caloric restriction of rhesus monkeys lowers oxidative damage in skeletal muscle. The FASEB Journal. 2000;14(12):1825–1836. doi: 10.1096/fj.99-0881com. [DOI] [PubMed] [Google Scholar]
- 235.Drew B, Dirks PA, Selman C, et al. Effects of aging and caloric restriction on mitochondrial energy production in gastrocnemius muscle and heart. American Journal of Physiology. 2003;284(2):R474–R480. doi: 10.1152/ajpregu.00455.2002. [DOI] [PubMed] [Google Scholar]
- 236.Lass A, Sohal BH, Weindruch R, Forster MJ, Sohal RS. Caloric restriction prevents age-associated accrual of oxidative damage to mouse skeletal muscle mitochondria. Free Radical Biology and Medicine. 1998;25(9):1089–1097. doi: 10.1016/s0891-5849(98)00144-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Bevilacqua L, Ramsey JJ, Hagopian K, Weindruch R, Harper ME. Long-term caloric restriction increases UCP3 content but decreases proton leak and reactive oxygen species production in rat skeletal muscle mitochondria. American Journal of Physiology. 2005;289(3):E429–E438. doi: 10.1152/ajpendo.00435.2004. [DOI] [PubMed] [Google Scholar]
- 238.Hamilton ML, Van Remmen H, Drake JA, et al. Does oxidative damage to DNA increase with age? Proceedings of the National Academy of Sciences of the United States of America. 2001;98(18):10469–10474. doi: 10.1073/pnas.171202698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Usuki F, Yasutake A, Umehara F, Higuchi I. Beneficial effects of mild lifelong dietary restriction on skeletal muscle: prevention of age-related mitochondrial damage, morphological changes, and vulnerability to a chemical toxin. Acta Neuropathologica. 2004;108(1):1–9. doi: 10.1007/s00401-004-0844-0. [DOI] [PubMed] [Google Scholar]
- 240.Lee CM, Aspnes LE, Chung SS, Weindruch R, Aiken JM. Influences of caloric restriction on age-associated skeletal muscle fiber characteristics and mitochondrial changes in rats and mice. Annals of the New York Academy of Sciences. 1998;854:182–191. doi: 10.1111/j.1749-6632.1998.tb09901.x. [DOI] [PubMed] [Google Scholar]
- 241.Civitarese AE, Carling S, Heilbronn LK, et al. Calorie restriction increases muscle mitochondrial biogenesis in healthy humans. PLoS Medicine. 2007;4(3):p. e76. doi: 10.1371/journal.pmed.0040076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Sreekumar R, Unnikrishnan J, Fu A, et al. Effects of caloric restriction on mitochondrial function and gene transcripts in rat muscle. American Journal of Physiology. 2002;283(1):E38–E43. doi: 10.1152/ajpendo.00387.2001. [DOI] [PubMed] [Google Scholar]
- 243.Lopez-Lluch G, Hunt N, Jones B, et al. Calorie restriction induces mitochondrial biogenesis and bioenergetic efficiency. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(6):1768–1773. doi: 10.1073/pnas.0510452103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Lal SB, Ramsey JJ, Monemdjou S, Weindruch R, Harper ME. Effects of caloric restriction on skeletal muscle mitochondrial proton leak in aging rats. Journals of Gerontology A. 2001;56(3):B116–B122. doi: 10.1093/gerona/56.3.b116. [DOI] [PubMed] [Google Scholar]
- 245.Hancock CR, Han DH, Higashida K, Kim SH, Holloszy JO. Does calorie restriction induce mitochondrial biogenesis? A reevaluation. The FASEB Journal. 2011;25(2):785–791. doi: 10.1096/fj.10-170415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Desai VG, Weindruch R, Hart RW, Feuers RJ. Influences of age and dietary restriction on gastrocnemius electron transport system activities in mice. Archives of Biochemistry and Biophysics. 1996;333(1):145–151. doi: 10.1006/abbi.1996.0375. [DOI] [PubMed] [Google Scholar]
- 247.Bua E, McKiernan SH, Aiken JM. Calorie restriction limits the generation but not the progression of mitochondrial abnormalities in aging skeletal muscle. The FASEB Journal Biology. 2004;18(3):582–584. doi: 10.1096/fj.03-0668fje. [DOI] [PubMed] [Google Scholar]
- 248.McKiernan SH, Colman RJ, Lopez M, et al. Caloric restriction delays aging-induced cellular phenotypes in rhesus monkey skeletal muscle. Experimental Gerontology. 2011;46(1):23–29. doi: 10.1016/j.exger.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Gonzalez AA, Kumar R, Mulligan JD, Davis AJ, Weindruch R, Saupe KW. Metabolic adaptations to fasting and chronic caloric restriction in heart, muscle, and liver do not include changes in AMPK activity. American Journal of Physiology. 2004;287(5):E1032–E1037. doi: 10.1152/ajpendo.00172.2004. [DOI] [PubMed] [Google Scholar]
- 250.Shinmura K, Tamaki K, Bolli R. Short-term caloric restriction improves ischemic tolerance independent of opening of ATP-sensitive K+ channels in both young and aged hearts. Journal of Molecular and Cellular Cardiology. 2005;39(2):285–296. doi: 10.1016/j.yjmcc.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 251.Wang ZQ, Floyd ZE, Qin J, et al. Modulation of skeletal muscle insulin signaling with chronic caloric restriction in cynomolgus monkeys. Diabetes. 2009;58(7):1488–1498. doi: 10.2337/db08-0977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Dirks AJ, Leeuwenburgh C. Aging and lifelong calorie restriction result in adaptations of skeletal muscle apoptosis repressor, apoptosis-inducing factor, X-linked inhibitor of apoptosis, caspase-3, and caspase-12. Free Radical Biology and Medicine. 2004;36(1):27–39. doi: 10.1016/j.freeradbiomed.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 253.Marzetti E, Lawler JM, Hiona A, Manini T, Seo AY, Leeuwenburgh C. Modulation of age-induced apoptotic signaling and cellular remodeling by exercise and calorie restriction in skeletal muscle. Free Radical Biology and Medicine. 2008;44(2):160–168. doi: 10.1016/j.freeradbiomed.2007.05.028. [DOI] [PubMed] [Google Scholar]
- 254.Phillips T, Leeuwenburgh C. Muscle fiber specific apoptosis and TNF-alpha signaling in sarcopenia are attenuated by life-long calorie restriction. The FASEB Journal. 2005;19(6):668–670. doi: 10.1096/fj.04-2870fje. [DOI] [PubMed] [Google Scholar]
- 255.Lee HY, Choi CS, Birkenfeld AL, et al. Targeted expression of catalase to mitochondria prevents age-associated reductions in mitochondrial function and insulin resistance. Cell Metabolism. 2010;12(6):668–674. doi: 10.1016/j.cmet.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Marzetti E, Lees HA, Wohlgemuth SE, Leeuwenburgh C. Sarcopenia of aging: underlying cellular mechanisms and protection by calorie restriction. BioFactors. 2009;35(1):28–35. doi: 10.1002/biof.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Payne AM, Dodd SL, Leeuwenburgh C. Life-long calorie restriction in Fischer 344 rats attenuates age-related loss in skeletal muscle-specific force and reduces extracellular space. Journal of Applied Physiology. 2003;95(6):2554–2562. doi: 10.1152/japplphysiol.00758.2003. [DOI] [PubMed] [Google Scholar]
- 258.McKiernan SH, Bua E, McGorray J, Aiken J. Early-onset calorie restriction conserves fiber number in aging rat skeletal muscle. The FASEB Journal. 2004;18(3):580–581. doi: 10.1096/fj.03-0667fje. [DOI] [PubMed] [Google Scholar]
- 259.Lagouge M, Argmann C, Gerhart-Hines Z, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1α . Cell. 2006;127(6):1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 260.Um JH, Park SJ, Kang H, et al. AMP-activated protein kinase-deficient mice are resistant to the metabolic effects of resveratrol. Diabetes. 2010;59(3):554–563. doi: 10.2337/db09-0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Baur JA, Pearson KJ, Price NL, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444(7117):337–342. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Timmers S, Konings E, Bilet L, et al. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metabolism. 2011;14(5):612–622. doi: 10.1016/j.cmet.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Pacholec M, Bleasdale JE, Chrunyk B, et al. SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. The Journal of Biological Chemistry. 2010;285(11):8340–8351. doi: 10.1074/jbc.M109.088682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Barger JL, Kayo T, Vann JM, et al. A low dose of dietary resveratrol partially mimics caloric restriction and retards aging parameters in mice. PLoS ONE. 2008;3(6):p. e2264. doi: 10.1371/journal.pone.0002264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Ungvari Z, Sonntag WE, de Cabo R, Baur JA, Csiszar A. Mitochondrial protection by resveratrol. Exercise and Sport Sciences Reviews. 2011;39(3):128–132. doi: 10.1097/JES.0b013e3182141f80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Murase T, Haramizu S, Ota N, Hase T. Suppression of the aging-associated decline in physical performance by a combination of resveratrol intake and habitual exercise in senescence-accelerated mice. Biogerontology. 2009;10(4):423–434. doi: 10.1007/s10522-008-9177-z. [DOI] [PubMed] [Google Scholar]
- 267.Jackson JR, Ryan MJ, Hao Y, Alway SE. Mediation of endogenous antioxidant enzymes and apoptotic signaling by resveratrol following muscle disuse in the gastrocnemius muscles of young and old rats. American Journal of Physiology. 2010;299(6):R1572–R1581. doi: 10.1152/ajpregu.00489.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Jackson JR, Ryan MJ, Alway SE. Long-term supplementation with resveratrol alleviates oxidative stress but does not attenuate sarcopenia in aged mice. The Journals of Gerontology A. 2011;66(7):751–764. doi: 10.1093/gerona/glr047. [DOI] [PMC free article] [PubMed] [Google Scholar]