

Abstract
Alzheimer’s disease is characterized in part by extracellular aggregation of the amyloid-β peptide in the form of diffuse and fibrillar plaques in the brain. Electron microscopy (EM) has made an important contribution in understanding of the structure of amyloid plaques in humans. Classical EM studies have revealed the architecture of the fibrillar core, characterized the progression of neuritic changes, and have identified the neurofibrillary tangles formed by paired helical filaments (PHF) in degenerating neurons. Clinical data has strongly correlated cognitive impairment in AD with the substantial synapse loss observed in these early ultrastructural studies. Animal models of AD-type brain amyloidosis have provided excellent opportunities to study amyloid and neuritic pathology in detail and establish the role of neurons and glia in plaque formation. Transgenic mice overexpressing mutant amyloid precursor protein (APP) alone with or without mutant presenilin 1 (PS1), have shown that brain amyloid plaque development and structure grossly recapitulate classical findings in humans. Transgenic APP/PS1 mice expressing human apolioprotein E isoforms also develop amyloid plaque deposition. However no ultrastructural data has been reported for these animals. Here we show results from detailed EM analysis of amyloid plaques in APP/PS1 mice expressing human isoforms of ApoE and compare these findings with EM data in other transgenic models and in human AD. Our results show that similar to other transgenic animals, APP/PS1 mice expressing human ApoE isoforms share all major cellular and subcellular degenerative features and highlight the identity of the cellular elements involved in Aβ deposition and neuronal degeneration.
Keywords: Alzheimer’s disease, electron microscopy, amyloid precursor protein, presenilin 1, ApoE
Introduction
Amyloid plaques are a defining feature of Alzheimer disease (AD) pathology. AD (senile) plaques are composed of extracellular deposits of amyloid-β (Aβ) and other plaque associated proteins and are surrounded by dystrophic neurites and glia. Robert Terry and Michael Kidd presented very detailed morphological studies of the plaque core, described the extensive neuritic changes as well as the accumulation of intraneuronal neurofibrillary tangles formed by paired helical filaments (PHF) [1-5]. In all cases of Alzheimer’s presenile dementia, Kidd et al., described the plaque’s structure as composed of a “central fibrillar core”, “cellular perikarya”, “axons and dendrites filled with an excess of neurofibrils”, and “cell processes filled with dense bodies” [4]. Almost no degenerating neuronal cell bodies were identified. In 1973 Wisniewski and Terry showed that the earliest sign of plaque formation was the appearance of few abnormal neurites, followed by “wisps” of deposited amyloid [6]. Later EM studies by Yamaguchi et al., characterized the structure of the “diffuse plaque” with the appearance of small amounts of amyloid fibrils and minimal neuritic dystrophy [7]. A number of ultrastructural studies in humans and also in aging primates documented a progressive decrease in synaptic numbers in cortical areas including the hippocampus [8-10]. Subsequent clinical studies have strongly correlated these findings with cognitive decline [11-13].
The generation of transgenic (tg) animals as models overexpressing mutant human APP and/or PS1 and PS2, tg APP mice expressing human ApoE isoforms, as well as Tau, has made substantial contributions to unraveling the diverse CNS changes developing in AD [14-18]. Tg and other genetically modified animals have been invaluable for revealing mechanisms of AD neuropathology [19-22], and while no mouse model has fully recapitulated the entire neuropathological spectrum of AD-like brain lesions, they have replicated major pathological features such as the diffuse and compact amyloid plaque, the massive neuritic dystrophy, synaptic deficits, and glial activation [23-26]. The first mouse models were based on the overexpression of single or multiple mutant molecules associated with familial AD (FAD), [18]. Masliah et al. in 1996 made a detailed comparison of the neurodegenerative pathology found in the PDAPP tg mouse line and in AD [23]. Although PHF were not found, the overproduction of mutant human APP was sufficient to cause extensive amyloid fibril deposition in the form of diffuse and mature amyloid plaques between 8 and 12 months of age. Axonal dystrophy, microglial and astroglial involvement have been reported as well [23,24,27]. Other APP tg models have all produced mature amyloid plaques [28-34]. In APP tg mice expressing human mutant forms of APP found in FAD, Masliah et al., Phiney et al., and Stalder et al., also showed evidence for extensive amyloid plaque formation, microglial cell process aggregation and a corona of dystrophic axons [23,30,35]. Interestingly, tg mice overexpressing the mutant PS1 alone have not been shown to accumulate Aβ in the brain [36] while, co-expression of mutant PS1 with APP has been always associated with robust amyloid deposition months earlier than APP tg mice alone. Kurt et al., provided a very detailed ultrastructural study on these mice [25]. They showed occasional Aβ deposits (plaques) as early as two months of age, with deposits gradually increasing in number at six months. Notably, none of the single transgenic (APP or PS1) animals made Aβ deposits at this timeframe. Electron microscopy characterized two main morphological plaque forms, those with an Aβ core and those without it. Both forms were present at the 12th week. Plaques were observed in the neuropil and also in the white matter. In mature plaques of 6 and 8 month old tg mice, the core was similarly surrounded by microglial cell processes; astroglial cell processes were also encountered, especially in older animals. Neuronal degeneration of the non-apoptotic type was registered close to the plaque in 8 month old double tg mice. In a similar model, Wirths et al., described neuritic plaques in the spinal cord and found ultrastructural evidence for massive axonal degeneration in white matter tracts structurally resembling Wallerian type degeneration [37]. Triple tg mouse models overexpressing mutant APP, PS1 and Tau developed amyloid plaques and tangle-like pathology [26,38].
The ε4 allele of apolipoprotein E (ApoE) is known so far to be the only firmly established risk factor for the more common sporadic forms of AD [39]. Astrocyte-specific expression of human ApoE3 and E4 isoforms in APP tg mice where the mouse apoE has been knocked out, restored Aβ and fibrillar Aβ deposition in the brain in vivo, suggesting their critical role in amyloid deposition with E4 resulting in greater Aβ pathology than E3 [40]. Pathways involved in ApoE and/or Aβ clearance have shown that overexpression of the low density lipoprotein receptor markedly inhibits in vivo amyloid deposition and in addition increases Aβ clearance [41]. However to date there are no ultrastructural studies to complement the light microscopic and functional data from tg animals expressing human apoE and no EM comparison is available with other models or AD. The aim of the current study is to provide a detailed ultrastructural investigation of amyloid plaques in APP/PS1 tg mice expressing each one of the three human apolipoprotein isoforms (ApoE2, ApoE3 or ApoE4) and to compare the neuritic plaque morphology to other existing tg animal models of AD.
Material and methods
Generation of human APOE isoform mice with APPswe/PS1(L166P) mutant transgenes
To determine the effect of human APOE2, APOE3 and APOE4 on amyloid deposition, we used APOE knock-in mouse models in which the endogenous murine APOE gene is replaced with the human APOE2, APOE3 or APOE4 gene. The generation of human APOE2, APOE3 and APOE4 knock-in mouse models has been reported previously and breeding pairs were gifts from Dr. Patrick Sullivan (Duke Universtity) [42,43]. APOE isoform mice were maintained on a C57BL/6J background. APPPS1-21 mice overexpress a human APP with Swedish mutation (KM670/671NL) and PS1 with L166P mutation under the control of Thy1 promoter. Breeding pairs were obtained as a gift from Dr. Mathias Jucker (University of Tubingen) [44]. To replace the murine APOE (mAPOE) gene with human APOE isoforms, we recently described the production of APPPS1-21/APOE knockin mice [45]. Briefly, APPPS1-21 mice were bred with APOE2/2, APOE3/3 or APOE4/4 knock-in mice. For example, APPPS1-21/APOE3/mAPOE mice and APOE3/mAPOE mice from the first generation were bred with each other to generate APPPS1-21/APOE3/3 and APOE3/3 mice. APPPS1-21/APOE3/3 and APOE3/3 mice from the second generation were bred to generate more APPPS1-21/APOE3/3 mice. After successful generation of APPPS1-21/APOE3/3 mice at the third generation, APPPS1-21/APOE3/3 mice were bred with APOE knockout (APOE-/-) mice on a C57BL/6J background (Stock number 002052, The Jackson Laboratory) [46]. APPPS1-21/APOE3/- mice from the fourth generation were bred with APOE3/3 mice to generate APPPS1-21/APOE3/3 mice. Littermates generated at the fifth generation were analyzed for Aβ accumulation, amyloid deposition, and microgliosis. Similarly, APPPS1-21/APOE2/2 and APPPS1-21/APOE4/4 mice were generated by five generations of breeding using the same mating strategy. All mice used in this study were maintained on a C57BL/6J background.
Transmission electron microscopy
Six months old mice (3 from each isoform-specific type and 3 age matched controls) were anesthetized with pentobarbital and perfused transcardially with a fixative composed of 1.5% glutaraldehyde and 1% paraformaldehyde in 0.1 M cacodylate buffer (pH 7.4). The brains were sliced into 1-mm-thick coronal sections and fixed for 24 hrs in 1.5% glutaraldehyde and then rinsed in cacodylate buffer. Sections were postfixed overnight in 1% osmium tetroxide. Tissue was dehydrated in graded solutions of ethanol from 40% to 100% (each for 10 min), cleared in propylene oxide, and flat-embedded in araldite. Ultrathin sections were cut with a Leica Ultracut UCT ultramicrotome. Sections were counterstained with saturated aqueous uranyl acetate, followed by lead citrate and analyzed with a JEOL 100CX transmission electron microscope.
Results
Overview of amyloid plaques
Investigation of semithin sections in all APP/PS1/HumanApoE knock-in animals revealed the presence of cortical (Figure 1) and subcorti-cal plaques. In all 6 months old APP/PS1/ HumanApoE knock-in mice, the vast majority of amyloid plaques fell in the category of the mature plaque. Ultrastructurally no gross morphological difference was detected between the three different knock-in transgenes, except that in APP/PS1/HumanApoE2 and E3 knock-in brains plaques had smaller core centers.
Figure 1.

Low and high magnification images of plastic sections showing cortical plaques in APP/PS1/Human ApoE knock-in tg mice. A and D – APP/PS1/ApoE2, B and E - APP/PS1/ApoE3, C and F - APP/PS1/ApoE4 tg mice. Scale bar: A, B and C – 100 μm; D, E and F – 20 μm.
Amyloid plaque epicenter
Ultrastructural investigation of amyloid plaques in ApoE2, ApoE3 and ApoE4 knock-in mice revealed that all mature plaques were represented by an epicenter of compacted Aβ fibrils, most of which were organized in parallel radial bundles giving the core a star-shape outline (Figure 2). In some, fibril bundles surrounded dark irregular and highly osmiophilic structures, probably representing terminally dystrophic cell processes (not shown). At high magnifications individual fibrils contained hollow lumina (Figure 3A, inset). Very rarely axonal terminals with small clear vesicles were observed (not shown), but in general, the core epicenter was surrounded by microglial cell processes (Figure 2 asterisks). Rarely single dystrophic myelinated axons and dystrophic dendritic profiles were observed.
Figure 2.

Panoramic view of a mature amyloid plaque in APP/PS1/ApoE4 tg mouse. Single black asterisks show processes of microglial cells surrounding the core epicenter, double asterisks show an astrocytic process. Scale bar – 5 μm.
Figure 3.

Electron micrograph showning microglial cell processes with numerous invaginations (infoldings). Infoldings contain bundles of amyliid fibrils (*). Inset in A. shows fibrils at higher magnification (arrow). B. Amyloid fibril-filled infoldings (*) seen very close to the RER (arrows) of a microglial cell. C. Microglial cell nuclear region (*) close to dystrophic myelinated neurite (DN). Scale bars: A and B - 0.3μm; C - 1 μm.
Microglial cells and their processes surround the amyloid plaque core
The immediate periphery of the plaque epicenter was surrounded by a crown of microglial cytoplasmic processes (Figure 2, single asterisks). These processes typically contained short cisternae of rough endoplasmic reticulum (RER) as well as free ribosomes (Figure 3B). A microglial cell nuclear region was very rarely observed (Figure 3C). The identification of microglial cells and their processes was made by their specific EM appearance and also after careful comparison of our data to images presented by Mattiace et al., and Stalder et al., where detailed EM immunohistocheimical characterization of these cells and their long processes has been presented [47,35]. Virtually all microglial cell processes had numerous invaginations filled by amyloid fibrils (Figure 3 asterisks, Figure 3A inset). Many of these fibril-filled spaces were very close to cisternae of RER (Figure 3B arrows). Lysosomes and phagolysosomes were frequently detected in these processes, while others were filled with debris from surrounding dystrophic neurites (Figure 4). Amyloid fibrils were arranged in loose parallel arrays similar to those detected by others in early diffuse plaques, other plaque deposits resembled a fibrillar lattice. The plaque core contained few astroglial cell processes as well (Figure 2), which in general had a lighter cellular matrix compared to microglial cells, and were devoid of rough endoplasmic reticulum and typically contained slender intermediate filament bundles (not shown).
Figure 4.
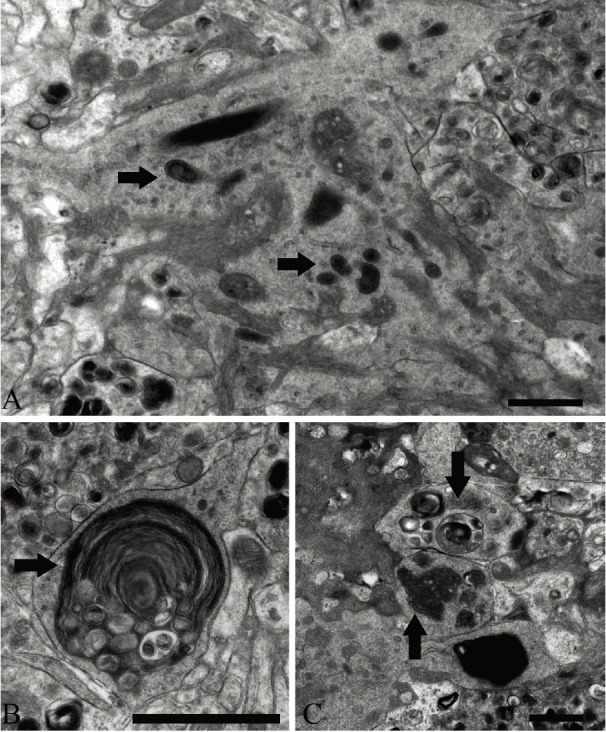
A. Electron micrograph showing processes of activated microglia exhibiting phagocytosis. Note also the deep invaginations filled with amyloid fibrils. Arrows point to lysosomes. B. Microglial process with internalized segment of a dystrophic myelinated axon (arrow). C. Microglial cell process with phagocytozed cellular debris (arrows). Scale bar: A and C – 500 nm, B – 1.5 μm.
Immediate periphery of the plaque core: numerous dystrophic neurites
All mature plaques presented with numerous dystrophic neurites with dense bodies and vacuoles filled with lamellar material. They were found predominantly in the immediate periphery surrounding the microglial processes (Figures 2, 3, 4 and 5). The majority were in an advanced stage of degeneration (Figure 5C-I). Almost all were non-myelinated (Figure 2 and 5). Neuritic processes with signs of mild dystrophy contained combinations of clear synaptic vesicles and few large dense core vesicles, in others normal single or multiple mitochondria were also observed (Figure 5A). Profiles showing moderate signs of dystrophy exhibited autophagic vacuoles filled with lamellar material and/or degenerating mitochondria (Figure 5A inset 5C). A number of degenerating mitochondria were frequently seen in profiles with developed dystrophy, where lysosomes were also identified (Figure 5A inset, 5B and 5C). In neuritic processes with more advanced degeneration there were abundant autophagic vacuoles, phagolysosomes and virtually no normal organelles (Figure 5D-H). In a number of neuritic processes we were not able to detect a continuous cytoplasmic membrane (Figure 5I, asterisk) and borders between these processes and the neighboring neuropil was not discernable (Figure 5I, arrows). Occasionally single Hirano-like bodies were observed in dystrophic profiles (Figure 9). Interestingly we were also able to detect a number of dystrophic processes, which maintained morphologically distinct synapses on seemingly intact dendritic spines or shafts (Figure 6A-C). They were identified as axonal terminals. While the degree of degeneration in these terminals varied, in all, the pre-synaptic and post-synaptic densities as well as the synaptic cleft maintained normal ultrastructural appearance (Figure 6B and 6C, asterisk). As in areas close to the plaque epicenter, in the immediate plaque periphery of all transgenic animals we detected a number of dystrophic myelinated axonal profiles with compacted organelles and thinning of the myelin sheath (not shown), in others there was a visible transition from a myelinated to a non-myelinated segment (Figure 7A). Swollen axonal profiles with compaction of neurofilaments and signs of dystrophy and edema of the oligodendrocytic cytoplasm was also observed (Figure 7C, asterisks), in others a corona of compacted neurofilaments surrounding a center of organelle compaction was frequently detected (Figure 7D).
Figure 5.

Electron micrographs showing dystrophic neurites at different stages of degeneration; A and B – early stage, C – mid stage, D-H - late stage. Asterisk in A shows accumulation of normal mitochondria, inset in addition shows degenerating mitochondrion (arrow) and autophagic vacuoles. Asterisk in G shows a dystrophic neurite with very condensed axoplasmic matrix and accumulation of autophagic vacuoles. Autophagic vacuoles are also abundant in D, E, F, H. Micrograph in I shows a neurite (*) in very advanced stage of dystrophy with no limiting cytoplasmic membrane (arrows). Scale bars: – 500 nm.
Figure 9.

Hirano body (HB) in a dystrophic neurite (*). Inset shows many equally spaced parallel filaments seen in the HB. Scale bar – 500 nm.
Figure 6.

Micrographs of axonal terminals at different stages of degeneration showing morphologically normal synapses. A. Moderately degenerated terminal (arrow) and non-degenerated terminal (double arrow) synapsing on the same dendritic shaft (*), APP/PS1/ApoE3 tg mouse. B. Grossly degenerated axonal terminal synapsing on a normal dendritic spine (*), APP/PS1/ApoE4 tg mouse. C. Grossly degenerated axonal terminal synapsing on a dendritic spine (*) showing a normal mitochondrion (m), APP/PS1/ApoE2 tg mouse. Even at this high magnification no abnormal features in the structure of the synaptic membranes are observed. Scale bars: – 1 μm.
Figure 7.

A. Micrograph showing a transition from a myelinated (white arrow) to a non-myelinated (black arrow) segment in a longitudinally sectioned axonal profile. B. Micrograph showing a white matter myelinated axon with morphological signs of degeneration of the Wallerian type. C. Micrograph showing a swollen axonal profile with compaction of neurofillaments and edema of the olygodendrocitic cytoplasm (asterisks); arrow pointing to the node of Ranvier. D. Micrograph of a very swollen myelinated axon - a corona of compacted neurofilaments surrounds a center of organelle aggregation. Scale bar: A – 1 μm, B, C and D – 2 μm.
Wider periphery of the plaque core and white matter
In areas very close to cortical or hippocampal plaques, single degenerating neuronal cell perikarya and their dendrites revealing very electron dense nuclear and cytoplasmic matrices, were frequently observed (Figure 8A and 8B). However no apoptotic morphology was detected in these degenerating neurons. The cytoplasm of these dying cells was filled with dense fibrillar material (Figure 8C, 8D and 8E) and in many cases cytoplasmic membranes were not visible. In the latter cases amyloid fibrils were in direct apposition to the extracellular matrix (Figure 8E, arrows). Careful examination of cross and longitudinal sections through the major white matter tracts such as the external capsule and the axonal bundle of stratum lacunosum in the hippocampus and hippocampal commisure revealed many single dystrophic myelinated and non-myelinated axons. Many myelinated axons showed dystrophic features similar to Wallerian degeneration (Figure 7B), while others showed similar degeneration changes seen in dystrophic plaque neurites.
Figure 8.

Micrographs of degenerating neuronal cell bodies (A and B) and neuronal cell processes (C, D and E) revealing grossly condensed matrices. Asterisks show accumulation of amyloid fibrils in the dystrophic cytoplasm of degenerating cells. Arrow in E indicates an interface between a degenerating cell process and the surrounding neuropil where the cell membrane is not detected and amyloid fibrils can be identified. E is a high magnification of D rotated at 90°. Scale bars – A and B -1 μm, C, D and E – 1 μm.
Discussion
In this study, the compact amyloid epicenter (core), the dystrophic cellular elements directly adjacent or close to the amyloid core were examined at the ultrastructural level in APP/PS1 tg mice expressing one of the three human ApoE isoforms - ApoE2, ApoE3 and ApoE4 respectively.
The plaque epicenter was almost exclusively occupied by amyloid fibrils and largely devoid of cellular elements. We found that the amyloid fibrillar star’s periphery resided within deep cytoplamic infoldings of microglial cell processes which separated the amyloid epicenter from the dystrophic neuropil surrounding it. This plaque architecture is similar to many other mouse models [23,25,35,48-50]. The existence of a microglial core periphery was not surprising, since animal studies have shown that microglial response around plaques occurs often and relatively soon [51,52]. Historically, in many human and animal studies the relationship between amyloid deposition/turnover and microglia has been discussed in great detail [2,23,25,35,48-50,53-55] and is beyond the scope of this study. Nevertheless it has to be noted that to date this relationship remains poorly understood although many have argued that microglia cells may also be considered to be a driving force in depositing or clearing fibrillar amyloid [54,56-58]. Interestingly, after careful ultrastructural examination of serial sections with subsequent 3-dimentional amyloid plaque reconstruction of APP23 tg mouse brains, no evidence of amyloid phagocytosis and/or amyloid secretion by microglia has been detected [48]. In addition, Grathworth et al., in an elegant and well designed in vivo study, have shown that the formation and maintenance of the amyloid plaque can occur in almost complete absence of microglia and that neuritic dystrophy is a process that may develop independent of microglia [59].
Although smaller in number, we were able to observe astrocytic processes in the plaque core, some of them filled with phagocytosed debris similar to that of microglia. Various ultrastructural studies have detected astrocytic processes in human AD plaques [23,57], and were also shown to exhibit β-amyloid immunoreactivity with gold probes [45]. Although we were not able to clearly distinguish between dormant and hypertrophic astroglia, their constant presence around the plaque epicenter and around dystrophic axons warrants further investigation.
Similar to single APP tg, double APP/PS1 tg and triple APP/PS1/Tau tg mice [23,25,26,30,60], our studies revealed numerous neuritic profiles at different stages of swelling and degeneration which surrounded the microglial processes ring. Surviving synaptic figures in many of these dystrophic profiles argue in favor of the interpretation that the majority of the dystrophic neurites surrounding amyloid plaques are axonal (presynaptic) in origin. These suggestions are supported also by the fact that many single neuritic profiles close to the amyloid territory, and showing exactly the same dystrophic morphology, were myelinated. Importantly, they were very frequently seen in the white matter plaques as well and argue in favor of long axon degeneration. Likewise, many EM studies in AD [12,62] support the predominant axonal origin of the plaque’s dystrophic neurites.
We also found that degenerating axons undergo extensive cytoskeletal changes in addition to the compaction of normal and dystrophic organelles. Similar alterations have been observed in APP tg mice, in tg mice with human four-repeat tau proteins and in mice expressing mutant human tau [23,25,30,61,63,64,65]. Early extracellular Aβ deposits in transgenic mice expressing wild-type human tau have shown to induce increased tau phosphorylation and coexisted with neuritic and cytoskeletal pathology that was strikingly similar to our observations [61]. The above studies have also concluded that a long exposure to Aβ in vivo is insufficient to drive the formation of PHF. At the same time double tg mice lacking the human APP transgene failed to develop extracellular plaques,no intracellular Aβ immunoreactivity was found and no physiological features of synaptic dysfunction was recorded [26]. Axonal swelling and accumulation of dystrophic organelles, such as mitochondria and autophagic vacuoles and the apparent cytoskeletal pathology, while indicative of progressive axonal degeneration, may also represent gross disturbances in anterograde or retrograde transport [66,65,61]. Changes in the axoplasmic cytoskeleton in association with organelle compaction has been consistently observed by us and others after experimental traumatic brain injury [67,68,69] and these features have been classically interpreted as axonal transport impairment [70]. In APP/PS1 human ApoE isoform knock-in animals, we have consistently encountered similar changes in long fiber tract axons. The latter exhibited not only “classical” plaque neuritic pathology, but in addition dystrophic changes similar to the Wallerian type of axonal degeneration, a phenomenon observed also by Wirths et al., (2006) in the spinal cord in APP/PS1 tg mice [37] as well as in AD [1]. Impaired axonal transport appears to be a major pathological alteration in later stages of the AD progression in humans and the axonal pathology described by us and others recapitulates the cardinal features of axonal impairment and possible chronic deficits. Hirano bodies have been observed in some cases of AD, amyotrophic lateral sclerosis/parkinsonism-dementia complex of Guam and in many tg models [23,61,71]. Different studies indicate the presence of actin, tropomyosin and vinculine [72,73] but currently, Hirano body formation is considered to be a non-specific lesion in AD-type neuropathologies and is thought to reflect alterations of the actin network in dystrophic axons [71]. Our studies, in agreement with other studies in tg animals, found no evidence of PHF.
As in APP/PS1 tg animals, APP/PS1/Human ApoE knock-in mice which develop mature amyloid plaques reveal a number of neurons which show classical EM signs of non-apoptotic cell death. However, at this postnatal age, no indication of autophagic or apoptotic cell death has been detected ultrastructurally, although many axonal processes exhibited a pathology consistent with the engagement of autophagic vacuoles. Our data are similar to data from APP/PS1 tg mice where the lysosomal pathology was prevailing in degenerating neurons of older animals [25,74]. In our single time-point study, it is difficult to argue that axonal pathology and neuronal cell death are directly related since we have found much less neuronal cell body degeneration compared to the massive axonal dystrophy throughout the brain and especially within the neuritic plaque. Nonetheless, non-apoptotic type of cell death can be substantial given the protracted nature of the pathology observed in these animals. Mitochondrial dysfunction and oxidative stress in degenerating neurites as causative factors for the progressive cell death occurring in APP/PS1 tg animals has also been discussed [75]. Although apoptosis has been implicated in the neuronal cell death in aging APP/PS mice [75,76] based on a number of activated caspase 3 immunoreactive cells and ultrastructural morphology (believed to be the best morphological method for distinguishing apoptosis from other types of neuronal cell degeneration), the involvement of this cell death type in tg animals remains uncertain [77]. The involvement of apoptotic cell death is also unclear in human AD cases [78]. Implicating the potential role of autophagic neuronal cell death, Yang et al., (2008) have presented evidence for a cross talk between apoptosis and autophagy by showing robust activated caspase 3 staining in autophagic vacuoles in dystrophic neurites of APP/PS1 tg mice [76]. Interestingly, in AD brains certain hippocampal neurons have also shown features of granulovacuolar degeneration with the expression of activated caspase 3 [79]. Taken together these findings reinforce the idea of possible connection between elements of apoptotic and autophagic degeneration in some experimental models, and also in cases of AD. However, further investigation is needed to resolve this issue.
In summary, our studies show that like in senile primate AD models, single tg APP mice, double tg APP/PS1 mice, and triple tg APP/PS1/Tau mice, APP/PS1 mice expressing human ApoE isoforms develop mature neuritic plaques with similar ultrastructural components and share the cardinal features of the senile plaque. Therefore this model may be useful for the study of AD pathogenesis and treatment strategies in the context of human ApoE isoforms.
Acknowledgments
Supported by NIH grant AG13956 (DMH).
References
- 1.Terry RD. The Fine Structure of Neurofibrillary Tangles in Alzheimer’s Disease. J Neuropathol Exp Neurol. 1963;22:629–42. doi: 10.1097/00005072-196310000-00005. [DOI] [PubMed] [Google Scholar]
- 2.Terry RD, Gonatas NK, Weiss M. Ultrastructural Studies in Alzheimer’s Presenile Dementia. Am J Pathol. 1964;44:269–97. [PMC free article] [PubMed] [Google Scholar]
- 3.Kidd M. Paired helical filaments in electron microscopy of Alzheimer’s disease. Nature. 1963;197:192–3. doi: 10.1038/197192b0. [DOI] [PubMed] [Google Scholar]
- 4.Kidd M. Alzheimer’s Disease--an Electron Microscopical Study. Brain. 1964;87:307–20. doi: 10.1093/brain/87.2.307. [DOI] [PubMed] [Google Scholar]
- 5.Ikeda K, Haga C, Kosaka K. Light and electron microscopic examination of amyloid-rich primitive plaques: comparison with diffuse plaques. J Neurol. 1990;237:88–93. doi: 10.1007/BF00314668. [DOI] [PubMed] [Google Scholar]
- 6.Wisniewski H, Terry R. Re-examination of the pathogenesis of the senile plaque. In: Zimmerman HM (ed) Progress in neuropathology, NY and London: Grune and Stratton. 1973;2:26. [Google Scholar]
- 7.Yamaguchi H, Hirai S, Shoji M, Harigaya Y, Okamoto Y, Nakazato Y. Alzheimer type dementia: diffuse type of senile plaques demonstrated by beta protein immunostaining. Prog Clin Biol Res. 1989;317:467–74. [PubMed] [Google Scholar]
- 8.Terry RD, Masliah E, Salmon DP, Butters N, De-Teresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–80. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 9.Martin LJ, Pardo CA, Cork LC, Price DL. Synaptic pathology and glial responses to neuronal injury precede the formation of senile plaques and amyloid deposits in the aging cerebral cortex. Am J Pathol. 1994;145:1358–81. [PMC free article] [PubMed] [Google Scholar]
- 10.Scheff SW, Price DA. Synaptic pathology in Alzheimer’s disease: a review of ultrastructural studies. Neurobiol Aging. 2003;24:1029–46. doi: 10.1016/j.neurobiolaging.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 11.DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol. 1990;27:457–64. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- 12.Masliah E, Terry R. The role of synaptic proteins in the pathogenesis of disorders of the central nervous system. Brain Pathol. 1993;3:77–85. doi: 10.1111/j.1750-3639.1993.tb00728.x. [DOI] [PubMed] [Google Scholar]
- 13.DeKosky ST, Scheff SW, Styren SD. Structural correlates of cognition in dementia: quantification and assessment of synapse change. Neurodegeneration. 1996;5:417–21. doi: 10.1006/neur.1996.0056. [DOI] [PubMed] [Google Scholar]
- 14.Janus C, Chishti MA, Westaway D. Transgenic mouse models of Alzheimer’s disease. Biochim Biophys Acta. 2000;1502:63–75. doi: 10.1016/s0925-4439(00)00033-8. [DOI] [PubMed] [Google Scholar]
- 15.van Leuven F. Single and multiple transgenic mice as models for Alzheimer’s disease. Prog Neurobiol. 2000;61:305–12. doi: 10.1016/s0301-0082(99)00055-6. [DOI] [PubMed] [Google Scholar]
- 16.Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius LJ, Mackey B, Olney J, Mckeel D, Wozniak D, Paul SM. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA. 2000;97:2892–7. doi: 10.1073/pnas.050004797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gotz J. Tau and transgenic animal models. Brain Res Brain Res Rev. 2001;35:266–86. doi: 10.1016/s0165-0173(01)00055-8. [DOI] [PubMed] [Google Scholar]
- 18.Crews L, Rockenstein E, Masliah E. APP transgenic modeling of Alzheimer’s disease: mechanisms of neurodegeneration and aberrant neurogenesis. Brain Struct Funct. 2010;214:111–26. doi: 10.1007/s00429-009-0232-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hutton M, Hardy J. The presenilins and Alzheimer’s disease. Hum Mol Genet. 1997;6:1639–46. doi: 10.1093/hmg/6.10.1639. [DOI] [PubMed] [Google Scholar]
- 20.Cruts M, Van Broeckhoven C. Molecular genetics of Alzheimer’s disease. Ann Med. 1998;30:560–5. doi: 10.3109/07853899809002605. [DOI] [PubMed] [Google Scholar]
- 21.Rocchi A, Pellegrini S, Siciliano G, Murri L. Causative and susceptibility genes for Alzheimer’s disease: a review. Brain Res Bull. 2003;61:1–24. doi: 10.1016/s0361-9230(03)00067-4. [DOI] [PubMed] [Google Scholar]
- 22.Pastor P, Goate AM. Molecular genetics of Alzheimer’s disease. Curr Psychiatry Rep. 2004;6:125–33. doi: 10.1007/s11920-004-0052-6. [DOI] [PubMed] [Google Scholar]
- 23.Masliah E, Sisk A, Mallory M, Mucke L, Schenk D, Games D. Comparison of neurodegenerative pathology in transgenic mice overexpressing V717F beta-amyloid precursor protein and Alzheimer’s disease. J Neurosci. 1996;16:5795–811. doi: 10.1523/JNEUROSCI.16-18-05795.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Masliah E, Sisk A, Mallory M, Games D. Neurofibrillary pathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. J Neuropathol Exp Neurol. 2001;60:357–68. doi: 10.1093/jnen/60.4.357. [DOI] [PubMed] [Google Scholar]
- 25.Kurt MA, Davies DC, Kidd M, Duff K, Rolph SC, Jennings KH, Howlett DR. Neurodegenerative changes associated with beta-amyloid deposition in the brains of mice carrying mutant amyloid precursor protein and mutant presenilin-1 transgenes. Exp Neurol. 2001;171:59–71. doi: 10.1006/exnr.2001.7717. [DOI] [PubMed] [Google Scholar]
- 26.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–21. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 27.Mucke L, Yu GQ, McConlogue L, Rockenstein EM, Abraham CR, Masliah E. Astroglial expression of human alpha(1)-antichymotrypsin enhances alzheimer-like pathology in amyloid protein precursor transgenic mice. Am J Pathol. 2000;157:2003–10. doi: 10.1016/s0002-9440(10)64839-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Bürki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci USA. 1997;94:13287–92. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moechars D, Dewachter I, Lorent K, Reversé D, Baekelandt V, Naidu A, Tesseur I, Spittaels K, Haute CV, Checler F, Godaux E, Cordell B, Van Leuven F. Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J Biol Chem. 1999;274:6483–92. doi: 10.1074/jbc.274.10.6483. [DOI] [PubMed] [Google Scholar]
- 30.Phinney AL, Deller T, Stalder M, Calhoun ME, Frotscher M, Sommer B, Staufenbiel M, Jucker M. Cerebral amyloid induces aberrant axonal sprouting and ectopic terminal formation in amyloid precursor protein transgenic mice. J Neurosci. 1999;19:8552–9. doi: 10.1523/JNEUROSCI.19-19-08552.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bornemann KD, Staufenbiel M. Transgenic mouse models of Alzheimer’s disease. Ann N Y Acad Sci. 2000;908:260–6. doi: 10.1111/j.1749-6632.2000.tb06653.x. [DOI] [PubMed] [Google Scholar]
- 32.Rockenstein E, Mallory M, Mante M, Sisk A, Masliah E. Early formation of mature amyloidbeta protein deposits in a mutant APP transgenic model depends on levels of Abeta(1-42) J Neurosci Res. 2001;66:573–82. doi: 10.1002/jnr.1247. [DOI] [PubMed] [Google Scholar]
- 33.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 34.Borchelt DR, Ratovitski T, van Lare J, Lee MK, Gonzales V, Jenkins NA, Copeland NG, Price DL, Sisodia SS. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19:939–45. doi: 10.1016/s0896-6273(00)80974-5. [DOI] [PubMed] [Google Scholar]
- 35.Stalder M, Phinney A, Probst A, Sommer B, Staufenbiel M, Jucker M. Association of microglia with amyloid plaques in brains of APP23 transgenic mice. Am J Pathol. 1999;154:1673–84. doi: 10.1016/S0002-9440(10)65423-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O’Campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med. 1998;4:97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- 37.Wirths O, Weis J, Szczygielski J, Multhaup G, Bayer TA. Axonopathy in an APP/PS1 transgenic mouse model of Alzheimer’s disease. Acta Neuropathol. 2006;111:312–9. doi: 10.1007/s00401-006-0041-4. [DOI] [PubMed] [Google Scholar]
- 38.Samura E, Shoji M, Kawarabayashi T, Sasaki A, Matsubara E, Murakami T, Wuhua X, Tamura S, Ikeda M, Ishiguro K, Saido TC, Westaway D, St George Hyslop P, Harigaya Y, Abe K. Enhanced accumulation of tau in doubly transgenic mice expressing mutant betaAPP and presenilin-1. Brain Res. 2006;1094:192–9. doi: 10.1016/j.brainres.2005.12.134. [DOI] [PubMed] [Google Scholar]
- 39.Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE. Systematic meta-analyses of Alzheimer disease genetic association: the Alz-Gene database. Nat Genet. 2007;39:7–23. doi: 10.1038/ng1934. [DOI] [PubMed] [Google Scholar]
- 40.Holtzman DM, Fagan AM, Mackey B, Tenkova T, Sartorius L, Paul SM, Bales K, Ashe KH, Irizarry MC, Hyman BT. Apolipoprotein E facilitates neuritic and cerebrovascular plaque formation in an Alzheimer’s disease model. Ann Neurol. 2000;47:739–47. [PubMed] [Google Scholar]
- 41.Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer’s disease. Neuron. 2009;63:287–303. doi: 10.1016/j.neuron.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Knouff C, Hinsdale ME, Mezdour H, Altenburg MK, Watanabe M, Quarfordt SH, Sullivan PM, Maeda N. Apo E structure determines VLDL clearance and atherosclerosis risk in mice. J Clin Invest. 1999;103:1579–86. doi: 10.1172/JCI6172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sullivan PM, Mezdour H, Aratani Y, Knouff C, Najib J, Reddick RL, Quarfordt SH, Maeda N. Targeted replacement of the mouse apolipoprotein E gene with the common human APOE3 allele enhances diet-induced hypercholesterolemia and atherosclerosis. J Biol Chem. 1997;272:17972–80. doi: 10.1074/jbc.272.29.17972. [DOI] [PubMed] [Google Scholar]
- 44.Radde R, Bolmont T, Kaeser SA, Coomaraswamy J, Lindau D, Stoltze L, Calhoun ME, Jäggi F, Wolburg H, Gengler S, Haass C, Ghetti B, Czech C, Hölscher C, Mathews PM, Jucker M. Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 2006;7:940–6. doi: 10.1038/sj.embor.7400784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim J, Jiang H, Park S, Eltorai AE, Stewart FR, Yoon H, Basak JM, Finn MB, Holtzman DM. Haploinsufficiency of human APOE reduces amyloid deposition in a mouse model of amyloid-beta amyloidosis. J Neurosci. 2011;31:18007–12. doi: 10.1523/JNEUROSCI.3773-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Piedrahita JA, Zhang SH, Hagaman JR, Oliver PM, Maeda N. Generation of mice carrying a mutant apolipoprotein E gene inactivated by gene targeting in embryonic stem cells. Proc Natl Acad Sci USA. 1992;89:4471–5. doi: 10.1073/pnas.89.10.4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mattiace LA, Davies P, Dickson DW. Detection of HLA-DR on microglia in the human brain is a function of both clinical and technical factors. Am J Pathol. 1990;136:1101–14. [PMC free article] [PubMed] [Google Scholar]
- 48.Stalder M, Deller T, Staufenbiel M, Jucker M. 3D-Reconstruction of microglia and amyloid in APP23 transgenic mice: no evidence of intracellular amyloid. Neurobiol Aging. 2001;22:427–34. doi: 10.1016/s0197-4580(01)00209-3. [DOI] [PubMed] [Google Scholar]
- 49.Wegiel J, Wang KC, Imaki H, Rubenstein R, Wronska A, Osuchowski M, Lipinski WJ, Walker LC, LeVine H. The role of microglial cells and astrocytes in fibrillar plaque evolution in transgenic APP(SW) mice. Neurobiol Aging. 2001;22:49–61. doi: 10.1016/s0197-4580(00)00181-0. [DOI] [PubMed] [Google Scholar]
- 50.Nagele RG, Wegiel J, Venkataraman V, Imaki H, Wang KC. Contribution of glial cells to the development of amyloid plaques in Alzheimer’s disease. Neurobiol Aging. 2004;25:663–74. doi: 10.1016/j.neurobiolaging.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 51.Koenigsknecht-Talboo J, Meyer-Luehmann M, Parsadanian M, Garcia-Alloza M, Finn MB, Hyman BT, Bacskai BJ, Holtzman DM. Rapid microglial response around amyloid pathology after systemic anti-Abeta antibody administration in PDAPP mice. J Neurosci. 2008;28:14156–64. doi: 10.1523/JNEUROSCI.4147-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, de Calignon A, Rozkalne A, Koenigsknecht-Talboo J, Holtzman DM, Bacskai BJ, Hyman BT. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature. 2008;451:720–4. doi: 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wisniewski HM, Wegiel J, Wang KC, Kujawa M, Lach B. Ultrastructural studies of the cells forming amyloid fibers in classical plaques. Can J Neurol Sci. 1989;16:535–42. doi: 10.1017/s0317167100029887. [DOI] [PubMed] [Google Scholar]
- 54.Perlmutter LS, Barron E, Chui HC. Morphologic association between microglia and senile plaque amyloid in Alzheimer’s disease. Neurosci Lett. 1990;119:32–6. doi: 10.1016/0304-3940(90)90748-x. [DOI] [PubMed] [Google Scholar]
- 55.Wegiel J, Imaki H, Wang KC, Wegiel J, Rubenstein R. Cells of monocyte/microglial lineage are involved in both microvessel amyloidosis and fibrillar plaque formation in APPsw tg mice. Brain Res. 2004;1022:19–29. doi: 10.1016/j.brainres.2004.06.058. [DOI] [PubMed] [Google Scholar]
- 56.Wisniewski HM, Wegiel J, Wang KC, Lach B. Ultrastructural studies of the cells forming amyloid in the cortical vessel wall in Alzheimer’s disease. Acta Neuropathol. 1992;84:117–27. doi: 10.1007/BF00311383. [DOI] [PubMed] [Google Scholar]
- 57.Wegiel J, Wang KC, Tarnawski M, Lach B. Microglia cells are the driving force in fibrillar plaque formation, whereas astrocytes are a leading factor in plague degradation. Acta Neuropathol. 2000;100:356–64. doi: 10.1007/s004010000199. [DOI] [PubMed] [Google Scholar]
- 58.Lee CY, Landreth GE. The role of microglia in amyloid clearance from the AD brain. J Neural Transm. 2010;117:949–60. doi: 10.1007/s00702-010-0433-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Grathwohl SA, Kälin RE, Bolmont T, Prokop S, Winkelmann G, Kaeser SA, Odenthal J, Radde R, Eldh T, Gandy S, Aguzzi A, Staufenbiel M, Mathews PM, Wolburg H, Heppner FL, Jucker M. Formation and maintenance of Alzheimer’s disease beta-amyloid plaques in the absence of microglia. Nat Neurosci. 2009;12:1361–3. doi: 10.1038/nn.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kurt MA, Davies DC, Kidd M. beta-Amyloid immunoreactivity in astrocytes in Alzheimer’s disease brain biopsies: an electron microscope study. Exp Neurol. 1999;158:221–8. doi: 10.1006/exnr.1999.7096. [DOI] [PubMed] [Google Scholar]
- 61.Boutajangout A, Authelet M, Blanchard V, Touchet N, Tremp G, Pradier L, Brion JP. Characterisation of cytoskeletal abnormalities in mice transgenic for wild-type human tau and familial Alzheimer’s disease mutants of APP and presenilin-1. Neurobiol Dis. 2004;15:47–60. doi: 10.1016/j.nbd.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 62.Masliah E, Hansen L, Albright T, Mallory M, Terry RD. Immunoelectron microscopic study of synaptic pathology in Alzheimer’s disease. Acta Neuropathol. 1991;81:428–33. doi: 10.1007/BF00293464. [DOI] [PubMed] [Google Scholar]
- 63.Probst A, Gotz J, Wiederhold KH, Tolnay M, Mistl C, Jaton AL, Hong M, Ishihara T, Lee VM, Trojanowski JQ, Jakes R, Crowther RA, Spillantini MG, Burki K, Goedert M. Axonopathy and amyotrophy in mice transgenic for human four-repeat tau protein. Acta Neuropathol. 2000;99:469–81. doi: 10.1007/s004010051148. [DOI] [PubMed] [Google Scholar]
- 64.Lin WL, Lewis J, Yen SH, Hutton M, Dickson DW. Ultrastructural neuronal pathology in transgenic mice expressing mutant (P301L) human tau. J Neurocytol. 2003;32:1091–105. doi: 10.1023/B:NEUR.0000021904.61387.95. [DOI] [PubMed] [Google Scholar]
- 65.Lin WL, Zehr C, Lewis J, Hutton M, Yen SH, Dickson DW. Progressive white matter pathology in the spinal cord of transgenic mice expressing mutant (P301L) human tau. J Neurocytol. 2005;34:397–410. doi: 10.1007/s11068-006-8726-0. [DOI] [PubMed] [Google Scholar]
- 66.Tsukita S, Ishikawa H. The movement of membranous organelles in axons. Electron microscopic identification of anterogradely and retrogradely transported organelles. J Cell Biol. 1980;84:513–30. doi: 10.1083/jcb.84.3.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mac Donald CL, Dikranian K, Bayly P, Holtzman D, Brody D. Diffusion tensor imaging reliably detects experimental traumatic axonal injury and indicates approximate time of injury. J Neurosci. 2007;27:11869–76. doi: 10.1523/JNEUROSCI.3647-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rodriguez-Paez AC, Brunschwig JP, Bramlett HM. Light and electron microscopic assessment of progressive atrophy following moderate traumatic brain injury in the rat. Acta Neuropathol. 2005;109:603–16. doi: 10.1007/s00401-005-1010-z. [DOI] [PubMed] [Google Scholar]
- 69.Kelley BJ, Farkas O, Lifshitz J, Povlishock JT. Traumatic axonal injury in the perisomatic domain triggers ultrarapid secondary axotomy and Wallerian degeneration. Exp Neurol. 2006;198:350–60. doi: 10.1016/j.expneurol.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 70.Buki A, Povlishock JT. All roads lead to disconnection? Traumatic axonal injury revisited. Acta Neurochir (Wien) 2006;148:181–93. doi: 10.1007/s00701-005-0674-4. discussion 193-4. [DOI] [PubMed] [Google Scholar]
- 71.Perl DP. Neuropathology of Alzheimer’s disease. Mt Sinai J Med. 2010;77:32–42. doi: 10.1002/msj.20157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Galloway PG, Perry G, Kosik KS, Gambetti P. Hirano bodies contain tau protein. Brain Res. 1987;403:337–40. doi: 10.1016/0006-8993(87)90071-0. [DOI] [PubMed] [Google Scholar]
- 73.Goldman JE. The association of actin with Hirano bodies. J Neuropathol Exp Neurol. 1983;42:146–52. doi: 10.1097/00005072-198303000-00004. [DOI] [PubMed] [Google Scholar]
- 74.Cataldo AM, Peterhoff CM, Schmidt SD, Terio NB, Duff K, Beard M, Mathews PM, Nixon RA. Presenilin mutations in familial Alzheimer disease and transgenic mouse models accelerate neuronal lysosomal pathology. J Neuropathol Exp Neurol. 2004;63:821–30. doi: 10.1093/jnen/63.8.821. [DOI] [PubMed] [Google Scholar]
- 75.Blanchard V, Moussaoui S, Czech C, Touchet N, Bonici B, Planche M, Canton T, Jedidi I, Gohin M, Wirths O, Bayer TA, Langui D, Duyckaerts C, Tremp G, Pradier L. Time sequence of maturation of dystrophic neurites associated with Abeta deposits in APP/PS1 transgenic mice. Exp Neurol. 2003;184:247–63. doi: 10.1016/s0014-4886(03)00252-8. [DOI] [PubMed] [Google Scholar]
- 76.Yang DS, Kumar A, Stavrides P, Peterson J, Peterhoff CM, Pawlik M, Levy E, Cataldo AM, Nixon RA. Neuronal apoptosis and autophagy cross talk in aging PS/APP mice, a model of Alzheimer’s disease. Am J Pathol. 2008;173:665–81. doi: 10.2353/ajpath.2008.071176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Selznick LA, Holtzman DM, Han BH, Gokden M, Srinivasan AN, Johnson EM, Roth KA Jr. In situ immunodetection of neuronal caspase-3 activation in Alzheimer disease. J Neuropathol Exp Neurol. 1999;58:1020–6. doi: 10.1097/00005072-199909000-00012. [DOI] [PubMed] [Google Scholar]
- 78.Roth KA. Caspases, apoptosis, and Alzheimer disease: causation, correlation, and confusion. J Neuropathol Exp Neurol. 2001;60:829–38. doi: 10.1093/jnen/60.9.829. [DOI] [PubMed] [Google Scholar]
- 79.Stadelmann C, Deckwerth TL, Srinivasan A, Bancher C, Brück W, Jellinger K, Lassmann H. Activation of caspase-3 in single neurons and autophagic granules of granulovacuolar degeneration in Alzheimer’s disease. Evidence for apoptotic cell death. Am J Pathol. 1999;155:1459–66. doi: 10.1016/S0002-9440(10)65460-0. [DOI] [PMC free article] [PubMed] [Google Scholar]