

Abstract
A series of functionalized congeners of 1,3-dialkylxanthines has been prepared as adenosine receptor antagonists. On the basis of the high potency of 8-(p-hydroxyphenyl)-1,3-dialkylxanthines, the parent compounds were 8-[4-[(carboxymethyl)oxy]phenyl] derivatives of theophylline and 1,3-dipropylxanthine. A series of analogues including esters of ethanol and N-hydroxysuccinimide, amides, a hydrazide, an acylurea, and anilides were prepared. The potency in blocking A1-adenosine receptors (inhibition of binding of N6-[3H]cyclohexyladenosine to brain membranes) and A2-adenosine receptors (inhibition of 2-chloroadenosine-elicited accumulations of cyclic AMP in brain slices) was markedly affected by structural changes distal to the primary pharmacophore (8-phenyl-1,3-dialkylxanthine). Potencies in the dipropyl series at the A1 receptor ranged from K1 values of 1.2 nM for a congener with a terminal amidoethyleneamine moiety to a K1 value of 58 nM for the parent carboxylic acid to a K1 of 96 nM for the bulky ureido congener. Certain congeners were up to 145-fold more active at A1 receptors than at A2 receptors. Various derivatives of the congeners should be useful as receptor probes and for radioidodination, avidin binding, and preparation of affinity columns.
Alkylxanthines, with theophylline (1) as the prototype, represent the major class of antagonists for adenosine receptors.1 Although theophylline and caffeine are relatively weak adenosine antagonists, with affinity constants in the 10–50 µM range, they undoubtedly owe many of their pharmacological effects to blockade of adenosine-mediated functions. Two classes of adenosine receptors have been characterized.1 The A1-adenosine receptor is inhibitory to adenylate cyclase and appears involved in antilipolytic, cardiac depressant, and central depressant effects of adenosine analogues. The A2-adenosine receptor is stimulatory to adenylate cyclase and appears involved in hypotensive, vasodilatory, antithrombotic, and endocrine effects of adenosine analogues. Xanthines, by blockade of adenosine binding at such receptors, would have the opposite effects. Some xanthines, such as 3-iso-butyl-1-methylxanthine, not only block adenosine receptors but also have potent inhibitory effects on phosphodiesterases.2a The presence of an 8-phenyl group greatly enhances the potency of theophylline as an adenosine antagonist.2a,b Even more potent antagonists result from the replacement of the 1,3-methyl groups of 8-phenyl-theophylline with n-propyl groups and by situating uncharged electron-donating para substituents on the 8-phenyl ring. Affinity constants (Ki) of less than 1 nM at A1-adenosine receptors have been attained.3,22 In addition to high potency, some selectivity toward the A1 subclass of adenosine receptors has been obtained with 1,3-dipropyl-8-phenylxanthines.4
1,3-Dipropyl-8-(p-hydroxyphenyl)xanthine (2b) was chosen as a suitable lead compound in an effort to develop potent and selective functionalized congeners as antagonists for adenosine receptors. An N6-phenyladenosine was chosen as a suitable lead compound for a parallel effort to develop potent and selective agonists for A1-adenosine receptors.5 In the design of active, covalent conjugates of drugs, the goals of the functionalized congener approach are several, including increasing the potency, prolonging the duration of action, and/or changing the specificity. This approach has been applied to isoproterenol and has lead to analogues with activity at β-adrenergic receptors as much as 4 orders of magnitude greater than that of isoproterenol.6–8 Nontherapeutic applications of active, functionalized drugs include receptor probes,5,9,10 immobilized ligands for affinity chromatography,11 and radio-labeled analogues.
We report here the synthesis of a series of highly potent analogues of theophylline and 1,3-dipropylxanthine, some of which contain groups designed for radiolabeling through the introduction of radioisotopes of iodine. The functionalized congeners are also suitable for the preparation of affinity columns. A biotinylated conjugate is intended for use as a molecular probe through linkage to avidin.12a Recently, biotinylated derivatives of peptide hormones have been described.12b,c
Results and Discussion
Synthetic Methods
The carboxylic acid congener of theophylline (8a) and the dipropyl analogue (8b) were synthesized by a standard approach4,13,14 to xanthines as shown in Figure 1. 5,6-Diamino-1,3-dimethyluracil (3a) was commercially available, but the 1,3-dipropyl compound (3b) was prepared with dipropylurea and cyanoacetic acid as the starting material as described previously.13,14 The imidazole ring was formed by oxidative closure of the benzylidene adduct 6 derived from the appropriate diaminouracil and the substituted benzaldehyde 4. The latter was the product of alkylation of p-hydroxybenzaldehyde by iodoacetate.
Figure 1.

Synthetic approach to functionalized xanthine derivatives.
Ring closure of the benzylidene adduct 6 was effected by heating with substoichiometric amounts of anhydrous ferric chloride.14 In the case of the dipropyl derivative 6b, considerable ethyl ester 10 formed in this reaction when ethanol was the solvent. To obviate separating the mixture of acid and ethyl ester, the esterification was brought to completion by prolonged heating of the reaction mixture in the presence of 1 equiv of ferric chloride. In contrast, use of trifluoroethanol as the solvent during ring closure produced 8b exclusively. Compound 8b also was prepared by basic hydrolysis of the ester 10.
Coupling of the carboxylic acid congeners to amines using carbodiimides presented problems due to limited solubility. Attempts to couple 8b to various polar amines using carbodiimides in dimethylformamide often resulted in isolation of the N-acylurea 11 derived from the acid and the coupling reagent. Compound 8a was coupled in low yield to p-toluidine. In an alternate approach to amide formation (Figure 2), the N-hydroxysuccinimide ester 12 of the carboxylic acid congener was prepared and proved to be readily separable from the N-acylisourea by crystallization. The N-hydroxysuccinimide ester 12 of the carboxylic acid congeners provided an activated form of the drug for coupling to amines (see below) or to bio-polymers such as proteins, the latter then serving as drug carriers.
Figure 2.

Methods of coupling and chain elongation of functionalized xanthines.
As an alternative to coupling the preformed xanthine congeners, the amide bond was introduced on the substituted benzaldehyde (e.g., 5, Figure 1) prior to formation of the imidazole ring as above.
The ethyl ester 9 could be aminolyzed by excess unhindered amines in dimethylformamide to form amides 13 as shown in Figure 2. Aminolysis by alkyldiamines produced the functionalized amino congeners 13c,d. Compound 13c, derived from ethylenediamine, was the basis for further chain elongation. The primary amine was readily acylated by N-hydroxysuccinimide esters to form amides 14. A tertiary amine 16 was made via reductive amination sequentially with p-hydroxybenzaldehyde and acetaldehyde. The iminium salt 15 was isolated and reduced with sodium cyanoborohydride (Figure 3).
Figure 3.

Reactions of amino-functionalized xanthines.
Biological Activity
8-(p-Hydroxyphenyl)theophylline and 1,3-dipropyl-8-(p-hydroxyphenyl)theophylline, the model compounds chosen for functionalization, have been shown in earlier studies to be potent antagonists of A1- and A2-adenosine receptors.3,4 8-(p-Hydroxyphenyl)theophylline (2a) is 280-fold more potent than theophylline in displacing [3H]cyclohexyladenosine from A1-adenosine receptors in rat cerebral cortical membranes and is 107-fold more potent than theophylline in antagonizing A2-adenosine receptor mediated activation of cyclic AMP generation by 2-chloroadenosine in guinea pig cerebral cortical slices.4 Replacement of the 1,3-dimethyl groups of the theophylline analogue 2a with n-propyl groups yields 1,3-dipropyl-8-(p-hydroxyphenyl)xanthine (2b). This analogue is an extremely potent A1-adenosine antagonists with a Ki value vs. [3H]cyclohexyladenosine binding in rat cerebral cortical slices of 2.9 nM. The change in the alkyl residues, thus, has increased potency at A1 receptors by about 17-fold. The change in alkyl residues also increases potency at A2 receptors but to a much lesser extent (2.6-fold), yielding a somewhat selective A1 antagonist.
Functionalization of these two xanthines was based on the presence of a p-(carboxymethyl)oxy residue on the 8-phenyl ring. This functionalization permitted facile synthesis of a wide variety of amides. In the case of the 8-phenyltheophyllines, the p-(carboxymethyl)oxy compound 8a has a 10-fold lower potency than the p-hydroxy compound at A1 receptors and a 3.3-fold lower activity at A2 receptors (Table I). It appears likely that the presence of the anionic carboxyl group is not favorable to high affinity binding to either receptor. A similar result was reported for a series of carboxy-functionalized adenosine receptor agonists.5 The derivative of 8-phenyltheophylline having an anionic p-carboxyl group directly on the phenyl ring was even less potent with Ki values of 3000 nM at A1 receptors and 2500 nM at A2 receptors.4 Blocking the carboxylate as an amide as in compound 9a enhanced receptor potency at both receptor subtypes.
Table I.
Effect of Functionalization on the Activity of 1,3-Dialkyl-8-(p-hydroxyphenyl)xanthines at A1- and A2-Adenosine Receptors
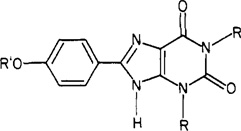 | ||||
|---|---|---|---|---|
|
Ki,a nM |
||||
| compd | R′ | A1 receptor | A2 receptors | ratio A1/A2 |
| R = CH3 | ||||
| 2a | H | 50 ± 20 | 130 ± 30 | 2.6 |
| 8a | HO2CCH2 | 500 ± 200 | 430 ± 80 | 0.86 |
| 9a | 26 ± 5 | 20 ± 7 | 0.77 | |
| R = n-C3H7 | ||||
| 2b | H | 2.9 ± 0.8 | 50 ± 10b | 17 |
| 8b | HO2CCH2 | 58 ± 3 | 34 ± 13 | 0.59 |
| 9b | 36 ± 23 | 750 ± 370 | 21 | |
| 9c | 4.1 ± 1.5 | 62 ± 37 | 15 | |
| 10 | CH3CH2OCOCH2 | 42 ± 3 | 30 ± 12 | 0.71 |
| 11 |  |
96 ± 25 | > 1000 (30% inhibn) | > 10 |
| 12 |  |
9.0 ± 0.7 | 40 ± 14 | 4.4 |
| 13a | H2NCOCH2 | 6.0 ± 1.0 | 47 ± 2 | 7.8 |
| 13b | (CH3)2 NCOCH2 | 32 ± 7 | 68 ± 39 | 2.1 |
| 13c | H2N(CH2)2NHCOCH2 | 1.2 ± 0.5 | 49 ± 17 | 41 |
| 13d | H2N( CH2)8NHCOCH2 | 5 ± 2 | 470 ± 170 | 94 |
| 13e | H2NNHCOCH2 | 59 ± 0.7 | 32 ± 4 | 0.54 |
| 14a | CH3CONH(CH2)2NHCOCH2 | 24 ± 3.5 | 62 ± 3 | 2.6 |
| 14b | 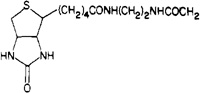 |
54 ± 2 | 180 ± 80 | 3.3 |
| 16 | 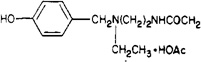 |
2.2 ± 0.7 | 320 ± 70 | 145 |
IC50 values for A1 receptors were obtained from antagonism of binding of 1 nM [3H]cyclohexyladenosine to rat cerebral cortical membranes. IC50 values for A1 receptors were obtained from antagonism of 3H-labeled cyclic AMP accumulation elicited by 15 µM 2-chloroadenosine in [3H]adenine-labeled guinea pig cerebral cortical slices. Ki values were calculated from the equation Ki = IC50/(1 + concentration of adenosine analogue/Ka for adenosine analogue). Values are means ± SEM for two to four experiments. Each experiment contained triplicate determinations.
Values from ref 4.
Further syntheses of functionalized congeners were based on the anticipation that the higher potency and selectivity of 1,3-dipropyl-8-(p-hydroxyphenyl)xanthine relative to the l,3-dimethyl homologue would be paralleled by higher activity in the series of p-(carboxymethyl)oxy derivatives. The p-(carboxymethyl)oxy compound 8b, itself, was 20-fold less potent than the p-hydroxy compound 2b at A1 receptors. At A2 receptors the p-(carboxymethyl)oxy compound was nearly equipotent with the p-hydroxy compound. Again it appears likely that the presence of the anionic carboxy group mitigates against high affinity at A1 receptors. In an earlier study,4 8-(p-carboxyphenyl)-1,3-dipropylxanthine was found to be about 60-fold less potent than the p-hydroxy compound at A1 receptors, while being only 2-fold less active at A2 receptors.4 The ethyl ester 10 has potency at A1 and A2 receptors nearly identical with that of the anionic carboxylic acid 8b, a finding that does not support the proposal that the carboxylate moiety is responsible for the diminished affinity. The ester of N-hydroxysuccinimide 12 is 6.4-fold more potent than the anionic carboxylate 8b at the A1 receptor, while being similar in activity to 8b at the A2 receptor. The carboxamide 13a has high affinity at A1 receptors and is moderately selective, being 8-fold more potent at A1 receptors than at A2 receptors. Remarkably, the p-toluide 9b was no more potent than the acid at A1 receptors, while being 22-fold less active than the acid at A2 receptors. This finding stands in direct contrast to results obtained with the analogous compounds in the theophylline (1,3-dimethyl) series. In that series the p-toluide 9a was about 20-fold more potent than the acid 8a both at A1 receptors and at A2 receptors. Obviously, contributions to affinity afforded by the 1,3-dialkyl substituents and by para substituents on the 8-phenyl ring are not independent and can greatly influence each other in either a positive or a negative manner. This fact has been noted in an earlier study on 8-phenyl-1,3-dialkylxanthines.4 The p-hydroxy anilide 9c was nearly 10-fold more potent than the p-toluide 9b at both A1 and A2 receptors. This illustrates the potential contributions of minor structural modifications distant from the primary pharmacophore (in this case the 8-phenylxanthine) to the biological activity.
Long-chain compounds with terminal amino groups had both enhanced A1 binding and selectivity over the A2 receptor. The (aminoethyl)amide 13c was synthesized with a view of increasing water solubility and also of providing a key intermediate for preparation of affinity columns, fluorescent probes, and a biotin-containing xanthine. The (aminoethyl)amide was very potent at A1-adenosine receptors with a Ki value of 1.2 nM. It was some 40-fold less potent at A2-adenosine receptors. The solubility of compound 13c in 0.1 M sodium phosphate, pH 7.2, was 90 µM, compared to 26 µM for compound 13a and 3.2 µM for compound 2b. The presence of ethyl and hydroxybenzyl substituents on the terminal amino group (16) had little effect on the potency at A1 receptors, while reducing potency at A2 receptors by over 4-fold. This represents the most selective A1 antagonist (145-fold) in the present series. The analogues in which a functionalized congener is linked to a phenolic moiety were designed for radioiodination. These two compounds are representative of a general approach to radiopharmaceuticals through the attachment of functional moieties into which radioisotopes can be easily introduced.5
The acylated compounds 14a and 14b were relatively nonselective antagonists for A1- and A2-adenosine receptors in contrast to the parent amine, which exhibits a 40-fold selectivity for A1 receptors. Although less potent than the parent amine, the potencies of the acylated compounds suggest that affinity columns prepared through acyl coupling to the amino compound could be effective in isolation of solubilized A1 and A2 receptors and/or xanthine-binding sites. The biotinylated analogue is being investigated as a histochemical probe for adenosine receptors.12a
A systematic analysis of the effect of chain length on activity of (aminoalkyl)amides was not carried out. The (aminooctyl) amide 13d was only 4-fold less potent than the (aminoethyl)amide 13c at A1 receptors, while being 10-fold less potent at A2 receptors. Thus the use of longer spacer chains appear feasible for preparation of affinity columns if the (aminoethyl)amide proves unsatisfactory. Moreover, the high potency of amino congeners of adenosine receptor agonists resembles previous findings of subnanomolar affinity of an adenosine agonist bearing a distal primary amine. This may be due to a favorable interaction with the A1 receptor, in which an ionic interaction of the distal protonated amine with an anionic moiety of the receptor enhances binding affinity.
One acylurea 11 was prepared and found to have relatively low activity at both A1 and A2 receptors. This bulky dicyclohexylurea derivative may exceed the steric limitations for efficient binding to the receptor.
Conclusions
In summary, the functionalized congener approach to xanthine antagonists for adenosine receptors has yielded a series of potent compounds that in some cases are moderately selective for A1 receptors. The effects on biological activities caused by modifications of functions distal from the primary pharmcophore in some cases are quite impressive. Similar effects were noted for congeners and conjugates of isoproterenol.6–8
Affinities of congeners and derived analogues for the A1 receptors seem somewhat more sensitive to distal modifications than affinities for the A2 receptor. As yet no selective A2-receptor antagonists have been discovered in this study or in earlier studies,4 and as yet no specific A1-receptor antagonists are available. The present set of functionalized xanthines should prove useful in delineating the role of A1- and A2-adenosine receptor blockade in the antiasthmatic,15 diuretic,16 respiratory stimulant,17 central stimulant,18 cardiac stimulant,19 analgesic adjuvant,20 and antiinflammatory21 activities of theophylline and caffeine.
A further benefit of applying the congener approach to xanthines is the opportunity to increase water solubility. The series of very potent 8-phenylxanthines reported by Bruns et al.22 was highly nonpolar with aqueous solubility very often falling below 10 µM.4 By increasing water solubility through the attachment of highly polar groups it should be possible to overcome undesirable binding to plasma proteins and partition into lipids. This could lead to improved pharmacokinetics of xanthine-derived adenosine antagonists.
Experimental Section
Thin-layer chromatography was carried out with use of Analtech silica gel GF plates with mixtures of chloroform/methanol/acetic acid (v/v/v; A, 50/50/5; B, 94/4/2). Reagent grade dimethylformamide (DMF, Aldrich gold label) was stored over 3-Å molecular sieves. Dicyclohexylcarbodiimide (DCC) was purchased from Sigma. 5,6-Diamino-1,3-dimethyluracil hydrate was purchased from Aldrich. The benzylidene adducts of the diaminouracils were prepared by the representative method given for compound 6b.
Proton NMR spectra were taken on a Varian 220-MHz instrument in the Fourier transform mode. Elemental analyses and mass spectrometry (Finnigan 1015D) were carried out by P. Parisius and N. Whittaker of the Section on Microanalytical Services and Instrumentation of the NIADDK Laboratory of Chemistry. The larger xanthine analogues that did not form molecular ions by chemical-ionization mass spectrometry were identified by Drs. L. Pannell and H. Fales of NHLBI/NIH using californium plasma desorption mass spectrometry.
4-[(Carboxymethyl)oxy]benzaldehyde [(4-Formylphenoxy)acetic Acid, 4]
To a solution of p-hydroxybenzaldehyde (49 g, 0.40 mol) in 250 mL of DMF were added iodoacetic acid (75 g, 0.40 mol) and anhydrous potassium carbonate (120 g), and the mixture was stirred at 60 °C for 3 days. The resulting solid was dispersed mechanically in a mixture of ethyl acetate (400 mL) and water (600 mL). The mixture was neutralized cautiously with phosphoric acid. After the dissolution of the solid mass, the neutral aqueous layer was withdrawn. The organic layer was extracted repeatedly with a concentrated solution of dibasic sodium phosphate to remove additional acidic organic material. The aqueous extracts were combined, filtered through glass wool, and acidified to pH 1 with 6 N HCl. This solution was placed in the refrigerator overnight, and a product of tan crystals (21.9 g) was collected. Unreacted p-hydroxybenzaldehyde was recovered upon evaporation of the organic layer. The yield based on recovery of starting material was 60%.
4[[(p-Toluidinocarbonyl)methyl]oxy]benzaldehyde (5a)
Dicyclohexylcarbodiimide (DCC; 1.32 g, 6.4 mmol) was added to a solution of compound 4 (1.15 g, 6.4 mmol) in tetrahydrofuran (50 mL). After the mixture was stirred for 10 min, p-toluidine (0.7 g, 6.5 mmol) was added. After 1 h, the precipitate was removed by filtration, and the filtrate was reduced in volume by evaporation. A crystalline product (1.09 g) was obtained by recrystallization of the concentrated filtrate from ethyl acetate-/petroleum ether. In order to obtain analytically pure material, it was necessary to remove a higher Rf impurity by thin-layer chromatography (solvent B). This impurity proved to be the imine adduct of the product with p-toluidine (CHN analysis). NMR of 5a (ppm, Me2SO-d6): 9.90 (s, 1 H, CHO), 7.91 and 7.15 (each d, 2 H, O-phenyl, J = 8.7 Hz), 7.51 and 7.20 (each d, 2 H, N-phenyl, J = 8.8 Hz), 4.85 (8, 2 H, CH2), 2.27 (s, 3 H, CH3).
4-[[(4-Hydroxyanilino)carbonyl]methoxy]benzaldehyde (5b)
Compound 4 (1.80 g, 10 mmol) was dissolved in 25 mL of tetrahydrofuran containing 20% DMF. To this solution were added DCC (2.06 g, 10 mmol) and after 10 min a solution of p-aminophenol hydrochloride (1.46 g, 10 mmol) and triethylamine (0.78 g, 10 mmol) in DMF (10 mL). After 2 h the precipitate was removed by filtration and washed with tetrahydrofuran. The combined filtrates were evaporated and triturated with water. A yellow oil separated and crystallized, providing 2.40 g of 5b. Recrystallization from ethanol/petroleum ether gave an analytical sample.
6-Amino-1,3-dipropyl-5-[[4-[(carboxymethyl)oxy]-benzylidene]amino]uracil (6b)
Compound 4 (1.51 g, 8.37 mmol) was dissolved in a mixture of methanol (35 mL) and acetic acid (5 mL) in a 250-mL boiling flask on a steam bath. To this was added a methanolic solution (60 mL) of freshly synthesized 5,6-diamino-1,3-dipropyluraci110 (3b). After the mixture was heated for 15 min, the volume was reduced by evaporation until crystallization occurred. Ether (40 mL) was added and the nearly white solid was collected to give 2.80 g. Mass spectrum (CI-NH3), m/z 389 (s), 331 (w), 329 (w), 227 (s), 212 (s), 167 (9).
8-[4-[(Carboxymethyl)oxy]phenyl]-1,3-dimethylxanthine (8a)
The benzylidene adduct between compound 4 (0.609 g, 3.38 mmol) and 5,6-diamino-1,3-dimethyluracil hydrate (0.58 g, 3.4 mmol) was prepared as above for the synthesis of 6b. Tan crystals (0.963 g, 85.7%) were obtained upon cooling the reaction mixture overnight in the refrigerator. The benzylidene adduct (98 mg), used without further purification, was dissolved in warm DMF (7 mL), treated with ferric oxide (20 mg), and heated on the steam bath for 4 h. After addition of an equal volume of ethanol, the precipitate was collected and dried to give 76 mg of 8a (67% overall yield from 4).
8-[4-[(Carboxymethyl)oxy]phenyl]-1,3-dipropylxanthine (8b)
Method A
The benzylidene adduct 6b (191 mg, 0.49 mmol) was suspended in trifluoroethanol (15 mL) and dissolved by refluxing on a steam bath. Anhydrous ferric chloride (20 mg) was added, and heating was continued for 2 h. Ether was added to complete the precipitation of product, which was collected and dried in vacuo. The crude product, 0.17 g, was recrystallized from DMF/methanol/ether to give chromatographically pure 8b.
Method B
The ethyl ester 10 (114 mg, 0.28 mmol) was dissolved in DMF (5 mL) and treated with aqueous 0.1 N sodium carbonate (5 mL). The mixture was heated on the steam bath for 0.5 h. The solvent was evaporated, leaving a white film, which was triturated with dilute HCl. The resulting white precipitate was collected and washed with water and dried in vacuo. This material (105 mg) was homogeneous by TLC (solvent B; Rf 0.42) and identical with 8b prepared by method A.
8-[4-[[(p-Toluidinocarbonyl)methyl]oxy]phenyl]-1,3-dipropylxanthine (9b)
The p-toluide of the carboxylic acid congener 8b was prepared by the method described below for compound 9c, except that the reaction was continued overnight.
8-[4-[[[(4-Hydroxyanilino)carbonyl]methyl]oxy]-phenyl]-1,3-dipropylxanthine(9c)
The benzylidene adduct between freshly prepared compound 3b13,14 (0.385 mmol) and the substituted benzaldehyde 5b (88 mg, 0.325 mmol) was formed according to the method described for compound 6b. The solid adduct (0.14 g, 90% yield) was dissolved in hot absolute ethanol (10 mL), treated with ferric chloride (20 mg), and heated on the steam bath until the product precipitated (30 min). Ether was added, and the product (93 mg, 60% overall yield from compounds 3b and 4) was collected by filtration.
8-[4-[(Carboxymethyl)oxy]phenyl]-1,3-dipropylxanthine Ethyl Ester (10)
Compound 6b (1.69 g, 4.3 mmol) was suspended in 100 mL of absolute ethanol. Anhydrous ferric chloride (0.70 g, 4.3 mmol) was added, and the mixture was refluxed on a steam bath for 1 day. The slow conversion of the free acid (identical with compound 8b, Rf 0.35) to the ethyl ester (Rf 0.78) was followed by TLC on silica gel using solvent B. The reaction mixture was evaporated in vacuo to a small volume, and dry diethyl ether was added. The bulky crystalline mass was collected by filtration, washed with ether, and dried in vacuo to give 1.27 g of 10.
N-[[[[4-(1,3-Dipropyl-8-xanthinyl)phenyl]oxy]methyl]-carbonyl]-N,N'-dicyclohexylurea (11) and 8-[4-[(Carboxymethyl)oxy]phenyl]-1,3-dipropylxanthine N-Hydroxysuccinimide Ester (12)
The carboxylic acid congener 8b (18.4 mg, 0.048 mmol) was dissolved in DMF (5 mL), cooled in an ice bath, and treated with N-hydroxysuccinimide (6 mg) and DCC (11 mg). After the mixture was stirred for 1 day at room temperature, the urea was removed by filtration. Upon addition of water a white solid precipitated and was collected. Recrystallization from DMF/water provided 11.1 mg of pure 12. A side product removed by crystallization proved to be the N-acylurea 11.
The identical urea 11 was isolated as an undesired product in the attempted DCC coupling in DMF/water of the carboxylic acid 6b with highly polar amines.
8-[4[[[[(2-Aminoethyl)amino]carbonyl]methyl]oxy]-phenyl]-1,3-dipropylxanthine (13c)
Compound 10 (57.5 mg, 0.14 mmol) was dissolved in warm DMF (1.0 mL). When the solution reached room temperature, ethylenediamine (1.0 mL) was added. After the solution was stirred overnight, most of the solvent was evaporated under a stream of nitrogen. The resulting oil was triturated with methanol. After crystallization began, ether was added, and the product was collected and dried to give 59 mg of 13c, homogeneous by TLC (solvent system A): UV peaks at 309 and 248 nm.
8-[4-[[[[[2-(Biotinylamino)ethyl]amino]carbonyl]-methyl]oxy]phenyl]-1,3-dipropylxanthine (14b)
Compound 13c (24.1 mg, 0.056 mmol) was suspended in 1 mL of DMF. N-Succinimidyl-d-biotin (Sigma, 23.6 mg, 0.069 mmol) was added with stirring. A solution formed after several minutes, and a precipitate appeared soon thereafter. After 1 day methanol (1 mL) and diethyl ether were added. The precipitate was collected and dried to give 26.6 mg of 14b.
8-[4-[[[[[2-[(4-Hydroxybenzylidene)ammonio]ethyl]-amino]carbonyl]methyl]oxy]phenyl]-1,3-dipropylxanthine Acetate (15)
Compound 13c (56 mg, 0.13 mmol) and 4-hydroxybenzaldehyde (19 mg, 0.16 mmol) were dissolved in warm acetic acid (5%) in ethanol (2 mL) and heated on a steam bath for 2 h. The solvent was evaporated and the residue triturated with ether to give 15 as a tan solid: NMR (ppm, Me2SO-d6) 8.15 (s, 1 H, CH=N), 8.05 and 7.04 (each d, 2 H, 8-phenyl, J = 8.9 Hz), 7.55 and 6.78 (each d, 2 H, phenol, J = 8.5 Hz), 4.56 (s, 2 H, CH2O), 3.59 (CH2N), 1.91 (s, 3 H, acetate), and signals from propyl groups (Table III).
Table III.
NMR Dataa for Xanthines
 | ||||
|---|---|---|---|---|
| compd | Ab | Bb | Cc | R |
| 8a | 8.07 | 7.05 | 4.77 | |
| 8b | 8.05 | 7.05 | 4.78 | |
| 9a | 8.12 | 7.14 | 4.79 | 7.52,b 7.14,b 2.27 (s, 3 H, CH3) |
| 9b | 8.10 | 7.14 | 4.78 | 7.53,b 7.14,b 2.27 (s, 3 H, CH3) |
| 9c | 8.13 | 7.17 | 4.77 | 7.45,b 6.76b |
| 10 | 8.07 | 7.08 | 4.88 | 4.12 (q, 2 H, CH2), 1.23 (t, 3 H, CH3) |
| 11 | 8.03 | 6.86 | 4.77 | 5.57 (1 H), 3.5 (1 H), 1.2–1.6 (20 H) |
| 12 | 8.09 | 7.14 | 5.43 | 2.84 (s, 4 H) |
| 13a | 8.07 | 7.07 | 4.51 | 7.58 (s, 1 H, NH), 7.17 (s, 1 H, NH) |
| 13b | 8.05 | 7.04 | 4.91 | 3.02 (s, 3 H), 2.86 (s, 3 H) |
| 13c | 8.05 | 7.05 | 4.55 | 3.20 (2 H, CH2, 2.68 (2 H, CH2) |
| 13d | 8.00 | 6.98 | 4.53 | 3.12 (2 H, CH2), 2.77 (2 H, CH2), 1.2–1.6 (12 H) |
| 13e | 8.05 | 7.08 | 4.56 | 9.35 (1 H, NH) |
| 14a | 8.08 | 7.09 | 4.55 | 3.1–3.2 (4 H, (CH2)2), 1.79 (s, 3 H, CH3) |
| 14b | 8.07 | 7.04 | 4.55 | 6.40d and 6.34d (each s, 1 H, NH), 4.3d and 4.1d (each 1 H, CH2NH), 3.17 (4 H, CH2NH), 3.05d (1 H, CHS), 2.6d and 2.8d (each 1 H, CH2S), 2.04d (t, 2 H, CH2CO), 1.3–1.5d (6 H) |
| 16 | 8.06 | 7.1 | 4.53 | 7.1 (2 H, Ar), 6.68 (2 H, Ar), 4.34 (s, 2 H, CH2Ar), 3.44, 3.22, and 2.43 (each 2 H, CH2), 1.0 (CH3) |
In Me2SO-d6, δ from Me4Si. Signals from the 1,3-dialkyl groups are not included. Typical values are as follows: 3.51 (s, 3 H), 3.28 (s, 3 H) for compound 9a; 4.06 (t, 2 H, CαH2), 3.91 (t, 2 H, CαH2), 1.78 (2 h, CβH2), 1.63 (2 H, CβH2), 0.90 (6 H, CH3) for compound 9b.
Doublet, 2 H, aromatic, J = 8–9 Hz
Singlet, 2 H.
Biotin moiety.
8-[4-[[[[[2-[Ethyl(4-hydroxybenzyl)ammonio]ethyl]-amino]carbonyl]methyl]oxy]phenyl]-1,3-dipropylxanthine Acetate (16)
Compound 15 (8.7 mg, 0.015 mmol) was suspended in methanol (1 mL) and treated with excess sodium cyanoborohydride (20 mg, 0.32 mmol). The mixture was warmed at 60 °C to form a solution and treated with acetaldehyde (0.03 mL). After 2 h the solvent was evaporated and the residue was chromatographed on LH-20 eluting with methanol. Evaporation of the solvent left a clear film of 16 (5.9 mg, 61%). The product was chromatographically pure (Rf 0.45, Analtech RPS-F, 75% MeOH/5% HOAc/H2O, positive Pauly reaction, unreactive toward ninhydrin). An average molecular weight of 563 was determined by californium plasma desorption mass spectroscopy.
Biochemical Assays
Inhibition of binding of 1 nM N6-[3H]cyclohexyladenosine to A1-adenosine receptors in rat cerebral cortical membranes was assayed as described.24 Inhibition of binding by a range of concentrations of each xanthine was assessed in triplicate for at least two separate experiments. Inhibition of 2-chloroadenosine-stimulated cyclic AMP accumulation in [3H]adenine-labeled guinea pig cerebral cortical slices was assayed essentially as described.24 In the present experiments 10 µg/mL of adenosine deaminase was present in incubations with slices to prevent effects of endogenous adneosine, and 30 µM 4-[3(cyclopentyloxy)-4-methoxyphenyl]-2-pyrrolidone (rolipram, ZK 62711)25 was present to inhibit phosphodiesterases. Under these conditions 2-chloroadenosine elicited a maximal 10–20-fold increase in levels of radioactive cyclic AMP in guinea pig cortical slices with an EC50 of aboout 8 µmol (data not shown). Inhibition of the response to 15 µM 2-chloroadenosine by a range of concentrations of each xanthine was assessed in triplicate in at least two separate experiments.
Table II.
Analytical Data
| compd | method | % yield | mp, °C | formula | anal. | MSf |
|---|---|---|---|---|---|---|
| 4 | a | 60 | 191–193 | C9H8O4 | C, H | 209, 181, 135g |
| 5a | b | 63 | 124–126 | C16H15NO3 | H, N; Cl | 270g |
| 5b | b | 89 | 185–186 | C15H13NO4 | C, H, N | 272g |
| 6b | a | 86 | 179–180 | C19H24N4O5 | C, H, N | 389 |
| 8a | c | 67 | >310 | C15H14N4O5·1/2Me2SO | C, H, N | 331, 272 |
| 8b | c | 77 | 283–285 | C19H22N4O5 | C, H, N | 387, 329 |
| 9a | a | 22 | 287–290 | C22H21N5O4·1/2H2O | C,H, N | 420, 272 |
| 9b | c | 32 | >300 | C26H29N5O4·5/4H2O | C, H, N | 476i |
| 9c | c | 60 | >320 | C25H27N5O5·H2O | C, H, N | 478i |
| 10 | a | 71 | 243–244 | C21H26N4O5 | C, H, N | 415g |
| 11 | a | 63 | d < 190j | C32H44N6O5·1/2H2O | C, H, N | 593i |
| 12 | a | 48 | 241–245 | C23H25N5O7·1/2H2O | C, H, N | 386,h 329 |
| 13a | d | 62k | 301–303 | C19H23N5O4 | H, N; Cm | 386, 329 |
| 13b | d | 68k | 227–231 | C21H27N5O4 | n | 414, 329 |
| 13c | d | 99 | 218–220d | C21H28N6O4·5/6H2O | C, H, N | 429, 411g |
| 13d | d | 14k | > 300 | C27H40N6O4 | o | 513i |
| 13e | d | 90 | >310 | C19H24N6O4·7/4H2O | C, H, N | 401, 328 |
| 14a | e | 51 | 309–312 | C23H30N6O5·5/4H2O | C, H, N | 471i |
| 14b | e | 50 | 262–264 | C31H42N8O6S·3/2H2O | C, H, N | 655i |
| 15 | a | 75 | 186–190 | C30H36N6O7 | C, H, N |
Refer to Experimental Section.
Carbodiimide coupling as in 5b
Benzylidene formation and oxidation (overall yield given). Compound 8a was recrystallized from Me2SO/ether.
Aminolysis as in 13d.
N-Hydroxysuccinimide coupling
Chemical ionization, NH3 gas, unless noted.
CH4 gas.
Converted to compound 13a by ionizing gas.
Californium plasm desoprtion mass spectrometry,24 positive ions.
Raise temperature rapidly.
Chromatographically homogeneous product obtained after preparative TLC (solvent B).
C: calcd, 71.36; found, 70.80; accurate mass; calcd, 269.1052; found, 269.1040.
C: calcd, 59.21; found, 58.07; accurate mass; calcd, 385.1750; found, 385.1730.
Accurate mass; calcd, 413.2063; found, 413.2076.
Noncrystalline solid; could not be recrystallized.
Acknowledgment
We thank Dr. M. Shamim for helpful discussions.
Registry No. 3b, 81250-34-2; 4, 22042-71-3; 5a, 17172-57-5; 5b, 96865-80-4; 6a, 96865-95-1; 6b, 96865-81-5; 7b, 96865-96-2; 8a, 96865-82-6 8b, 96865-83-7; 9a, 96865-84-8; 9b, 96865-85-9; 9c, 96865-86-0; 10, 96865-87-1; 11, 96865-88-2; 12, 96865-89-3; 13a, 96865-90-6; 13b, 96865-91-7; 13c, 96865-92-8; 13d, 96865-93-9; 13e, 96865-94-0; 14a, 96896-78-5; 14b, 96896-79-6; 15, 96896-81-0; 16, 96896-82-1; H2N(CH2)8NH2, 373-44-4; p-hydroxybenzaldehyde, 123-08-0; iodoacetic acid, 64-69-7; p-toluidine, 106-49-0; p-aminophenol, 123-30-8; 5,6-diamino-1,3-dimethyluracil, 5440-00-6; ethylenediamine, 107-15-3; N-succinimidyl-d-biotin, 35013-72-0; adenosine, 58-61-7.
References
- 1.(a) Daly JW. J. Med. Chem. 1982;25:197. doi: 10.1021/jm00345a001. [DOI] [PubMed] [Google Scholar]; (b) Daly JW. Adv. Cyclic Nucleotide Res. in press. [PubMed] [Google Scholar]
- 2.(a) Smellie FW, Davis CW, Daly JW, Wells JN. Life Sci. 1979;24:2475. doi: 10.1016/0024-3205(79)90458-2. [DOI] [PubMed] [Google Scholar]; (b) Bruns RF. Biochem. Pharmacol. 1981;30:325. doi: 10.1016/0006-2952(81)90062-9. [DOI] [PubMed] [Google Scholar]
- 3.Bruns RF, Daly JW, Snyder SH. Proc. Natl. Acad. Sci. U.S.A. 1980;77:5547. doi: 10.1073/pnas.77.9.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Daly JW, Padgett W, Shamim MT, Butts-Lamb P, Waters J. J. Med. Chem. 1985;28:487. doi: 10.1021/jm00382a018. [DOI] [PubMed] [Google Scholar]
- 5.Jacobson KA, Kirk KL, Padgett WL, Daly JW. Functionalized congeners of adenosine: preparation of analogues with high affinity for A1-adenosine receptors. J. Med. Chem. 1985;28:1341–1346. doi: 10.1021/jm00147a039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Jacobson KA, Marr-Leisy D, Rosenkranz RP, Verlander MS, Melmon KL, Goodman M. J. Med. Chem. 1983;26:492. doi: 10.1021/jm00358a007. [DOI] [PubMed] [Google Scholar]; (b) Rosenkranz RP, Hoffman BB, Jacobson KA, Verlander MS, Klevans L, O’Donnell M, Goodman M, Melmon KL. Mol. Pharmacol. 1983;24:429. [PubMed] [Google Scholar]
- 7.Jacobson KA, Rosenkranz RP, Verlander MS, Melmon KL, Goodman M. In: Peptides 1982. Bláha K, Malorň P, editors. Berlin: Walter de Gruyter; 1983. p. 337. [Google Scholar]
- 8.Verlander MS, Jacobson KA, Rosenkranz RP, Melmon KL, Goodman M. Biopolymers. 1983;22:531. doi: 10.1002/bip.360220166. [DOI] [PubMed] [Google Scholar]
- 9.Simons SS, Thompson EB, Johnson DF. Biochemistry. 1979;18:4915. doi: 10.1021/bi00589a020. [DOI] [PubMed] [Google Scholar]
- 10.Jacobson AE, Bajwa BS, Streaty RA, Klee WA, Rice KC. Life. Sci. 1983;33(Suppl I):159. doi: 10.1016/0024-3205(83)90468-x. [DOI] [PubMed] [Google Scholar]
- 11.Pfeuffer T, Gaugler B, Metzger H. FEBS Lett. 1983;164:154. doi: 10.1016/0014-5793(83)80040-4. [DOI] [PubMed] [Google Scholar]
- 12.(a) Jacobson KA, Kirk KL, Padgett W, Daly JW. FEES. Lett. 1985;184:30. doi: 10.1016/0014-5793(85)80646-3. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lavielle S, Chassaing G, Marquet A. Biochim. Biophys. Acta. 1983;759:270. doi: 10.1016/0304-4165(83)90323-9. [DOI] [PubMed] [Google Scholar]; (c) Hoffman K, Zhang WJ, Romovacek H, Finn FM, Bothner-By AA, Mishra PK. Biochemistry. 1984;23:2547. doi: 10.1021/bi00307a002. [DOI] [PubMed] [Google Scholar]
- 13.Papesch V, Schroeder EH. J. Org. Chem. 1951;16:1879. [Google Scholar]
- 14.Bariana DS. Can. J. Chem. 1968;46:3413. [Google Scholar]
- 15.Fredholm BB, Brodin K, Strandberg K. Acta Pharmacol. Toxicol. 1979;45:336. doi: 10.1111/j.1600-0773.1979.tb02402.x. [DOI] [PubMed] [Google Scholar]
- 16.Osswald H. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1979;288:79. doi: 10.1007/BF00501815. [DOI] [PubMed] [Google Scholar]
- 17.Stroud MW, Lambertsen CJ, Ewing JH, Kough RH, Gould RA, Schmidt CF. J. Pharmacol. Exp. Ther. 1955;114:461. [PubMed] [Google Scholar]
- 18.Snyder SH, Katims JJ, Annau Z, Bruns RF, Daly JW. Proc. Natl. Acad. Sci. U.S.A. 1981;78:3260. doi: 10.1073/pnas.78.5.3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ogilvie RI, Fernandez PG, Winsberg F. Eur. J. Clin. Pharmacol. 1977;17:409. doi: 10.1007/BF00561059. [DOI] [PubMed] [Google Scholar]
- 20.Laska EM, Sunshine A, Zighelboim I, Roure I, Marrero J, Wanderling J, Olson N. Clin. Pharmacol. Ther. 1983;33:498. doi: 10.1038/clpt.1983.68. [DOI] [PubMed] [Google Scholar]
- 21.Dunlap MK, Kaplar RJ, Rosenberg EW. Clin. Res. 1982;30:582A. [Google Scholar]
- 22.Bruns RF, Daly JW, Synder SH. Proc. Natl. Acad. Sci. U.S.A. 1983;80:2077. doi: 10.1073/pnas.80.7.2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.MacFarlane RD. Anal. Chem. 1983;55:1247A. [Google Scholar]
- 24.Daly JW, Butts-Lamb P, Padgett W. Cell. Mol. Neurobiol. 1983;3:6. doi: 10.1007/BF00734999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwabe U, Miyake M, Ohga Y, Daly JW. Mol. Pharmacol. 1976;12:900. [PubMed] [Google Scholar]