

Abstract
Focal brain ischemia leads to a slow type of neuronal death in the penumbra that starts several hours after ischemia and continues to mature for days. During this maturation period, blood flow, cellular ATP and ionic homeostasis are gradually recovered in the penumbral region. In striking contrast, protein synthesis is irreversibly inhibited. This study used a rat focal brain ischemia model to investigate whether or not irreversible translational inhibition is due to abnormal aggregation of translational complex components, i.e. the ribosomes and their associated nascent polypeptides, protein synthesis initiation factors and co-translational chaperones. Under electron microscopy, most rosette-shaped polyribosomes were relatively evenly distributed in the cytoplasm of sham-operated control neurons, but clumped into large abnormal aggregates in penumbral neurons subjected to 2 h of focal ischemia followed by 4 h of reperfusion. The abnormal ribosomal protein aggregation lasted until the onset of delayed neuronal death at 24–48 h of reperfusion after ischemia. Biochemical study further suggested that translational complex components, including small ribosomal subunit protein 6 (S6), large subunit protein 28 (L28), eukaryotic initiation factors 2α, 4E and 3g, and co-translational chaperone heat-shock cognate protein 70 (HSC70) and co-chaperone Hdj1, were all irreversibly clumped into large abnormal protein aggregates after ischemia. Translational complex components were also highly ubiquitinated. This study clearly demonstrates that focal ischemia leads to irreversible aggregation of protein synthesis machinery that contributes to neuronal death after focal brain ischemia.
Keywords: brain ischemia, chaperone, heat-shock cognate protein 70, protein aggregation, protein synthesis, ribosomal proteins, stress granule, ubiquitin
Although substantial progress has been made, cellular and molecular mechanisms underlying focal ischemic neuronal damage are still incompletely understood. Focal brain ischemia leads to acute cell death in the core area and a slow type of neuronal death in the penumbra that can begin hours after ischemia and continue to mature for days (Siesjo et al. 1995; Lee et al. 2002; Wang et al. 2004). During this maturation period, protein synthesis is persistently inhibited in these penumbral neurons destined to die (Mies et al. 1991; Hossmann 1993; Kokubo et al. 2003). Activation of PKR-like ER eIF-2α kinase (PERK) may be responsible, at least in part, for the increase in eukaryotic initiation factor 2a (eIF2α) phosphorylation and for the transient suppression of protein synthesis early in reperfusion after transient brain ischemia. However, the nature of the transient change in phosphorylation of PERK and eIF2α may suggest that eIF2α phosphorylation cannot account for irreversible inhibition of protein synthesis after brain ischemia (Hu and Wieloch 1993; Burda et al. 1994; Althausen et al. 2001; DeGracia 2004; Owen et al. 2005).
Cellular proteins in non-native states, i.e. those newly synthesized, misfolded, denatured or damaged, expose sticky hydrophobic surfaces and are highly toxic in cells (Bukau et al. 1996; Eggers et al. 1997; Fink 1999; Hardesty et al. 1999; Taylor et al. 2002). To avoid cellular proteotoxicity, proteins in non-native states are normally protected and processed by cellular protein quality control systems (Hartl and Hayer-Hartl 2002). Molecular chaperones can shield hydrophobic surfaces of unfolded proteins, thus preventing their misfolding and aggregation. Irreparably damaged proteins must be quickly degraded by the ubiquitin-proteasome system (Hu et al. 2004). Large protein aggregates and damaged subcellular structures may also be eliminated by the machinery of autophagy (Shintani and Klionsky 2004).
Newly synthesized proteins are the major sources of unfolded proteins in normal cells (Hartl and Hayer-Hartl 2002). Numerous nascent polypeptides normally emerge from their parent polyribosomes at any given moment. Without chaperone protection, partially or newly synthesized polypeptide chains on polyribosomes expose hydrophobic segments; they are highly prone to intramolecular misfolding and intermolecular aggregation driven by the exposed hydrophobic force in the extremely crowded intracellular milieu (Hartl and Hayer-Hartl 2002). Therefore, protein folding during mRNA translation, i.e. co-translational folding, generally requires co-operative interaction among nascent polypeptides, assistant chaperone protein heat-shock cognate protein 70 (HSC70), its co-chaperone Hdj1 [a member of the heat-shock protein 40 (HSP40) family] and a cellular ATP supply (Frydman 2001; Hartl and Hayer-Hartl 2002). If nascent chains cannot be folded successfully, they must be quickly tagged by ubiquitin and degraded by proteasomes (Murata et al. 2003).
We previously reported abnormal protein aggregation in ischemic penumbral neurons after focal brain ischemia (Hu et al. 2001). However, the molecular composition of protein aggregates was unknown. In this study, we analyzed the molecular composition of protein aggregates and clearly demonstrated that translational complex components are major elements of protein aggregates after focal brain ischemia. Irreparable damage of protein synthesis machinery by abnormal aggregation of translational complex components leads to irreversible malfunction of protein synthesis, thus contributing to neuronal death after focal brain ischemia.
Materials and methods
Focal brain ischemia model
Male Wistar rats (Charles River, Wilmington, MA, USA) weighing 260–310 g were fasted overnight with free access to water. All experimental procedures were approved by the Animal Care and Use Committee, the University of Miami School of Medicine, and were performed in compliance with the National Institutes of Health guidelines on the ethical use of animals. All efforts were made to minimize animal suffering and to reduce the number of animal used. Anesthesia was induced by inhalation of 3% halothane in N2O : O2 (70% : 30%) and was maintained at 0.7–1.5% during the surgical procedures. The tail artery was cannulated to monitor blood gases, pH, blood glucose and blood pressure. Heparin (0.1 mL, 300 U/mL) was administered through the tail artery to avoid formation of thrombosis distal to the occluding filament. A surgical incision was made to expose the right common carotid artery (CCA), internal carotid artery (ICA) and external carotid artery (ECA). The ECA was ligated proximal to the origin of any branches, such as the occipital artery. The proximal CCA was then ligated and temporarily closed proximal to the carotid bifurcation by a microvascular clip. A small incision was made in the CCA. The occlusion filament was inserted into the ICA through the CCA 19–21 mm distal from the bifurcation to produce the middle cerebral artery occlusion (MCAO). The filament was prepared from monofilament fishing line and covered with a distal cylinder of silicon rubber (diameter 0.31–0.32 mm). After the MCAO had been performed, the animals were extubated and allowed to resume spontaneous breathing. Two hours after induction of ischemia, the filament was withdrawn while the animals were breathing halothane in N2O : O2 via a face mask. During the operation, the temperature was maintained at 37 ± 0.5°C by a heating pad.
Three series of 2 h MCAO animals were prepared for biochemical analysis, electron microscopy (EM) and confocal microscopy. They were re-anesthetized at 1, 4 or 24 h of reperfusion (n = 3 in each experimental group). For biochemical studies, brains were obtained by freezing them in situ with liquid nitrogen while the animals were artificially ventilated (Pontén et al. 1973). The penumbral tissue samples (about 50 mg) were dissected at – 12°C in a cold room, according to the procedure described previously (Hu et al. 2001). Figure 1 illustrates a coronal brain plane between the striatal and hippocampal levels: region 1 is the striatal ischemic core, region 2 is the neocortical ischemia core, region 3 represents ischemic penumbra and region 4 is the contralateral neocortical region (for details, see Hu et al. 2001). The penumbral tissue samples were collected for biochemical analyses. For EM, brains were perfusion-fixed with ice-cold 2% paraformaldehyde and 2.5% glutaraldehyde in 0.1 mol/L cacodylate buffer. For laser-scanning confocal microscopy, brains were perfusion-fixed with ice-cold 4% paraformaldehyde in phosphate-buffered saline (PBS) and processed for electron and confocal microscopic studies (see below). Sham-operated rats were subjected to the same surgical procedures but without induction of brain ischemia.
Fig. 1.
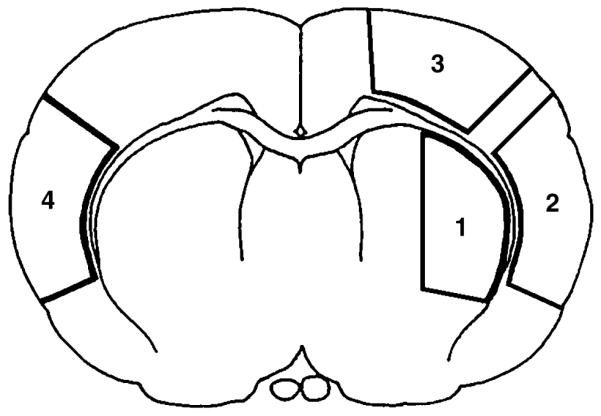
Schematic drawing of the four brain regions analyzed in this study. The coronal section was taken from Bregma 0.48. Regions 1–3 are in the ischemic hemisphere and region 4 is in the contralateral side.
Histopathology
Perfusion-fixed brains were cut at 20 μm coronally with a vibratome at the dorsal striatal level and stained with celestine blue and acid fuschin. Neuronal histopathology in brain regions 1–4 (Fig. 1) was examined according to the method of Smith et al. (1984).
Electron microscopy
Electron microscopy was carried out on brain tissue sections stained with the conventional osmium-uranyl-lead method, as described previously (Hu et al. 2001). Briefly, coronal brain sections were cut at a thickness of 120 μm with a vibratome, at the level between the striatal and dorsal hippocampal planes, and post-fixed for 1 h with 4% glutaraldehyde in 0.1 mol/L cacodylate buffer (pH 7.4). Brain sections were post-fixed for 2 h in 1% osmium tetroxide in 0.1 mol/L cacodylate buffer, rinsed in distilled water and stained with 1% aqueous uranyl acetate overnight. Tissue sections were dehydrated in an ascending series of ethanol to 100%, followed by dry acetone, and embedded in Durcopan ACM (Fluka Chemie AG, Bucks, Switzerland). Small squares from penumbral region 3 were dissected and mounted on resin blocks. Ultrathin sections (0.1 lm) were prepared and stained for 1 min with 3% lead citrate prior to examination with a transmission electron microscope (Zeiss 10C; Zeiss, Oberkochen, Germany).
Confocal microscopy
Confocal microscopy was performed on coronal brain sections (50 μm) from sham-operated control rats, and on rats subjected to 2 h of MCAO followed by 4 and 24 h of reperfusion. Brain sections were transferred to a 24-well microtiter plate half filled with 0.01 M citric acid/sodium citrate buffer (pH 6.0), heated five times for 5 s each time in a microwave oven set to 30% power, and then washed twice with 0.2% Triton X100 (TX100)/PBS for 10 min. Non-specific binding sites were blocked with 3% bovine serum albumin (BSA) in PBS/0.2% TX100 for 30 min. The brain sections were incubated overnight at 4°C with antibodies against ubiquitin (Cell Signaling, Beverly, MA, USA), eIF3η (SC-16377: Santa Cruz Biotechnology, Santa Cruz, CA, USA), small ribosomal subunit protein (S6) (2212: Cell Signaling) and large ribosomal subunit protein (L28) (SC-14151: Santa Cruz Biotechnology), or HSC70 (SPA-815: Stressgen Biotechnology, Victoria, Canada), all at dilutions of 1 : 150 in PBS containing 0.1% TX100. The sections were washed three times for 10 min each in PBS containing 0.1% TX100 at room temperature (23 ± 2°C) and incubated with a fluorescein-labeled secondary antibody (Jackson ImmunoResearch, West Grove, PA, USA). The sections were washed three times in PBS/0.1% TX100, mounted on glass slides and coverslipped using Gelvatol. Gelvatol was prepared by mixing 2.4 g polyvinyl alcohol with 6 g glycerol and 12 ml of 0.2 M Tris-base/HCl (pH 8.5), and heating the mixture to 50°C for 10 min, followed by centrifuge 7000 g for 15 min to remove undissolved particles, and then mixing DABCO (1.4.-diazabicyclo(2.2.2)octane, Sigma Cat No. D2522) to 2.5% of the solution. All reagents in Gelvatol were purchased from Sigma (Sigma-Aldrich Corp, St Louis, MO, USA). The slides were analyzed with a Zeiss 50 confocal microscope.
Sedimentation and immunoblot analyses of translational complex
The dorso-lateral neocortical tissue (about 100 mg) was homogenized with 10 volumes of ice-cold homogenization buffer (Hu et al. 2001). The homogenate was centrifuged at 10 000 g at 4°C for 10 min to obtain a pellet and a supernatant fraction. The pellet containing the translational complex was suspended with 10 volumes of ice-cold homogenization buffer containing 1% TX100 detergent and 400 mm KCl salt, sonicated five times for 5 s each, washed on a shaker overnight at 4°C, then centrifuged at 20 000 g for 10 min to obtain a TX/salt-insoluble fraction that contained protein aggregates formed after brain ischemia (Hu et al. 2000, 2001). The detergent/salt-insoluble fractions were suspended in PBS with a sonicator and layered onto pre-formed 10–55% linear sucrose gradients (made up in a buffer containing 20 mm Tris-HCl pH 8.0, 140 mm KCl, 5 mm MgCl2, 1 mm dithiothreitol) in 13 mL Beckman (Palo Alto, CA, USA) SW41 centrifuge tubes. Centrifugation was performed at 110 000 g. for 3.5 h with a Beckman SW41Ti rotor. Gradient fractions were collected from the bottom of the centrifuge tube. Thirty-six fractions, each with a volume of 365 μL, were collected with a fraction collector. Absorbance at an ultraviolet (UV) wavelength of 260 nm in each fraction was measured with a Beckman spectrometer. Aliquots (50 μL) of each fraction were analyzed by western blotting with 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE). Following electrophoresis, proteins were transferred to an Immobilon-P membrane (Millipore Corp., Billerica, MA, USA). Western blots were labeled with antibodies against the following translational complex components: S6 (molecular weight 32 kDa, Cat. no. 2212), eIF-2α (40 kDa, Cat. no. 9722) and eIF-4E (26 kDa, Cat. no. 9742) (all at dilution of 1 : 1000; Cell Signaling Technology); L28 (15.7 kDa, Cat. no. SC-14151) and eIF-3η (115 kDa, Cat. no. SC-16377) (all at dilution of 1 : 700; Santa Cruz Biotechnology); HSC70 (73 kDa, Cat. no. SPA-815, 1 : 10 000) and Hdj1 (40 kDa, Cat. no. SPA-400, 1 : 1000 (Stressgen Biotech); and ubiquitinated proteins (> 90 kDa, Cat. no. 3936, 1 : 1000; Cell Signaling Technology). The blots were developed with an enhanced chemiluminescence (ECL) detection method (Amersham Bioscience, Piscataway, NJ, USA).
Statistical analysis
Immunoblot bands were scanned into a computer and analyzed with Kodak ID image analysis software (Eastman Kodak Company, Rochester, NY, USA). Each immunoblot band was calculated as the mean intensity value subtracted from the background value. The optical densities of immunoblot bands in each translational complex component peak fractions were quantified using Kodak 1D gel analysis software, and then the optical densities were added together. Data are expressed as mean ± SD. One-way anova followed by Fisher’s protected least significant difference (PLSD) post-hoc test was used to assess statistical significance (p < 0.05).
Results
Histopathology
Neuronal damage in brain regions after focal ischemia was examined on vibratome sections stained with acid fuchsin and celestin blue. Substantial neuronal death was already present at 1 h of reperfusion in the striatal core region (Fig. 1). Some small infarct foci were found at 1 h of reperfusion in the cortical core region. By 4–24 h of reperfusion, selective neuronal death was seen in the neocortical penumbral region (Fig. 1, region 3). No neuronal death was observed in the contralateral hemisphere or in sham-operated controls. All these data are consistent with our previous study (Hu et al. 2001).
Ultrastructural evidence of translational complex aggregation after focal brain ischemia
Ultrastructure of penumbral neocortical neurons in the dorsolateral neocortical region from sham-operated control rats and rats subjected to 2 h of focal ischemia followed by 24 h of reperfusion is shown in Fig. 2. Brain neurons, particularly pyramidal neurons, have unique nuclei and dendrites distinguishable from other brain cell types under EM (Hu et al. 2000; Liu and Hu 2004; Liu et al. 2005a). Ribosomal rosettes were normally scattered in the cytoplasm or associated with the rough endoplasmic reticulum (ER) of sham-operated neocortical neurons (Fig. 2a, arrows). The ER and mitochondria (M) were seen in the cytoplasm (Fig. 2a). After ischemia, ribosomal rosettes and ER-associated ribosomal studs were greatly reduced, whereas single ribosomes (Fig. 2b, arrowheads) and large ribosome-like aggregates (Fig. 2b, arrows) appeared in the cytoplasm. Protein aggregate-containing penumbral neurons had intact cell membranes and relatively normal nuclear morphology (Fig. 2b, N), suggesting that ribosomal aggregate-containing neurons were still alive but would die slowly (Mies et al. 1991; Hu et al. 2001). Higher magnification of the neocortical neuron is shown in Fig. 1(a and b). Ribosomal rosettes and ER-associated ribosomal studs were normally distributed in sham-operated neurons (Fig. 2c, arrows). After ischemia, ribosomes were abnormally clumped into large clusters (Fig. 2d, arrows). Some large ribosomal clusters were also associated with the mitochondrial membranes (Fig. 2d, M).
Fig. 2.

(a, b) Electron micrographs of neuronal soma in the penumbral regions from a sham-operated control rat (a) and a rat subjected to 2 h MCAO followed by 24 h of reperfusion (b). The ER, mitochondria (M), nucleus (N) and ribosomal rosettes (arrows) were normally distributed in neurons from a sham-operated brain. After ischemia, ribosomal rosettes and ER-associated ribosomal studs were either dissociated into single ribosomes (b, arrowheads) or clumped into large aggregates (b, arrows). Scale bar = 0.5 μm. (c, d) Higher magnification of ribosomal aggregation after focal ischemia. Brain sections were from the same tissues as in Fig. 2 (a,b). Ribosomal rosettes (c, arrows) and ER-associated ribosomal studs (c, arrowheads) were distributed normally in a sham-operated control neuron. After ischemia, ribosomes were abnormally clumped into large clusters or aggregates (d, arrows). Some aggregates were associated with mitochondria (M). Scale bar = 0.1 μm.
Biochemical analysis of translational complex aggregation
The above EM studies suggest that ribosomes are aggregated in penumbral neurons after focal ischemia. To study ribosomal protein aggregation further, we carried out sedimentation analysis of translational complex components in a sucrose density gradient, in conjunction with immunoblotting (Figs 3 and 4). When proteins become aggregated, their detergent/salt solubility decreases dramatically and they cannot be dissolved in a 1% Triton X100 and 400 mm KCl solution (Kabakov and Gabai 1994; Kazantsev et al. 1999; Hazeki et al. 2000; Stockel and Hartl 2001). We therefore isolated protein aggregate-containing fractions from rat brains after focal brain ischemia with a buffer solution containing 1% Triton X100 and 400 mm KCl. Dorsolateral neocortical tissues from ischemic hemispheres collected from rats subjected to either sham-surgery or 2 h MCAO followed by 4 and 24 h of reperfusion, as described previously (see Hu et al. 2001), were used for this sedimentation analysis. The detergent/salt-insoluble fractions were resolved on 10–55% sucrose gradients by centrifugation, and the sedimentation profiles were recorded at the UV wavelength of 260 nm (UV-260) (Figs 3a and 4a). Thirty-six fractions were collected. The major sedimentation peak in sham-control samples was located between fractions 10 and 14 in the gradient. However, the major sedimentation peak of post-ischemic samples shifted to higher densities located between fractions 16 and 21 at 4 h of reperfusion, and shifted further to between fractions 27 and 33 at 24 h of reperfusion (Figs 3a and 4a).
Fig. 3.

(a) Sedimentation, (b) immunoblots and (c, d) optical density analyses of translational complex components in sucrose density gradient fractions. Rats were subjected either to sham surgery or 2 h of MCAO followed by 4 and 24 h of reperfusion. The detergent/salt-insoluble protein aggregate-containing fractions were resolved in 10–55% sucrose gradients by ultracentrifugation. (a) One major UV-260 nm absorbance peak between density fractions 11 and 14 from the sham-control sample was resolved in the density gradient (sham, arrow). This peak shifted to higher density fractions 16–21 at 4 h (4 h, arrow, dashed line) and fractions 27–33 at 24 h of reperfusion (24 h, arrow, solid line), respectively. (b) L28 (kDa, immunoblot between 10 and 25 kDa is shown), S6 (between 25 and 45 kDa), eIF2α (30–55 kDa), eIF3η (between 75 and 150 kDa) and eIF4E (between 15 and 35 kDa) were shifted into higher densities after ischemia (4 and 24 h). (c, d) Changes in optical densities of protein bands of L28, S6, eIF2α, eIF4E and eIF3η were evaluated with Kodak 1D image software. The levels of L28 and S6 are presented as folds of controls. The levels of eIFs were undetectable in controls. Therefore, they are presented as optical densities. Data are expressed as mean ± SD (n = 3); *denotes p < 0.05 between control and experimental conditions, one-way anova followed by Fisher’s PLSD post-hoc test.
Fig. 4.

Immunoblot analysis of HSC70, Hdj1, ubi-proteins, HSP70 and HSC/HSP70 interacting protein (HIP) in the density gradient fractions. The UV-260 profiles in the gradient fractions were the same as in Fig. 3a. (b) Relative to sham-operated controls, HSC70 (immunoblot membrane between 55 and 90 kDa is shown), Hdj1 (between 30 and 50 kDa) and ubi-proteins (ubi-, > 90 kDa) were shifted into higher densities after ischemia (4 and 24 h). Inducible HSP70 (between 55 and 90 kDa) was absent from the sham-operated control and 4 h of reperfusion but deposited into protein aggregate-containing fractions at 24 h of reperfusion, whereas HIP (between 45 and 75 kDa) was present in the control and gradually disappeared from the protein aggregate-containing fractions after focal ischemia. (c, d) Changes in protein bands of HSC70, Hdj1, Ubi-proteins, HSP70 and HIP from three different individual rat samples were evaluated with Kodak 1D image software. Data are expressed as mean ± SD (n = 4); *denotes p < 0.05 between control and experimental conditions, one-way anova followed by Fisher’s PLSD post hoc test.
All 36 fractions from sham-operated control and postischemic samples were subjected to SDS–PAGE and analyzed by western blot with antibodies against translational complex components L28, S6, eIF2α, eIF3η and eIF4E (Fig. 3b). Among these translational complex components, S6 is a 40S ribosomal protein, L28 is a 60S ribosomal protein, and eIF2α, eIF3η and eIF4E are eukaryotic protein synthesis initiation factors. L28, S6 and eIF3η were resolved within the sedimentation peak of sham-operated control samples (Fig. 3b, Sham), suggesting that the control UV-260 peak contained the detergent/salt-washed ribosomal protein translational components. Protein translational complex components eIF2α and eIF4E were undetectable in the sedimentation peak of sham-operated control samples (Fig. 3b, Sham), indicating that they were washed from the translational complex fractions by the detergent/salt treatment. Relative to control samples, much larger quantities of all these translational complex components were deposited into higher density protein aggregate-containing fractions in post-ischemic samples (Fig. 3). The optical densities of the immunoblot bands in each fraction peak were quantified and added together using Kodak 1D gel analysis software. Quantitative analysis indicates that the optical density ratio of western blots of S6 versus L28 in the sedimentation peak was 3.87 ± 0.28 (mean ± SD) in sham-operated non-ischemic brains, suggesting that this sedimentation peak contained an imbalanced stoichiometry of 40S and 60S ribosomal subunits. However, the optical density ratios of S6 versus L28 were changed to 0.91 ± 0.26 and 0.64 ± 0.04 in the sedimentation peaks of brains subjected to 2 h of focal ischemia followed by 4 and 24 h of reperfusion, respectively. Relative to those of sham-operated controls, the levels of large ribosomal protein L28 and eIF3η were increased two-to eightfold in the higher density protein aggregate-containing fractions at 4 and 24 h of reperfusion, whereas the level of small ribosomal protein S6 was increased in the higher density peaks mainly at 24 h of reperfusion (Figs 3b, c and d). The protein levels of eIF2α and eIF4E were also increased significantly and continuously in the higher density protein aggregate-containing fractions at 4 and 24 h of reperfusion (Figs 3b and d).
To study further whether or not co-translational chaperones were also aggregated with translational complex proteins after focal brain ischemia, we utilized the same sucrose gradient fractions as in Fig. 3 to analyze the nascent polypeptide-associated chaperones HSC70 and Hdj1 (HSP40), as well as ubiquitin-conjugated proteins (ubiproteins) (Fig. 4). Similar to ribosomal proteins and protein synthesis initiation factors, the co-translational chaperones were highly accumulated, mainly in the higher density protein aggregate-containing fractions at 4 and 24 h of reperfusion (Figs 4b and c). High molecular weight ubiproteins, which may represent ubiquitin-conjugated unfolded proteins in the translational complex or unprotected nascent peptides, were also concomitantly increased in the higher density protein aggregate-containing fractions at 4 and 24 h of reperfusion after focal ischemia (Figs 4b and c). Quantitative analysis further confirmed that the levels of HSC70, Hdj1 and ubi-proteins were increased five to 80-fold in the protein aggregate-containing fractions at 4 and 24 h of reperfusion after ischemia (Fig. 4c).
HSP70 chaperone protein is an inducible member of the HSP70 family. Its protein expression was not induced until 24 h of reperfusion, and it was highly accumulated in the higher density protein aggregate-containing fractions after focal brain ischemia (Figs 4b and d). In contrast, heat-shock interacting protein (HIP) was detected in the gradient peak fractions from sham-operated controls, but was greatly reduced at 4 h of reperfusion and disappeared at 24 h of reperfusion from the protein aggregate-containing fractions after focal ischemia (Figs 4b and d).
Confocal microscopic study of translational complex aggregation
To study the cellular distribution of translational complex aggregation, we performed confocal microscopy of translational complex components in brain sections from shamoperated control rats and rats subjected to 2 h of MCAO followed by 4 and 24 h of reperfusion (Fig. 5). Brain sections were immunolabeled with antibodies against translational complex components eIF3η, S6, L28 and HSC70 (HSC), and ubiquitin (Ubi), and they were examined by confocal microscopy. Ubi-proteins and eIF3η immunostainings were relatively evenly distributed in sham-operated control neurons (Fig. 5), but appeared in aggregation patterns in the cytoplasm at 4 and 24 h of reperfusion in the cortical neurons after focal ischemia (Fig 5, 4 and 24h, arrows). At 24 h of reperfusion after ischemia, some neurons are dead and therefore devoid of ubiquitin immunoreactivity (Fig. 5, 4 and 24 h, arrowheads; Hu et al. 2000, 2001). Relative to the sham-operated control, S6, L28 and HSC70 immunoreactivities were greatly reduced at both 4 and 24 h of reperfusion after focal ischemia (Fig. 5).
Fig. 5.

Confocal microscopic images of the penumbral region stained with antibodies against ubiquitin, eIF3η, ribosomal S6 protein, ribosomal L28 protein and HSC70, respectively. Sections are shown from a sham-operated control rat and from rats subjected to 2 h of MCAO followed by 4 and 24 h of reperfusion. The ubiquitin and eIF3η shows aggregation patterns after ischemia (arrows), whereas S6, L28 and HSC70 immunoreactivities were decreased during the post-ischemic phase.
Discussion
Focal ischemia leads to acute cell death within hours during ischemia in the core region, and delayed neuronal death in hours, days or even months in penumbral neurons after ischemia. The cell death in the core region is mostly due to cell swelling and membrane rupture to release cell contents, i.e. necrosis. In contrast, penumbral neurons die in a delayed fashion (Siesjo et al. 1995). During this delay period, the light microscopic morphology of penumbral neurons destined to die is virtually normal. Key cell survival factors of cerebral blood flow, cellular ATP and ionic homeostasis are gradually recovered in penumbral neurons during reperfusion. In striking contrast, protein synthesis is irreversibly inhibited only in penumbral neurons destined to die (Mies et al. 1991). Irreversible inhibition of protein synthesis has long been considered as a hallmark of delayed neuronal death after focal ischemia, but the underlying mechanism is unknown (Mies et al. 1991; Hossmann 1993; Mengesdorf et al. 2002). This study demonstrates for the first time that irreversible inhibition of protein synthesis is probably caused by the irreversible aggregation of translational machinery after focal brain ischemia. EM study shows that ribosomes are clumped into large abnormal aggregates in the penumbral neurons after ischemia. Translational complex components consisting of ribosomal small subunit protein S6, large subunit protein L28, co-translational chaperone HSC70 and its co-chaperone Hdj1, and translational initiation factor eIF2α, eIF3η and eIF4E, are all aggregated into higher densities of a sucrose gradient after focal ischemia. Translational complex components are also highly ubiquitinated during the post-ischemic phase. Confocal microscopy further suggests that immunoreactivities of translational complex components are either clustered or greatly reduced after focal brain ischemia. Aggregation of ubi-proteins is an irreversible process (Angelidis et al. 1999; Hu et al. 2000, 2001; Ohtsuka and Hata 2000). The results indicate that translational complex components are irreversibly clumped into large abnormal protein aggregates, resulting in irreparable damage to protein synthesis machinery after focal brain ischemia.
The present study has confirmed our original hypothesis that protein aggregation is a major pathological event contributing to neuronal death after focal brain ischemia (Hu et al. 2000, 2001; Liu et al. 2005a,b). In this study, we refine our hypothesis to suggest that it is the aggregation of protein synthesis machinery, in particular, that leads to irreversible inhibition of protein biosynthesis in neurons destined to die after focal ischemia. Neurons containing EM-visible ribosomal aggregates have intact cell membranes, relatively normal mitochondria and nuclei, suggesting that translational complex aggregation takes place in living neurons (Fig. 2). However, these penumbral neurons are destined to die in a slow fashion after focal brain ischemia (Mies et al. 1991). Confocal microscopic results demonstrate that eIF3η and ubi-proteins form clustered patterns in morphologically normal neurons after focal ischemia. Protein ubiquitination is a strictly ATP-dependent process and takes place only in living neurons (Hu et al. 2000, 2001). Dead neurons lost their ubiquitin immunoreactivity completely (Fig. 5). Ubi-proteins are greatly increased in the protein aggregate fractions after ischemia (Figs 3 and 4). Protein synthesis machinery is virtually completely inhibited in penumbral living neurons after focal ischemia (Mies et al. 1991). Several lines of evidence indicate that the translational complex aggregation occurs in living neurons after focal ischemia.
The objective of this study was to investigate irreversible protein aggregation of protein translational machinery in neurons after focal brain ischemia. Fractions that contained protein translational complex aggregates were isolated from detergent and salt-washed pellets of initial tissue homogenates, followed by sucrose gradient centrifugation. Therefore, the sedimentation peak does not represent usual sucrose gradient isolated ribosomal fractions Rather, it may be composed of a mixture of detergent and salt-washed ribosomal subcomplex components, i.e. a large portion of 40S and a small portion of either 60S or 80S ribosomal complexes in sham-operated non-ischemic brains. This assumption is supported by evidence that the sedimentation peaks of sham-operated control brains appear to contain an imbalanced stoichiometry of 40S and 60S ribosomal components, i.e. high levels of 40S ribosomal S6 protein and eIF3η, and a low level of 60S ribosomal L28 protein (Fig. 3). The optical density ratio of S6 versus L28 was 3.87 ± 0.28 (mean ± SD) in the sedimentation peaks from sham-operated non-ischemic brains, but changed to 0.91 ± 0.26 and 0.64 ± 0.04 in the sedimentation peaks from brains subjected to ischemia followed by 4 and 24 h of reperfusion, respectively. The presence of eIF3 in the control sedimentation peaks is likely because native 40S subunits in the cytoplasm are stably associated with eIF3, which prevents association of 40S with 60S subunits (Freienstein and Blobel 1975; Thompson et al. 1977; Kolupaeva et al. 2005). eIF2 and eIF4 were not found in the non-ischemic sedimentation peak, probably because they were washed from 40S ribosomes by detergent and salt during sample preparation. After brain ischemia, however, the sedimentation peaks shifted to higher density fractions, most likely because they are composed of translational complex protein aggregates, as evidenced by the fact that the post-ischemic sedimentation peaks contained higher levels of all the protein translational components investigated in this study, including ribosomal S6 and L28 proteins, protein synthesis initiation factors and co-translational chaperones. These data strongly suggest that, unlike those of control brain tissues, detergent and salt are no longer able to dissociate ribosomes or polysomes of postischemic brain tissues into protein translational complex components, probably because of irreversible aggregation of protein synthesis machinery after brain ischemia.
The size of EM-visible protein aggregates is in the narrow range of 100–250 nm, as indicated by the scale bar in Fig. 2 and in several previous publications (Hu et al. 2000, 2001; Liu and Hu 2004, Liu et al. 2005a). They are about, or less than, 1/10th of the mitochondria in diameter under EM. Therefore, it is very hard to see individual protein aggregates by light microscopy, unless these aggregates form membrane-associated clusters. In comparison with EM-visible protein aggregates, the ubiquitin and eIF3 immunostaining cluster patterns seen in Fig. 5 must exceed 500–1000 nm in order to be viewed by confocal microscopy. In our previous studies, the ubiquitin-immunostained clusters shown in Fig. 5 were also observed in neurons at early periods of reperfusion after brain ischemia in both global and focal ischemia models (Hu et al. 2000, 2001). However, there are several differences between the immunostained clusters viewed by confocal microscopy and EM-visible protein aggregates after ischemia: (i) the time course is different, i.e. the immunostained clusters appear before 1 h of reperfusion, but EM-visible protein aggregates are not seen until 2–4 h of reperfusion after ischemia (Hu et al. 2000, 2001); (ii) the regional distribution is different, i.e. the immunostained clusters can be seen in both the CA1 region and dentate gyrus (DG) area, whereas EM-visible protein aggregates are observed only in the CA1 neurons after ischemia. The CA1 neurons will die in a delayed fashion, whereas DG neurons will survive after a short period of cerebral ischemia (Hu et al. 2000); and (iii) the reversibility is different, i.e. EM-visible protein aggregate formation is irreversible and progressively increases until neuronal death takes place in CA1 neurons, whereas ubiquitin-immunostained clusters can also be found transiently in DG surviving neurons after a short period of cerebral ischemia (Hu et al. 2000, 2001). In all cases, neither ubiquitin-immunostained clusters nor EM-visible protein aggregates were observed in control brains. The evidence seems to suggest that the immunostained clusters appear during the early period of reperfusion and may or may not transform into EM-visible protein aggregates after brain ischemia.
Protein translational complex aggregation has not been reported after focal brain ischemia. The following evidence suggests that protein translational complex aggregation after focal ischemia may damage neurons. (1) During reperfusion after focal brain ischemia, blood flow and cellular ATP level are soon recovered, but inhibition of overall rate of protein synthesis persists only in penumbral neurons destined to die after focal brain ischemia (Mies et al. 1991). As shown in this study, irreversible inhibition of protein synthesis is likely caused by translational complex aggregation after focal ischemia. (2) Measures that induce expression of molecular chaperones protect neurons from ischemic damage (Sharp et al. 1999; Yenari et al. 2001). The major function of molecular chaperones is to shield hydrophobic surfaces of misfolded proteins to prevent protein aggregation-induced toxicity (Giffard et al. 2004). Ischemic pre-conditioning preventing neuronal protein aggregation also protects neurons from ischemic damage (Liu et al. 2005b). (3) Measures such as hyperthermia that induce protein aggregation increase neuronal death (Li et al. 1995; Ginsberg and Busto 1998; Lepock 2003). (4) Protein aggregate-containing neurons will eventually die in most pathological conditions, the so-called conformational diseases (Gow and Sharma 2003). Therefore, protein aggregation or proteotoxicity may contribute to neuronal death after focal ischemia.
In summary, brain ischemia depletes ATP and changes intracellular homeostasis, thus disabling ATP-dependent protein quality control systems including molecular chaperones and folding enzymes, and ubiquitin-proteasomal and autophagic degradation during the post-ischemic phase (Hu et al. 2000, 2001). The common result of these cellular alterations can lead to overload of unfolded proteins on protein synthesis machinery in neurons. Consequently, unfolded proteins and unprocessed nascent polypeptides, ribosomes and their associated folding chaperones, are stacked and gradually aggregated during the post-ischemic phase. Protein aggregation is virtually an irreversible process (Hu et al. 2004). Therefore, abnormal aggregation of protein translational complexes may irreparably damage protein synthesis machinery and eventually contribute to neuronal death in penumbral neurons after focal brain ischemia.
Acknowledgements
This work was supported by National Institutes of Health grants NS040407 and NS36810.
Abbreviations used
- eIF
eukaryotic initiation factor
- HSC
heat-shock cognate protein
- HSP
heat-shock protein
- L28
large ribosomal subunit protein 28
- MCAO
middle cerebral artery occlusion
- PBS
phosphate-buffered saline
- S6
small ribosomal subunit protein 6
- SDS–PAGE
sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- TX100
Triton X100
- ubi-proteins
ubiquitin-conjugated proteins
References
- Althausen S, Mengesdorf T, Mies G, Olah L, Nairn AC, Proud CG, Paschen W. Changes in the phosphorylation of initiation factor eIF-2alpha, elongation factor eEF-2 and p70, S6 kinase after transient focal cerebral ischaemia in mice. J. Neurochem. 2001;78:779–787. doi: 10.1046/j.1471-4159.2001.00462.x. [DOI] [PubMed] [Google Scholar]
- Angelidis CE, Lazaridis I, Pagoulatos GN. Aggregation of hsp70 and hsc70 in vivo is distinct and temperature-dependent and their chaperone function is directly related to non-aggregated forms. Eur. J. Biochem. 1999;259:505–512. doi: 10.1046/j.1432-1327.1999.00078.x. [DOI] [PubMed] [Google Scholar]
- Bukau B, Hesterkamp T, Luirink J. Growing up in a dangerous environment: a network of multiple targeting and folding pathways for nascent polypeptides in the cytosol. Trends Cell Biol. 1996;6:480–486. doi: 10.1016/0962-8924(96)84946-4. [DOI] [PubMed] [Google Scholar]
- Burda J, Martin ME, Garcia A, Alcazar A, Fando JL, Salinas M. Phosphorylation of the alpha subunit of initiation factor 2 correlates with the inhibition of translation following transient cerebral ischaemia in the rat. Biochem. J. 1994;302:335–338. doi: 10.1042/bj3020335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGracia DJ. Acute and persistent protein synthesis inhibition following cerebral reperfusion. J. Neurosci. Res. 2004;77:771–776. doi: 10.1002/jnr.20225. [DOI] [PubMed] [Google Scholar]
- Eggers DK, Welch WJ, Hansen WJ. Complexes between nascent polypeptides and their molecular chaperones in the cytosol of mammalian cells. Mol. Biol. Cell. 1997;8:1559–1573. doi: 10.1091/mbc.8.8.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink AL. Chaperone-mediated protein folding. Physiol. Rev. 1999;79:425–449. doi: 10.1152/physrev.1999.79.2.425. [DOI] [PubMed] [Google Scholar]
- Freienstein C, Blobel G. Nonribosomal proteins associated with eukaryotic native small ribosomal subunits. Proc. Natl Acad. Sci. USA. 1975;72:3392–3396. doi: 10.1073/pnas.72.9.3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frydman J. Folding of newly translated proteins in vivo: the role of molecular chaperones. Annu. Rev. Biochem. 2001;70:603–647. doi: 10.1146/annurev.biochem.70.1.603. [DOI] [PubMed] [Google Scholar]
- Giffard RG, Xu L, Zhao H, Carrico W, Ouyang Y, Qiao Y, Sapolsky R, Steinberg G, Hu B, Yenari MA. Chaperones, protein aggregation, and brain protection from hypoxic/ischemic Injury. J. Exp. Biol. 2004;207:3213–3220. doi: 10.1242/jeb.01034. [DOI] [PubMed] [Google Scholar]
- Ginsberg MD, Busto R. Combating hyperthermia in acute stroke: a significant clinical concern. Stroke. 1998;29:529–534. doi: 10.1161/01.str.29.2.529. [DOI] [PubMed] [Google Scholar]
- Gow A, Sharma R. The unfolded protein response in protein aggregating diseases. Neuromol. Med. 2003;4:73–94. doi: 10.1385/NMM:4:1-2:73. [DOI] [PubMed] [Google Scholar]
- Hardesty B, Tsalkova T, Kramer G. Co-translational folding. Curr. Opin. Struct. Biol. 1999;9:111–114. doi: 10.1016/s0959-440x(99)80014-1. [DOI] [PubMed] [Google Scholar]
- Hartl FU, Hayer-Hartl M. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science. 2002;295:1852–1858. doi: 10.1126/science.1068408. [DOI] [PubMed] [Google Scholar]
- Hazeki N, Tukamoto T, Goto J, Kanazawa I. Formic acid dissolves aggregates of an N-terminal huntingtin fragment containing an expanded polyglutamine tract: applying to quantification of protein components of the aggregates. Biochem. Biophys. Res. Commun. 2000;277:386–393. doi: 10.1006/bbrc.2000.3682. [DOI] [PubMed] [Google Scholar]
- Hossmann K-A. Disturbances of cerebral protein synthesis and ischemic cell death. Prog. Brain Res. 1993;96:167–177. doi: 10.1016/s0079-6123(08)63265-3. [DOI] [PubMed] [Google Scholar]
- Hu BR, Wieloch T. Stress-induced inhibition of protein synthesis initiation: modulation of initiation factor 2 and guanine nucleotide exchange factor activities following transient cerebral ischemia in the rat. J. Neurosci. 1993;13:1830–1838. doi: 10.1523/JNEUROSCI.13-05-01830.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu BR, Martone ME, Jones YZ, Liu CL. Protein aggregation after transient cerebral ischemia. J. Neurosci. 2000;20:3191–3199. doi: 10.1523/JNEUROSCI.20-09-03191.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu BR, Janelidze S, Ginsberg MD, Busto R, Perez-Pinzon M, Sick TJ, Siesjo BK, Liu CL. Protein aggregation after focal brain ischemia and reperfusion. J. Cereb. Blood Flow Metab. 2001;21:865–875. doi: 10.1097/00004647-200107000-00012. [DOI] [PubMed] [Google Scholar]
- Hu BR, Martone ME, Liu CL. In: Protein aggregation, unfolded protein response and delayed neuronal death after brain ischemia, in Maturation Phenomenon in Cerebral Ischemia. Buchan A, Ito I, editors. Vol. V. Springer-Verlag; Berlin, Heidelberg: 2004. pp. 225–237. [Google Scholar]
- Kabakov AE, Gabai VL. Stress-induced insolubilization of certain proteins in ascites tumor cells. Arch. Biochem. Biophys. 1994;309:247–253. doi: 10.1006/abbi.1994.1109. [DOI] [PubMed] [Google Scholar]
- Kazantsev A, Preisinger E, Dranovsky A, Goldgaber D, Housman D. Insoluble detergent-resistant aggregates form between pathological and nonpathological lengths of polyglutamine in mammalian cells. Proc. Natl Acad. Sci. USA. 1999;96:11 404–11 409. doi: 10.1073/pnas.96.20.11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokubo Y, Liu J, Rajdev S, Kayama T, Sharp FR, Weinstein PR. Differential cerebral protein synthesis and heat shock protein 70 expression in the core and penumbra of rat brain after transient focal ischemia. Neurosurgery. 2003;53:186–190. doi: 10.1227/01.neu.0000069023.01440.d6. [DOI] [PubMed] [Google Scholar]
- Kolupaeva VG, Unbehaun A, Lomakin IB, Hellen CU, Pestova TV. Binding of eukaryotic initiation factor 3 to ribosomal 40S subunits and its role in ribosomal dissociation and anti association. RNA. 2005;11:470–486. doi: 10.1261/rna.7215305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Zipfel GJ, Park KH, He YY, Hsu CY, Choi DW. Zinc translocation accelerates infarction after mild transient focal ischemia. Neuroscience. 2002;115:871–878. doi: 10.1016/s0306-4522(02)00513-4. [DOI] [PubMed] [Google Scholar]
- Lepock JR. Cellular effects of hyperthermia: relevance to the minimum dose for thermal damage. Int. J. Hyperthermia. 2003;19:252–566. doi: 10.1080/0265673031000065042. [DOI] [PubMed] [Google Scholar]
- Li GC, Mivechi NF, Weitzel G. Heat shock proteins, thermotolerance, and their relevance to clinical hyperthermia. Int. J. Hyperthermia. 1995;11:459–488. doi: 10.3109/02656739509022483. [DOI] [PubMed] [Google Scholar]
- Liu CL, Hu BR. Alterations of N-ethylmaleimide-sensitive atpase following transient cerebral ischemia. Neuroscience. 2004;128:767–774. doi: 10.1016/j.neuroscience.2004.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CL, Ge P, Zhang F, Hu BR. Co-translational protein aggregation after transient cerebral ischemia. Neuroscience. 2005a;134:1273–1284. doi: 10.1016/j.neuroscience.2005.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Chen S, Kamme F, Hu BR. Ischemic preconditioning prevents protein aggregation after transient cerebral ischemia. Neuroscience. 2005b;134:69–80. doi: 10.1016/j.neuroscience.2005.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengesdorf T, Proud CG, Mies G, Paschen W. Mechanisms underlying suppression of protein synthesis induced by transient focal cerebral ischemia in mouse brain. Exp. Neurol. 2002;177:538–546. doi: 10.1006/exnr.2002.8002. [DOI] [PubMed] [Google Scholar]
- Mies G, Ishimaru S, Xie Y, Seo K, Hossmann KA. Ischemic thresholds of cerebral protein synthesis and energy state following middle cerebral artery occlusion in rat. J. Cereb. Blood Flow Metab. 1991;11:753–761. doi: 10.1038/jcbfm.1991.132. [DOI] [PubMed] [Google Scholar]
- Murata S, Chiba T, Tanaka K. CHIP: a quality-control E3 ligase collaborating with molecular chaperones. Int. J. Biochem. Cell Biol. 2003;35:572–578. doi: 10.1016/s1357-2725(02)00394-1. [DOI] [PubMed] [Google Scholar]
- Ohtsuka K, Hata M. Molecular chaperone function of mammalian Hsp70 and Hsp40. Int. J. Hyperthermia. 2000;16:231–245. doi: 10.1080/026567300285259. [DOI] [PubMed] [Google Scholar]
- Owen CR, Kumar R, Zhang P, McGrath BC, Cavener DR, Krause GS. PERK is responsible for the increased phosphorylation of eIF2alpha and the severe inhibition of protein synthesis after transient global brain ischemia. J. Neurochem. 2005;94:1235–1242. doi: 10.1111/j.1471-4159.2005.03276.x. [DOI] [PubMed] [Google Scholar]
- Pontén U, Ratcheson RA, Salford L, Siesjo BK. Optimal freezing conditions for cerebral metabolites in rats. J. Neurochem. 1973;21:1127–1138. doi: 10.1111/j.1471-4159.1973.tb07567.x. [DOI] [PubMed] [Google Scholar]
- Sharp FR, Massa SM, Swanson RA. Heat-shock protein protection. Trends Neurosci. 1999;22:97–99. doi: 10.1016/s0166-2236(98)01392-7. [DOI] [PubMed] [Google Scholar]
- Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siesjo BK, Katsura K, Zhao Q, Folbergrova J, Pahlmark K, Siesjo P, Smith ML. Mechanisms of secondary brain damage in global and focal ischemia: a speculative synthesis. J. Neurotrauma. 1995;12:943–956. doi: 10.1089/neu.1995.12.943. [DOI] [PubMed] [Google Scholar]
- Smith ML, Auer RN, Siesjo BK. The density and distribution of ischemic brain injury in the rat following 2–10 min of forebrain ischemia. Acta Neuropathol. (Berl.) 1984;64:319–332. doi: 10.1007/BF00690397. [DOI] [PubMed] [Google Scholar]
- Stockel J, Hartl FU. Chaperonin-mediated de novo generation of prion protein aggregates. J. Mol. Biol. 2001;313:861–872. doi: 10.1006/jmbi.2001.5085. [DOI] [PubMed] [Google Scholar]
- Taylor JP, Hardy J, Fischbeck KH. Toxic proteins in neurodegenerative disease. Science. 2002;296:1991–1995. doi: 10.1126/science.1067122. [DOI] [PubMed] [Google Scholar]
- Thompson HA, Sadnik I, Scheinbuks J, Moldave K. Studies on native ribosomal subunits from rat liver. Purification and characterization of ribosome dissociation factors. Biochemistry. 1977;16:2221–2230. doi: 10.1021/bi00629a028. [DOI] [PubMed] [Google Scholar]
- Wang W, Redecker C, Bidmon HJ, Witte OW. Delayed neuronal death and damage of GDNF family receptors in CA1 following focal cerebral ischemia. Brain Res. 2004;1023:92–101. doi: 10.1016/j.brainres.2004.07.034. [DOI] [PubMed] [Google Scholar]
- Yenari MA, Dumas TC, Sapolsky RM, Steinberg GK. Gene therapy for treatment of cerebral ischemia using defective herpes simplex viral vectors. Ann. N Y Acad. Sci. 2001;939:340–357. doi: 10.1111/j.1749-6632.2001.tb03643.x. [DOI] [PubMed] [Google Scholar]