

Abstract
STIM1 and CRACM1 (or Orai1) are essential molecular components mediating store-operated Ca2+ entry (SOCE) and Ca2+ release-activated Ca2+ (CRAC) currents. Although STIM1 acts as a luminal Ca2+ sensor in the endoplasmic reticulum (ER), the function of STIM2 remains unclear. Here we reveal that STIM2 has two distinct modes of activating CRAC channels: a store-operated mode that is activated through depletion of ER Ca2+ stores by inositol 1,4,5-trisphosphate (InsP3) and store-independent activation that is mediated by cell dialysis during whole-cell perfusion. Both modes are regulated by calmodulin (CaM). The store-operated mode is transient in intact cells, possibly reflecting recruitment of CaM, whereas loss of CaM in perfused cells accounts for the persistence of the store-independent mode. The inhibition by CaM can be reversed by 2-aminoethoxydiphenyl borate (2-APB), resulting in rapid, store-independent activation of CRAC channels. The aminoglycoside antibiotic G418 is a highly specific and potent inhibitor of STIM2-dependent CRAC channel activation. The results reveal a novel bimodal control of CRAC channels by STIM2, the store dependence and CaM regulation, which indicates that the STIM2/CRACM1 complex may be under the control of both luminal and cytoplasmic Ca2+ levels.
Keywords: STIM1, calmodulin, store-operated calcium entry
Changes in intracellular free calcium concentration ([Ca2+]i) represent one of the most widespread and important signaling events, regulating a plethora of cellular responses. Many cell types employ store-operated Ca2+ entry (SOCE) as their principal pathway for Ca2+ influx (1–3). This mechanism is engaged after Ca2+ release from stores, where the depleted stores lead to activation of Ca2+ release-activated Ca2+ (CRAC) channels (4 – 6). Recent work (7–11) has identified STIM1 and CRACM1 (or Orai1) as essential components for functional SOCE. When overexpressed jointly, but not individually, STIM1 and CRACM1 reconstitute large CRAC currents (11–14). In mammals, there exist several homologs of these proteins: STIM1 and STIM2 in the endoplasmic reticulum (ER) and CRACM1, CRACM2, and CRACM3 in the plasma membrane. On Ca2+ depletion of stores, STIM1 translocates into junctional structures close to the plasma membrane (8, 15–17), where it may bind to and activate CRACM1 (18, 19). Mutational analysis revealed that several key amino acids in CRACM1 determine the selectivity of CRAC currents, demonstrating that CRACM1 forms the ion-selective pore of the CRAC channel (18 –20).
All three CRACM homologs represent functional store-operated channels with differential properties (12, 21, 22). The role of STIM2 in SOCE appears to be complex and remains incompletely understood. When overexpressed alone, STIM2 inhibits SOCE (16), yet coexpressed with CRACM1, STIM2 causes constitutive rather than store-dependent Ca2+ entry that is strongly enhanced by 2-aminoethoxydiphenyl borate (2-APB; ref. 14). When expressed in HEK293 cells, this protein resides exclusively in the ER, but unlike STIM1, it does not redistribute into junctions after store depletion, unless STIM1 is also overexpressed (16). To gain insight into the functional properties of STIM2, we performed a careful analysis of how it regulates CRAC channels.
MATERIALS AND METHODS
Subcloning and overexpression of CRACM and STIM
Full-length human CRACM1, CRACM2, and CRACM3 were subcloned as described previously (18, 22). For electrophysiological analysis, CRACM proteins were overexpressed in HEK293 cells stably expressing STIM1 and STIM2 (16) using lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). Experiments were performed 24 – 48 h post-transfection. For the experiments with HEK293 cells stably expressing STIM2, cells were grown for several days or weeks in the absence or presence of G418 (500 μg/ml). Green cells were analyzed for the effects of CRACM overexpression.
Electrophysiology
Patch-clamp experiments were performed in the tight-seal whole-cell configuration at 21–25°C. High-resolution current recordings were acquired using the EPC-9 (HEKA). Voltage ramps of 50 ms duration spanning a range of − 100 to + 100 mV were delivered from a holding potential of 0 mV at a rate of 0.5 Hz over a period of 100 –700 s. All voltages were corrected for a liquid junction potential of 10 mV (3 mV with Cl− as main internal anion). Currents were filtered at 2.9 kHz and digitized at 100 μs intervals. Capacitive currents were determined and corrected before each voltage ramp. Extracting the current amplitude at −80 mV from individual ramp current records assessed the low-resolution temporal development of currents. Where applicable, statistical errors of averaged data are mean ± SE with n determinations. Standard external solutions were as follows (in mM): 120 NaCl, 10 CsCl, 2.8 KCl, 2 MgCl2, 10 CaCl2, 10 TEA-Cl, 10 HEPES, and 10 glucose, pH 7.2 with NaOH, 300 mOsm. In some experiments, 2, 5, or 50 μM 2-APB or 10 μM G418 was added to the standard external solution and applied through wide-tipped puffer pipettes. Standard internal solutions were as follows (in mM): 120 Cs-glutamate, 8 NaCl, 20 Cs·BAPTA, 3 MgCl2, 10 HEPES, and 0.02 inositol 1,4,5-trisphosphate (InsP3), pH 7.2 with CsOH, 300 mOsm. As indicated in the figure legends, for some experiments [Ca2+]i was buffered to 150 or 100 nM by 20 mM Cs·BAPTA and 8 mM CaCl2 or 10 mM Cs·EGTA and 3.6 mM CaCl2, respectively. For passive-depletion experiments, the internal solution was supplemented with Cs·BAPTA in the absence of InsP3 and Ca2+. In some experiments, Cs-glutamate and Cs·BAPTA were equimolarly replaced by KCl and K·BAPTA. In others, G418, Na·ATP and Na·GTP, or calmodulin (CaM) were added to intracellular solutions as specified in the figure legends. All chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Fluorescence measurements
Cells grown on coverslips were placed in external solution containing (in mM): 107 NaCl, 7.2 KCl, 1 CaCl2, 1.2 MgCl2, 11.5 glucose, and 10 HEPES, pH 7.2 with NaOH, and loaded with fura-2 acetoxymethylester (2 μM) for 30 min at 20°C. Cells were washed, and dye was allowed to de-esterify for a minimum of 30 min at 20°C. Approximately 95% of the dye was confined to the cytoplasm as determined by the signal remaining after saponin permeabilization. Cells on coverslips were placed in external solution in the absence or presence of 1 mM CaCl2. Ca2+ measurements were made using an InCyt dual-wavelength fluorescence imaging system (Intracellular Imaging Inc., Cincinnati, OH, USA). Fluorescence emission at 505 nm was monitored with excitation at 340 and 380 nm; intracellular Ca2+ measurements are shown as 340/380 nm ratios obtained from groups (35– 45) of single cells. The details of these Ca2+ measurements were described previously (23). All measurements shown represent minimum of three independent experiments.
RESULTS
Initial experiments, in which we compared CRAC currents in HEK293 cells stably overexpressing STIM1 or STIM2, confirmed that both proteins slightly modify endogenous ICRAC. In response to Ca2+ store depletion by InsP3, STIM2-expressing cells exhibited reduced ICRAC (~0.2 pA/pF) and STIM1-expressing cells had slightly increased ICRAC (~0.7 pA/pF) compared to wild-type cells (~0.5 pA/pF; see Fig. 1A; ref. 13). To investigate the functional interaction of STIM proteins with CRACM1, HEK293 cells stably overexpressing STIM1 or STIM2 were transiently transfected with CRACM1 and ICRAC was measured in response to InsP3. Consistent with our previous observations (13, 14), STIM1 cells generated large CRAC currents, whereas STIM2 cells did not (Fig. 1B). Although we did not observe readily discernable constitutive CRAC channel activity corresponding to the previously described Ca2+ entry (14), a small basal CRAC current of <1 pA/pF at break-in could have gone unnoticed. However, a small CRAC current developed very slowly, on average amounting to approximately −1 pA/pF at −80 mV. Under identical experimental conditions, despite the lack of InsP3-induced CRACM1 currents, external application of 2-APB (2 μM) induced large CRACM1 currents with the typical inwardly rectifying current-voltage (I/V) relationships (Fig. 1B, C). 2-APB is a compound that has facilitatory effects on CRAC currents at low doses (≤5 μM) but inhibits them at high doses (≥10 μM; refs. 23–26). In cells expressing STIM2 and CRACM1, 2-APB applied at 50 μM caused a rapid, large, and transient activation of CRAC current (Fig. 1D). In these experiments, we omitted InsP3 from the patch pipette and also buffered [Ca2+]i to 150 nM to keep stores repleted, so the 2-APB effect observed here did not require store depletion.
Figure 1.

Store-independent activation of CRACM1 by STIM2 through 2-APB. In all experiments (except black trace in A and green traces in E and F), data are from HEK293 cells grown in the presence of 500 μg/ml G418. A) Average CRAC current densities at −80 mV in wild-type HEK293 cells (black, n=14) and in cells stably overexpressing STIM1 (blue, n=14) or STIM2 (red, n=8) in response to 20 μM InsP3. [Ca2+]i was clamped to near zero with 20 mM BAPTA. B) Average CRAC current densities induced by 20 μM InsP3 + 20 mM BAPTA in STIM1 + CRACM1 cells (black, n=15), STIM2 + CRACM1 cells (blue, n=10), and STIM2 + CRACM1 cells stimulated by 2 μM 2-APB (red, n=9). C) Representative current-voltage (I/V) relationships of CRAC currents in STIM1 + CRACM1-expressing cells or 2 μM 2-APB-induced currents in STIM2 + CRACM1-expressing cells shown in B. D) Average CRAC current densities in HEK293 cells expressing STIM2 alone (purple, n=5; trace is flat around 0 pA/pF and masked by blue trace), CRACM1 alone (black, n=3, trace is flat around 0 pA/pF and masked by blue trace), STIM1 + CRACM1 (blue, n=8), and STIM2 + CRACM1 (red, n=9). In all cells, [Ca2+]i was buffered to 150 nM using 20 mM BAPTA and 8 mM CaCl2. E) High-resolution average CRAC currents at −80 mV in STIM2 + CRACM1 cells grown in the presence (red, n=7) or absence (green, n=5) of G418 induced by 50 μM 2-APB as in D. F) Comparison of activation kinetics of InsP3- and 2-APB-induced gating of CRAC channels. Data sets are taken from B and E and plotted on same time scale to illustrate speed of STIM2-dependent and 2-APB-induced activation relative to STIM1-dependent and store-operated activation of CRACM1.
To better resolve the kinetics of this response, we performed high-resolution recordings of the 2-APB effect at a fixed membrane potential of −80 mV. Figure 1E shows that the large 2-APB-induced current peaked within ~8 s and disappeared to almost zero within a further 10 s. This 2-APB-induced effect was specific to cells overexpressing STIM2 and CRACM1, since it was not observed in HEK293 cells that overexpressed either STIM2 or CRACM1 alone or a combination of STIM1 and CRACM1 (Fig. 1D). The 2-APB-induced activation seems to be too rapid to be mediated by store-dependent translocation of STIM2 to couple to CRACM1. We therefore surmise that a significant portion of CRACM1 channels must somehow be in a readily activated state, possibly precoupled in a complex with STIM2. Immunofluorescence analysis reveals that STIM2 is distributed in ER structures throughout the cell but is also found close to the plasma membrane, where it partially overlaps with CRACM1 localization (Fig. 2B). Store depletion had no effect on this distribution of STIM2. This is in contrast to STIM1, which redistributes into CRACM1-associated junctions after store depletion (Fig. 2A). Interestingly, coimmunoprecipitation experiments revealed that not only STIM1 but also STIM2 can physically interact with CRACM1 (Fig. 2C), which indicates that both STIM proteins interact with CRACM1. If STIM2 and CRACM1 exist coupled without store depletion and can generate large CRAC currents in response to 2-APB, what prevents the CRACM1 channel from being open? As described here, it appears that an endogenous inhibitor prevents activation of the channel and that 2-APB reverses the action of this inhibitor.
Figure 2.

Immunolocalization of STIM and CRACM proteins. Images were obtained by confocal microscopy, while colocalization (yellow) was demonstrated by merging the images using ImageJ software (NIH). A) Cells stably expressing CFP-CRACM1 (green) were transfected with STIM1 (red), treated with 2 μM thapsigargin for 10 min, and then fixed, permeabilized, and blocked. Cells were sequentially double-stained with sheep STIM1-specific and rabbit anti-GFP antibodies (Invitrogen), followed by corresponding 2° antibodies. B) Cells stably expressing STIM2 (red) were transfected with CFP-CRACM1 (green), treated with 2 μM thapsigargin for 10 min, and then fixed, permeabilized, and blocked. Cells were sequentially double-stained with sheep STIM2-specific and rabbit anti-GFP antibodies (Invitrogen), followed by corresponding 2° antibodies. C) Binding of STIM proteins to CRACM1 was assessed after transient transfection of STIM1 or STIM2 into HEK293 cells stably expressing CRACM1. Approximate molecular masses of proteins were 85 kDa for STIM1, 105 kDa for STIM2, and 55 kDa for CFP-CRACM1, the glycosylated form of which is ~70 kDa. After transfection, cells were lysed and the proteins were quantified. For immunoprecipitations, 200 μg of protein/sample were incubated with protein G beads coated with rabbit anti-GFP antibodies (Invitrogen) for 2 h followed by separation with SDS-PAGE and transfer to PVDF membranes. Protein expression was confirmed by loading 5 μg of untransfected lysate. STIM proteins were detected by Western blot using an antibody cross-reacting with both STIM1 and STIM2 (BD Biosciences, San Jose, CA, USA), while pulldown and expression of CFP-CRACM1 was demonstrated by stripping and reprobing a goat-anti-GFP antibody (Abcam). D) STIM2-expressing cells were incubated for 3 wk in the presence or absence of G418. Cells were lysed and the proteins were quantified. Proteins (5 μg/sample) were separated by SDS-PAGE and transferred to PVDF membranes. Levels of STIM expression were demonstrated by Western blot with an antibody cross-reacting to both STIM1 and STIM2 (BD Biosciences).
Interestingly, the distinct effect of STIM2 could be selectively modified by the aminoglycoside antibiotic G418. G418 is routinely present at 500 μg/ml (720 μM) in the growth medium to maintain selection pressure on cells stably transfected with STIM1 or STIM2. HEK293 cells grown for several days in the absence of G418 exhibited enhanced responses to 2-APB and strikingly different responses to store depletion, yet there was no significant decrease in protein levels (Fig. 2D). As shown in Fig. 1E, these cells responded with even faster kinetics and with larger current amplitudes to 50 μM 2-APB. To appreciate the speed of this response, Fig. 1F superimposes the fastest possible store-operated activation of CRACM1 through STIM1 using InsP3 as a stimulus and the 2-APB-mediated activation of CRACM1 through STIM2, demonstrating that full activation of CRACM1 in STIM2-expressing cells is complete before the activation of STIM1-mediated CRAC currents even begins. These results suggest that the aminoglycoside inhibits store-operated gating of CRAC channels through STIM2 and that 2-APB can overcome this inhibition. This effect of G418 was specific to STIM2, since STIM1-expressing cells, which were also routinely grown in G418, produced large CRACM1-mediated currents (see Fig. 1B) with similar properties as those produced by transient coexpression of STIM1 and CRACM1 in HEK293 cells that remained unexposed to G418 (13, 18).
The removal of G418 provided an important condition under which to analyze STIM2 activation in response to store depletion. Thus, in cells not exposed to G418, InsP3 perfusion now generated amplified CRAC currents (Fig. 3A). These CRAC currents were characterized by a biphasic activation pattern consisting of an initial fast activation phase followed by a slower phase that set in after ~100 s and rarely reached steady state within 300 s. The relative proportion of the two phases was somewhat variable across experimental days and may reflect the different coupling mechanisms of STIM2 populations (see below). Typical current amplitudes at − 80 mV obtained 300 s into the experiment were approximately − 15 pA/pF and had the signature I/V curves typical of CRAC currents (Fig. 3B).
Figure 3.
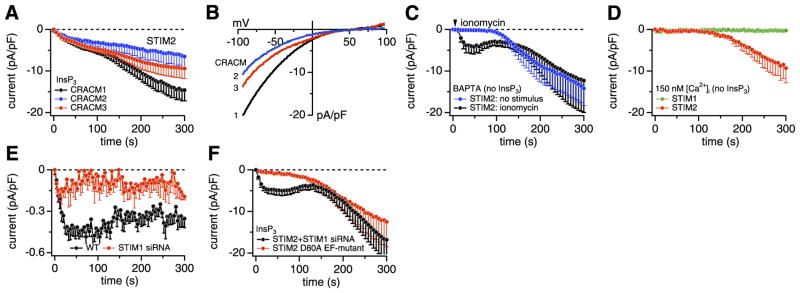
Store-operated activation of CRACM1 through STIM2. A–F) STIM2-expressing HEK293 cells were grown without G418. A) Average CRAC current densities induced by 20 μM InsP3 in STIM2-expressing HEK293 cells transfected with CRACM1 (black, n=34), CRACM2 (blue, n=5), and CRACM3 (red, n=5) with [Ca2+]i clamped to near zero with 20 mM BAPTA. B) Average current-voltage (I/V) relationships of CRAC currents extracted from representative STIM2 + CRACM1, 2, and 3 cells shown in A at 300 s into experiment. Data represent leak-subtracted currents evoked by 50 ms voltage ramps from −100 to + 100 mV, normalized to cell capacitance (pA/pF). C) Average CRAC current densities in STIM2-expressing HEK293 cells transfected with CRACM1, where [Ca2+]i was clamped to near zero with 20 mM BAPTA to induce passive store depletion (blue, n=7). In other cells (black, n=6), active store depletion was induced by brief (2 s) application of 2 μM ionomycin (indicated by the arrow). D) Average CRAC current densities in STIM1- (green, n=6) or STIM2-expressing HEK293 cells (red, n=7) transfected with CRACM1, where store depletion was prevented by omission of InsP3 and buffering [Ca2+]i to 150 nM with 20 mM BAPTA + 8 mM CaCl2. E) Average CRAC current densities at − 80 mV in wt HEK293 cells (black, n=14) or cells treated with STIM1-specific siRNA (red, n=6) in response to 20 μM InsP3. [Ca2+]i was clamped to near zero with 20 mM BAPTA. Data demonstrate that STIM1 siRNA treatment was effective and suppressed native CRAC currents. F) Average CRAC current densities in STIM2 + CRACM1 cells that were cotransfected with siRNA against STIM1 (black, n=5) in parallel and exactly as in E. Average CRAC current densities in CRACM1-expressing cells that were cotransfected with the D80A EF-hand mutant of STIM2 (red, n=5). In both experimental sets, store depletion was induced by 20 μM InsP3 and [Ca2+]i buffered to near zero by 20 mM BAPTA.
Since STIM1 can activate all three CRACM homologs (22), we examined whether STIM2 could also couple to CRACM2 and CRACM3. Figure 3A demonstrates that this is indeed the case. Coexpression of STIM2 with any of the three homologs produced biphasic currents, although the secondary phase of activation was less pronounced in CRACM2- and CRACM3-expressing cells. The I/V relationships of these currents were very similar and exhibited typical CRAC inward rectification (Fig. 3B), suggesting that both STIM homologs can couple to any of the CRACM homologs to activate store-operated currents.
It is important to ascertain that the InsP3-induced currents observed in Fig. 3A developed as a consequence of store depletion. Therefore, we performed additional experiments in which store depletion was induced independently of InsP3. We first examined whether CRAC currents were activated when preventing store refilling with 20 mM BAPTA in the pipette. This also produced large CRAC-like currents but with a significant delay (Fig. 3C, blue trace), which would be consistent with the time needed to passively deplete stores through leak pathways. We then performed the same experiments but used the Ca2+ ionophore ionomycin to rapidly release Ca2+ from intracellular stores (Fig. 3C, black trace). With this protocol, we observed biphasic activation of CRACM1 currents that were similar in both amplitude and kinetics to those activated by InsP3. If both phases of CRAC current were store-dependent, we should have been able to suppress current activation entirely by perfusing cells with an InsP3-free solution and buffering [Ca2+]i to 150 nM to avoid store depletion. As can be seen in Fig. 3D (green trace), these conditions indeed prevent store-dependent activation of CRAC currents in STIM1- and CRACM1-expressing cells. Surprisingly, however, the same experimental conditions still generated the slow secondary phase in STIM2- and CRACM1-expressing cells (Fig. 3D, red trace). These results indicate that the first phase of the CRAC current is indeed caused by store depletion, but the second phase develops regardless of the filling state of intracellular stores and is store independent. As we reveal below, this secondary, store-independent phase of current activation appears to be caused by the diffusional escape of Ca2+-CaM into the patch pipette during perfusion of cells in patch-clamp recordings.
Although the above experiments suggest that store depletion via InsP3 or ionomycin results in activation of CRAC channels, the store dependence of STIM2 could have resulted from its comigration with STIM1 into junctions. We therefore examined whether STIM2 can serve as a Ca2+ sensor and activate CRAC channels in a store-dependent manner when STIM1 expression was suppressed by small interfering RNA (siRNA). We confirmed the efficacy of siRNA treatment in wild-type (wt) HEK293 cells, where endogenous CRAC currents were almost completely suppressed after siRNA treatment (Fig. 3E). However, in parallel experiments performed in STIM2- and CRACM1-expressing cells, STIM1 knockdown did not significantly affect the bi-phasic CRAC currents (Fig. 3F), suggesting that STIM2 has the intrinsic ability to respond to store depletion. We surmised that the Ca2+-sensing function of STIM2 might be mediated by an EF-hand motif located in the N terminus of the protein facing the lumen of the ER, since such a motif is also found in STIM1 and has been shown to serve this function (8, 17, 27). To test this idea, we introduced a point mutation into the EF-hand motif of the STIM2 protein (D80A) that would be predicted to disrupt Ca2+ binding. The equivalent mutation in STIM1 (D76A) has been shown to abolish its Ca2+-sensing function and results in constitutive activation of SOCE (8, 17, 27). As illustrated in Fig. 3F, the D80A mutant of STIM2 failed to support the rapid, store-operated activation phase of CRAC channels, whereas the delayed, store-independent activation phase remained unaffected. Together, these results suggest that STIM2 can mediate both store-operated and store-independent activation of CRAC channels independent of STIM1 and that the Ca2+-sensing function of STIM2 resides within its luminal EF-hand motif.
Since the cells grown in the absence of G418 responded to InsP3-induced store depletion, we were able to assess the inhibitory effect of this antibiotic on STIM2-mediated activation of CRACM1 by perfusing cells with defined concentrations of G418. Figure 4A illustrates the dose-dependent inhibition of both phases of the InsP3-induced CRACM1 currents by in- creasing concentrations of G418, resulting in a half-maximal inhibitory concentration of ~0.6 μM (Fig. 4B). Under identical experimental conditions, where 10 μM G418 was perfused intracellularly, it did not modify InsP3-induced CRAC currents in STIM1- and CRACM1-coexpressing cells (Fig. 4C). Although the most potent effect of G418 appears to be mediated from the intracellular space and specifically affects STIM2-expressing cells, we also observed some small inhibition of the aminoglycoside when it was applied at 10 μM from the extracellular side. This effect, however, did not seem to be specific for STIM2, since it was similar in both STIM1- and STIM2-expressing cells (Fig. 4C, D), suggesting that it might be a pharmacological effect on CRACM1. The specific and potent inhibition of STIM2-mediated CRAC currents caused by intracellular G418 (and possibly other aminoglycoside antibiotics) thus may provide a powerful pharmacological tool to assess STIM2 function in native cell systems.
Figure 4.

Effects of G418 on STIM1- and STIM2-mediated activation of CRACM1. A) Average CRAC current densities in STIM2 + CRACM1 cells in the absence of G418 (closed circles, n=34) or presence of 300 nM (open circles, n=9), 1 μM (closed squares, n=11), 3 μM (open squares, n=8), or 10 μM (closed triangles, n=8) of G418 in the pipette. B) Dose-response relationship of average CRAC current densities as a function of G418 concentration extracted at 300 s from the recordings shown in E. Data were fitted with a dose-response curve, yielding an IC50 value of 640 nM and a Hill coefficient of 2.6. C) Average CRAC currents in STIM1 + CRACM1 cells (n=7) grown in the presence of G418. Pipette solutions contained InsP3 (20 μM) and 10 μM G418, demonstrating that these cells are largely insensitive to intracellular G418. Extracellular application of 10 μM G418 caused a small and reversible reduction in CRAC current. D) Effects of extracellularly applied G418 on STIM2 + CRACM1. Average CRAC currents in STIM2 + CRACM1 cells grown without G418. Pipette solutions contained InsP3 (20 μM) to activate biphasic CRAC currents. Bar indicates extracellular application of various concentrations of G418. These caused a small and reversible reduction in CRAC current: 300 nM (filled circles, n=3), 1 μM (open circles, n=4), and 10 μM (filled squares, n=5).
Figure 3 shows CRAC currents that developed during active store depletion by InsP3, which caused the typical biphasic activation of CRACM1 currents, with the first phase being store-operated, whereas the second phase developed independent of the filling state of stores. Hence, the secondary phase is either activated by some ingredient of our pipette filling solution or it is caused by the loss of some cytosolic factor that constitutively suppresses CRAC channels. We tested for these possibilities by exchanging the major ingredients of the standard pipette filling solution. However, the substitution of the primary intracellular salt Cs-glutamate with KCl (n=5, data not shown) and the replacement of BAPTA with EGTA (see Fig. 5B) were both ineffective in suppressing the secondary current component. Thus, we conclude that the secondary CRAC current phase is likely to result from the washout of a cellular factor. To substantiate this idea, we performed experiments in which we varied pipette tip diameters and analyzed biphasic currents. Smaller pipette tips (with higher series resistance) would reduce the rate of diffusional escape of the cytosolic inhibitor, and Fig. 5A demonstrates that this indeed delayed the development of the secondary phase. The cytosolic factor does not appear to be either of the two major cytosolic nucleotides, since adding physiological concentrations of ATP (3 mM) and GTP (0.3 mM) failed to suppress the secondary phase (n=5; data not shown).
Figure 5.
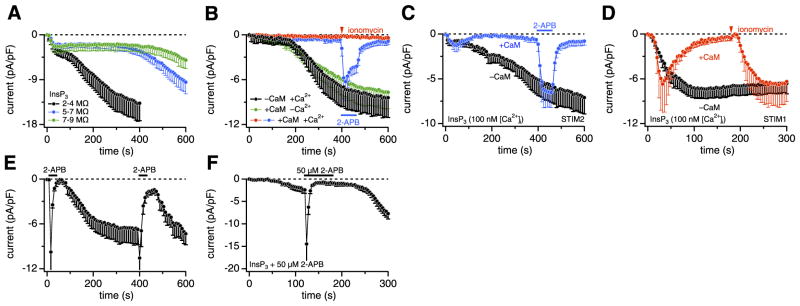
STIM2 causes transient store-operated activation of CRACM1. A–F) STIM2-expressing HEK293 cells were grown without G418. A) Average CRAC current densities in STIM2 + CRACM1 cells, where store depletion was induced by 20 μM InsP3 + 20 mM BAPTA. Traces represent binned data of experiments in which pipette series resistances were within the following ranges: 2– 4 MΩ (black, n=17), 5–7 MΩ (blue, n=7), or 7–9 MΩ (green, n=7). B) Average CRAC current densities in STIM2 + CRACM1 cells. CRAC currents developed normally both in the absence of added CaM with [Ca2+]i buffered to 100 nM with 10 mM EGTA + 3.6 mM CaCl2 (black, n=5) or in its presence (100 μM) with [Ca2+]i buffered to 0 with 10 mM EGTA and no added Ca2+ (green, n=9). However, CRAC currents were suppressed by the combined presence of 100 μM CaM and 100 nM [Ca2+]i (blue, n=7; red, n=4). Application of 2 μM ionomycin for 3 s (red) or 5 μM 2-APB for 60 s (blue) is indicated in graph. C) Average CRAC currents in STIM2 + CRACM1 cells induced by 20 μM InsP3 with [Ca2+]i buffered to 100 nM with 10 mM EGTA + 3.6 mM CaCl2 in the absence (black, n=7) or additional presence of 100 μM CaM (blue, n=7) in pipette. Application of 5 μM 2-APB for 60 s to CaM-treated cells (blue) is indicated in graph. D) Average CRAC currents in STIM1 + CRACM1 cells induced by 20 μM InsP3 with [Ca2+]i buffered to 100 nM using 10 mM EGTA + 3.6 mM CaCl2. CRAC currents developed in the absence of added CaM (black, n=9) but were rapidly suppressed by 100 μM CaM in pipette (red, n=6). Application of 2 μM ionomycin for 3 s (red) reversed the CaM-mediated inhibition of CRAC currents. E) Average CRAC current densities in STIM2-expressing HEK293 cells transfected with CRACM1, where [Ca2+]i was clamped to near zero with 20 mM BAPTA. 2-APB (50 μM) was applied for 30 s as indicated in graph (n=7). F) Average CRAC current densities at − 80 mV in STIM2 + CRACM1 cells in response to 20 μM InsP3 + 20 mM BAPTA. Pipette solution additionally contained 50 μM 2-APB. At the indicated time, 50 μM 2-APB was applied extracellularly.
We next considered CaM as a possible factor, since CaM can regulate numerous ion channels (28), including CRAC channels (29, 30). We performed experiments in which [Ca2+]i was buffered to near resting levels of ~100 nM using EGTA as chelator and CaM was either omitted or included in the pipette filling solution. In the absence of CaM, with [Ca2+]i buffered to resting levels, the CRAC current activated after a delay (Fig. 5B, black trace), similar to the experiments described in Fig. 3D (red trace), where BAPTA was used as chelator. However, additional inclusion of CaM (100 μM) essentially prevented the store-independent activation of CRAC currents (Fig. 5B, red and blue traces), suggesting that CaM may indeed represent the sought cytosolic inhibitor. Subsequent exposure of cells to ionomycin (Fig. 5B, red trace) to deplete stores was ineffective in activating CRAC currents, but channels remained activable by 5 μM 2-APB (Fig. 5B, blue trace). The inhibitory effect of CaM required some [Ca2+]i, since the same concentration of CaM failed to suppress CRAC current activation when [Ca2+]i was clamped to near zero with 10 mM EGTA and no added Ca2+ (Fig. 5B, green trace). Thus, STIM2-mediated CRAC currents are inhibited by Ca2+-bound CaM but not by resting [Ca2+]i alone or Ca2+-free apo-CaM. We further confirmed that CaM can inhibit store-operated CRAC channel activation by perfusing cells with both InsP3 and CaM and [Ca2+]i buffered to 100 nM (Fig. 5C, blue trace). This resulted in a transient CRAC current, presumably due to fast InsP3-mediated store depletion and STIM2-mediated activation of CRACM1, which was then curtailed by inhibition through CaM as it accumulated intracellularly. CaM also prevented the secondary, store-independent CRAC current phase, normally seen in the absence of CaM (Fig. 5C, black trace). Nevertheless, the CRAC channels remained available for activation by 5 μM 2-APB and were suppressed again by CaM on washout of 2-APB.
The inhibitory effect of CaM could occur at various points in the signal transduction process of SOCE, including InsP3 receptors, STIM sensors, and/or CRAC channels. We examined this issue by performing experiments in STIM1- and CRACM1-expressing cells under identical experimental conditions as those for STIM2 shown in Fig. 5C. With [Ca2+]i buffered to near resting levels of ~100 nM and CaM absent from the pipette filling solution, InsP3 induced a sustained CRAC current with monophasic activation kinetics (Fig. 5D, black trace). Addition of 100 μM CaM resulted in a similar activation of CRAC current, but the current was rapidly inactivated (Fig. 5D, red trace). This transient CRAC current response was similar to that observed with STIM2; however, in contrast to STIM2-expressing cells (see Fig. 5B, green trace), subsequent ionomycin application in STIM1-expressing cells completely restored CRAC currents (Fig. 5D, red trace). This result suggests that the CaM-induced inhibition of CRAC currents does not occur at the level of either STIM1 or CRACM1 but instead appears to be mediated by store refilling. Indeed, CaM has been demonstrated to inhibit InsP3 receptors (31, 32), and this mechanism would enable stores to refill even in the presence of InsP3. Together, these data suggest that the CaM-mediated inhibition of CRAC currents can occur indirectly through inhibition of InsP3 receptors and more directly through inhibition of STIM2-mediated CRAC currents. The latter mechanism is specific for STIM2, since CRAC currents produced through STIM1 remain unaffected by CaM if stores are emptied in an InsP3-independent fashion.
The above data suggest that CaM acts as a specific inhibitor of STIM2 coupled to CRAC channels and dialysis of cells in whole-cell recordings leads to wash-out of CaM and disinhibition of CRAC channels. By supplementing the pipette solution with CaM, the washout is counteracted and CRAC channels are suppressed; however, 2-APB can still activate the CaM-inhibited channels. A simple interpretation of this result would be that 2-APB might displace the inhibitory CaM from the STIM2/CRACM1 complex. We obtained support for this hypothesis from experiments in which we probed the effects of 2-APB before and after the secondary phase had fully developed. Figure 5E illustrates that the same concentration of 2-APB (50 μM) produced almost identical peak amplitudes of approximately − 10 pA/pF of STIM2-dependent CRAC current when applied 10 s after whole-cell establishment and after the CRAC current reached a plateau, with the second application timed when a significant amount of the endogenous CaM had washed out of the cell. Thus, the absolute efficacy was essentially the same as during the first application, whereas the relative efficacy of the second 2-APB application in facilitating the CRAC current was lower after the inhibitor had washed out when compared to the first application. This result would be compatible with the hypothesis that 2-APB interferes with CaM binding to the STIM2/ CRACM1 complex and such a mechanism would presumably occur at an intracellular site. However, we could not obtain direct and conclusive evidence for an intracellular effect of 2-APB. Inclusion of 50 μM 2-APB in the patch pipette failed to activate CRAC currents by itself, although InsP3-induced CRAC currents appeared to be delayed and slightly reduced (Fig. 5F). Subsequent extracellular application of 50 μM 2-APB, however, remained fully effective in first facilitating and then blocking CRAC currents. Hence, the exact mechanism and site of action of 2-APB remains to be investigated.
Unlike the situation in whole-cell recordings, the endogenous CaM cannot escape from intact cells, and, therefore, one would expect the initial store-operated activation of CRAC channels by STIM2 to be followed by inhibition, resulting in a transient SOCE phase. We tested this hypothesis in intact cells loaded with the Ca2+ indicator Fura-2 and stimulated store-operated Ca2+ influx through muscarinic receptors using carbachol to rapidly deplete stores and thapsigargin to prevent store refilling. This experiment was performed both in the presence and absence of extracellular Ca2+ and in stable STIM2-expressing HEK293 cells that were transiently transfected with an empty vector or with CRACM1. For comparison, we also included STIM1-and CRACM1-expressing cells. As illustrated in Fig. 6A–C, all cell populations produced a similar transient increase in [Ca2+]i when Ca2+ was absent, due to InsP3-mediated release of Ca2+ from intracellular stores. As expected, the presence of extracellular Ca2+ produced a plateau phase of elevated [Ca2+]i that was likely due to SOCE in all cell populations. However, the CRACM1-expressing cells exhibited a more pronounced shoulder after the release transient due to store-operated activation by STIM1 or STIM2. To better appreciate the differences in Ca2+ entry between the two populations, we subtracted the signals obtained in the absence of Ca2+ from those in the presence of Ca2+ to arrive at the net influx components in the cell populations. As can be seen in Fig. 6D, the STIM1-expressing cells produced a sustained increase in [Ca2+]i, whereas STIM2-expressing cells produced a transient increase in SOCE that decayed. This transient increase in SOCE reflects the SOCE induced by STIM2 coupling to CRACM1 channels after store depletion and presumably the subsequent inhibition by CaM. The slightly increased basal [Ca2+]i of the STIM2- and CRACM1-expressing cells may result from a low level of constitutively open CRAC channels.
Figure 6.
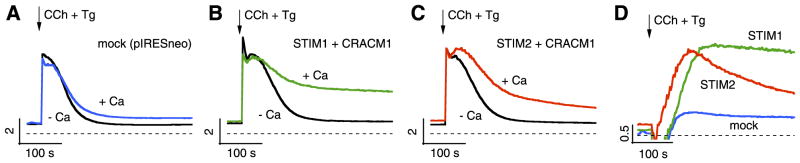
STIM2 causes transient SOCE in intact cells. A) Changes in [Ca2+]i measured as ratios of fura-2 fluorescence excited at 340 and 380 nm in STIM2 cells transfected with empty vector in the absence (black) or presence (blue) of 1 mM extracellular Ca2+. Carbachol (100 μM) and thapsigargin (2 μM) were applied as indicated by arrow and maintained for duration of experiment. B) Identical experimental conditions as in A, but for STIM1 + CRACM1 cells. C) Identical experimental conditions as in A, but for STIM2 + CRACM1 cells. D) These traces represent subtracted traces from A–C, where [Ca2+]i signal in Ca2+-free solution was subtracted from that obtained with Ca2+ to yield net Ca2+ entry.
DISCUSSION
Taken together, the above results provide compelling evidence for both store-dependent and store-independent activation of CRAC channels by STIM2. Our data suggest that in stably transfected HEK293 cells, STIM2 exists in at least two functional states, with most of the STIM2 molecules coupling to CRAC channels in a store-independent manner (constitutively coupled) and a smaller population of STIM2 molecules remaining available to activate CRAC channels after store depletion (store coupled). Store-dependent activation can be demonstrated both in patch-clamp experiments when cells are perfused with InsP3 (Fig. 3A) or when store depletion is caused by ionomycin (Fig. 3C), which both result in rapid activation of CRAC currents. SOCE is also evident in [Ca2+]i imaging experiments in intact cells when cells are stimulated with an InsP3-mobilizing agonist and blocking store refilling with thapsigargin (Fig. 6). The main difference between CRAC in patch-clamp experiments and SOCE in intact cells is that the latter manifests itself as a transient increase in [Ca2+]i (Fig. 6), whereas whole-cell recordings exhibit a more persistent CRAC current that is followed by an even larger, secondary activation of CRAC channel activity (Fig. 3A, C, F). The transient nature of STIM2-mediated SOCE in intact cells may also explain why the store-operated mechanism remained undetected in typical assays of SOCE (14, 16), where thapsigargin-induced store depletion in zero extracellular Ca2+ is followed by Ca2+ readmission after several hundreds of seconds to probe SOCE. During the thapsigargin/zero Ca2+ exposure, any STIM2-mediated SOCE would remain undetectable and channel activity would have subsided by the time Ca2+ is readmitted.
The second population of STIM2 molecules is not coupled to store depletion (14). It is observed in patch-clamp experiments as a large CRAC current that typically activates after ~100 –200 s under a variety of experimental conditions, regardless of the filling state of intracellular stores. Although the store-operated population can be suppressed experimentally in maintaining the stores filled with Ca2+, the store-independent population can only be delayed by reducing the rate of diffusion through the pipette and the cytosol (Fig. 5A), which effectively slows down the washout and loss of a cytosolic factor that normally inhibits the constitutive activity of STIM2 and CRACM1. We propose that this cytosolic factor is CaM, since inclusion of CaM in the pipette filling solution completely suppresses activation of the slow CRAC current phase (Fig. 5B). This inhibition requires at least resting levels of [Ca2+]i approximately ~100 nM, as apo-CaM is ineffective when [Ca2+]i is buffered to near zero (Fig. 5B). In the case of store-operated activation of CRACM1 by STIM2, we surmise that the CaM is initially absent, so activation can take place. Subsequently, possibly mediated by the increase in [Ca2+]ii, Ca2+-CaM is recruited to the STIM2/CRACM1 complex and inhibits channel activity to cause transient SOCE. In addition, CaM appears to also have a more general effect on SOCE that is mediated through inhibition of InsP3 receptor activity (31, 32). This effect allows stores to refill even in the presence of high levels of InsP3 and results in suppression of CRAC channel activity independently of whether CRAC channels are activated through STIM1 or STIM2. Sidestepping this InsP3-dependent regulation by enforcing store depletion through ionomycin reveals that the inhibitory effect of CaM at the STIM/CRAC complex is specific to STIM2, as it is not observed with STIM1 (Fig. 5C, D).
Similar to CaM, G418 appears to be an effective and potent inhibitor of STIM2-mediated CRAC channel activation. In cells grown in the presence of G418, both populations of STIM2 molecules are blocked by the antibiotic at the intracellular side (Fig. 4A, B). An attractive hypothesis that remains to be substantiated would be that G418 acts as a CaM mimetic and occupies the same inhibitory binding site as CaM on the STIM2/ CRACM1 complexes. Consistent with this notion is the finding that regardless of whether the STIM2/ CRACM1 complexes are suppressed by G418 or the endogenous CaM, they can be rapidly activated by 2-APB. This activation is so rapid that it cannot possibly result from store depletion. The fastest store-dependent activation of CRAC currents by InsP3 is typically characterized by ~4 – 6 s delay and then proceeds over ~60 s to reach full activation. However, the 2-APB-mediated activation occurs with a half-time of ~2 s and is virtually complete within 5 s (Fig. 1E, F), i.e., at a time when store-dependent activation just begins. This rapid, store-independent gating of CRAC channels appears to be mediated by some close interaction of STIM2 and CRACM1, as it is not observed in cells overexpressing CRACM1 alone or in combination with STIM1. It is tempting to speculate that agonist-mediated signals might recruit these precoupled STIM2/ CRAC complexes to activate CRAC channels even without store depletion and with very fast kinetics. In this context, we note that the carbachol-induced activation of Ca2+ influx illustrated in Fig. 6 develops considerably more quickly in STIM2-expressing cells than in STIM1 expressors (see Fig. 6D).
At this point, it remains unclear how 2-APB can achieve such fast gating of CRAC channels and even overcome the apparent block by G418 and CaM. If we accept that STIM2 activates CRACM1 via direct binding in a conformational coupling manner, the simplest interpretation would be that 2-APB simply displaces G418 or CaM from its binding site on the STIM2/ CRACM1 complex. This would imply an intracellular action 2-APB; however, this issue remains unresolved, since both facilitatory and inhibitory effects of 2-APB are only seen when the compound is applied extracellularly (26). We confirmed that intracellularly applied 2-APB (50 μM) is indeed ineffective in activating or suppressing CRAC currents, whereas subsequent extracellular application of 50 μM 2-APB first activates and then inhibits CRAC currents (Fig. 5F). However, lipophilic compounds are often ineffective when applied intracellularly in patch-clamp experiments and extracellularly applied 2-APB has been demonstrated to be membrane permeable and suppress InsP3 receptor function (33), suggesting that, in principle, the compound might gain access to the cytosol when applied extracellularly.
Taken together, our data provide compelling evidence for a bimodal functional coupling between STIM2 to CRACM channels, with both store-dependent and -independent components. Since both STIM1 and STIM2 are widely expressed (34), both may serve distinct, but important, roles in SOCE. Relative differences in ER Ca2+ sensing by the two proteins may provide a broader sensitivity to store emptying. STIM2 is additionally activable in a store-independent manner, with the rapid action of 2-APB revealing that a substantial component of STIM2 appears to be in a precoupled configuration. Identifying physiological mechanisms that regulate this process would be an important future area of investigation. Indeed, the revelation that both components of STIM2-mediated channel activation are modified by CaM provides an important clue to the role of a potentially powerful cytosolic Ca2+-mediated control process in the activation of CRAC channels, complementing the clear role of luminal Ca2+ in activating the channels.
Acknowledgments
We thank M. Bellinger and A. Love for help with cell culture and transfections. Supported in part by National Institutes of Health (NIH) grants R01-AI050200 and R01-NS040927 (R.P.) and AI058173 (D.L.G.). C.P. was supported by a fellowship from the Deutsche Forschungsgemeinschaft (PE-1478/1–1).
References
- 1.Parekh AB, Penner R. Store depletion and calcium influx. Physiol Rev. 1997;77:901–930. doi: 10.1152/physrev.1997.77.4.901. [DOI] [PubMed] [Google Scholar]
- 2.Parekh AB, Putney JW., Jr Store-operated calcium channels. Physiol Rev. 2005;85:757– 810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- 3.Putney JW., Jr Capacitative calcium entry revisited. Cell Calcium. 1990;11:611– 624. doi: 10.1016/0143-4160(90)90016-n. [DOI] [PubMed] [Google Scholar]
- 4.Hoth M, Penner R. Calcium release-activated calcium current in rat mast cells. J Physiol. 1993;465:359–386. doi: 10.1113/jphysiol.1993.sp019681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoth M, Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 1992;355:353–356. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- 6.Zweifach A, Lewis RS. Mitogen-regulated Ca2+ current of T lymphocytes is activated by depletion of intracellular Ca2+ stores. Proc Natl Acad Sci U S A. 1993;90:6295– 6299. doi: 10.1073/pnas.90.13.6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- 8.Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE, Jr, Meyer T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol. 2005;15:1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, Velicelebi G, Stauderman KA. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169:435– 445. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vig M, Peinelt C, Beck A, Koomoa DL, Rabah D, Koblan-Huberson M, Kraft S, Turner H, Fleig A, Penner R, Kinet JP. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science. 2006;312:1220–1223. doi: 10.1126/science.1127883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang SL, Yeromin AV, Zhang XH, Yu Y, Safrina O, Penna A, Roos J, Stauderman KA, Cahalan MD. Genome-wide RNAi screen of Ca2+ influx identifies genes that regulate Ca2+ release-activated Ca2+ channel activity. Proc Natl Acad Sci U S A. 2006;103:9357–9362. doi: 10.1073/pnas.0603161103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mercer JC, Dehaven WI, Smyth JT, Wedel B, Boyles RR, Bird GS, Putney JW., Jr Large store-operated calcium selective currents due to co-expression of Orai1 or Orai2 with the intracellular calcium sensor, Stim1. J Biol Chem. 2006;281:24979–24990. doi: 10.1074/jbc.M604589200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peinelt C, Vig M, Koomoa DL, Beck A, Nadler MJ, Koblan-Huberson M, Lis A, Fleig A, Penner R, Kinet JP. Amplification of CRAC current by STIM1 and CRACM1 (Orai1) Nat Cell Biol. 2006;8:771–773. doi: 10.1038/ncb1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soboloff J, Spassova MA, Tang XD, Hewavitharana T, Xu W, Gill DL. Orai1 and STIM reconstitute store-operated calcium channel function. J Biol Chem. 2006;281:20661–20665. doi: 10.1074/jbc.C600126200. [DOI] [PubMed] [Google Scholar]
- 15.Wu MM, Buchanan J, Luik RM, Lewis RS. Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J Cell Biol. 2006;174:803– 813. doi: 10.1083/jcb.200604014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Soboloff J, Spassova MA, Hewavitharana T, He LP, Xu W, Johnstone LS, Dziadek MA, Gill DL. STIM2 is an inhibitor of STIM1-mediated store-operated Ca2+ entry. Curr Biol. 2006;16:1465–1470. doi: 10.1016/j.cub.2006.05.051. [DOI] [PubMed] [Google Scholar]
- 17.Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, Stauderman KA, Cahalan MD. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437:902–905. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vig M, Beck A, Billingsley JM, Lis A, Parvez S, Peinelt C, Koomoa DL, Soboloff J, Gill DL, Fleig A, Kinet JP, Penner R. CRACM1 multimers form the ion-selective pore of the CRAC channel. Curr Biol. 2006;16:2073–2079. doi: 10.1016/j.cub.2006.08.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yeromin AV, Zhang SL, Jiang W, Yu Y, Safrina O, Cahalan MD. Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature. 2006;443:226–229. doi: 10.1038/nature05108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A, Hogan PG. Orai1 is an essential pore subunit of the CRAC channel. Nature. 2006;443:230–233. doi: 10.1038/nature05122. [DOI] [PubMed] [Google Scholar]
- 21.Gwack Y, Srikanth S, Feske S, Cruz-Guilloty F, Oh-Hora M, Neems DS, Hogan PG, Rao A. Biochemical and functional characterization of Orai family proteins. J Biol Chem. 2007;282:16232–16243. doi: 10.1074/jbc.M609630200. [DOI] [PubMed] [Google Scholar]
- 22.Lis A, Peinelt C, Beck A, Parvez S, Monteilh-Zoller M, Fleig A, Penner R. CRACM1, CRACM2, and CRACM3 are store-operated Ca2+ channels with distinct functional properties. Curr Biol. 2007;17:794– 800. doi: 10.1016/j.cub.2007.03.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma HT, Venkatachalam K, Parys JB, Gill DL. Modification of store-operated channel coupling and inositol trisphosphate receptor function by 2-aminoethoxydiphenyl borate in DT40 lymphocytes. J Biol Chem. 2002;277:6915– 6922. doi: 10.1074/jbc.M107755200. [DOI] [PubMed] [Google Scholar]
- 24.Braun FJ, Broad LM, Armstrong DL, Putney JW., Jr Stable activation of single Ca2+ release-activated Ca2+ channels in divalent cation-free solutions. J Biol Chem. 2001;276:1063–1070. doi: 10.1074/jbc.M008348200. [DOI] [PubMed] [Google Scholar]
- 25.Hermosura MC, Monteilh-Zoller MK, Scharenberg AM, Penner R, Fleig A. Dissociation of the store-operated calcium current ICRAC and the Mg-nucleotide-regulated metal ion current MagNuM. J Physiol. 2002;539:445– 458. doi: 10.1113/jphysiol.2001.013361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prakriya M, Lewis RS. Potentiation and inhibition of Ca2+ release-activated Ca2+ channels by 2-aminoethyldiphenyl borate (2-APB) occurs independently of IP3 receptors. J Physiol. 2001;536:3–19. doi: 10.1111/j.1469-7793.2001.t01-1-00003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spassova MA, Soboloff J, He LP, Xu W, Dziadek MA, Gill DL. STIM1 has a plasma membrane role in the activation of store-operated Ca2+ channels. Proc Natl Acad Sci U S A. 2006;103:4040– 4045. doi: 10.1073/pnas.0510050103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Levitan IB. It is calmodulin after all! Mediator of the calcium modulation of multiple ion channels. Neuron. 1999;22:645–648. doi: 10.1016/s0896-6273(00)80722-9. [DOI] [PubMed] [Google Scholar]
- 29.Moreau B, Straube S, Fisher RJ, Putney JW, Jr, Parekh AB. Ca2+-calmodulin-dependent facilitation and Ca2+ inactivation of Ca2+ release-activated Ca2+ channels. J Biol Chem. 2005;280:8776– 8783. doi: 10.1074/jbc.M409619200. [DOI] [PubMed] [Google Scholar]
- 30.Vaca L. Calmodulin inhibits calcium influx current in vascular endothelium. FEBS Lett. 1996;390:289–293. doi: 10.1016/0014-5793(96)00675-8. [DOI] [PubMed] [Google Scholar]
- 31.Adkins CE, Morris SA, De Smedt H, Sienaert I, Torok K, Taylor CW. Ca2+-calmodulin inhibits Ca2+ release mediated by type-1, -2 and -3 inositol trisphosphate receptors. Biochem J. 2000;345(Pt 2):357–363. [PMC free article] [PubMed] [Google Scholar]
- 32.Patel S, Morris SA, Adkins CE, O’Beirne G, Taylor CW. Ca2+-independent inhibition of inositol trisphosphate receptors by calmodulin: redistribution of calmodulin as a possible means of regulating Ca2+ mobilization. Proc Natl Acad Sci U S A. 1997;94:11627–11632. doi: 10.1073/pnas.94.21.11627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maruyama T, Kanaji T, Nakade S, Kanno T, Mikoshiba K. 2APB, 2-aminoethoxydiphenyl borate, a membrane-penetrable modulator of Ins(1,4,5)P3-induced Ca2+ release. J Biochem. 1997;122:498–505. doi: 10.1093/oxfordjournals.jbchem.a021780. [DOI] [PubMed] [Google Scholar]
- 34.Williams RT, Manji SS, Parker NJ, Hancock MS, Van Stekelenburg L, Eid JP, Senior PV, Kazenwadel JS, Shandala T, Saint R, Smith PJ, Dziadek MA. Identification and characterization of the STIM (stromal interaction molecule) gene family: coding for a novel class of transmembrane proteins. Biochem J. 2001;357:673– 685. doi: 10.1042/0264-6021:3570673. [DOI] [PMC free article] [PubMed] [Google Scholar]