

Abstract
Background
In acute coronary syndromes, C‐reactive protein (CRP) strongly relates to subsequent death, but surprisingly not to recurrent myocardial infarction. Other biomarkers may reflect different processes related to these outcomes. We assessed 8 inflammatory and vascular biomarkers and the risk of death and recurrent nonfatal cardiovascular events in the 16 weeks after an acute coronary syndrome.
Methods and Results
We measured blood concentrations of CRP, serum amyloid A (SAA), interleukin‐6 (IL‐6), soluble intercellular adhesion molecule (ICAM), soluble vascular cell adhesion molecule (VCAM), E‐selectin, P‐selectin, and tissue plasminogen activator antigen (tPA) 24 to 96 hours after presentation with acute coronary syndrome in 2925 subjects participating in a multicenter study. Biomarkers were related to the risk of death, and recurrent nonfatal acute coronary syndromes (myocardial infarction or unstable angina) over 16 weeks using Cox proportional hazard models. On univariate analyses, baseline CRP (P=0.006), SAA (P=0.012), and IL‐6 (P<0.001) were related to death, but not to recurrent nonfatal acute coronary syndromes. VCAM and tPA related to the risk of death (P<0.001, P=0.021, respectively) and to nonfatal acute coronary syndromes (P=0.021, P=0.049, respectively). Adjusting for significant covariates reduced the strength of the associations; however, CRP and SAA continued to relate to death.
Conclusions
In acute coronary syndromes, the CRP inflammatory axis relates to the risk of death and may reflect myocardial injury. VCAM and tPA may have greater specificity for processes reflecting inflammation and thrombosis in the epicardial arteries, which determine recurrent coronary events.
Keywords: acute coronary syndromes, biomarkers, CRP, death, nonfatal events, risk
Introduction
Inflammation and thrombosis are key components of both myocardial necrosis and disrupted coronary plaques in acute coronary syndromes.1 Shortly after these events, plasma concentration of the hepatic acute phase reactant C‐reactive protein (CRP) increases several fold.2–4 Although high concentrations of CRP relate to subsequent mortality over several months after an acute coronary syndrome, CRP is not related to recurrent myocardial infarction.5–13 Some authors speculate that in the acute coronary syndrome setting, elevated CRP predominantly reflects the extent of myocardial injury, which determines early mortality, rather than inflammation in the coronary arterial system, which may drive recurrent nonfatal coronary events.14
Support for this hypothesis could come from several previously unexplored lines of evidence. These include whether there are similar relationships between mortality and recurrent nonfatal acute coronary syndromes with other biomarkers that either stimulate CRP (eg, interleukin‐6) or track closely with it (eg, SAA) and whether other markers that are more closely linked to arterial inflammation and thrombosis have different relationships to these clinical end points. Inflammatory biomarkers, which reflect processes more specific to endothelial and coronary artery thrombosis, include soluble selectins, cellular adhesion molecules, and endogenous tissue plasminogen activator.
The Myocardial Ischemia Reduction with Aggressive Cholesterol Lowering (MIRACL) study provided an opportunity to measure multiple biomarkers simultaneously for this purpose. We aimed to assess how various baseline biomarkers of inflammation, sampled 24 to 96 hours after hospital admission for an acute coronary syndrome, associated with different clinical outcomes up to 4 months after an acute coronary syndrome.
Methods
Study Population
The MIRACL study enrolled 3086 subjects admitted with unstable angina or non‐Q‐wave myocardial infarction to 122 hospitals in Europe, North America, South Africa, and Australia.15 These diagnoses required chest discomfort lasting ≥15 minutes within the 24 hours preceding hospitalization and representing a change in the usual pattern of angina. The diagnosis of unstable angina required new or dynamic ST‐wave or T‐wave changes in ≥2 contiguous ECG leads or a new wall‐motion or myocardial perfusion abnormality. The diagnosis of myocardial infarction required elevation of serum creatine kinase, its MB fraction, or troponin to a level exceeding 2 times the upper limit of normal. All patients provided informed consent. The protocol was approved by local institutional review boards.
Subjects were excluded if coronary revascularization was planned or anticipated. Subjects were randomized 24 to 96 hours after admission to atorvastatin 80 mg daily or matching placebo for 16 weeks. The combined primary end point for the MIRACL study was a recurrent ischemic event, defined as death, nonfatal myocardial infarction, recurrent unstable angina, or resuscitated cardiac arrest. The end points for this analysis were all‐cause death and recurrent nonfatal acute coronary syndromes (ie, acute myocardial infarction and recurrent unstable angina requiring emergency hospitalization and with objective evidence of myocardial ischemia as defined above). Patients with recurrent nonfatal acute coronary syndromes who subsequently died during the 16‐week follow‐up were counted as deaths only.
Measurement of Biomarkers
Blood was collected into serum and EDTA tubes 24 to 96 hours after hospital admission for an acute coronary syndrome to measure baseline levels of biomarkers. The tubes were centrifuged on site, and the serum or plasma was shipped to the core laboratory for storage at −70°C. Samples were then sent to the inflammatory markers core laboratory, where they were measured in batches.16–18 High‐sensitivity CRP and serum amyloid A (SAA) were measured by high‐sensitivity immunonephelometry (Dade Behring, Deerfield, IL). Interleukin‐6 (IL‐6), soluble intercellular adhesion molecule (ICAM), soluble vascular cell adhesion molecule (VCAM), soluble E‐selectin, and soluble P‐selectin were measured by ELISA (R&D Systems, Minneapolis, MN), and endogenous tissue plasminogen activator antigen (tPA) was measured by ELISA (American Diagnostica, Greenwich, CT). Troponin I was measured using an ACS:180 Chemiluminescence cTNI Immunoassay (Bayer Diagnostics, Tarrytown, NY). The coefficients of variation for reproducibility of the assays were excellent: CRP, 4.5% at 12.6 mg/L and 5.2% at 48 mg/L; SAA, 6.2% at 14.8 mg/L; IL‐6, 11.6% at 1.1 pg/mL and 7.0% at 4.7 pg/mL; ICAM, 7.4% at 117 ng/mL and 6.1% at 453 ng/mL; VCAM, 8.5% at 24.9 ng/mL and 8.9% at 49.6 ng/mL; E‐selectin, 8.8% at 20.4 ng/mL and 7.4% at 115 ng/mL; P‐selectin, 9.9% at 94 ng/mL and 7.9% at 730 ng/mL; tPA, 5.5% at 6.0 ng/mL and 4.9% at 15.0 ng/mL.
Subgroup With Known Baseline Ejection Fraction
In 910 subjects, baseline left ventricular ejection fraction (LVEF) was documented from echocardiography, angiography, or a nuclear medicine study. In this subgroup we explored the relationship between abnormal LVEF (defined as <55%) and the biomarkers and clinical outcomes.
Statistics
This report focuses on the subjects in the MIRACL study who had baseline levels of inflammatory markers measured prior to randomization, including 2925 (95% of all MIRACL subjects) for CRP, 2852 (92%) for IL‐6, 2923 (95%) for SAA, 2858 (93%) for ICAM, 2860 (93%) for VCAM, 2633 (85%) for E‐selectin, 2858 (93%) for P‐selectin, and 2780 (90%) for tPA. As the distributions of inflammatory markers were skewed, biomarkers are described using median and interquartile ranges (25% to 75%) and models used nonparametric quartile analysis. We used chi‐square tests for categorical variables and Kruskall–Wallis tests for continuous variables to compare baseline variables and the continuous distribution of biomarkers among subjects who died, had recurrent nonfatal acute coronary syndromes, or had no adverse events on follow‐up. Cox proportional hazards models were used to assess the relative risk (hazard ratio) for events during the 16 weeks of the study over quartiles of biomarkers, with the lowest quartile as the reference category. A linear trend test was also included to assess whether the risk of clinical outcome was monotonically increasing or decreasing over quartiles of biomarker level. As there were no significant treatment‐by‐biomarker interaction effects in the models, results for both treatment groups (atorvastatin and placebo) were combined with treatment as a covariate. Multivariable models were used, adjusting for treatment assignment and other significant covariates related to recurrent events (see Table 1). Because of the exploratory nature of this study, 2‐sided nominal (uncorrected for multiple testing) P values are reported for all statistical comparisons based on our prespecified analysis plan.
Table 1.
Baseline Characteristics of Subjects Who Died, Had a Nonfatal ACS, or Did Not Have an Event Over the 16 Weeks of the Study (P Value From Kruskall–Wallis)
| Death (n=127) | Nonfatal ACS (n=348) | No Event (n=2450) | P Value | |
|---|---|---|---|---|
| Age, mean (SD) | 75 (10) | 68 (12) | 65 (12) | <0.001 |
| Male (%) | 81 (64) | 232 (67) | 1591 (65) | 0.81 |
| Past MI (%) | 56 (44) | 114 (33) | 558 (23) | <0.001 |
| Prior coronary revascularization | 20 (16) | 43 (12) | 238 (10) | 0.04 |
| Hypertension (%) | 82 (65) | 184 (53) | 1326 (54) | 0.058 |
| Current smoker (%) | 14 (11) | 81 (23) | 723 (30) | <0.001 |
| Diabetes (%) | 44 (35) | 91 (26) | 530 (22) | <0.001 |
| Body mass index (kg/m2), mean (SD) | 26.9 (8.5) | 27.4 (4.4) | 27.6 (5.2) | 0.052 |
| Admission to randomization (h), mean (SD) | 68 (45) | 63 (23) | 63 (23) | 0.45 |
| LVEF (%), median (25%, 75%)* | 45 (33, 55) | 50 (42, 61) | 56 (47, 65) | <0.001 |
| Total cholesterol (mg/dL), mean (SD) | 200 (48) | 204 (41) | 206 (37) | 0.12 |
| HDL (mg/dL), mean (SD) | 45 (13) | 45 (11) | 47 (13) | 0.002 |
| LDL (mg/dL), mean (SD) | 122 (40) | 123 (34) | 124 (33) | 0.61 |
| Triglycerides (mg/dL), mean (SD) | 166 (69) | 186 (83) | 183 (92) | 0.09 |
| Troponin I, ng/mL (%) | 64 (50) | 140 (40) | 1042 (43) | 0.10 |
| CRP (mg/L), median (25%, 75%) | 20.1 (6.0, 52.9) | 10.2 (4.5, 33.1) | 10.2 (4.2, 35.0) | 0.005 |
| IL‐6 (pg/mL), median (25%, 75%) | 10.8 (5.5, 20.8) | 7.1 (3.7, 14.1) | 6.7 (3.7, 14.6) | <0.001 |
| SAA (mg/dL), median (25%, 75%) | 2.5 (0.7, 19.7) | 1.4 (0.5, 8.6) | 1.6 (0.6, 8.2) | 0.02 |
| VCAM (ng/L), median (25%, 75%) | 959 (674, 1231) | 870 (656, 1069) | 808 (619, 1020) | <0.001 |
| ICAM (ng/L), median (25%, 75%) | 307 (259, 366) | 302 (263, 366) | 299 (252, 358) | 0.17 |
| P‐Selectin (ng/L), median (25%, 75%) | 65 (46, 96) | 59 (42, 90) | 59 (41, 93) | 0.56 |
| E‐Selectin (ng/L) median (25%, 75%) | 47 (34, 63) | 47 (33, 62) | 48 (35, 62) | 0.63 |
| tPA (ng/L), median (25%, 75%) | 15.1 (10.7, 24.3) | 14.6 (9.6, 21.5) | 13.5 (8.5, 20.3) | 0.003 |
ACS indicates acute coronary syndrome; MI, myocardial infarction; LVEF, left ventricular ejection fraction; HDL, high‐density lipoprotein; LDL, low‐density lipoprotein; CRP, C‐reactive protein; IL‐6, interleukin‐6; SAA, serum amyloid A; VCAM, vascular cell adhesion molecule; ICAM, intercellular adhesion molecule; tPA, tissue plasminogen activator antigen.
LVEF measured in 910 subjects.
Results
Of the 2925 subjects with CRP measured in the MIRACL study, 127 (4%) died, 205 (7%) had recurrent myocardial infarction, and 215 (7%) had recurrent unstable angina with objective evidence of ischemia over the 16 weeks of the study.
Table 1 compares baseline descriptive data on subjects who had an event (death, nonfatal MI, or unstable angina) and those who remained event free. Patients who had an event were older (P<0.001); were more likely to have a history of past myocardial infarct (P<0.001), prior revascularization (P=0.04), or diabetes (P<0.001); were less likely to be current smokers (P<0.001); and had lower high‐density lipoprotein (HDL) cholesterol (P=0.002) and lower LVEF (P<0.001). Their baseline concentrations of CRP, IL‐6, SAA, VCAM, and tPA were significantly higher, and ICAM tended to be higher (Table 1).
In the following univariate and multivariable models, atorvastatin associated with a 3% to 9% reduction in the risk of death and a 19% to 22% reduction in the risk of recurrent nonfatal acute coronary syndromes. However, as there were no significant treatment‐by‐biomarker interactions, we combined the atorvastatin and placebo groups and included treatment as a covariate. Also, we chose to assess the relationship of biomarkers to outcomes using test‐of‐trend statistics, which reflect the continuous relationship of biomarkers to outcomes. However, our multivariable models also showed hazard ratios for each quartile compared with the reference (first) quartile.
CRP, IL‐6, and SAA
Univariate analyses showed strong correlations between death and increasing quartiles of baseline CRP, IL‐6, and SAA (Figure 1). Interestingly, these CRP axis biomarkers did not correlate with recurrent nonfatal acute coronary events (Figure 1).
Figure 1.
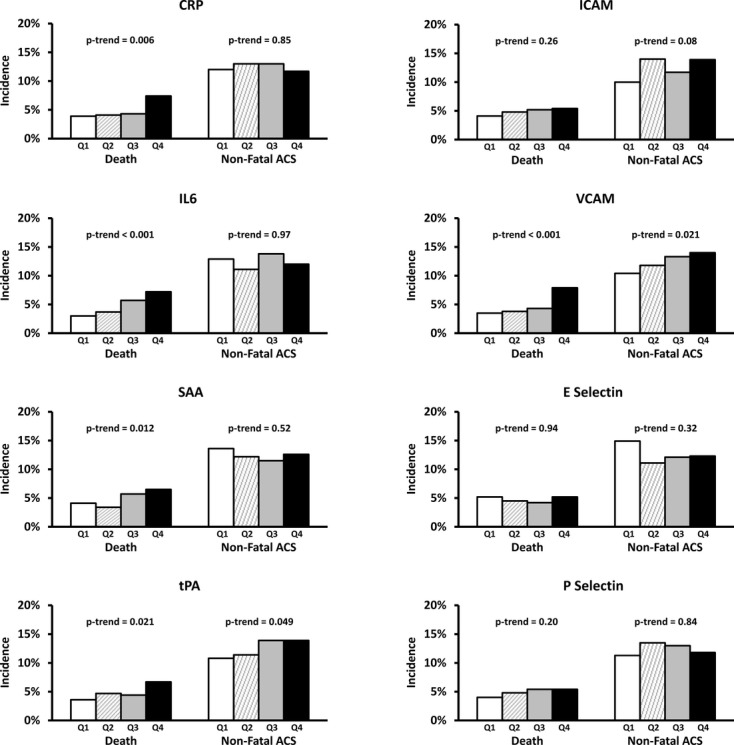
Incidence rates and test‐of‐trend for the risk of death and recurrent nonfatal acute coronary syndromes (ACS) of myocardial infarction or unstable angina, associated with quartiles of each biomarker. CRP indicates C‐reactive protein; ICAM, intercellular adhesion molecule; IL‐6, interleukin‐6; VCAM, vascular cell adhesion molecule; SAA, serum amyloid A; tPA, tissue plasminogen activator antigen.
VCAM, tPA, and Other Biomarkers
In univariate analyses, baseline quartiles of VCAM and tPA concentrations associated with death as well as with recurrent nonfatal coronary events (Figure 1). None of the other biomarkers bore any relationship either to death or to recurrent nonfatal acute coronary syndromes (Figure 1).
Biomarkers and Risk After Adjustment for Other Covariates
Adjustment for covariates in multivariable models attenuated the statistically significant hazard ratios. For example, for fatal events, multivariable adjustment reduced the hazard ratio for the fourth compared with the first quartile for CRP (from 1.92 to 1.69), IL‐6 (from 2.40 to 1.61), SAA (from 1.61 to 1.41), VCAM (from 2.36 to 1.12), and tPA (from 1.90 to 1.27). Similarly, multivariable adjustment attenuated the hazard ratios for nonfatal acute coronary syndromes for the fourth compared with the first quartile for VCAM (from 1.41 to 1.12) and tPA (1.31 to 1.20). In the multivariable models, CRP and SAA associated with a higher risk of death (P=0.034 and P=0.049, respectively; Table 2). However, no other biomarkers were related to death, and none of the other biomarkers were related to nonfatal acute coronary syndromes in multivariable models (Table 2).
Table 2.
Hazard Ratios for Quartiles of Biomarkers at Baseline and Death and Recurrent Nonfatal ACS of Myocardial Infarction or Unstable Angina
| Quartile 1, Reference | Quartile 2, HR (95% CI) | Quartile 3, HR (95% CI) | Quartile 4, HR (95% CI) | Test‐of‐Trend | |
|---|---|---|---|---|---|
| CRP | |||||
| Death | 1.00 | 0.97 (0.54 to 1.75) | 1.08 (0.61 to 1.91) | 1.69 (0.99 to 2.90) | 0.034 |
| Nonfatal ACS | 1.00 | 1.06 (0.78 to 1.44) | 1.09 (0.80 to 1.49) | 0.98 (0.70 to 1.36) | 0.941 |
| IL‐6 | |||||
| Death | 1.00 | 1.05 (0.56 to 1.98) | 1.29 (0.71 to 2.35) | 1.61 (0.90 to 2.90) | 0.060 |
| Nonfatal ACS | 1.00 | 0.82 (0.60 to 1.13) | 0.94 (0.69 to 1.27) | 0.84 (0.61 to 1.16) | 0.439 |
| SAA | |||||
| Death | 1.00 | 0.66 (0.36 to 1.21) | 1.14 (0.66 to 1.97) | 1.41 (0.82 to 2.43) | 0.045 |
| Nonfatal ACS | 1.00 | 0.87 (0.65 to 1.18) | 0.85 (0.62 to 1.16) | 0.96 (0.70 to 1.33) | 0.783 |
| ICAM | |||||
| Death | 1.00 | 1.21 (0.69 to 2.10) | 1.26 (0.73 to 2.19) | 1.53 (0.89 to 2.62) | 0.131 |
| Nonfatal ACS | 1.00 | 1.41 (1.03 to 1.94) | 1.18 (0.84 to 1.65) | 1.48 (1.07 to 2.05) | 0.064 |
| VCAM | |||||
| Death | 1.00 | 0.83 (0.45 to 1.52) | 0.74 (0.40 to 1.34) | 1.12 (0.65 to 1.94) | 0.484 |
| Nonfatal ACS | 1.00 | 1.09 (0.79 to 1.51) | 1.11 (0.80 to 1.54) | 1.12 (0.81 to 1.55) | 0.531 |
| E‐selectin | |||||
| Death | 1.00 | 1.01 (0.59 to 1.73) | 0.92 (0.52 to 1.62) | 1.26 (0.74 to 2.13) | 0.486 |
| Nonfatal ACS | 1.00 | 0.77 (0.56 to 1.06) | 0.85 (0.62 to 1.16) | 0.87 (0.64 to 1.20) | 0.489 |
| P‐selectin | |||||
| Death | 1.00 | 1.40 (0.79 to 2.45) | 1.69 (0.98 to 2.93) | 1.71 (0.99 to 2.95) | 0.043 |
| Nonfatal ACS | 1.00 | 1.30 (0.95 to 1.77) | 1.29 (0.94 to 1.77) | 1.13 (0.82 to 1.56) | 0.508 |
| tPA | |||||
| Death | 1.00 | 1.17 (0.67 to 2.03) | 0.83 (0.46 to 1.48) | 1.27 (0.75 to 2.16) | 0.574 |
| Nonfatal ACS | 1.00 | 1.01 (0.73 to 1.41) | 1.16 (0.84 to 1.59) | 1.20 (0.88 to 1.65) | 0.178 |
Hazard ratios are adjusted by treatment group, age, past myocardial infarction, prior coronary revascularization, diabetes mellitus, current smoking, HDL cholesterol, and positive troponin. ACS indicates acute coronary syndrome; CRP, C‐reactive protein; IL‐6, interleukin‐6; SAA, serum amyloid A; ICAM, intercellular adhesion molecule; VCAM, vascular cell adhesion molecule; tPA, tissue plasminogen activator antigen.
Subgroup With Known Baseline Ejection Fraction
Among the 910 subjects with LVEF assessed at baseline, 402 (44%) had an abnormal LVEF of <55%. Compared with normal LVEF, abnormal LVEF associated with higher median CRP (9.2 versus 14.6 mg/L, P<0.0001), IL‐6 (5.9 versus 8.6 pg/L, P<0.0001), SAA (1.3 versus 2.4 mg/L, P<0.0001), and VCAM (765 versus 857 ng/L, P=0.0027), but was unrelated to the other baseline biomarkers. Abnormal LVEF was also related to death (HR 3.14, 95% CI 1.56 to 6.34, P=0.0014) and recurrent nonfatal acute coronary syndromes (HR 1.55, 95% CI 1.05 to 2.30, P=0.03).
Discussion
Our analysis of the MIRACL study provides several novel findings relating biomarkers to the early risk of death or recurrent ischemic coronary events after an acute non‐ST‐segment elevation coronary syndrome. We have extended the previously described association between CRP and death to other members of the CRP axis, namely, SAA and IL‐6. We have also extended the previously described absence of a relationship between CRP and nonfatal events to SAA and IL‐6. In contrast, our study found that VCAM and tPA, markers of endothelial activation and thrombosis, were related both to mortality and to recurrent acute coronary syndromes. Recurrent myocardial infarction and unstable angina share a similar biology (plaque disruption and thrombosis) and justify combining these end points. A major strength of our study was the simultaneous comparison of multiple biomarkers, beyond CRP, within the same cohort with a large sample size.
CRP, SAA, and IL‐6
IL‐6 is released in relatively small concentrations into the bloodstream from numerous cell types associated with inflamed plaques.19 In the liver, this molecule promotes the production of CRP and SAA, which are released into the bloodstream in far greater concentrations.19 As IL‐6 is the primary driver of CRP and SAA production, these biomarkers align in a common pathway. Our study demonstrates that they relate to cardiovascular risk in a similar manner. A novel feature of this analysis was the evaluation of several biomarkers in this inflammatory axis.
In other studies of acute coronary syndromes, higher CRP at presentation relates to death in the short term11,13,20–21 and in follow‐up exceeding 6 months after the initial event.5,20,22–24 However, CRP fails to relate consistently to the wider range of cardiovascular events including recurrent myocardial infarction.6,13 With longer follow‐up after an acute coronary syndrome or in patients with chronic coronary heart disease, CRP does relate both to fatal and nonfatal MI25–26and reflects known relationships between low‐grade inflammation in chronic coronary artery disease and the long‐term risk of cardiovascular events. Our study provides further evidence that levels of CRP and its related biomarkers SAA and IL‐6 do not relate to the short‐term risk of nonfatal cardiovascular outcomes, but do relate to death.
Why this inflammatory axis relates only to death but not to other cardiovascular end points remains unclear. One hypothesis is that higher CRP in the setting of an acute coronary syndrome reflects a greater burden of myocardial ischemia and tissue injury.14 Most studies of acute coronary syndromes, including MIRACL, support this concept, as subjects with higher troponins also have higher concentrations of CRP.11,16,22,27 Some earlier reports did not find a relationship between CRP and troponin, but this disparity may have been a result of the use of less sensitive troponin assays.5,28
Our subgroup analysis of patients with known LVEF at baseline also supports this concept. Abnormal LVEF associated with higher concentrations of CRP, SAA, and IL6 and to a higher risk of death. This suggests that these biomarkers reflect greater myocardial damage or less reserve, which drives the strong relationship of these biomarkers to death.
In other smaller reports, higher CRP concentrations shortly after an acute coronary syndrome related to more severe wall‐motion abnormalities on echocardiography, a higher incidence of heart failure,21 and a higher risk of cardiac rupture or aneurysm formation after a Q‐wave MI.29 Our study shows consistent relationships to short‐term outcomes across this inflammatory axis and suggests that CRP, SAA, and IL‐6 reflect a more severe and complicated consequence of acute coronary syndrome that relates to death.
Cellular Adhesion Molecules, Selectins, and tPA
In contrast to the CRP axis, VCAM and tPA related both to death and to recurrent nonfatal acute coronary syndromes of myocardial infarction or unstable angina. Activated endothelial cells express E‐selectin, VCAM, and ICAM, adhesion molecules implicated in leukocyte recruitment and migration into the vessel wall.30–31 P‐selectin and tPA also reflect platelet activation and the balance of fibrin formation and fibrinolysis.17,32 Thus, these markers relate more directly to biological mechanisms that promote arterial thrombus and subsequent fatal and nonfatal cardiovascular events.
Biomarkers and Risk After Multivariable Adjustment
After adjusting for known significant covariates, most of the hazard ratios for biomarkers were lower, and some no longer remained statistically significant. This was expected, as most biomarkers reflect inflammatory and thrombotic processes that are exacerbated by covariates such as age, HDL cholesterol, diabetes, and degree of myocardial injury (troponin). In other words, the biomarkers assess the causal pathway through which upstream risk factors (our covariates) exert their adverse effect on outcomes; any relationship of biomarkers to outcome will be diluted by models that include more proximal factors in these processes. The implication of this attenuation in risk is that the biomarkers provide valuable insights into the mechanisms relating clinical outcomes to myocardial injury, arterial inflammation, and thrombosis, but may not substantially enhance clinical risk prediction models.
Limitations
The prespecified analysis plan made no correction for multiple comparisons because of the exploratory nature of this analysis, and the data on VCAM and tPA are hypothesis generating and require confirmation in a separate study. As many significance tests are reported, nominal P values should be interpreted with caution. The biomarkers in our study may have different long‐term relationships to outcomes beyond the 16 weeks of this study. Also, we could not assess the affect of sampling biomarkers at different times after the onset of acute coronary syndrome. However, peak levels of CRP and SAA occur 1 to 3 days after an acute inflammatory insult, which was within the time frame of the baseline biomarkers.19
Conclusions
Our analysis of the MIRACL study shows a strong relationship between early levels of CRP and the closely related markers of IL‐6 and SAA with death following an acute coronary syndrome. These biomarkers are not associated with recurrent nonfatal coronary events. In contrast, early levels of VCAM and tPA are related to both mortality and recurrent nonfatal coronary syndromes and may have greater specificity for processes reflecting inflammation and thrombosis in the epicardial arteries, which determine recurrent coronary events.
Sources of Funding
This study was supported by an unrestricted grant from Pfizer, Inc.
Disclosures
Payman Zamani, Gregory G. Schwartz, and Nader Rifai report no conflicts. Anders G. Olsson reports research support from Pfizer, Amgen, Karobio, MSD, Sanofi, and Takeda; consulting for Artery Therapeutics, AstraZeneca, Genzyme, Karobio, MSD, Pfizer, Roche, and Sanofi; Weihang Bao is an employee of Pfizer; Peter Libby is a consultant for and on the Steering Committee for Pfizer‐sponsored Precision trial (uncompensated position); Peter Ganz is a consultant for Pfizer, Genentech, and Gilead; Scott Kinlay reports research support from Novartis and Medtronic to the VA Cardiovascular Division. Dr Scott Kinlay and Dr Peter Ganz had full access to the data and take responsibility for the integrity of the data and the accuracy of the data analysis.
References
- 1.Libby P, Theroux P. Pathophysiology of coronary artery disease. Circulation. 2005; 111:3481-3488 [DOI] [PubMed] [Google Scholar]
- 2.de Beer FC, Hind CR, Fox KM, Allan RM, Maseri A, Pepys MB. Measurement of serum C‐reactive protein concentration in myocardial ischaemia and infarction. Br Heart J. 1982; 47:239-243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith SJ, Bos G, Esseveld MR, Van Eijk HG, Gerbrandy J. Acute‐phase proteins from the liver and enzymes from myocardial infarction: a quantitative relationship. Clin Chim Acta. 1977; 81:75-85 [DOI] [PubMed] [Google Scholar]
- 4.Pietila K, Harmoinen A, Poyhonen L, Koskinen M, Heikkila J, Ruosteenoja R. Intravenous streptokinase treatment and serum C‐reactive protein in patients with acute myocardial infarction. Br Heart J. 1987; 58:225-229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heeschen C, Hamm CW, Bruemmer J, Simoons ML. Predictive value of C‐reactive protein and troponin T in patients with unstable angina: a comparative analysis. CAPTURE Investigators. Chimeric c7E3 AntiPlatelet Therapy in Unstable angina REfractory to standard treatment trial. J Am Coll Cardiol. 2000; 35:1535-1542 [DOI] [PubMed] [Google Scholar]
- 6.James SK, Armstrong P, Barnathan E, Califf R, Lindahl B, Siegbahn A, Simoons ML, Topol EJ, Venge P, Wallentin L. Troponin and C‐reactive protein have different relations to subsequent mortality and myocardial infarction after acute coronary syndrome: a GUSTO‐IV substudy. J Am Coll Cardiol. 2003; 41:916-924 [DOI] [PubMed] [Google Scholar]
- 7.Biasucci LM, Liuzzo G, Grillo RL, Caligiuri G, Rebuzzi AG, Buffon A, Summaria F, Ginnetti F, Fadda G, Maseri A. Elevated levels of C‐reactive protein at discharge in patients with unstable angina predict recurrent instability. Circulation. 1999; 99:855-860 [DOI] [PubMed] [Google Scholar]
- 8.Liuzzo G, Biasucci LM, Gallimore JR, Grillo RL, Rebuzzi AG, Pepys MB, Maseri A. The prognostic value of C‐reactive protein and serum amyloid a protein in severe unstable angina. N Engl J Med. 1994; 331:417-424 [DOI] [PubMed] [Google Scholar]
- 9.Berk BC, Weintraub WS, Alexander RW. Elevation of C‐reactive protein in “active” coronary artery disease. Am J Cardiol. 1990; 65:168-172 [DOI] [PubMed] [Google Scholar]
- 10.Ferreiros ER, Boissonnet CP, Pizarro R, Merletti PF, Corrado G, Cagide A, Bazzino OO. Independent prognostic value of elevated C‐reactive protein in unstable angina. Circulation. 1999; 100:1958-1963 [DOI] [PubMed] [Google Scholar]
- 11.Morrow DA, Rifai N, Antman EM, Weiner DL, McCabe CH, Cannon CP, Braunwald E. C‐reactive protein is a potent predictor of mortality independently of and in combination with troponin T in acute coronary syndromes: a TIMI 11A substudy. Thrombolysis in Myocardial Infarction. J Am Coll Cardiol. 1998; 31:1460-1465 [DOI] [PubMed] [Google Scholar]
- 12.Oltrona L, Ardissino D, Merlini PA, Spinola A, Chiodo F, Pezzano A. C‐reactive protein elevation and early outcome in patients with unstable angina pectoris. Am J Cardiol. 1997; 80:1002-1006 [DOI] [PubMed] [Google Scholar]
- 13.Toss H, Lindahl B, Siegbahn A, Wallentin L. Prognostic influence of increased fibrinogen and C‐reactive protein levels in unstable coronary artery disease. FRISC Study Group. Fragmin during Instability in Coronary Artery Disease. Circulation. 1997; 96:4204-4210 [DOI] [PubMed] [Google Scholar]
- 14.De Servi S, Mariani M, Mariani G, Mazzone A. C‐reactive protein increase in unstable coronary disease cause or effect? J Am Coll Cardiol. 2005; 46:1496-1502 [DOI] [PubMed] [Google Scholar]
- 15.Schwartz GG, Olsson AG, Ezekowitz MD, Ganz P, Oliver MF, Waters D, Zeiher A, Chaitman BR, Leslie S, Stern T. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes: the MIRACL study: a randomized controlled trial. JAMA. 2001; 285:1711-1718 [DOI] [PubMed] [Google Scholar]
- 16.Kinlay S, Schwartz GG, Olsson AG, Rifai N, Leslie SJ, Sasiela WJ, Szarek M, Libby P, Ganz P. High‐dose atorvastatin enhances the decline in inflammatory markers in patients with acute coronary syndromes in the MIRACL study. Circulation. 2003; 108:1560-1566 [DOI] [PubMed] [Google Scholar]
- 17.Kinlay S, Schwartz GG, Olsson AG, Rifai N, Bao W, Libby P, Ganz P. Endogenous tissue plasminogen activator and risk of recurrent cardiac events after an acute coronary syndrome in the MIRACL study. Atherosclerosis. 2009; 206:551-555 [DOI] [PubMed] [Google Scholar]
- 18.Kinlay S, Schwartz GG, Olsson AG, Rifai N, Sasiela WJ, Szarek M, Ganz P, Libby P. Effect of atorvastatin on risk of recurrent cardiovascular events after an acute coronary syndrome associated with high soluble CD40 ligand in the Myocardial Ischemia Reduction with Aggressive Cholesterol Lowering (MIRACL) Study. Circulation. 2004; 110:386-391 [DOI] [PubMed] [Google Scholar]
- 19.Gabay C, Kushner I. Acute‐phase proteins and other systemic responses to inflammation. N Engl J Med. 1999; 340:448-454 [DOI] [PubMed] [Google Scholar]
- 20.Mueller C, Buettner HJ, Hodgson JM, Marsch S, Perruchoud AP, Roskamm H, Neumann FJ. Inflammation and long‐term mortality after non‐ST elevation acute coronary syndrome treated with a very early invasive strategy in 1042 consecutive patients. Circulation. 2002; 105:1412-1415 [DOI] [PubMed] [Google Scholar]
- 21.Suleiman M, Aronson D, Reisner SA, Kapeliovich MR, Markiewicz W, Levy Y, Hammerman H. Admission C‐reactive protein levels and 30‐day mortality in patients with acute myocardial infarction. Am J Med. 2003; 115:695-701 [DOI] [PubMed] [Google Scholar]
- 22.James SK, Lindahl B, Siegbahn A, Stridsberg M, Venge P, Armstrong P, Barnathan ES, Califf R, Topol EJ, Simoons ML, Wallentin L. N‐terminal pro‐brain natriuretic peptide and other risk markers for the separate prediction of mortality and subsequent myocardial infarction in patients with unstable coronary artery disease: a Global Utilization of Strategies To Open occluded arteries (GUSTO)‐IV substudy. Circulation. 2003; 108:275-281 [DOI] [PubMed] [Google Scholar]
- 23.Lindahl B, Toss H, Siegbahn A, Venge P, Wallentin L. Markers of myocardial damage and inflammation in relation to long‐term mortality in unstable coronary artery disease. FRISC Study Group. Fragmin during Instability in Coronary Artery Disease. N Engl J Med. 2000; 343:1139-1147 [DOI] [PubMed] [Google Scholar]
- 24.Zairis MN, Adamopoulou EN, Manousakis SJ, Lyras AG, Bibis GP, Ampartzidou OS, Apostolatos CS, Anastassiadis FA, Hatzisavvas JJ, Argyrakis SK, Foussas SG. The impact of hs C‐reactive protein and other inflammatory biomarkers on long‐term cardiovascular mortality in patients with acute coronary syndromes. Atherosclerosis. 2007; 194:397-402 [DOI] [PubMed] [Google Scholar]
- 25.Mulvihill NT, Foley JB, Murphy RT, Curtin R, Crean PA, Walsh M. Risk stratification in unstable angina and non‐Q wave myocardial infarction using soluble cell adhesion molecules. Heart. 2001; 85:623-627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haverkate F, Thompson SG, Pyke SD, Gallimore JR, Pepys MB. Production of C‐reactive protein and risk of coronary events in stable and unstable angina. European Concerted Action on Thrombosis and Disabilities Angina Pectoris Study Group. Lancet. 1997; 349:462-466 [DOI] [PubMed] [Google Scholar]
- 27.Kennon S, Price CP, Mills PG, Ranjadayalan K, Cooper J, Clarke H, Timmis AD. The effect of aspirin on C‐reactive protein as a marker of risk in unstable angina. J Am Coll Cardiol. 2001; 37:1266-1270 [DOI] [PubMed] [Google Scholar]
- 28.Zairis MN, Lyras AG, Bibis GP, Patsourakos NG, Makrygiannis SS, Kardoulas AD, Glyptis MP, Prekates AA, Cokkinos DV, Foussas SG. Association of inflammatory biomarkers and cardiac troponin I with multifocal activation of coronary artery tree in the setting of non‐ST‐elevation acute myocardial infarction. Atherosclerosis. 2005; 182:161-167 [DOI] [PubMed] [Google Scholar]
- 29.Anzai T, Yoshikawa T, Shiraki H, Asakura Y, Akaishi M, Mitamura H, Ogawa S. C‐reactive protein as a predictor of infarct expansion and cardiac rupture after a first Q‐wave acute myocardial infarction. Circulation. 1997; 96:778-784 [DOI] [PubMed] [Google Scholar]
- 30.Li AC, Glass CK. The macrophage foam cell as a target for therapeutic intervention. Nat Med. 2002; 8:1235-1242 [DOI] [PubMed] [Google Scholar]
- 31.Szmitko PE, Wang CH, Weisel RD, de Almeida JR, Anderson TJ, Verma S. New markers of inflammation and endothelial cell activation: Part I. Circulation. 2003; 108:1917-1923 [DOI] [PubMed] [Google Scholar]
- 32.Blann AD, Nadar SK, Lip GY. The adhesion molecule P‐selectin and cardiovascular disease. Eur Heart J. 2003; 24:2166-2179 [DOI] [PubMed] [Google Scholar]