

Summary
In this perspective, we revise the historic notion that cancer is a disease of mitochondria. We summarize recent findings on the function and rewiring of central carbon metabolism in melanoma. Metabolic profiling studies using stable isotope tracers show that glycolysis is decoupled from the tricarboxylic acid (TCA) cycle. This decoupling is not ‘dysfunction’ but rather an alternate wiring required by tumor cells to remain metabolically versatile. In large part, this requirement is met by glutamine feeding the TCA cycle as an alternative source of carbon. Glutamine is also used in non-conventional ways, like traveling in reverse through the TCA flux to feed fatty acid biosynthesis. The biosynthetic networks linked with non-essential amino acids alanine, serine, arginine, and proline are also significantly impacted by the use of glutamine as an alternate carbon source.
Keywords: metabolism, mitochondria, glutamine, systems biology, NMR
Introduction
The metabolism of cancer has long been of interest because it differs from the metabolism of normal cells. The hope is that these metabolic differences can be exploited to selectively kill cancer cells. The concepts of cancer metabolism have undergone a substantial revision since their initial framing 90 yr ago (Warburg, 1923). Aerobic glycolysis, an enhanced consumption of glucose-yielding lactate as the main end product, is a metabolic signature of many tumors including melanoma (Table 1). This metabolic feature, commonly known as the Warburg effect, is considered a direct consequence of perturbations in signaling cascades triggered by mutations or changes of expression in tumor suppressors and oncogenes. It is currently believed that enhanced glycolysis is both a hallmark of malignant transformation and a potential therapeutic target (Hanahan and Weinberg, 2011). Some have interpreted Otto Warburg’s observation to mean that inefficient fermentative conversion of glucose to ATP is caused by defects in mitochondrial oxidative metabolism (Fosslien, 2008). In other words, it is commonly perceived that tumors manifesting aerobic glycolysis do so because the mitochondrial oxidative phosphorylation is somehow impaired. However, this view is likely an overinterpretation because recent studies show that the reliance of tumor cells on glycolysis is not explained by malfunctioning mitochondria, but rather by the relentless drive of these cells to grow and proliferate (Koppenol et al., 2011).
Table 1.
Metabolic signature of melanoma
| Metabolic phenotype | Metabolic re-wiring in melanoma | General advantage for tumor survival |
|---|---|---|
| Warburg effect: enhanced glycolysis in the presence of oxygen (normoxia) | Increased glucose consumption; larger flux to lactate versus oxidative phosphorylation in the TCA cycle | Robust metabolism: supports proliferation by yielding biosynthetic building blocks and energy without excessive oxidative flux |
| Pasteur effect: enhanced glycolysis in the absence of oxygen (hypoxia) | Additional, hypoxia-induced re-routing of flux from glucose to lactate, and not to TCA cycle | Metabolic flexibility: effective adaptation to variable environmental conditions during tumor progression (metastasis) |
| Glutaminolysis: enhanced use of glutamine as carbon and energy source; forward oxidative TCA cycle flux | Increased glutamine consumption; TCA cycle dominated by oxidative flux from glutamine | Metabolic versatility: utilization of alternative nutrients such as glutamine (commonly a nitrogen source) |
| Reductive carboxylation: reverse TCA cycle flux by converting oxoglutarate to isocitrate | Increased contribution of reductive versus oxidative TCA flux to acetyl-CoA and fatty acid synthesis; stronger effect in absence of oxygen | Metabolic versatility: recruitment of alternative metabolic routes to produce building blocks (and maintain redox balance) |
| Alanine synthesis: enhanced production and excretion of alanine | Increased flux from glucose to alanine | Metabolic flexibility: disposal of excessive ammonia generated by glutaminolysis |
| Serine synthesis: enhanced production of serine | Increased flux to serine from glucose versus salvage | Robust metabolism: supports enhanced need for nucleotides (C1/folate metabolism) |
| Arginine auxotrophy: requirement for exogenous arginine | Repressed synthesis of arginine replaced by salvage from the media | Unknown: limiting arginine degradation and NO synthesis may be a protection mechanism |
| Proline synthesis: enhanced production of proline | Increased flux from glutamine to proline versus usage of external proline or synthesis from arginine | Metabolic flexibility: balancing flux to account for arginine deficiency and enhanced glutamine utilization |
TCA, tricarboxylic acid.
This revision of Warburg’s hypothesis suggests that the massive consumption of glucose by cancer cells could allow them to meet the needs of other biosynthetic pathways by diverting carbon flux from glycolysis. These peripheral metabolic pathways, like the pentose phosphate pathway, serine-glycine synthesis, and the folate cycle, appear to play a critical role in proliferation by ensuring the balanced production of cellular building blocks (Mazurek, 2011). This diversion is implemented via decoupling of glycolysis from the mitochondrial tricarboxylic acid (TCA) cycle. However, in contrast to earlier perceptions about ‘mitochondrial dysfunction’ as a key driver of such glycolytic decoupling, a growing body of evidence shows that in fact oxidative phosphorylation is preserved in many cancer cells including melanoma (Cai et al., 2012; Scott et al., 2011). Moreover, the alternative carbon source glutamine was implicated in melanoma and several other types of cancer as a major input to the TCA cycle providing cells with an additional important carbon and energy source (DeBerardinis et al., 2007). The interest in Myc as a ubiquitous oncogene was heightened by its role in mitochondrial glutaminase regulation and glutamine catabolism (Gao et al., 2009; Wise et al., 2008). The remarkable glutamine-fueled metabolic activity of mitochondria appears to have a strong favorable impact on the biosynthetic budget, energy, and redox homeostasis in tumor cells. This emerging paradigm of cancer metabolism is illustrated below by examples from recent studies on melanoma.
Stable isotope tracing reveals decoupling of glycolysis from TCA cycle in melanoma
High glucose uptake and high lactate production probably caused by perturbations in signal transformation have been identified as metabolic hallmarks of melanoma (Belhocine et al., 2006; Govindarajan et al., 2007). Until recently though, few studies have actually quantified other aspects of metabolism in melanoma. In the last few years, quantification of cellular metabolic flux was made possible by the use of stable isotopes as tracers in combination with bioanalytical platforms like nuclear magnetic resonance spectroscopy, gas- or liquid-chromatography-mass spectrometry (Filipp et al., 2012; Lemons et al., 2010; Yang et al., 2008). These approaches can be combined with models of metabolic networks to deduce metabolic flux and achieve a comprehensive picture of the dynamic aspects of metabolism. In other cancers, it has been particularly insightful to compare metabolic profiles of cancer cell lines with non-transformed or less malignant lines (Frezza et al., 2011; Richardson et al., 2008; Yang et al., 2007). We recently used a similar approach to study glucose and glutamine metabolism in a panel of melanoma cell lines and melanocytes (Scott et al., 2011). This analysis revealed previously unknown aspects of melanoma metabolism. For example, melanocytes and melanoma cells exhibited increased fermentation of glucose to lactate under oxygen limitation (the Pasteur effect). However, this effect was stronger in melanoma cell lines indicating that they have a better ability to adapt to hypoxic conditions, and only melanoma cells consumed more glucose and produced more lactate than melanocytes irrespective of the availability of oxygen (the Warburg effect) (Figure 1). Another observation beyond the Warburg effect was that the metabolism of melanoma lines was not strictly glycolytic, as their TCA cycle functions even under hypoxia (Scott et al., 2011). The alternative source of carbon for the TCA cycle was found to be glutamine. So, the ability to switch from glucose to glutamine as an alternative source of carbon allows decoupling of mitochondrial TCA activity from cytosolic glycolysis. This decoupling appears to provide cancer cells with a robust, flexible, and versatile metabolic machinery (Table 1).
Figure 1.

Glutamine compensates for metabolic decoupling of mitochondria in melanoma. Metabolism in melanoma (right-hand panels) is characterized by high glucose uptake, but also by high glucose-to-lactate conversion despite the presence of oxygen (upper panels), typical features of the Warburg effect. In addition, glutamine import and decoupling of mitochondrial metabolism even in the presence of oxygen provides a robust metabolic phenotype with the flexibility to respond to physiological challenges like hypoxia in tumor tissues (lower panels). Normal cells (left-hand panels) are able to exhibit the Pasteur effect by increasing lactate production under hypoxia. In the presence of oxygen melanocytes break glycolytic carbon down in oxidative phosphorylation. However, they fail to maintain mitochondrial TCA cycle activity in the absence of oxygen.
Mitochondrial metabolism in melanoma
That mitochondrial metabolism may be impaired in tumorigenesis is not in doubt as multiple TCA cycle enzymes have been found to be mutated in various tumor tissues. In these examples, somatic mutations of TCA enzymes, such as fumarate hydratase and succinate dehydrogenase, can lead to metabolic bypasses using the TCA cycle as a metabolic hub rather than as an uninterrupted cycle (Frezza et al., 2011; Janeway et al., 2011). However, jumping to the conclusion that the mitochondria of melanoma are dysfunctional would be premature. When assessing TCA cycle activity in melanoma, two distinct features have to be addressed: On the one hand, which TCA cycle enzymes are found to be mutated in melanoma and what is the metabolic consequence of such mutations? On the other hand, considering its decoupling from glycolysis, are there any differences in TCA cycle activity in melanoma?
Mutations in isocitrate dehydrogenases only occur in a small percentage of melanomas
The TCA cycle enzyme isocitrate dehydrogenase (IDH) has been identified to be mutated in a large percentage of progressive gliomas and in a subset of acute myeloid lymphoma. These mutations are heterozygous and occur at R132 in the cytosolic IDH1 or at R172 in the homologous mitochondrial IDH2 (Parsons et al., 2008). To test whether melanoma shared this genetic feature, 78 human melanoma samples were analyzed for IDH mutation status. A somatic, heterozygous R132C mutation in IDH1 was identified in one human melanoma metastasis to the lung (Lopez et al., 2010). A different set of 39 samples confirmed the minor fraction of IDH mutations in melanoma with four samples with mutations (two each for IDH1 and IDH2) (Shibata et al., 2011). In glioma and leukemia, IDH1 R132 and IDH2 R172 mutations had attracted interest because they gave rise to a novel enzymatic activity that formed 2-hydroxyglutarate by reduction but not carboxylation of oxoglutarate (Dang et al., 2009; Ward et al., 2010). Mutant IDHs producing 2-hydroxyglutarate interfere with DNA and histone methylation, thereby contributing to oncogenesis (Lu et al., 2012; Turcan et al., 2012). While glioma cells carry up to 95% mutant IDH, in melanoma, the few detected cases of mutant IDH argue against a strong selective pressure in progression of the disease (Lopez et al., 2010). Nonetheless, the IDH1 R132 mutation identified in melanoma appeared to confer a growth advantage (Lopez et al., 2010; Shibata et al., 2011). This suggests that a 2-hydroxyglutarate-mediated shift in the methylation pattern is a potential mechanism for melanoma, too. In the panel of melanoma lines we have studied, we have seen little to no production of 2-hydroxyglutarate indicating the wild-type allele of IDH is predominant in these samples (unpublished observations). So far, IDH remains the only TCA cycle enzyme identified as having mutations in melanoma.
Fatty acid production is supported by reverse TCA cycle flux from glutamine
The acquired ability of melanoma to effectively utilize glutamine has consequences for the carbon flux through the TCA cycle. Oxidation of glutamine’s carbon backbone in the mitochondria is a major metabolic fate of glutamine and a primary source of energy for proliferating cells. In this way, glutamine can be routed through the TCA cycle to maintain oxidative phosphorylation, a process called glutaminolysis (Table 1). Glutaminolysis can potentially provide carbon to feed into fatty acid synthesis (DeBerardinis et al., 2007), but this depends on conversion of the TCA cycle intermediate malate into pyruvate by malic enzyme, followed by production of acetyl-CoA from pyruvate by pyruvate dehydrogenase. Melanoma cells demonstrated weak labeling of lactate (a proxy for pyruvate) from glutamine (Scott et al., 2011). This suggests low malic enzyme activity in melanoma in contrast to other cancer cell types (Metallo et al., 2012). Additionally, pyruvate dehydrogenase activity is low under hypoxia, as glucose is almost quantitatively converted to lactate, and there is little flux from pyruvate into the TCA cycle (Scott et al., 2011). However, profiling studies of melanoma showed that glutamine was a precursor for fatty acid synthesis and that this occurred via a reversal in the conventional direction of the TCA cycle (Figure 2) (Filipp et al., 2012; Scott et al., 2011). This reverse TCA flux involves reductive carboxylation of oxoglutarate to isocitrate by IDH (Table 1) allowing melanoma cells to synthesize fatty acids from glutamine while glucose is mainly diverted into glycolytic products. Fatty acid synthase (FAS) is the only enzyme that can synthesize palmitate, and it is necessary for tumor cell survival. FAS is overexpressed in a variety of human cancers, including cutaneous melanoma, creating a great need for its substrate acetyl-CoA (Carvalho et al., 2008; Zecchin et al., 2011). AS forward glutaminolysis cannot provide carbon to feed into fatty acid synthesis, together with limited flux from glycolysis into the acetyl-CoA pool in melanoma, this creates a need to run the TCA cycle in reverse (Figure 2). We included in our profiling efforts a characterization of different melanoma cell lines and demonstrated reverse TCA cycle flux operates in these cells irrespective of which oncogenes (B-RAF or N-RAS) are mutated (Filipp et al., 2012). Suppression of the mitochondrial IDH2-dependent route through mitochondria caused partial suppression of fatty acid synthesis, and combined knock down of IDH2 and its cytosolic analogue IDH1 completely shut down this pathway (Filipp et al., 2012). Similar results were found in an independent study on osteosarcoma cells (Mullen et al., 2012), although there have been some differences in findings about the relative importance of IDH1 and/or IDH2 to the process in other systems (Metallo et al., 2012; Wise et al., 2011). Studies on tumor cell lines with mutant IDHs, particularly in glioma, exhibited a similar metabolic flux. It is noteworthy to mention that IDH mutations so far have never occurred homozygously. Exclusive formation of 2-hydroxyglutarate in the reductive direction would limit the capacity to produce acetyl-CoA under hypoxia or when mitochondrial function is otherwise impaired. Overall, the rerouting of carbon from glutamine to supply acetyl-CoA provides a perfect illustration of the metabolic versatility of cancer cells.
Figure 2.

Reverse tricarboxylic acid (TCA) cycle flux feeds carbon from glutamine into fatty acid synthesis. (A) Mitochondrial metabolism is rewired in melanoma. The different carbon sources for TCA input are marked in red for glucose and blue for glutamine. Incoming acetyl-CoA units from glycolysis are boxed in red. Flux from glutamine can run in two directions. In the conventional forward direction (clockwise, lighter arrow) two carbons of glutamine get decarboxylated in one complete round of the TCA cycle. This forward path from glutamine is called glutaminolysis and cannot generate acetyl-CoA or pyruvate from the carbon skeleton of glutamine in melanoma. Carbon can also travel through the TCA cycle in reverse (counterclockwise, blue arrow). This reverse TCA path includes a reversible reaction called reductive carboxylation. Glutamine enters the TCA cycle as oxoglutarate and undergoes reductive carboxylation by IDHs to form isocitrate and then citrate thereby making the carbon available for lipogenic acetyl-CoA (boxed in blue). This reverse (reductive) flux is more important than initially anticipated, especially under hypoxia where glycolytic acetyl-CoA is limited and glutamine becomes the major lipogenic carbon source for lipids. (B) The percentage of acetyl-CoA in fatty acids derived from substrate measured in WM35 melanoma cells in normoxia (21% oxygen) and hypoxia (1% oxygen) after stable isotope tracing with uniform 13C-labeled substrate. Under hypoxia the percentage of acetyl-CoA derived from glutamine in fatty acids increases by more than twofold. The direct comparison of glucose and glutamine-derived acetyl-CoA shows that the source of carbon for fatty acid synthesis switches between normoxic and hypoxic conditions from glucose to glutamine. (C) The mass distribution of citrate labeling reveals reverse directionality of TCA flux in WM35 melanoma cells. Reverse TCA cycle flux preserves the stable isotope imprint from glutamine and leaves a substantial fraction of +5-mass labeled citrate (reverse blue arrow). In forward TCA cycle flux under glutaminolysis citrate is +4-mass labeled.
Alanine biosynthesis bridges glycolytic and glutaminolytic metabolism
Among other examples of metabolic flexibility and versatility largely associated with glutamine-fueled mitochondrial activity are the changes in metabolism of non-essential amino acids alanine, serine, arginine, and proline observed in melanoma. Like any other amino acid, alanine serves as an important anabolic endpoint feeding protein synthesis. Although alanine is likely available in tumor tissues or in media supplements in cell culture experiments and therefore could be readily derived from external sources, it is actively produced in melanoma cells compared with melanocytes (Scott et al., 2011). This finding suggests that alanine is a bridging metabolite between cytosolic and mitochondrial metabolism, given that alanine aminotransferase (or glutamic pyruvic transaminase, GPT) catalyzes the transfer of an amino group from glutamate to pyruvate, with the products of this reversible transamination reaction being alanine and oxoglutarate (Figure 3). While glutamine-derived oxoglutarate supplies the TCA cycle with extra carbon, excess nitrogen is excreted in the form of alanine, suggesting alanine production as a potential therapeutic target ancillary to other targets within the domain of glutamine utilization.
Figure 3.
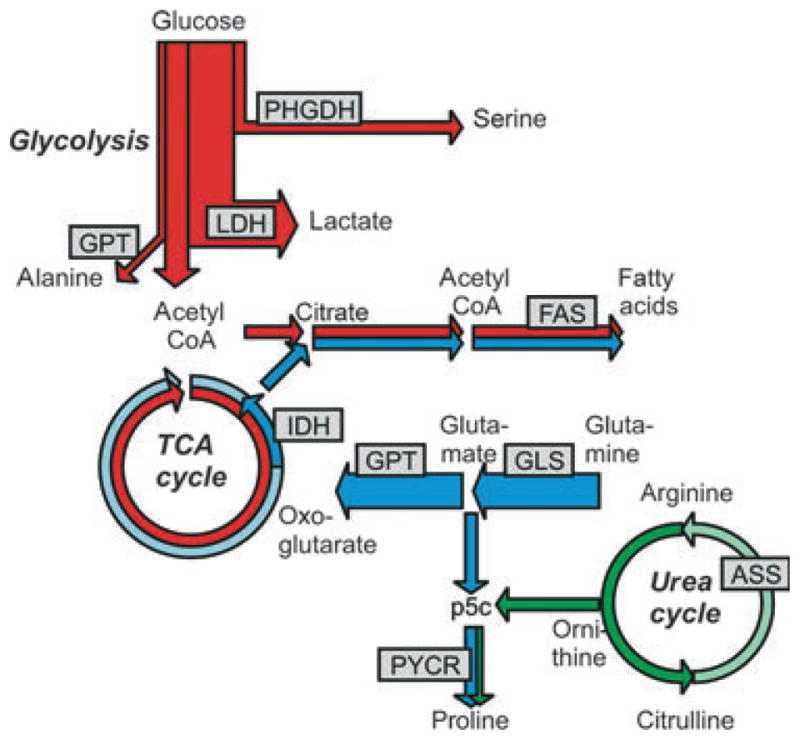
Metabolic rewiring in melanoma engages more carbon than just glucose. Metabolic pathway map of melanoma highlighting major cellular carbon sources. Glucose (red) is mainly converted to lactate. A fraction of the glycolytic flux is diverted into serine and alanine. Glutamine (blue) is the major precursor of tricarboxylic acid (TCA) cycle metabolites. Oxoglutarate formed from glutamine traverses the TCA cycle in the conventional forward direction (clockwise, light blue arrow, oxidative glutaminolysis). Carbon from glutamine also runs in reverse (counterclockwise, blue arrow, reductive carboxylation), providing a route for incorporation of carbon from glutamine into fatty acids (for experimental measurement see Figure 2). Arginine (green) is a potential precursor for proline via ornithine and pyrroline-5-carboxylate (p5c) (green arrows). However, in melanoma, arginine cannot be synthesized via the urea cycle due to a lack of expression of arginosuccinate synthetase. Enzymes mentioned in the text are mapped on the metabolic pathway map of melanoma: PHGDH, phosphoglycerate dehydrogenase; LDH, lactate dehydrogenase; FAS, fatty acid synthase; IDH, isocitrate dehydrogenase; GPT, glutamate pyruvic transaminase; GLS, glutaminase; PYCR, pyrroline-5-carboxylate reductase.
Serine biosynthesis diverts glycolytic flux and contributes to oncogenesis
Serine synthesis (Table 1) kept two surprises buried for melanoma researchers. Despite the availability of serine in the plasma, glycolytic carbon is diverted into synthesis of this metabolite in melanoma cells (Scott et al., 2011). Further, the gatekeeping enzyme phosphoglycerate dehydrogenase (PHGDH) was found to be upregulated and amplified in melanoma (Locasale et al., 2011; Possemato et al., 2011). In about 40% of melanomas, the PHGDH gene on chromosome 1p12 had amplified copy numbers (Locasale et al., 2011; Possemato et al., 2011). Intriguingly, although the PHGDH gene was essential in melanoma cell lines where it was amplified, gene amplification was not related to cell line serine synthesis, which varied widely between cell lines (Locasale et al., 2011). Undoubtedly, serine is an important metabolite for growing cells, not only as an amino acid, but also as an essential intermediary metabolite in the homeostasis of the one-carbon folate pool which is needed for the synthesis of purines and thymidylate in nucleic acids. Despite the essentiality of PHGDH, its mode of action in melanoma is less clear. Cell viability upon knockdown was not affected by the presence of serine in the culture medium (Locasale et al., 2011). In addition, data on the potential role of the aminotransferase in the serine synthesis pathway for production of the TCA cycle intermediate oxoglutarate are contradictory (Mullarky et al., 2011; Possemato et al., 2011) (Locasale et al., 2011). While alanine has been suggested to link with glutamine metabolism (Scott et al., 2011) and aspartate transaminase is also important for mitochondrial metabolism likely through its role in the malate-aspartate shuttle (Thornburg et al., 2008), targeting the serine synthesis pathway via knockdown of PHGDH in many copy number–amplified cell lines that are sensitive to PHGDH knockdown did not result in a decrease in the levels of TCA cycle intermediates (Mullarky et al., 2011). The lack of metabolic rescue indicated that the serine gatekeeper PHGDH might have a growth-mediating advantage beyond its metabolic function, which needs to be understood in greater detail.
Arginine auxotrophy creates an opportunity to starve melanoma therapeutically
In contrast to other non-essential amino acids, melanomas are auxotrophic for arginine (Table 1). Melanomas do not synthesize arginine because of a lack of expression of the urea cycle enzyme arginosuccinate synthetase (ASS) (Figure 3) (Dillon et al., 2004) and therefore require an external source of arginine (Delage et al., 2010). This deficiency has motivated clinical trials of an arginine-degrading enzyme (arginine deimidase, modified by pegylation to reduce immunogenicity and improve serum half-life) as an agent to effectively starve melanoma of arginine. These trials have been partially effective with a small number of patients with advanced melanoma (Delage et al., 2010). However, studies with melanoma cell lines have shown that arginine deimidase treatment can induce ASS expression and therefore arginine synthesis, which may be a resistance mechanism in vivo to arginine deimidase therapy (Manca et al., 2011). That arginine auxotrophy may not be an unique feature for melanoma was indicated by low metabolite release rates of ornithine and citrulline for all tested cancer lines with the exception of leukemia cells (Jain et al., 2012). Leukemia cells have active ASS, whereas low ASS activity is a common feature for more cancers than melanoma such as hepatocarcinoma, prostate cancer, and renal cancer (Delage et al., 2010). A recent paper has indicated that ASS is induced by c-Myc stabilization (Tsai et al., 2012). This opens up the possibility that combining inhibition of this signaling pathway with treatments to degrade arginine could be an effective combination therapy for melanoma.
Proline synthesis from glutamine tracks with melanoma progression
Arginine auxotrophy has important consequences for metabolic pathways connecting arginine, ornithine, glutamine, glutamate, and proline. There are two possible routes for proline synthesis (Table 1, Figure 3), from arginine through ornithine or from glutamine through glutamate. Both routes converge at the last intermediate, pyrroline-5-carboxylate. The lack of ASS limits the amount of arginine that can feed into proline synthesis, because the majority of arginine has to be used for protein production. Therefore, proline synthesis vitally depends on the glutamine route. Increased proline synthesis has been observed in cellular models of breast cancer (Richardson et al., 2008) and melanoma progression (Scott et al., 2011). In a detailed study of melanoma cell lines, we deciphered the subcellular locations and kinetic characteristics of three isoforms of human pyrroline-5-carboxylate reductase (PYCR), the final enzyme in the proline synthesis pathway (manuscript in preparation). Only one of these isoforms was previously described. Using 13C-glutamine, 13C-arginine, or 13C-ornithine as tracers, we established the relative importance of the isozymes in proline synthesis originating from either glutamine or arginine/ornithine. Mitochondrial PYCR2 functions exclusively in the glutamate route and cytosolic PYCRL functions only in the ornithine route, while mitochondrial PYCR1 is potentially involved in both pathways. PYCR1 and 2 are abundant in melanoma cells but not in melanocytes. In contrast, PYCRL is expressed to a similar degree in melanocytes and melanoma cell lines. P5CS, the enzyme that converts glutamate to P5C, is also expressed in melanoma but is not detected in melanocytes. In conclusion, therefore, the mitochondrial glutamate-dependent route is upregulated in melanoma, thereby connecting proline synthesis to glutamine but decoupling it from arginine. The key question remains of why melanoma cells synthesize proline even in the presence of an adequate supply of extracellular proline, and indeed whether it is proline synthesis per se that is the requirement or whether PYCRs, which are NAD(P)-utilizing enzymes, have a role in maintaining redox balance as part of a network of proline-metabolizing enzymes (Liu et al., 2012).
What comes next in melanoma metabolism research?
While recent studies on melanoma metabolism have demonstrated the avid consumption of alternative nutrients, further work needs to be carried out to determine how robust or indispensable the glucose or glutamine utilization pathways are in melanoma and to evaluate the suitability of metabolic enzymes as potential drug targets. Glioblastoma cells have shown a capacity to grow without glucose and switch glutamine utilization pathways when glucose is lacking (Yang et al., 2009) and also switch to alternate anaplerotic substrates when glutamine utilization is suppressed (Cheng et al., 2011). Glucose-independent growth may be less likely in melanoma, as melanoma cells showed much less glutamine conversion to lactate through glutaminolysis compared with glioblastoma cells (Scott et al., 2011; Yang et al., 2009). This suggests they may also be relatively deficient in the connected pathway of gluconeogenesis necessary for survival in the absence of glucose and may be relatively susceptible to therapeutics targeting glucose utilization. A metabolic profiling study comparing different cancer cell lines showed unique differences in adenosine and inosine excretion in melanoma cells (Jain et al., 2012), which suggests purine synthesis as a specific therapeutic target in melanoma. Another crucial future direction should be an effort to merge research on metabolism and melanoma-driving signaling mutations. Additionally, study of mutations in core metabolic enzymes has not been exhausted, and it is expected that next-generation sequencing efforts will identify significant genetic alterations in melanoma samples. Pioneering work using this technology has already identified modifications in genes for two metabolite transporters (Wei et al., 2011).
Melanoma profiling reveals ubiquitous themes of cancer metabolism
The rewiring of various pathways contributes to three distinct features of melanoma metabolism: metabolic robustness, flexibility, and versatility. The predominant metabolic phenotype of melanoma, upregulated glycolysis, yields lactate as the main product and constitutes the widely known Warburg effect. The Warburg effect contributes to metabolic robustness by diverting carbon flux from glucose toward the synthesis of building blocks to support proliferation, while still contributing to energy production. Next, melanoma cell lines exhibit a stronger adaptive glycolytic shift under hypoxia, known as the Pasteur effect, as compared to melanocytes. This is an example of metabolic flexibility, which allows cancer cells to seamlessly adapt to changing environmental conditions in the course of tumor progression. Finally, the metabolic versatility of cancer cells is best illustrated by the acquired ability of melanoma to effectively utilize glutamine as an alternative source of carbon. These three features of cancer metabolism have to be understood and considered for successful therapeutic targeting of melanoma metabolism.
Significance.
Stable isotope-enhanced research has brought new insights into the basic understanding of metabolism in melanoma, particularly on the role of glutamine-fueled mitochondrial metabolism. Metabolic rewiring affords melanoma a metabolic robustness, flexibility, and versatility. Interfering with the rewiring of central carbon metabolism in melanoma is a new therapeutic opportunity.
Acknowledgments
This work was supported by NIH grants CA154887 to F.V.F. and CA128814 to J.W.S. and A.L.O.
References
- Belhocine TZ, Scott AM, Even-Sapir E, Urbain JL, Essner R. Role of nuclear medicine in the management of cutaneous malignant melanoma. J Nucl Med. 2006;47:957–967. [PubMed] [Google Scholar]
- Cai Q, Lin T, Kamarajugadda S, Lu J. Regulation of glycolysis and the Warburg effect by estrogen-related receptors. Oncogene 2012 doi: 10.1038/onc.2012.221. ????, ????–???? [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho MA, Zecchin KG, Seguin F, et al. Fatty acid synthase inhibition with Orlistat promotes apoptosis and reduces cell growth and lymph node metastasis in a mouse melanoma model. Int J Cancer. 2008;123:2557–2565. doi: 10.1002/ijc.23835. [DOI] [PubMed] [Google Scholar]
- Cheng T, Sudderth J, Yang C, Mullen AR, Jin ES, Mates JM, DeBerardinis RJ. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc Natl Acad Sci USA. 2011;108:8674–8679. doi: 10.1073/pnas.1016627108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA. 2007;104:19345–19350. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delage B, Fennell DA, Nicholson L, McNeish I, Lemoine NR, Crook T, Szlosarek PW. Arginine deprivation and argininosuccinate synthetase expression in the treatment of cancer. Int J Cancer. 2010;126:2762–2772. doi: 10.1002/ijc.25202. [DOI] [PubMed] [Google Scholar]
- Dillon BJ, Prieto VG, Curley SA, Ensor CM, Holtsberg FW, Bomalaski JS, Clark MA. Incidence and distribution of argininosuccinate synthetase deficiency in human cancers: a method for identifying cancers sensitive to arginine deprivation. Cancer. 2004;100:826–833. doi: 10.1002/cncr.20057. [DOI] [PubMed] [Google Scholar]
- Filipp FV, Scott DA, Ronai ZA, Osterman AL, Smith JW. Reverse TCA cycle flux through isocitrate dehydrogenases 1 and 2 is required for lipogenesis in hypoxic melanoma cells. Pigment Cell Melanoma Res. 2012;25:375–383. doi: 10.1111/j.1755-148X.2012.00989.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fosslien E. Cancer morphogenesis: role of mitochondrial failure. Ann Clin Lab Sci. 2008;38:307–329. [PubMed] [Google Scholar]
- Frezza C, Zheng L, Folger O, et al. Haem oxygenase is synthetically lethal with the tumour suppressor fumarate hydratase. Nature. 2011;477:225–228. doi: 10.1038/nature10363. [DOI] [PubMed] [Google Scholar]
- Gao P, Tchernyshyov I, Chang TC, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458:762–765. doi: 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindarajan B, Sligh JE, Vincent BJ, et al. Overexpression of Akt converts radial growth melanoma to vertical growth melanoma. J Clin Invest. 2007;117:719–729. doi: 10.1172/JCI30102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Jain M, Nilsson R, Sharma S, Madhusudhan N, Kitami T, Souza AL, Kafri R, Kirschner MW, Clish CB, Mootha VK. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science. 2012;336:1040–1044. doi: 10.1126/science.1218595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janeway KA, Kim SY, Lodish M, et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci USA. 2011;108:314–318. doi: 10.1073/pnas.1009199108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325–337. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- Lemons JM, Feng XJ, Bennett BD, Legesse-Miller A, Johnson EL, Raitman I, Pollina EA, Rabitz HA, Rabinowitz JD, Coller HA. Quiescent fibroblasts exhibit high metabolic activity. PLoS Biol. 2010;8:e1000514. doi: 10.1371/journal.pbio.1000514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Le A, Hancock C, Lane AN, Dang CV, Fan TW, Phang JM. Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c-MYC. Proc Natl Acad Sci USA. 2012;109:8983–8988. doi: 10.1073/pnas.1203244109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locasale JW, Grassian AR, Melman T, et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet. 2011;43:869–874. doi: 10.1038/ng.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez GY, Reitman ZJ, Solomon D, Waldman T, Bigner DD, McLendon RE, Rosenberg SA, Samuels Y, Yan H. IDH1(R132) mutation identified in one human melanoma metastasis, but not correlated with metastases to the brain. Biochem Biophys Res Commun. 2010;398:585–587. doi: 10.1016/j.bbrc.2010.06.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Ward PS, Kapoor GS, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manca A, Sini MC, Izzo F, et al. Induction of arginosuccinate synthetase (ASS) expression affects the antiproliferative activity of arginine deiminase (ADI) in melanoma cells. Oncol Rep. 2011;25:1495–1502. doi: 10.3892/or.2011.1220. [DOI] [PubMed] [Google Scholar]
- Mazurek S. Pyruvate kinase type M2: a key regulator of the metabolic budget system in tumor cells. Int J Biochem Cell Biol. 2011;43:969–980. doi: 10.1016/j.biocel.2010.02.005. [DOI] [PubMed] [Google Scholar]
- Metallo CM, Gameiro PA, Bell EL, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2012;481:380–384. doi: 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullarky E, Mattaini KR, Vander Heiden MG, Cantley LC, Locasale JW. PHGDH amplification and altered glucose metabolism in human melanoma. Pigment Cell Melanoma Res. 2011;24:1112–1115. doi: 10.1111/j.1755-148X.2011.00919.x. [DOI] [PubMed] [Google Scholar]
- Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS, DeBerardinis RJ. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2012;481:385–388. doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Possemato R, Marks KM, Shaul YD, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011;476:346–350. doi: 10.1038/nature10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson AD, Yang C, Osterman A, Smith JW. Central carbon metabolism in the progression of mammary carcinoma. Breast Cancer Res Treat. 2008;110:297–307. doi: 10.1007/s10549-007-9732-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DA, Richardson AD, Filipp FV, Knutzen CA, Chiang GG, Ronai ZA, Osterman AL, Smith JW. Comparative metabolic flux profiling of melanoma cell lines: beyond the Warburg effect. J Biol Chem. 2011;286:42626–42634. doi: 10.1074/jbc.M111.282046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata T, Kokubu A, Miyamoto M, Sasajima Y, Yamazaki N. Mutant IDH1 confers an in vivo growth in a melanoma cell line with BRAF mutation. Am J Pathol. 2011;178:1395–1402. doi: 10.1016/j.ajpath.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornburg JM, Nelson KK, Clem BF, Lane AN, Arumugam S, Simmons A, Eaton JW, Telang S, Chesney J. Targeting aspartate aminotransferase in breast cancer. Breast Cancer Res. 2008;10:R84. doi: 10.1186/bcr2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai WB, Aiba I, Long Y, Lin HK, Feun L, Savaraj N, Kuo MT. Activation of Ras/PI3K/ERK Pathway Induces c-Myc Stabilization to Upregulate Argininosuccinate Synthetase, Leading to Arginine Deiminase Resistance in Melanoma Cells. Cancer Res. 2012;72:2622–2633. doi: 10.1158/0008-5472.CAN-11-3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turcan S, Rohle D, Goenka A, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483:479–483. doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. Versuche am überlebenden Karzinomgewebe. Biochem Z. 1923;142:317–333. [Google Scholar]
- Ward PS, Patel J, Wise DR, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225–234. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, Walia V, Lin JC, et al. Exome sequencing identifies GRIN2A as frequently mutated in melanoma. Nat Genet. 2011;43:442–446. doi: 10.1038/ng.810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise DR, DeBerardinis RJ, Mancuso A, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci USA. 2008;105:18782–18787. doi: 10.1073/pnas.0810199105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise DR, Ward PS, Shay JE, Cross JR, Gruber JJ, Sachdeva UM, Platt JM, DeMatteo RG, Simon MC, Thompson CB. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci USA. 2011;108:19611–19616. doi: 10.1073/pnas.1117773108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Richardson AD, Smith JW, Osterman A. Comparative metabolomics of breast cancer. Pac Symp Biocomput. 2007;18:1–192. [PubMed] [Google Scholar]
- Yang C, Richardson A, Osterman A, Smith J. Profiling of central metabolism in human cancer cells by two-dimensional NMR, GC-MS analysis, and isotopomer modeling. Metabolomics. 2008;4:13–29. [Google Scholar]
- Yang C, Sudderth J, Dang T, Bachoo RM, McDonald JG, DeBerardinis RJ. Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or Akt signaling. Cancer Res. 2009;69:7986–7993. doi: 10.1158/0008-5472.CAN-09-2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecchin KG, Alberici LC, Riccio MF, Eberlin MN, Vercesi AE, Graner E, Catharino RR. Visualizing inhibition of fatty acid synthase through mass spectrometric analysis of mitochondria from melanoma cells. Rapid Commun Mass Spectrom. 2011;25:449–452. doi: 10.1002/rcm.4875. [DOI] [PubMed] [Google Scholar]