

Abstract
Helicobacter pylori is highly adapted for colonization of the human stomach and is present in about half of the human population. When present, H. pylori is usually the numerically dominant gastric microorganism. H. pylori typically does not cause any adverse effects, but is associated with an increased risk of non-cardia gastric adenocarcinoma, gastric lymphoma and peptic ulcer. Disorders such as esophageal diseases and childhood-onset asthma have been recently reported to occur more frequently in individuals who lack H. pylori, compared with H. pylori-positive persons. In this review, we discuss biologic factors that allow H. pylori to colonize the human stomach, mechanisms by which H. pylori increases the risk for peptic ulcer disease and non-cardia gastric adenocarcinoma, and potential benefits that H. pylori might confer to humans.
H. pylori as a member of the normal human microbiota
From birth to death, humans are in contact with microbes, either transiently or persistently. Virtually every mucosal and cutaneous surface in the human body is colonized by persistent residential microbes 1–5 (Figure 1). In most niches of the human body, including the oral cavity, esophagus, colon and skin, many bacterial species are present and no single species predominates. The distribution of the microbes is not accidental; each niche is colonized by microbes that are either conserved among most humans or host-specific. It has been presumed that the conserved microbiota have specific adaptations that permit persistence at particular locales.
Figure 1.
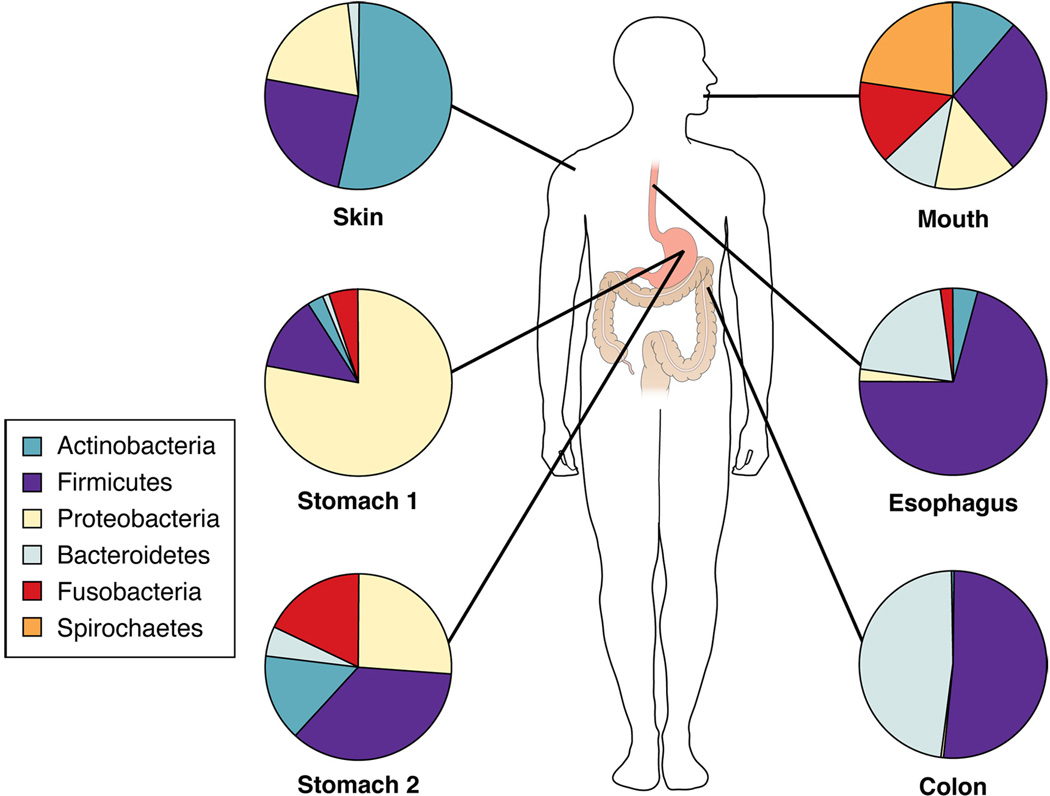
Major bacterial phyla in the stomach and at diverse anatomical sites 1, 2, 4, 5, 11. Stomach 1 depicts a stomach in which H. pylori is detected by conventional methods and Stomach 2 depicts a stomach in which H. pylori is not detected.
What is known about bacterial colonization of the human stomach? Most studies of this topic have focused on Helicobacter pylori. Several points can be summarized:
Natural colonization by H. pylori is restricted to humans and possibly several other primates (although the latter is not certain).
The stomach is the major habitat of H. pylori. There may be extension of the H. pylori habitat into the proximal duodenum or distal esophagus, usually in the presence of gastric metaplasia in those sites 6, 7. H. pylori also has been found overlying ectopic gastric epithelium in Meckel’s diverticulum, but this is an uncommon circumstance 8. H. pylori genetic sequences have been identified in oral and colonic contents, but it is not clear whether these organisms are transient or residential.
H. pylori gastric colonization is acquired early in life (almost always before the age of 10 years), and in the absence of antibiotic therapy, generally persists for life 9, 10.
When present, H. pylori usually is the numerically dominant gastric microorganism. Studies of the bacterial flora of the human stomach, based on PCR amplification of 16S rRNA sequences, show that H. pylori represents a high proportion (70%–95%) of the clones identified 11, 12. The human stomach is occasionally colonized by “Candidatus Helicobacter heilmannii” 13, which is closely related to H. pylori, but such colonization is relatively uncommon. Colonization of the human stomach by a single dominant species is similar to the bacterial colonization pattern sometimes observed in the human vagina 14. However, in the vagina, the dominant organism may be one of several Lactobacillus species, whereas in the stomach, only a single species (H. pylori) is typically present. Thus, H. pylori can be considered as the dominant microbiota of the human stomach.
H. pylori in human populations
H. pylori is present in human populations throughout the world. Phylogeographic studies indicate that humans have been colonized by H. pylori for at least 58,000 years, since before the most recent (but pre-historic) out-of-Africa migration 15. As humans traveled around the world populating new geographic regions, they carried their ancestral H. pylori with them 16. Based on the presence of gastric Helicobacter species (but not H. pylori) in other mammals (reviewed in 17), it is possible that gastric helicobacters are ancestral in mammals, and we may have carried the ancestors of present-day H. pylori before we evolved into humans. Unlike most other residential microbiota of which we are aware, H. pylori is becoming less common in human populations with socioeconomic development; this clearly has been happening over the course of the 20th century in Western countries 18, 19. Since humans are the only natural host for H. pylori, the decreasing prevalence can be attributed to diminished transmission among humans and perhaps a decreased duration of gastric colonization. Contributing factors include improved sanitation, smaller family sizes, and the frequent use of antibiotics during childhood. Thus, H. pylori is a major human residential organism that is becoming increasingly less common.
Tropism of H. pylori for the human stomach
H. pylori is highly adapted to colonize the human stomach, whereas most other bacteria cannot persistently colonize this niche. The major factors that limit bacterial colonization of the human stomach are: (i) acidity, (ii) peristalsis, (iii) nutrient availability, (iv) host innate and adaptive immunity, and (v) competing microbes. Specific features of H. pylori allow it to resist each of these stresses (Table 1). H. pylori resists acid by hydrolyzing urea to yield ammonia, and by regulating gene expression to respond to changes in pH 20–22. H. pylori expresses multiple paralogous outer membrane proteins, many of which are phase-variable; several of these appear to bind to receptors on the surface of gastric epithelial cells and could diminish the rate of bacterial wash-out due to peristalsis 23, 24. H. pylori has numerous mechanisms to obtain nutrients, including the induction of tissue inflammation and the presence of systems that facilitate transport and uptake of nutrients. Innate and adapative host immune responses are limited by several secreted H. pylori proteins (discussed in the next section) and multiple systems counteract the actions of reactive oxygen and nitrogen species 25, 26. H. pylori produces anti-bacterial peptides 27 that might reduce competition from other microbes.
Table 1.
Examples of H. pylori adaptations that facilitate gastric colonization
| Adaptation | Function | Reference |
|---|---|---|
| Spiral shape | Hydrodynamic movement | 42 |
| Polar flagella | Motility in the gastric niche | 42 |
| Flagellin structure | Modification of TLR5 recognition site | 44 |
| Urease | Resistance to gastric acidity | 20 |
| LPS structure | Lipid A with low bioactivity | 43 |
| LPS Lewis antigens | Mimicry of host cell molecules | 45 |
| Natural competence | Ability to adapt to changing gastric conditions | 69 |
| Multiple adhesins | Attachment to epithelium resists peristalsis | 23, 24 |
| VacA | Inhibition of T cell activities | 48–50, 53 |
| Products of cag PAI | Signaling within gastric epithelium | 35, 37, 83 |
Studies in rodent models have provided further insight into the H. pylori constituents required for gastric colonization. Approaches such as signature-tagged mutagenesis and microarray tracking of transposon mutants have led to the identification of more than 100 bacterial genes required for gastric colonization 28–30. The expression of several of these genes is upregulated during growth of H. pylori in the gastric environment 31.
H. pylori exclusively colonizes gastrointestinal sites overlying gastric mucosa 6–8. The development of atrophic gastritis late in life (characterized by thinning of the gastric mucosa and loss of gastric acidity) diminishes or eliminates H. pylori colonization 32. Potential reasons for the specific association of H. pylori with normal gastric mucosal epithelium include a low pH requirement for metabolic processes; dependence on specific nutrients, mucins or cell-surface components that are specific features of gastric epithelium; or the inability to compete in environments where other microbes are more abundant. The reasons that H. pylori variants have not arisen that can breech the requirement for gastric epithelium are not known, but we speculate that the biologic cost of the necessary adaptations to increase the host tissue niche exceeds the benefit to the organism, in terms of transmission to new hosts, consistent with a Nash equilibrium 33. At least in the past, when H. pylori was so successful at colonizing humans, niche expansion was not necessary, and possibly deleterious. In the future, with an increasingly narrow bottleneck for H. pylori transmission, there could be selection for variants that colonize a broader range of epithelial surfaces; such variants might be more readily transmitted to new hosts.
Biologic factors that promote the co-existence of H. pylori and humans
Most H. pylori localize within the gastric mucus layer and do not directly interact with host cells. However, some organisms adhere to gastric epithelial cells and occasionally are internalized by these cells 34. Adherence of H. pylori to gastric epithelial cells stimulates numerous signaling pathways 35, and many H. pylori strains secrete toxins or other effector molecules 36, 37. H. pylori elicits a humoral immune response 38, and tissue infiltration by mononuclear and polymorphonuclear leukocytes occurs in all humans who are persistently colonized 39. The host inflammatory response to H. pylori is relatively weak in comparison to the response to many transient bacterial pathogens, but the host response to H. pylori is more substantial and complex than that which occurs in response to other intestinal luminal bacteria. Despite causing numerous alterations in the gastric environment and eliciting a host immune response, H. pylori persistently colonizes the human stomach for long time periods and usually does not have adverse effects 40, 41.
What factors contribute to the stability of the H. pylori-host equilibrium? One salient factor is the localization of H. pylori within the gastric mucus layer, without any substantial invasion of host tissue 42. Another factor is the synthesis of H. pylori components that are highly adapted to reduce the intensity of the host immune response. H. pylori lipopolysaccharide (LPS) is characterized by modifications of the lipid A component that make it less proinflammatory than LPSs from other gram-negative bacterial species 43. H. pylori flagella are poorly recognized by TLR5 (a component of the innate immune recognition system), due to modifications in the TLR5 recognition site 44. Many H. pylori strains express LPS O antigens that are structurally related to Lewis blood group antigens found on human cells 45. This molecular mimicry could permit H. pylori LPS to be recognized as a self antigen. Incorporation of a modified form of cholesterol into H. pylori membranes and the coating of H. pylori with host molecules such as plasminogen might represent additional types of antigenic disguise 46, 47.
H. pylori produces several factors that target host immune cells. For example, many H. pylori strains secrete a protein (VacA) that targets human CD4+ T cells, inhibiting the transcription factor NFAT and inhibiting T cell proliferation 48–51. VacA targets not only CD4+ T cells, but also inhibits antigen presentation by B cells 52 and disrupts the normal functions of CD8+ T cells, macrophages and mast cells 53–56. Two other H. pylori proteins (arginase and gamma-glutamyl transferase (GGT)) are also reported to cause alterations in T cells 57, 58, and H. pylori arginase downregulates production of inducible nitric oxide synthase by macrophages 59. In addition to the targeting of immune cells by the H. pylori proteins described above, H. pylori causes numerous additional effects on immune cells via mechanisms that have not yet been elucidated 41. By targeting host immune cells, H. pylori can potentially downregulate host responses and thereby maximize its persistence.
Heterogeneity among H. pylori
H. pylori strains isolated from unrelated individuals exhibit a high level of genetic diversity (reviewed in 60, 61). Nucleotide sequences of conserved genes are 92%–99% identical among different H. pylori strains, but several H. pylori genes are more highly diverse in sequence 62–64. In addition to variation in the sequences of individual genes among H. pylori strains, there is considerable variation in gene content. One study analyzed genomic DNA from 56 different H. pylori strains using array hybridization methods and identified 1150 genes that were present in all of the strains tested (thus representing a “core” genome) 65. In contrast, 25% of the 1531 genes analyzed were absent from at least one of the 56 strains, indicating the extensive plasticity of the H. pylori genome.
H. pylori has evolved highly effective systems for generating diversity 61, 66. Mechanisms include lack of mismatch DNA repair to maximize variation 67, use of repetitive DNA for intragenomic recombination to change phenotypes, and natural competence for DNA uptake to facilitate acquisition of new genetic sequences. Gastric colonization with more than one distinct H. pylori strain is common; this multiplicity of infection provides substrate for acquisition of new genetic sequences and recombination events, which occur commonly 68. Efficient systems for generating genetic diversity allow H. pylori to adapt to changing conditions within individual human stomachs and also permit bacterial adaptation to the gastric environments of new hosts 69, 70. The simultaneous presence in the stomach of multiple H. pylori strains that can recombine could permit the emergence of the most flexible and robust bacterial populations; conversely, a decrease in the multiplicity of strains in the stomach, associated with socioeconomic advancement, might accelerate the loss of H. pylori 61, 66.
One of the most striking differences among H. pylori strains is the presence or absence of a 40-kb region of chromosomal DNA known as the cag pathogenicity island (PAI) 71. One gene in the H. pylori cag PAI encodes an effector protein (CagA) whereas others encode proteins that assemble into a type IV secretion apparatus that translocates CagA into gastric epithelial cells 37, 72. Within gastric epithelial cells, CagA is phosphorylated by host cell kinases 73. Both phosphorylated CagA and non-phosphorylated CagA cause numerous alterations in gastric epithelial cells, including activation of the phosphatase SHP-2 and dephosphorylation of cellular proteins 74, alterations in cell morphology and cell motility 75, 76, alterations of tight junctions 77, alterations in cell scattering and proliferation 78, activation of β-catenin 79, and perturbation of epithelial cell differentiation and polarity 80, 81. In addition to the effects on gastric epithelial cells that result from actions of CagA, products of the cag PAI contribute to CagA-independent alterations in gastric epithelial cells, including stimulation of the synthesis of interleukin (IL)-8, a proinflammatory cytokine 82, 83.
Most H. pylori strains secrete a protein known as VacA via an autotransporter mechanism (reviewed in 84). The VacA protein was originally identified based on its capacity to cause vacuolation in cultured human epithelial cells 36, but multiple other activities of this protein have subsequently been identified 84. All H. pylori strains contain a vacA gene, but there is marked variation among strains in vacA nucleotide sequences. vacA alleles have been classified into separate families based on diversity at several loci (designed s, i, m); variations in sequence are associated with variations in VacA functional activity in cell culture assays 62, 85–87. Active forms of VacA cause detectable alterations in gastric epithelial cells and immune cells whereas inactive forms of VacA (predominantly type s2) lack activity in most in vitro cell culture assays62, 88, 89. Effects of active VacA on gastric epithelial cells include alterations of late endocytic compartments 90, increased plasma membrane permeability 91, increased mitochondrial membrane permeability 92, 93 and apoptosis 94 (reviewed in 84). Most VacA-induced alterations are attributable to insertion of VacA into cell membranes, oligomerization, and formation of anion-selective channels 91, 95–97. Since the inactive s2 form of VacA is well-conserved, it is likely to have a functional role that it not yet understood.
Individual H. pylori strains differ considerably in the expression and binding properties of outer membrane proteins (OMP) that function as adhesins. In particular, there are differences among strains in the expression and binding properties of BabA (an OMP that binds the fucosylated Lewis b receptor on gastric epithelial cells) and SabA (an OMP that binds to sialyl Lewis X receptors) 23, 24. These differences among strains in adhesin expression result in strain-specific variations in binding of H. pylori to gastric epithelial cells. There also are differences among strains in the expression of the outer membrane OipA (HopH) due to phase variation, which could result in strain-specific variations in H. pylori-induced signaling in gastric epithelial cells 98.
H. pylori strains can be broadly categorized into 2 groups: strains that express multiple factors that interact with host tissue (including proteins encoded by the cag PAI, active forms of VacA, and outer membrane proteins such as BabA) and strains that lack these factors 62, 99, 100. Strains with intermediate properties have been identified, although less frequently than expected than if the distribution of H. pylori virulence factors were completely random. Recent studies indicate that CagA and active forms of VacA have reciprocal or antagonistic actions; consequently, there may be selection for strains that encode both of these factors or strains that lack both factors 101–103.
H. pylori strains that express multiple ‘interaction factors’ (CagA+, s1-VacA+, BabA+ strains) are predicted to be highly interactive with the host, whereas strains that lack these factors would be relatively non-interactive (Figure 2). Concordant with these predictions, CagA+, s1-VacA+, BabA+ strains are associated with increased gastric mucosal inflammatory cell infiltration and increased gastric epithelial injury, compared to strains that do not express these factors 99, 104. In addition, the colonization density of CagA+, s1-VacA+, BabA+ strains is typically higher than that of strains that do not express these factors 105.
Figure 2.

Interactions of H. pylori with human gastric mucosa. Within the gastric mucosa, most H. pylori localize within the gastric mucus layer and do not directly adhere to gastric epithelial cells. VacA, secreted by non-adherent bacteria, can cause alterations in several cell types, including gastric epithelial cells and T cells 84. Binding of H. pylori to gastric epithelial cells is mediated by several bacterial adhesins, including BabA and SabA23, 24. Adherent H. pylori assemble a type IV secretion apparatus (comprised of proteins encoded by genes in the cag pathogenicity island), which translocates the CagA protein into gastric epithelial cells 37, 72. Within gastric epithelial cells, CagA is phosphorylated by host cell kinases; both phosphorylated and non-phosphorylated CagA can cause numerous cellular alterations. Strain-specific variations in the expression of these bacterial factors are an important determinant of interactions between H. pylori and the human host.
H. pylori strains expressing multiple interaction factors and strains that lack these factors might occupy different niches in the gastric environment or each could have selective advantages at different times during prolonged colonization. Currently, people in developing countries are predominantly colonized by cagA+ strains, whereas those in many developed countries are colonized by an almost equal proportion of cagA+ and cagA− strains 100, 106. This suggests that there is an accelerated loss of cagA+ strains from some populations 18. cagA+ strains induce the production of beta-defensin 2 and other antimicrobial effectors to a greater extent than cagAstrains 107, which might render cagA+ strains more susceptible to eradication from the host. In addition, cagA+ strains seem to be more efficiently eradicated by antibiotics than are cagAstrains 108.
H. pylori and gastroduodenal disease
Although H. pylori typically colonizes the human stomach for many decades without adverse consequences, the presence of H. pylori is associated with an increased risk for several diseases, including peptic ulcers, non-cardia gastric adenocarinoma, and gastric MALT lymphoma (reviewed in 109, 110). What factors account for the development of these diseases in subsets of people who harbor H. pylori?
The risks of peptic ulcer disease and non-cardia gastric adenocarcinoma are determined in part by characteristics of the H. pylori strain with which an individual is colonized. Most of the H. pylori polymorphisms associated with varying disease risk are found in genes that encode bacterial products that interact with host tissue. Numerous studies, particularly in Western countries, have shown that cag PAI-positive H. pylori strains are associated with an increased risk of peptic ulcer disease, premalignant gastric lesions and gastric cancer, compared to strains that lack the cag PAI 100, 111–114. Moreover, the number of tyrosine phosphorylation (EPIYA) motifs in CagA proteins correlates with gastric cancer risk 115, 116. Strains that express forms of VacA that are active in vitro (for example, s1/i1/m1) are associated with a higher risk of disease than those that express inactive forms of VacA 62, 85, 112, 117. Similarly, strains that express BabA and OipA (HopH) outer membrane proteins are also associated with a higher risk of disease than strains that lack these factors 99, 118. Based on data from human epidemiologic studies, it is difficult to determine which of these bacterial factors is most closely linked to adverse disease outcomes, since these interaction factors tend to cluster together in H. pylori strains 62, 99, 117.
Studies involving gerbil and transgenic mouse models suggest that products of the cag PAI (including CagA) have an important role in contributing to adverse disease outcome 119–122. Some studies in rodents suggest that products of the cag PAI and VacA enhance the ability of H. pylori contribute to colonize the stomach 123, 124, but other studies have not reached the same conclusions 125. Notably, there are limitations of various animal models in replicating the human host environment. For example, some H. pylori factors (such as those encoded by the cag PAI) are present on genetically metastable elements that are commonly deleted during colonization of mice 126, 127. Furthermore, human T cells are susceptible to VacA, whereas mouse T cells are not 51, 128.
Host and environmental factors also are important determinants of H. pylori-associated disease risk. For example, male gender, specific IL-1β haplotypes, and various other proinflammatory gene polymorphisms are associated with an increased risk of non-cardia adenocarcinoma 129, 130. There might be synergy between bacterial and host polymorphisms in determining disease risk 112. Environmental factors that may influence the risk of gastric cancer include the level of dietary salt intake, intake of fresh fruit and vegetables, and the presence of various parasitic infections 131–133.
The use of antibiotics to eradicate H. pylori has dramatically altered the incidence and natural history of peptic ulcer disease 134. H. pylori resistance to macrolides, fluoroquinolones, and nitroimidzaoles is gradually increasing due to the widespread use of these antibiotics in the community for multiple indications. Increasing antibiotic resistance decreases the efficacy of current triple-drug treatment regimens for H. pylori and may portend future difficulties in our ability to treat peptic ulceration with antibiotics.
Potential benefits of H. pylori
H. pylori colonization is associated with many biological costs to the host; conversely, a growing body of literature suggests that the absence of H. pylori might also be associated with an increased risk of various diseases. An absence of H. pylori could indicate that an individual was never colonized, or that the organism was present earlier in life and subsequently eradicated. The idea that H. pylori might actually confer benefits to humans has engendered considerably controversy among investigators, but we review here the current data and discuss the potential importance of health benefits that might be afforded by H. pylori. Not surprisingly, most of the potential benefits (as with the costs) come from cagA+ strains, which are the most interactive with their human hosts. In 1998, one of us used the term acagia to describe the absence of cagA+ H. pylori, a condition associated with disease risks that differ from those associated with the presence of cagA+ H. pylori 135.
Esophageal diseases
There are inverse associations between the presence of H. pylori (especially cagA+ strains) and disorders such as gastroesophageal reflux disease, Barrett’s esophagus and esophageal adenocarcinoma 136–140, suggesting a protective role of H. pylori. Depending on the study, the odds ratios for the presence of esophageal disorders in persons with cagA+ strains range as low as 0.2 (the inverse of an odds ratio of 5.0). One potential mechanism for this effect could be that H. pylori colonization diminishes gastric acidity; therefore, during reflux episodes, the acidic refluxate might be more damaging to the esophageal epithelium of H. pylori-negative than of H. pylori-positive persons. Another hypothesis is that H. pylori alters the expression of multi-functional gastric hormones that have effects on esophageal tissue 141, 142. The presence or absence of H. pylori might also affect other microbiota of the stomach 11, 12 and/or the distal esophagus 5, 143, which may have an effect on esophageal mucosal integrity. Better understanding of the mechanisms that underlie the inverse relationships between H. pylori and esophageal disorders will permit improved assessment of risk and could lead to new approaches for prevention of these diseases.
Asthma and allergic disorders
As H. pylori prevalence has declined, the incidence of asthma and related disorders, especially those that appear during childhood, has risen 144. Asthma is part of an allergy syndrome that can include rhinitis and cutaneous atopy (also called eczema) and is generally considered to arise from dysfunctional immune responses to common allergens. The absence of H. pylori is associated with an increased risk for allergies 145–147; this inverse association is specific for childhood-onset, but not later-onset, asthma, and is most pronounced for cagA+ H. pylori strains. The reduced incidence of colonization with cagA+ strains 18 (increased incidence of acagia) is consistent with increased incidences of asthma and allergic disorders 135. It is possible that the presence of cagA+ H. pylori in the stomach leads to gastric recruitment of T-cell populations, including regulatory T cells, that ultimately affect the activities of T-cells present in other mucosal and cutaneous sites 148, 149. Another hypothesis is that H. pylori-induced alterations in gastric hormone expression contribute to the pathogenesis of asthma and allergic disorders.
Infectious diseases
There has recently been interest in the hypothesis that H. pylori colonization might confer protection against various other infectious diseases. In support of this concept, a recent study demonstrated that another chronic infection (latent herpesvirus) conferred resistance to infection with two bacterial pathogens in a mouse model 150. Several studies have suggested that H. pylori protects against diarrheal diseases 151, 152, although this relationship has not been consistently observed 153. Mechanisms for protection might include production of antibacterial peptides by H. pylori or the host 27, 107, activating the immune system as an adjuvant 154, competition for niche, or hypergastrinemia leading to maintainence of gastric acidity throughout childhood. Recent studies in West Africa, where tuberculosis is endemic, have indicated that H. pylori-positive persons are less likely to reactivate latent tubercular infections 155. By providing partial protection against infectious diseases common in childhood, there would be strong selection for the presence of H. pylori. If H. pylori increased morbidity or mortality due to other infectious diseases, then there would have been a very powerful selection against its presence. As the incidence of childhood infectious diseases declines, so too would the positive selective pressure for maintenance of H. pylori in human populations. The introduction of clean water supplies, improved sanitation, and less crowding into human populations have resulted in a decreased incidence of lethal diarrheal diseases; these changes would be expected to result in reduced H. pylori transmission and reduced selection for maintenance of H. pylori.
Effects on metabolism
The mammalian stomach produces about 5%–10% of the body’s leptin and 60%–80% of ghrelin. Leptin and ghrelin are multi-functional hormones that help to regulate body weight 156. Is H. pylori involved in the physiologic regulation of these hormones? Multiple studies have shown that H. pylori-positive persons produce lower amounts of ghrelin than do H. pylori-negative persons 157, 158 and H. pylori eradication is associated with a subsequent increase in ghrelin production 159, 160. Since ghrelin has effects throughout the body, it is likely that the presence or absence of H. pylori will have substantial long-term metabolic consequences 161. The effects on leptin are less clear-cut, with apparently conflicting results 142, 162, 163 that could reflect many variables, such as a subject’s age, medications, and extent of gastric inflammation. Regardless of the specific findings, a generation of children is currently growing and developing without the contribution of H. pylori to gastric physiology; the consequent alterations in ghrelin and leptin production may affect overall energy homeostasis.
H. pylori as an indicator of changes in human microbiota
Are the apparent benefits associated with H. pylori colonization directly attributable to the presence of H. pylori, or is H. pylori simply a marker or “indicator organism” for exposure to other bacteria or foreign antigens that stimulate the immune system? Just as H. pylori is disappearing as a consequence of modern lifestyles (e.g. improved sanitation and exposure to antibiotics), other organisms (including those that are currently unknown or unappreciated by medical science) might be disappearing in parallel. H. pylori might be a marker or indicator organism for a more widespread change in human microecology 164. Some of the disease consequences associated with the lack of H. pylori (and specifically acagia) might reflect this phenomenon. At the very least, H. pylori is a marker for our changing (or disappearing) microbiota; at the most, its disappearance is central to these diseases. As our understanding of the broad effects associated with acagia continue to increase, it is likely that we will discover examples of phenotypes that are directly attributable to the absence of H. pylori, as well as phenotypes for which H. pylori is an indicator organism.
Conclusions
The rediscovery of gastric microbiota and the first sucessful culture of H. pylori in 1982 by Marshall and Warren opened a new chapter in human medicine 165. Early work, demonstrating a relationship between H. pylori and peptic ulcer disease, changed medical practice 134. The finding that H. pylori also increased the risk of gastric adenocarcinoma bolstered the view that H. pylori is a human pathogen. However, it is now becoming clear that the progressive disappearance of H. pylori in the 20th and 21st centuries, abetted by modern medical practices (including overuse of antibiotics in childhood), may have consequences. These may include an increased risk of gastroesophageal reflux disease and its sequelae, childhood asthma, and metabolic disorders. If continued studies confirm the findings reported thus far, then our medical approaches to H. pylori will need to change. These next years will be an exciting period in which the relationship between H. pylori and humans becomes more thoroughly understood.
Acknowledgments
Supported in part by the National Institutes of Health (R01 AI39657, R01 AI068009, R01GMB63270) and the Department of Veterans Affairs, and the Diane Belfer Program for Human Microbial Ecology.
Biographies


Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest disclosure: The authors are potential recipients of royalties for licensed products involving CagA or VacA. At present, there are no licensed commercial products involving CagA or VacA. This article does not discuss any commercial products.
REFERENCES
- 1.Kroes I, Lepp PW, Relman DA. Bacterial diversity within the human subgingival crevice. Proc Natl Acad Sci U S A. 1999;96:14547–14552. doi: 10.1073/pnas.96.25.14547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, Gordon JI, Relman DA, Fraser-Liggett CM, Nelson KE. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312:1355–1359. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gao Z, Tseng CH, Pei Z, Blaser MJ. Molecular analysis of human forearm superficial skin bacterial biota. Proc Natl Acad Sci U S A. 2007;104:2927–2932. doi: 10.1073/pnas.0607077104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pei Z, Bini EJ, Yang L, Zhou M, Francois F, Blaser MJ. Bacterial biota in the human distal esophagus. Proc Natl Acad Sci U S A. 2004;101:4250–4255. doi: 10.1073/pnas.0306398101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Talley NJ, Cameron AJ, Shorter RG, Zinsmeister AR, Phillips SF. Campylobacter pyloriand Barrett's esophagus. Mayo Clin Proc. 1988;63:1176–1180. doi: 10.1016/s0025-6196(12)65402-0. [DOI] [PubMed] [Google Scholar]
- 7.Wyatt JI, Rathbone BJ, Dixon MF, Heatley RV. Campylobacter pyloridis and acid induced gastric metaplasia in the pathogenesis of duodenitis. J Clin Pathol. 1987;40:841–848. doi: 10.1136/jcp.40.8.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ackerman Z, Peston D, Cohen P. Role of Helicobacter pylori infection in complications from Meckel's diverticulum. Dig Dis Sci. 2003;48:1068–1072. doi: 10.1023/a:1023704410208. [DOI] [PubMed] [Google Scholar]
- 9.Perry S, de la Luz Sanchez M, Yang S, Haggerty TD, Hurst P, Perez-Perez G, Parsonnet J. Gastroenteritis and transmission of Helicobacter pylori infection in households. Emerg Infect Dis. 2006;12:1701–1708. doi: 10.3201/eid1211.060086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brown LM. Helicobacter pylori: epidemiology and routes of transmission. Epidemiol Rev. 2000;22:283–297. doi: 10.1093/oxfordjournals.epirev.a018040. [DOI] [PubMed] [Google Scholar]
- 11.Bik EM, Eckburg PB, Gill SR, Nelson KE, Purdom EA, Francois F, Perez-Perez G, Blaser MJ, Relman DA. Molecular analysis of the bacterial microbiota in the human stomach. Proc Natl Acad Sci U S A. 2006;103:732–737. doi: 10.1073/pnas.0506655103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andersson AF, Lindberg M, Jakobsson H, Backhed F, Nyren P, Engstrand L. Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS ONE. 2008;3:e2836. doi: 10.1371/journal.pone.0002836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O'Rourke JL, Solnick JV, Neilan BA, Seidel K, Hayter R, Hansen LM, Lee A. Description of 'Candidatus Helicobacter heilmannii' based on DNA sequence analysis of 16S rRNA and urease genes. Int J Syst Evol Microbiol. 2004;54:2203–2211. doi: 10.1099/ijs.0.63117-0. [DOI] [PubMed] [Google Scholar]
- 14.Hyman RW, Fukushima M, Diamond L, Kumm J, Giudice LC, Davis RW. Microbes on the human vaginal epithelium. Proc Natl Acad Sci U S A. 2005;102:7952–7957. doi: 10.1073/pnas.0503236102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Linz B, Balloux F, Moodley Y, Manica A, Liu H, Roumagnac P, Falush D, Stamer C, Prugnolle F, van der Merwe SW, Yamaoka Y, Graham DY, Perez-Trallero E, Wadstrom T, Suerbaum S, Achtman M. An African origin for the intimate association between humans and Helicobacter pylori. Nature. 2007;445:915–918. doi: 10.1038/nature05562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Falush D, Wirth T, Linz B, Pritchard JK, Stephens M, Kidd M, Blaser MJ, Graham DY, Vacher S, Perez-Perez GI, Yamaoka Y, Megraud F, Otto K, Reichard U, Katzowitsch E, Wang X, Achtman M, Suerbaum S. Traces of human migrations in Helicobacter pyloripopulations. Science. 2003;299:1582–1585. doi: 10.1126/science.1080857. [DOI] [PubMed] [Google Scholar]
- 17.Solnick JV, Schauer DB. Emergence of diverse Helicobacter species in the pathogenesis of gastric and enterohepatic diseases. Clin Microbiol Rev. 2001;14:59–97. doi: 10.1128/CMR.14.1.59-97.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perez-Perez GI, Salomaa A, Kosunen TU, Daverman B, Rautelin H, Aromaa A, Knekt P, Blaser MJ. Evidence that cagA(+) Helicobacter pyloristrains are disappearing more rapidly than cagA(−) strains. Gut. 2002;50:295–298. doi: 10.1136/gut.50.3.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Banatvala N, Mayo K, Megraud F, Jennings R, Deeks JJ, Feldman RA. The cohort effect and Helicobacter pylori. J Infect Dis. 1993;168:219–221. doi: 10.1093/infdis/168.1.219. [DOI] [PubMed] [Google Scholar]
- 20.Marshall BJ, Barrett LJ, Prakash K, McCallum RW, Guerrant RL. Urea protects Helicobacter (Campylobacter) pylori from the bactericidal effect of acid. Gastroenterol. 1990;99:697–702. doi: 10.1016/0016-5085(90)90957-3. [DOI] [PubMed] [Google Scholar]
- 21.Pflock M, Finsterer N, Joseph B, Mollenkopf H, Meyer TF, Beier D. Characterization of the ArsRS regulon of Helicobacter pylori, involved in acid adaptation. J Bacteriol. 2006;188:3449–3462. doi: 10.1128/JB.188.10.3449-3462.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Merrell DS, Goodrich ML, Otto G, Tompkins LS, Falkow S. pH-regulated gene expression of the gastric pathogen Helicobacter pylori. Infect Immun. 2003;71:3529–3539. doi: 10.1128/IAI.71.6.3529-3539.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mahdavi J, Sonden B, Hurtig M, Olfat FO, Forsberg L, Roche N, Angstrom J, Larsson T, Teneberg S, Karlsson KA, Altraja S, Wadstrom T, Kersulyte D, Berg DE, Dubois A, Petersson C, Magnusson KE, Norberg T, Lindh F, Lundskog BB, Arnqvist A, Hammarstrom L, Boren T. Helicobacter pyloriSabA adhesin in persistent infection and chronic inflammation. Science. 2002;297:573–578. doi: 10.1126/science.1069076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ilver D, Arnqvist A, Ogren J, Frick IM, Kersulyte D, Incecik ET, Berg DE, Covacci A, Engstrand L, Boren T. Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science. 1998;279:373–377. doi: 10.1126/science.279.5349.373. [DOI] [PubMed] [Google Scholar]
- 25.Comtois SL, Gidley MD, Kelly DJ. Role of the thioredoxin system and the thiolperoxidases Tpx and Bcp in mediating resistance to oxidative and nitrosative stress in Helicobacter pylori. Microbiology. 2003;149:121–129. doi: 10.1099/mic.0.25896-0. [DOI] [PubMed] [Google Scholar]
- 26.Wang G, Alamuri P, Maier RJ. The diverse antioxidant systems of Helicobacter pylori. Mol Microbiol. 2006;61:847–860. doi: 10.1111/j.1365-2958.2006.05302.x. [DOI] [PubMed] [Google Scholar]
- 27.Putsep K, Branden CI, Boman HG, Normark S. Antibacterial peptide from Helicobacter pylori. Nature. 1999;398:671–672. doi: 10.1038/19439. [DOI] [PubMed] [Google Scholar]
- 28.Guo BP, Mekalanos JJ. Rapid genetic analysis of Helicobacter pylorigastric mucosal colonization in suckling mice. Proc Natl Acad Sci U S A. 2002;99:8354–8359. doi: 10.1073/pnas.122244899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kavermann H, Burns BP, Angermuller K, Odenbreit S, Fischer W, Melchers K, Haas R. Identification and characterization of Helicobacter pylorigenes essential for gastric colonization. J Exp Med. 2003;197:813–822. doi: 10.1084/jem.20021531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baldwin DN, Shepherd B, Kraemer P, Hall MK, Sycuro LK, Pinto-Santini DM, Salama NR. Identification of Helicobacter pylori genes that contribute to stomach colonization. Infect Immun. 2007;75:1005–1016. doi: 10.1128/IAI.01176-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Castillo AR, Woodruff AJ, Connolly LE, Sause WE, Ottemann KM. Recombinationbased In Vivo Expression Technology identifies Helicobacter pylori genes important for host colonization. Infect Immun. 2008;76:5632–5644. doi: 10.1128/IAI.00627-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karnes WE, Jr, Samloff IM, Siurala M, Kekki M, Sipponen P, Kim SW, Walsh JH. Positive serum antibody and negative tissue staining for Helicobacter pylori in subjects with atrophic body gastritis. Gastroenterology. 1991;101:167–174. doi: 10.1016/0016-5085(91)90474-y. [DOI] [PubMed] [Google Scholar]
- 33.Blaser MJ, Kirschner D. The equilibria that allow bacterial persistence in human hosts. Nature. 2007;449:843–849. doi: 10.1038/nature06198. [DOI] [PubMed] [Google Scholar]
- 34.Semino-Mora C, Doi SQ, Marty A, Simko V, Carlstedt I, Dubois A. Intracellular and interstitial expression of Helicobacter pylori virulence genes in gastric precancerous intestinal metaplasia and adenocarcinoma. J Infect Dis. 2003;187:1165–1177. doi: 10.1086/368133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guillemin K, Salama NR, Tompkins LS, Falkow S. Cag pathogenicity island-specific responses of gastric epithelial cells to Helicobacter pylori infection. Proc Natl Acad Sci U S A. 2002;99:15136–15141. doi: 10.1073/pnas.182558799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cover TL, Blaser MJ. Purification and characterization of the vacuolating toxin from Helicobacter pylori. J Biol Chem. 1992;267:10570–10575. [PubMed] [Google Scholar]
- 37.Odenbreit S, Puls J, Sedlmaier B, Gerland E, Fischer W, Haas R. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science. 2000;287:1497–1500. doi: 10.1126/science.287.5457.1497. [DOI] [PubMed] [Google Scholar]
- 38.Pérez-Pérez GI, Dworkin BM, Chodos JE, Blaser MJ. Campylobacter pylori antibodies in humans. Ann. Intern. Med. 1988;109:11–17. doi: 10.7326/0003-4819-109-1-11. [DOI] [PubMed] [Google Scholar]
- 39.Dooley CP, Cohen H, Fitzgibbons PL, Bauer M, Appleman MD, Perez-Perez GI, Blaser MJ. Prevalence of Helicobacter pylori infection and histologic gastritis in asymptomatic persons. N Engl J Med. 1989;321:1562–1566. doi: 10.1056/NEJM198912073212302. [DOI] [PubMed] [Google Scholar]
- 40.Blaser MJ, Atherton JC. Helicobacter pylori persistence: biology and disease. J Clin Invest. 2004;113:321–333. doi: 10.1172/JCI20925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Algood HM, Cover TL. Helicobacter pylori persistence: an overview of interactions between H. pylori and host immune defenses. Clin Microbiol Rev. 2006;19:597–613. doi: 10.1128/CMR.00006-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hazell SL, Lee A, Brady L, Hennessy W. Campylobacter pyloridis and gastritis: association with intercellular spaces and adaptation to an environment of mucus as important factors in colonization of the gastric epithelium. J Infect Dis. 1986;153:658–663. doi: 10.1093/infdis/153.4.658. [DOI] [PubMed] [Google Scholar]
- 43.Muotiala A, Helander IM, Pyhala L, Kosunen TU, Moran AP. Low biological activity of Helicobacter pylori lipopolysaccharide. Infection & Immunity. 1992;60:1714–1716. doi: 10.1128/iai.60.4.1714-1716.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Andersen-Nissen E, Smith KD, Strobe KL, Barrett SL, Cookson BT, Logan SM, Aderem A. Evasion of Toll-like receptor 5 by flagellated bacteria. Proc Natl Acad Sci U S A. 2005;102:9247–9252. doi: 10.1073/pnas.0502040102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aspinall GO, Monteiro MA. Lipopolysaccharides of Helicobacter pylori strains P466 and MO19: structures of the O antigen and core oligosaccharide regions. Biochemistry. 1996;35:2498–2504. doi: 10.1021/bi951853k. [DOI] [PubMed] [Google Scholar]
- 46.Jonsson K, Guo BP, Monstein HJ, Mekalanos JJ, Kronvall G. Molecular cloning and characterization of two Helicobacter pylori genes coding for plasminogen-binding proteins. Proc Natl Acad Sci U S A. 2004;101:1852–185. doi: 10.1073/pnas.0307329101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wunder C, Churin Y, Winau F, Warnecke D, Vieth M, Lindner B, Zahringer U, Mollenkopf HJ, Heinz E, Meyer TF. Cholesterol glucosylation promotes immune evasion by Helicobacter pylori. Nat Med. 2006;12:1030–1038. doi: 10.1038/nm1480. [DOI] [PubMed] [Google Scholar]
- 48.Gebert B, Fischer W, Weiss E, Hoffman R, Haas R. Helicobacter pylorivacuolating cytotoxin inhibits T lymphocyte activation. Science. 2003;301:1099–1102. doi: 10.1126/science.1086871. [DOI] [PubMed] [Google Scholar]
- 49.Sundrud MS, Torres VJ, Unutmaz D, Cover TL. Inhibition of primary human T cell proliferation by Helicobacter pylorivacuolating toxin (VacA) is independent of VacA effects on IL-2 secretion. Proc Natl Acad Sci U S A. 2004;101:7727–7732. doi: 10.1073/pnas.0401528101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boncristiano M, Paccani SR, Barone S, Ulivieri C, Patrussi L, Ilver D, Amedei A, D'Elios MM, Telford JL, Baldari CT. The Helicobacter pylori vacuolating toxin inhibits T cell activation by two independent mechanisms. J Exp Med. 2003;198:1887–1897. doi: 10.1084/jem.20030621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sewald X, Gebert-Vogal B, Prassl S, Barwig I, Weiss E, Fabbri M, Osicka R, Schiemann M, Busch DH, Semmrich M, Holzmann B, Sebo P, Haas R. CD18 is the T-lymphocyte receptor of the Helicobacter pylori vacuolating cytotoxin. Cell Host and Microbe. 2008 doi: 10.1016/j.chom.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 52.Molinari M, Salio M, Galli C, Norais N, Rappuoli R, Lanzavecchia A, Montecucco C. Selective inhibition of Ii-dependent antigen presentation by Helicobacter pyloritoxin VacA. J Exp Med. 1998;187:135–140. doi: 10.1084/jem.187.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Torres VJ, VanCompernolle SE, Sundrud MS, Unutmaz D, Cover TL. Helicobacter pylori vacuolating cytotoxin inhibits activation-induced proliferation of human T and B lymphocyte subsets. J Immunol. 2007;179:5433–5440. doi: 10.4049/jimmunol.179.8.5433. [DOI] [PubMed] [Google Scholar]
- 54.Supajatura V, Ushio H, Wada A, Yahiro K, Okumura K, Ogawa H, Hirayama T, Ra C. Cutting edge: VacA, a vacuolating cytotoxin of Helicobacter pylori , directly activates mast cells for migration and production of proinflammatory cytokines. J Immunol. 2002;168:2603–2607. doi: 10.4049/jimmunol.168.6.2603. [DOI] [PubMed] [Google Scholar]
- 55.de Bernard M, Cappon A, Pancotto L, Ruggiero P, Rivera J, Del Giudice G, Montecucco C. The Helicobacter pylori VacA cytotoxin activates RBL-2H3 cells by inducing cytosolic calcium oscillations. Cell Microbiol. 2005;7:191–198. doi: 10.1111/j.1462-5822.2004.00446.x. [DOI] [PubMed] [Google Scholar]
- 56.Zheng PY, Jones NL. Helicobacter pylori strains expressing the vacuolating cytotoxin interrupt phagosome maturation in macrophages by recruiting and retaining TACO (coronin 1) protein. Cell Microbiol. 2003;5:25–40. doi: 10.1046/j.1462-5822.2003.00250.x. [DOI] [PubMed] [Google Scholar]
- 57.Zabaleta J, McGee DJ, Zea AH, Hernandez CP, Rodriguez PC, Sierra RA, Correa P, Ochoa AC. Helicobacter pylori arginase inhibits T cell proliferation and reduces the expression of the TCR zeta-chain (CD3zeta) J Immunol. 2004;173:586–593. doi: 10.4049/jimmunol.173.1.586. [DOI] [PubMed] [Google Scholar]
- 58.Schmees C, Prinz C, Treptau T, Rad R, Hengst L, Voland P, Bauer S, Brenner L, Schmid RM, Gerhard M. Inhibition of T-cell proliferation by Helicobacter pylorigammaglutamyl transpeptidase. Gastroenterology. 2007;132:1820–1833. doi: 10.1053/j.gastro.2007.02.031. [DOI] [PubMed] [Google Scholar]
- 59.Gobert AP, McGee DJ, Akhtar M, Mendz GL, Newton JC, Cheng Y, Mobley HL, Wilson KT. Helicobacter pylori arginase inhibits nitric oxide production by eukaryotic cells: a strategy for bacterial survival. Proc Natl Acad Sci U S A. 2001;98:13844–13849. doi: 10.1073/pnas.241443798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Blaser MJ, Berg DE. Helicobacter pylori genetic diversity and risk of human disease. J Clin Invest. 2001;107:767–773. doi: 10.1172/JCI12672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Suerbaum S, Josenhans C. Helicobacter pylori evolution and phenotypic diversification in a changing host. Nat Rev Microbiol. 2007;5:441–452. doi: 10.1038/nrmicro1658. [DOI] [PubMed] [Google Scholar]
- 62.Atherton JC, Cao P, Peek RM, Jr, Tummuru MK, Blaser MJ, Cover TL. Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. Association of specific vacA types with cytotoxin production and peptic ulceration. J Biol Chem. 1995;270:17771–17777. doi: 10.1074/jbc.270.30.17771. [DOI] [PubMed] [Google Scholar]
- 63.Cao P, Cover TL. Two different families of hopQ alleles in Helicobacter pylori. J Clin Microbiol. 2002;40:4504–4511. doi: 10.1128/JCM.40.12.4504-4511.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ogura M, Perez JC, Mittl PR, Lee HK, Dailide G, Tan S, Ito Y, Secka O, Dailidiene D, Putty K, Berg DE, Kalia A. Helicobacter pylori evolution: lineage-specific adaptations in homologs of eukaryotic Sel1-like genes. PLoS Comput Biol. 2007;3:e151. doi: 10.1371/journal.pcbi.0030151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gressmann H, Linz B, Ghai R, Pleissner KP, Schlapbach R, Yamaoka Y, Kraft C, Suerbaum S, Meyer TF, Achtman M. Gain and loss of multiple genes during the evolution of Helicobacter pylori. PLoS Genet. 2005;1:e43. doi: 10.1371/journal.pgen.0010043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kang J, Blaser MJ. Bacterial populations as perfect gases: genomic integrity and diversification tensions in Helicobacter pylori. Nat Rev Microbiol. 2006;4:826–836. doi: 10.1038/nrmicro1528. [DOI] [PubMed] [Google Scholar]
- 67.Bjorkholm B, Sjolund M, Falk PG, Berg OG, Engstrand L, Andersson DI. Mutation frequency and biological cost of antibiotic resistance in Helicobacter pylori. Proc Natl Acad Sci U S A. 2001;98:14607–14612. doi: 10.1073/pnas.241517298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Suerbaum S, Smith JM, Bapumia K, Morelli G, Smith NH, Kunstmann E, Dyrek I, Achtman M. Free recombination within Helicobacter pylori. Proc Natl Acad Sci U S A. 1998;95:12619–12624. doi: 10.1073/pnas.95.21.12619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Baltrus DA, Guillemin K, Phillips PC. Natural transformation increases the rate of adaptation in the human pathogen Helicobacter pylori. Evolution. 2008;62:39–49. doi: 10.1111/j.1558-5646.2007.00271.x. [DOI] [PubMed] [Google Scholar]
- 70.Robinson K, Loughlin MF, Potter R, Jenks PJ. Host adaptation and immune modulation are mediated by homologous recombination in Helicobacter pylori. J Infect Dis. 2005;191:579–587. doi: 10.1086/427657. [DOI] [PubMed] [Google Scholar]
- 71.Censini S, Lange C, Xiang Z, Crabtree JE, Ghiara P, Borodovsky M, Rappuoli R, Covacci A. cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc. Natl. Acad. Sci. 1996;93:14648–14653. doi: 10.1073/pnas.93.25.14648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kwok T, Zabler D, Urman S, Rohde M, Hartig R, Wessler S, Misselwitz R, Berger J, Sewald N, Konig W, Backert S. Helicobacter exploits integrin for type IV secretion and kinase activation. Nature. 2007;449:862–866. doi: 10.1038/nature06187. [DOI] [PubMed] [Google Scholar]
- 73.Selbach M, Moese S, Hauck CR, Meyer TF, Backert S. Src is the kinase of the Helicobacter pyloriCagA protein in vitro and in vivo. J Biol Chem. 2002;277:6775–6778. doi: 10.1074/jbc.C100754200. [DOI] [PubMed] [Google Scholar]
- 74.Higashi H, Tsutsumi R, Muto S, Sugiyama T, Azuma T, Asaka M, Hatakeyama M. SHP-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science. 2002;295:683–686. doi: 10.1126/science.1067147. [DOI] [PubMed] [Google Scholar]
- 75.Segal ED, Cha J, Lo J, Falkow S, Tompkins LS. Altered states: involvement of phosphorylated CagA in the induction of host cellular growth changes by Helicobacter pylori. Proc Natl Acad Sci USA. 1999;96:14559–14564. doi: 10.1073/pnas.96.25.14559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Selbach M, Moese S, Hurwitz R, Hauck CR, Meyer TF, Backert S. The Helicobacter pylori CagA protein induces cortactin dephosphorylation and actin rearrangement by c-Src inactivation. Embo J. 2003;22:515–528. doi: 10.1093/emboj/cdg050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Amieva MR, Vogelmann R, Covacci A, Tompkins LS, Nelson WJ, Falkow S. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science. 2003;300:1430–1434. doi: 10.1126/science.1081919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mimuro H, Suzuki T, Tanaka J, Asahi M, Haas R, Sasakawa C. Grb2 is a key mediator of Helicobacter pylori CagA protein activities. Mol Cell. 2002;10:745–755. doi: 10.1016/s1097-2765(02)00681-0. [DOI] [PubMed] [Google Scholar]
- 79.Franco AT, Israel DA, Washington MK, Krishna U, Fox JG, Rogers AB, Neish AS, Collier-Hyams L, Perez-Perez GI, Hatakeyama M, Whitehead R, Gaus K, O'Brien DP, Romero-Gallo J, Peek RM., Jr Activation of {beta}-catenin by carcinogenic Helicobacter pylori. Proc Natl Acad Sci U S A. 2005;102:10646–10651. doi: 10.1073/pnas.0504927102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bagnoli F, Buti L, Tompkins L, Covacci A, Amieva MR. Helicobacter pylori CagA induces a transition from polarized to invasive phenotypes in MDCK cells. Proc Natl Acad Sci U S A. 2005;102:16339–16344. doi: 10.1073/pnas.0502598102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Saadat I, Higashi H, Obuse C, Umeda M, Murata-Kamiya N, Saito Y, Lu H, Ohnishi N, Azuma T, Suzuki A, Ohno S, Hatakeyama M. Helicobacter pylori CagA targets PAR1/MARK kinase to disrupt epithelial cell polarity. Nature. 2007;447:330–333. doi: 10.1038/nature05765. [DOI] [PubMed] [Google Scholar]
- 82.Fischer W, Puls J, Buhrdorf R, Gebert B, Odenbreit S, Haas R. Systematic mutagenesis of the Helicobacter pylori cag pathogenicity island: essential genes for CagA translocation in host cells and induction of interleukin-8. Mol Microbiol. 2001;42:1337–1348. doi: 10.1046/j.1365-2958.2001.02714.x. [DOI] [PubMed] [Google Scholar]
- 83.Viala J, Chaput C, Boneca IG, Cardona A, Girardin SE, Moran AP, Athman R, Memet S, Huerre MR, Coyle AJ, DiStefano PS, Sansonetti PJ, Labigne A, Bertin J, Philpott DJ, Ferrero RL. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cagpathogenicity island. Nat Immunol. 2004;5:1166–1174. doi: 10.1038/ni1131. [DOI] [PubMed] [Google Scholar]
- 84.Cover TL, Blanke SR. Helicobacter pylori VacA, a paradigm for toxin multifunctionality. Nat Rev Microbiol. 2005;3:320–332. doi: 10.1038/nrmicro1095. [DOI] [PubMed] [Google Scholar]
- 85.Rhead JL, Letley DP, Mohammadi M, Hussein N, Mohagheghi MA, Eshagh Hosseini M, Atherton JC. A new Helicobacter pylori vacuolating cytotoxin determinant, the intermediate region, is associated with gastric cancer. Gastroenterology. 2007;133:926–936. doi: 10.1053/j.gastro.2007.06.056. [DOI] [PubMed] [Google Scholar]
- 86.Pagliaccia C, de Bernard M, Lupetti P, Ji X, Burroni D, Cover TL, Papini E, Rappuoli R, Telford JL, Reyrat JM. The m2 form of the Helicobacter pylori cytotoxin has cell typespecific vacuolating activity. Proc Natl Acad Sci U S A. 1998;95:10212–10217. doi: 10.1073/pnas.95.17.10212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gangwer KA, Mushrush DJ, Stauff DL, Spiller B, McClain MS, Cover TL, Lacy DB. Crystal structure of the Helicobacter pylori vacuolating toxin p55 domain. Proc Natl Acad Sci U S A. 2007;104:16293–16298. doi: 10.1073/pnas.0707447104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.McClain MS, Cao P, Iwamoto H, Vinion-Dubiel AD, Szabo G, Shao Z, Cover TL. A 12-amino-acid segment, present in type s2 but not type s1 Helicobacter pylori VacA proteins, abolishes cytotoxin activity and alters membrane channel formation. J Bacteriol. 2001;183:6499–6508. doi: 10.1128/JB.183.22.6499-6508.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Letley DP, Rhead JL, Twells RJ, Dove B, Atherton JC. Determinants of non-toxicity in the gastric pathogen Helicobacter pylori. J Biol Chem. 2003;278:26734–26741. doi: 10.1074/jbc.M304071200. [DOI] [PubMed] [Google Scholar]
- 90.Papini E, de Bernard M, Milia E, Bugnoli M, Zerial M, Rappuoli R, Montecucco C. Cellular vacuoles induced by Helicobacter pylori originate from late endosomal compartments. Proc Natl Acad Sci U S A. 1994;91:9720–9724. doi: 10.1073/pnas.91.21.9720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Szabo I, Brutsche S, Tombola F, Moschioni M, Satin B, Telford JL, Rappuoli R, Montecucco C, Papini E, Zoratti M. Formation of anion-selective channels in the cell plasma membrane by the toxin VacA of Helicobacter pyloriis required for its biological activity. Embo J. 1999;18:5517–5527. doi: 10.1093/emboj/18.20.5517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Willhite DC, Cover TL, Blanke SR. Cellular vacuolation and mitochondrial cytochrome c release are independent outcomes of Helicobacter pylori vacuolating cytotoxin activity that are each dependent on membrane channel formation. J Biol Chem. 2003;278:48204–48209. doi: 10.1074/jbc.M304131200. [DOI] [PubMed] [Google Scholar]
- 93.Galmiche A, Rassow J, Doye A, Cagnol S, Chambard JC, Contamin S, de Thillot V, Just I, Ricci V, Solcia E, Van Obberghen E, Boquet P. The N-terminal 34 kDa fragment of Helicobacter pylori vacuolating cytotoxin targets mitochondria and induces cytochrome c release. Embo J. 2000;19:6361–6370. doi: 10.1093/emboj/19.23.6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cover TL, Krishna US, Israel DA, Peek RM., Jr Induction of gastric epithelial cell apoptosis by Helicobacter pylori vacuolating cytotoxin. Cancer Res. 2003;63:951–957. [PubMed] [Google Scholar]
- 95.McClain MS, Iwamoto H, Cao P, Vinion-Dubiel AD, Li Y, Szabo G, Shao Z, Cover TL. Essential role of a GXXXG motif for membrane channel formation by Helicobacter pylori vacuolating toxin. J Biol Chem. 2003;278:12101–12108. doi: 10.1074/jbc.M212595200. [DOI] [PubMed] [Google Scholar]
- 96.El-Bez C, Adrian M, Dubochet J, Cover TL. High resolution structural analysis of Helicobacter pylori VacA toxin oligomers by cryo-negative staining electron microscopy. J Struct Biol. 2005;151:215–228. doi: 10.1016/j.jsb.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 97.Czajkowsky DM, Iwamoto H, Cover TL, Shao Z. The vacuolating toxin from Helicobacter pylori forms hexameric pores in lipid bilayers at low pH. Proc Natl Acad Sci U S A. 1999;96:2001–2006. doi: 10.1073/pnas.96.5.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yamaoka Y, Kwon DH, Graham DY. A M(r) 34,000 proinflammatory outer membrane protein (oipA) from Helicobacter pylori. Proc Natl Acad Sci U S A. 2000;97:7533–7538. doi: 10.1073/pnas.130079797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gerhard M, Lehn N, Neumayer N, Boren T, Rad R, Schepp W, Miehlke S, Classen M, Prinz C. Clinical relevance of the Helicobacter pylori gene for blood-group antigenbinding adhesin. Proc Natl Acad Sci U S A. 1999;96:12778–12783. doi: 10.1073/pnas.96.22.12778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Van Doorn LJ, Figueiredo C, Megraud F, Pena S, Midolo P, Queiroz DM, Carneiro F, Vanderborght B, Pegado MD, Sanna R, De Boer W, Schneeberger PM, Correa P, Ng EK, Atherton J, Blaser MJ, Quint WG. Geographic distribution of vacA allelic types of Helicobacter pylori. Gastroenterology. 1999;116:823–830. doi: 10.1016/s0016-5085(99)70065-x. [DOI] [PubMed] [Google Scholar]
- 101.Yokoyama K, Higashi H, Ishikawa S, Fujii Y, Kondo S, Kato H, Azuma T, Wada A, Hirayama T, Aburatani H, Hatakeyama M. Functional antagonism between Helicobacter pylori CagA and vacuolating toxin VacA in control of the NFAT signaling pathway in gastric epithelial cells. Proc Natl Acad Sci U S A. 2005;102:9661–9666. doi: 10.1073/pnas.0502529102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Argent RH, Thomas RJ, Letley DP, Rittig MG, Hardie KR, Atherton JC. Functional association between the Helicobacter pylori virulence factors VacA and CagA. J Med Microbiol. 2008;57:145–150. doi: 10.1099/jmm.0.47465-0. [DOI] [PubMed] [Google Scholar]
- 103.Tegtmeyer N, Zabler D, Schmidt D, Hartig R, Brandt S, Backert S. Importance of EGF receptor, HER2/Neu and Erk1/2 kinase signaling for host cell elongation and scattering induced by the Helicobacter pylori CagA protein: antagonistic effects of the vacuolating cytotoxin VacA. Cell Microbiol. 2008 doi: 10.1111/j.1462-5822.2008.01269.x. [DOI] [PubMed] [Google Scholar]
- 104.Atherton JC, Peek RM, Jr, Tham KT, Cover TL, Blaser MJ. Clinical and pathological importance of heterogeneity in vacA, the vacuolating cytotoxin gene of Helicobacter pylori. Gastroenterology. 1997;112:92–99. doi: 10.1016/s0016-5085(97)70223-3. [DOI] [PubMed] [Google Scholar]
- 105.Atherton JC, Tham KT, Peek RM, Jr, Cover TL, Blaser MJ. Density of Helicobacter pylori infection in vivo as assessed by quantitative culture and histology. J Infect Dis. 1996;174:552–556. doi: 10.1093/infdis/174.3.552. [DOI] [PubMed] [Google Scholar]
- 106.Parsonnet J, Replogle M, Yang S, Hiatt R. Seroprevalence of CagA-positive strains among Helicobacter pylori-infected, healthy young adults. J Infect Dis. 1997;175:1240–1242. doi: 10.1086/593680. [DOI] [PubMed] [Google Scholar]
- 107.Hornsby MJ, Huff JL, Kays RJ, Canfield DR, Bevins CL, Solnick JV. Helicobacter pylori induces an antimicrobial response in rhesus macaques in a cag pathogenicity island-dependent manner. Gastroenterology. 2008;134:1049–1057. doi: 10.1053/j.gastro.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Broutet N, Marais A, Lamouliatte H, de Mascarel A, Samoyeau R, Salamon R, Megraud F. cagA Status and eradication treatment outcome of anti- Helicobacter pyloritriple therapies in patients with nonulcer dyspepsia. J Clin Microbiol. 2001;39:1319–1322. doi: 10.1128/JCM.39.4.1319-1322.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Suerbaum S, Michetti P. Helicobacter pylori infection. N Engl J Med. 2002;347:1175–1186. doi: 10.1056/NEJMra020542. [DOI] [PubMed] [Google Scholar]
- 110.Atherton JC. The pathogenesis of Helicobacter pylori-induced gastro-duodenal diseases. Annu Rev Pathol. 2006;1:63–96. doi: 10.1146/annurev.pathol.1.110304.100125. [DOI] [PubMed] [Google Scholar]
- 111.Blaser MJ, Perez-Perez GI, Kleanthous H, Cover TL, Peek RM, Chyou PH, Stemmermann GN, Nomura A. Infection with Helicobacter pyloristrains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res. 1995;55:2111–2115. [PubMed] [Google Scholar]
- 112.Figueiredo C, Machado JC, Pharoah P, Seruca R, Sousa S, Carvalho R, Capelinha AF, Quint W, Caldas C, van Doorn LJ, Carneiro F, Sobrinho-Simoes M. Helicobacter pylori and interleukin 1 genotyping: an opportunity to identify high-risk individuals for gastric carcinoma. J Natl Cancer Inst. 2002;94:1680–1687. doi: 10.1093/jnci/94.22.1680. [DOI] [PubMed] [Google Scholar]
- 113.Plummer M, van Doorn LJ, Franceschi S, Kleter B, Canzian F, Vivas J, Lopez G, Colin D, Munoz N, Kato I. Helicobacter pylori cytotoxin-associated genotype and gastric precancerous lesions. J Natl Cancer Inst. 2007;99:1328–1334. doi: 10.1093/jnci/djm120. [DOI] [PubMed] [Google Scholar]
- 114.Kuipers EJ, Perez-Perez GI, Meuwissen SG, Blaser MJ. Helicobacter pylori and atrophic gastritis: importance of the cagA status. J Natl Cancer Inst. 1995;87:1777–1780. doi: 10.1093/jnci/87.23.1777. [DOI] [PubMed] [Google Scholar]
- 115.Higashi H, Tsutsumi R, Fujita A, Yamazaki S, Asaka M, Azuma T, Hatakeyama M. Biological activity of the Helicobacter pylori virulence factor CagA is determined by variation in the tyrosine phosphorylation sites. Proc Natl Acad Sci U S A. 2002;99:14428–14433. doi: 10.1073/pnas.222375399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Basso D, Zambon CF, Letley DP, Stranges A, Marchet A, Rhead JL, Schiavon S, Guariso G, Ceroti M, Nitti D, Rugge M, Plebani M, Atherton JC. Clinical relevance of Helicobacter pylori cagA and vacA gene polymorphisms. Gastroenterology. 2008;135:91–99. doi: 10.1053/j.gastro.2008.03.041. [DOI] [PubMed] [Google Scholar]
- 117.van Doorn LJ, Figueiredo C, Sanna R, Plaisier A, Schneeberger P, de Boer W, Quint W. Clinical relevance of the cagA, vacA, and iceA status of Helicobacter pylori. Gastroenterology. 1998;115:58–66. doi: 10.1016/s0016-5085(98)70365-8. [DOI] [PubMed] [Google Scholar]
- 118.Yamaoka Y, Kikuchi S, el-Zimaity HM, Gutierrez O, Osato MS, Graham DY. Importance of Helicobacter pylori oipA in clinical presentation, gastric inflammation, and mucosal interleukin 8 production. Gastroenterology. 2002;123:414–424. doi: 10.1053/gast.2002.34781. [DOI] [PubMed] [Google Scholar]
- 119.Ogura K, Maeda S, Nakao M, Watanabe T, Tada M, Kyutoku T, Yoshida H, Shiratori Y, Omata M. Virulence factors of Helicobacter pylori responsible for gastric diseases in mongolian gerbil. J Exp Med. 2000;192:1601–1610. doi: 10.1084/jem.192.11.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Israel DA, Salama N, Arnold CN, Moss SF, Ando T, Wirth HP, Tham KT, Camorlinga M, Blaser MJ, Falkow S, Peek RM., Jr Helicobacter pylori strain-specific differences in genetic content, identified by microarray, influence host inflammatory responses. J Clin Invest. 2001;107:611–620. doi: 10.1172/JCI11450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ohnishi N, Yuasa H, Tanaka S, Sawa H, Miura M, Matsui A, Higashi H, Musashi M, Iwabuchi K, Suzuki M, Yamada G, Azuma T, Hatakeyama M. Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc Natl Acad Sci U S A. 2008 doi: 10.1073/pnas.0711183105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mimuro H, Suzuki T, Nagai S, Rieder G, Suzuki M, Nagai T, Fujita Y, Nagamatsu K, Ishijima N, Koyasu S, Haas R, Sasakawa C. Helicobacter pylori dampens gut epithelial self-renewal by inhibiting apoptosis, a bacterial strategy to enhance colonization of the stomach. Cell Host Microbe. 2007;2:250–263. doi: 10.1016/j.chom.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 123.Marchetti M, Rappuoli R. Isogenic mutants of the cag pathogenicity island of Helicobacter pylori in the mouse model of infection: effects on colonization efficiency. Microbiology. 2002;148:1447–1456. doi: 10.1099/00221287-148-5-1447. [DOI] [PubMed] [Google Scholar]
- 124.Salama NR, Otto G, Tompkins L, Falkow S. Vacuolating cytotoxin of Helicobacter pylori plays a role during colonization in a mouse model of infection. Infect Immun. 2001;69:730–736. doi: 10.1128/IAI.69.2.730-736.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wirth HP, Beins MH, Yang M, Tham KT, Blaser MJ. Experimental infection of Mongolian gerbils with wild-type and mutant Helicobacter pylori strains. Infect Immun. 1998;66:4856–4866. doi: 10.1128/iai.66.10.4856-4866.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sozzi M, Crosatti M, Kim SK, Romero J, Blaser MJ. Heterogeneity of Helicobacter pylori cag genotypes in experimentally infected mice. FEMS Microbiol Lett. 2001;203:109–114. doi: 10.1111/j.1574-6968.2001.tb10828.x. [DOI] [PubMed] [Google Scholar]
- 127.Philpott DJ, Belaid D, Troubadour P, Thiberge JM, Tankovic J, Labigne A, Ferrero RL. Reduced activation of inflammatory responses in host cells by mouse-adapted Helicobacter pylori isolates. Cell Microbiol. 2002;4:285–296. doi: 10.1046/j.1462-5822.2002.00189.x. [DOI] [PubMed] [Google Scholar]
- 128.Algood HM, Torres VJ, Unutmaz D, Cover TL. Resistance of primary murine CD4+ T cells to Helicobacter pylori vacuolating cytotoxin. Infect Immun. 2007;75:334–341. doi: 10.1128/IAI.01063-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.El-Omar EM, Carrington M, Chow WH, McColl KE, Bream JH, Young HA, Herrera J, Lissowska J, Yuan CC, Rothman N, Lanyon G, Martin M, Fraumeni JF, Jr, Rabkin CS. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature. 2000;404:398–402. doi: 10.1038/35006081. [DOI] [PubMed] [Google Scholar]
- 130.Machado JC, Figueiredo C, Canedo P, Pharoah P, Carvalho R, Nabais S, Castro Alves C, Campos ML, Van Doorn LJ, Caldas C, Seruca R, Carneiro F, Sobrinho-Simoes M. A proinflammatory genetic profile increases the risk for chronic atrophic gastritis and gastric carcinoma. Gastroenterology. 2003;125:364–371. doi: 10.1016/s0016-5085(03)00899-0. [DOI] [PubMed] [Google Scholar]
- 131.Joossens JV, Hill MJ, Elliott P, Stamler R, Lesaffre E, Dyer A, Nichols R, Kesteloot H. Dietary salt, nitrate and stomach cancer mortality in 24 countries. European Cancer Prevention (ECP) and the INTERSALT Cooperative Research Group. Int J Epidemiol. 1996;25:494–504. doi: 10.1093/ije/25.3.494. [DOI] [PubMed] [Google Scholar]
- 132.Chen VW, Abu-Elyazeed RR, Zavala DE, Ktsanes VK, Haenszel W, Cuello C, Montes G, Correa P. Risk factors of gastric precancerous lesions in a high-risk Colombian population.I. Salt. Nutr Cancer. 1990;13:59–65. doi: 10.1080/01635589009514045. [DOI] [PubMed] [Google Scholar]
- 133.Fox JG, Beck P, Dangler CA, Whary MT, Wang TC, Shi HN, Nagler-Anderson C. Concurrent enteric helminth infection modulates inflammation and gastric immune responses and reduces helicobacter-induced gastric atrophy. Nat Med. 2000;6:536–542. doi: 10.1038/75015. [DOI] [PubMed] [Google Scholar]
- 134.Marshall BJ, Goodwin CS, Warren JR, Murray R, Blincow ED, Blackbourn SJ, Phillips M, Waters TE, Sanderson CR. Prospective double-blind trial of duodenal ulcer relapse after eradication of Campylobacter pylori. Lancet. 1988;2:1437–1442. doi: 10.1016/s0140-6736(88)90929-4. [DOI] [PubMed] [Google Scholar]
- 135.Blaser MJ. Hypothesis: the changing relationships of Helicobacter pylori and humans: implications for health and disease. J Infect Dis. 1999;179:1523–1530. doi: 10.1086/314785. [DOI] [PubMed] [Google Scholar]
- 136.Vicari JJ, Peek RM, Falk GW, Goldblum JR, Easley KA, Schnell J, Perez-Perez GI, Halter SA, Rice TW, Blaser MJ, Richter JE. The seroprevalence of cagA-positive Helicobacter pylori strains in the spectrum of gastroesophageal reflux disease. Gastroenterology. 1998;115:50–57. doi: 10.1016/s0016-5085(98)70364-6. [DOI] [PubMed] [Google Scholar]
- 137.Vaezi MF, Falk GW, Peek RM, Vicari JJ, Goldblum JR, Perez-Perez GI, Rice TW, Blaser MJ, Richter JE. CagA-positive strains of Helicobacter pylori may protect against Barrett's esophagus. Am J Gastroenterol. 2000;95:2206–2211. doi: 10.1111/j.1572-0241.2000.02305.x. [DOI] [PubMed] [Google Scholar]
- 138.Peek RM, Jr, Vaezi MF, Falk GW, Goldblum JR, Perez-Perez GI, Richter JE, Blaser MJ. Role of Helicobacter pylori cagA (+) strains and specific host immune responses on the development of premalignant and malignant lesions in the gastric cardia. Int J Cancer. 1999;82:520–524. doi: 10.1002/(sici)1097-0215(19990812)82:4<520::aid-ijc9>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 139.Islami F, Kamangar F. Helicobacter pylori and esophageal cancer risk: a metal-analysis. Cancer Prev Res. 2008;1:329–338. doi: 10.1158/1940-6207.CAPR-08-0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ye W, Held M, Lagergren J, Engstrand L, Blot WJ, McLaughlin JK, Nyren O. Helicobacter pylori infection and gastric atrophy: risk of adenocarcinoma and squamous-cell carcinoma of the esophagus and adenocarcinoma of the gastric cardia. J Natl Cancer Inst. 2004;96:388–396. doi: 10.1093/jnci/djh057. [DOI] [PubMed] [Google Scholar]
- 141.Francois F, Roper J, Goodman AJ, Pei Z, Ghumman M, Mourad M, de Perez AZ, Perez-Perez GI, Tseng CH, Blaser MJ. The association of gastric leptin with oesophageal inflammation and metaplasia. Gut. 2008;57:16–24. doi: 10.1136/gut.2007.131672. [DOI] [PubMed] [Google Scholar]
- 142.Roper J, Francois F, Shue PL, Mourad MS, Pei Z, Olivares de Perez AZ, Perez-Perez GI, Tseng CH, Blaser MJ. Leptin and ghrelin in relation to Helicobacter pylori status in adult males. J Clin Endocrinol Metab. 2008;93:2350–2357. doi: 10.1210/jc.2007-2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Pei Z, Yang L, Peek RM, Levine SM, Jr, Pride DT, Blaser MJ. Bacterial biota in reflux esophagitis and Barrett's esophagus. World J Gastroenterol. 2005;11:7277–7283. doi: 10.3748/wjg.v11.i46.7277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Eder W, Ege MJ, von Mutius E. The asthma epidemic. N Engl J Med. 2006;355:2226–2235. doi: 10.1056/NEJMra054308. [DOI] [PubMed] [Google Scholar]
- 145.Chen Y, Blaser MJ. Helicobacter pylori colonization is inversely associated with childhood asthma. J Infect Dis. 2008;198:553–560. doi: 10.1086/590158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chen Y, Blaser MJ. Inverse associations of Helicobacter pylori with asthma and allergy. Arch Intern Med. 2007;167:821–827. doi: 10.1001/archinte.167.8.821. [DOI] [PubMed] [Google Scholar]
- 147.Blaser MJ, Chen Y, Reibman J. Does Helicobacter pylori protect against asthma and allergy? Gut. 2008;57:561–567. doi: 10.1136/gut.2007.133462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Harris PR, Wright SW, Serrano C, Riera F, Duarte I, Torres J, Pena A, Rollan A, Viviani P, Guiraldes E, Schmitz JM, Lorenz RG, Novak L, Smythies LE, Smith PD. Helicobacter pylori gastritis in children is associated with a regulatory T-cell response. Gastroenterology. 2008;134:491–499. doi: 10.1053/j.gastro.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 149.Robinson K, Kenefeck R, Pidgeon EL, Shakib S, Patel S, Polson RJ, Zaitoun AM, Atherton JC. Helicobacter pylori-induced peptic ulcer disease is associated with inadequate regulatory T cell responses. Gut. 2008;57:1375–1385. doi: 10.1136/gut.2007.137539. [DOI] [PubMed] [Google Scholar]
- 150.Barton ES, White DW, Cathelyn JS, Brett-McClellan KA, Engle M, Diamond MS, Miller VL, Virgin HWt. Herpesvirus latency confers symbiotic protection from bacterial infection. Nature. 2007;447:326–329. doi: 10.1038/nature05762. [DOI] [PubMed] [Google Scholar]
- 151.Rothenbacher D, Blaser MJ, Bode G, Brenner H. Inverse relationship between gastric colonization of Helicobacter pylori and diarrheal illnesses in children: results of a population-based cross-sectional study. J Infect Dis. 2000;182:1446–1449. doi: 10.1086/315887. [DOI] [PubMed] [Google Scholar]
- 152.Bode G, Rothenbacher D, Brenner H. Helicobacter pylori colonization and diarrhoeal illness: results of a population-based cross-sectional study in adults. Eur J Epidemiol. 2001;17:823–827. doi: 10.1023/a:1015618112695. [DOI] [PubMed] [Google Scholar]
- 153.Passaro DJ, Taylor DN, Meza R, Cabrera L, Gilman RH, Parsonnet J. Acute Helicobacter pylori infection is followed by an increase in diarrheal disease among Peruvian children. Pediatrics. 2001;108:E87. doi: 10.1542/peds.108.5.e87. [DOI] [PubMed] [Google Scholar]
- 154.Mattsson A, Lonroth H, Quiding-Jarbrink M, Svennerholm AM. Induction of B cell responses in the stomach of Helicobacter pylori-infected subjects after oral cholera vaccination. J Clin Invest. 1998;102:51–56. doi: 10.1172/JCI22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Perry S, Nguyen M, Yang S, Parsonnet J. Helicobacter pylori infection modifies immune response to M. tuberculosis infection. Abstract presented at the 42nd Annual Meeting of the Infectious Disease Society of America; 2004. p. 239. poster abstract #1072. [Google Scholar]
- 156.Inui A, Asakawa A, Bowers CY, Mantovani G, Laviano A, Meguid MM, Fujimiya M. Ghrelin, appetite, and gastric motility: the emerging role of the stomach as an endocrine organ. Faseb J. 2004;18:439–456. doi: 10.1096/fj.03-0641rev. [DOI] [PubMed] [Google Scholar]
- 157.Isomoto H, Nakazato M, Ueno H, Date Y, Nishi Y, Mukae H, Mizuta Y, Ohtsuru A, Yamashita S, Kohno S. Low plasma ghrelin levels in patients with Helicobacter pylori associated gastritis. Am J Med. 2004;117:429–432. doi: 10.1016/j.amjmed.2004.01.030. [DOI] [PubMed] [Google Scholar]
- 158.Osawa H, Nakazato M, Date Y, Kita H, Ohnishi H, Ueno H, Shiiya T, Satoh K, Ishino Y, Sugano K. Impaired production of gastric ghrelin in chronic gastritis associated with Helicobacter pylori. J Clin Endocrinol Metab. 2005;90:10–16. doi: 10.1210/jc.2004-1330. [DOI] [PubMed] [Google Scholar]
- 159.Nwokolo CU, Freshwater DA, O'Hare P, Randeva HS. Plasma ghrelin following cure of Helicobacter pylori. Gut. 2003;52:637–640. doi: 10.1136/gut.52.5.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Tatsuguchi A, Miyake K, Gudis K, Futagami S, Tsukui T, Wada K, Kishida T, Fukuda Y, Sugisaki Y, Sakamoto C. Effect of Helicobacter pylori infection on ghrelin expression in human gastric mucosa. Am J Gastroenterol. 2004;99:2121–2127. doi: 10.1111/j.1572-0241.2004.30291.x. [DOI] [PubMed] [Google Scholar]
- 161.Mera RM, Correa P, Fontham EE, Reina JC, Pradilla A, Alzate A, Bravo LE. Effects of a new Helicobacter pylori infection on height and weight in Colombian children. Ann Epidemiol. 2006;16:347–351. doi: 10.1016/j.annepidem.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 162.Azuma T, Suto H, Ito Y, Ohtani M, Dojo M, Kuriyama M, Kato T. Gastric leptin and Helicobacter pylori infection. Gut. 2001;49:324–329. doi: 10.1136/gut.49.3.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Cho I, Blaser MJ, Francois F, Mathew JP, Ye XY, Goldberg JD, Bini EJ. Helicobacter pylori and overweight status in the United States: data from the Third National Health and Nutrition Examination Survey. Am J Epidemiol. 2005;162:579–584. doi: 10.1093/aje/kwi237. [DOI] [PubMed] [Google Scholar]
- 164.Blaser MJ. Who are we? Indigenous microbes and the ecology of human diseases. EMBO Rep. 2006;7:956–960. doi: 10.1038/sj.embor.7400812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet. 1984;1:1311–1315. doi: 10.1016/s0140-6736(84)91816-6. [DOI] [PubMed] [Google Scholar]