

Abstract
The smoothened (SMO) receptor, a key signal transducer in the Hedgehog (Hh) signaling pathway is both responsible for the maintenance of normal embryonic development and implicated in carcinogenesis. The SMO receptor is classified as a class Frizzled (class F) G protein-coupled receptor (GPCR), although the canonical Hh signaling pathway involves the transcription factor Gli and the sequence similarity with class A GPCRs is less than 10%. Here we report the crystal structure at 2.5 Å resolution of the transmembrane domain of the human SMO receptor bound to the small molecule antagonist LY2940680. Although the SMO receptor shares the seven transmembrane helical (7TM) fold, most conserved motifs for class A GPCRs are absent, and the structure reveals an unusually complex arrangement of long extracellular loops stabilized by four disulfide bonds. The ligand binds at the extracellular end of the 7TM bundle and forms extensive contacts with the loops.
The smoothened (SMO) receptor is an essential component of the canonical Hedgehog (Hh) signaling pathway exerting a key role in the regulation of embryonic development in animals1,2. In vertebrates, the binding of Hh signaling protein to its receptor Patched3,4, a 12-transmbembrane protein which inhibits the activity of the SMO receptor5, results in translocation of the SMO receptor into cilia6. In cilia, the activated SMO receptor induces the modification of Gli transcription factors into the active form, thereby inhibiting the processing of Gli transcription factors into a repressor form. Subsequently, active Gli transcription factors translocate to the nucleus activating the transcription of Gli-targeted genes which control embryonic development and other processes2.
The SMO receptor shows a high sequence similarity with the Frizzled (FZD) receptors, which mediate the WNT signaling pathway7,8. Both SMO and FZD receptors contain an extracellular domain (ECD) composed of an extracellular cysteine rich domain (CRD) and a ECD linker domain (Supplementary Fig. 1), a seven-transmembrane helices (7TM) domain, and an intracellular C-terminal domain9. The CRD of the FZD receptors binds the endogenous lipoglycoprotein ligands, WNTs10,11; unfortunately, the function of the homologous SMO receptor CRD remains unclear. The 7TM domains of SMO and FZD receptors are reminiscent of class A G protein-coupled receptors (GPCRs), although their sequences are distinct from other GPCRs sharing less than 10% sequence identity. For this reason and because of their distinct signaling properties, the SMO and FZD receptors have been defined as the Frizzled class (class F)9,12.
The classification of the SMO receptor as a GPCR remains controversial principally due to the lack of GPCR-like features in the canonical Hh signaling pathway, although emerging evidence indicates that the SMO receptor can share some functional similarities with other classical GPCRs13. For example, activated SMO receptor can be phosphorylated by a GPCR kinase, leading to β-arrestin translocation and binding14. Moreover, the SMO receptor can couple to G proteins, particularly Gi15, controlling cell migration16. Finally, the function of the SMO receptor can be modulated by natural and synthetic small molecule agonists and antagonists, some of which are potential anti-tumor agents17.
Structure of the SMO receptor 7TM domain
An engineered construct of the human SMO receptor with a thermostabilized apocytochrome b562RIL (BRIL) fused to the N terminus of S190 and the C terminus truncated at Q555, which preserved the ligand binding property of wild type human SMO receptor, was expressed, purified, and crystallized in complex with the antagonist LY2940680 (refs 18,19) using a lipidic mesophase method20 (Supplementary Figs. 2–4). The structure was solved using a 3.5 Å single wavelength anomalous dispersion (SAD) dataset, followed by extending the resolution to 2.5 Å resolution using native data collected from five crystals (Supplementary Table 1).
The SMO receptor structure (Fig. 1) reveals a canonical GPCR 7TM-helical bundle fold with a short helix VIII packed parallel to the membrane bilayer. The ECD linker domain and long extracellular loops (ECLs) form intricate structures stabilized by four disulfide bonds: C193-C213, C217-C295, C314-C390, and C490-C507. The ligand LY2940680 binds in a pocket at the extracellular side of the receptor formed by the 7TM bundle and the ECLs. The receptor crystallizes as a parallel dimer in the crystallographic asymmetric unit (Fig. 1 and Supplementary Fig. 5), with the interface involving helices IV and V as observed for CXCR4 (ref 21). It has been reported that the SMO receptor forms a functionally important dimer, although it is unclear if the crystallographic dimer is the same as in the cell membrane22. Since the difference between the two protomers is small (Supplementary Fig. 5), we will focus on molecule A in the following discussion for brevity, except where otherwise noted.
Figure 1. Overall structure of the human SMO receptor 7TM domain in complex with LY2940680.

a, Overall structure of the SMO receptor bound to ligand LY2940680. The SMO receptor is crystallized as a dimer in an asymmetric unit with molecule A colored light blue and molecule B colored yellow. LY2940680 is shown in wheat carbon. The four disulfide bonds are shown in orange sticks. The lipids are shown in sticks with green carbons. Membrane boundaries are shown in dotted lines. b and c, Extracellular (EC) and intracellular (IC) view of the SMO receptor dimer.
7TM comparisons with class A GPCRs
Sequence similarity between the SMO receptor and class A GPCRs is very low (less than 10% sequence identity), and SMO and other class F receptors lack most of the conserved class A motifs, including D[E]R3.50Y in helix III, CWxP6.50 in helix VI and NP7.50xxY in helix VII. However, an overlay of the SMO receptor structure with previously solved class A GPCR structures shows relatively high spatial conservation of the 7TM bundle (Fig. 2a, b and Supplementary Fig. 6). Several intracellular structural features of class A GPCR 7TM bundles are also preserved, including a helical turn in the short intracellular loop 1 (ICL1), and a short intracellular helix VIII running parallel to the membrane surface, although it has distinct packing interface (residues T541, I544 and W545) with helix I (residues T251 and A254) (Fig. 2c). Structural similarity with class A GPCRs makes transplanting the Ballesteros and Weinstein (B&W) numbering23 system to class F receptors possible based upon structural superposition. In each helix, the following residues are assigned number 50: T2451.50, F2742.50, W3393.50, W3654.50, V4115.50, S4686.50, and I5307.50 (Supplementary Figs. 1, 2 and 6). The numbering of the other residues in each helix is counted relative to the X.50 position according to B&W numbering system23.
Figure 2. Comparison of the 7TM bundle of the SMO receptor with class A GPCRs.

a, Side view of superimposed structures of the SMO receptor (magenta) and class A GPCRs (light green). b, Extracellular view of the superimposed structures. c, Superposition of ICL1 and helix VIII. The residues involved in the packing interface of helix VIII and helix I in the SMO receptor are shown in sticks with magenta carbons. d–f, Superposition of Helix V–VII. The P5.50, P6.50 and P7.50 residues conserved in class A GPCRs are shown in sticks. In d, P4075.46 in the SMO receptor is shown in sticks. The α carbons of glycines in the SMO receptor are shown as yellow spheres. Structures of class A GPCRs used (PDB accessions): 1U19, 2RH1, 2YCW, 3RZE, 3PBL, 3UON, 4DAJ, 3EML, 3V2W, 3ODU, 4DJH, 4EA3, 4DKL, 4EJ4, 3VW7, 4GRV.
Despite the overall structural conservation, the 7TM fold of the SMO receptor has many distinct features. For example, when compared with class A GPCRs the extracellular tip of helix V is shifted towards the ligand binding cavity. Most importantly helices V, VI and VII of the SMO receptor lack the most conserved prolines, P5.50, P6.50 and P7.50, that play pivotal roles in the activation process of class A GPCRs. In the β2 adrenergic receptor, P5.50 has been shown to act as a local trigger of GPCR activation along with I3.40 and F6.44 (ref 24). Instead of P5.50 in helix V of class A GPCRs, the SMO receptor has P4075.46 in an adjacent helical turn (Fig. 2d). In helix VI, P6.50 induces a kink in class A GPCRs, which facilitates the large movement of the intracellular segment of helix VI during activation. The SMO receptor has no proline in helix VI, and thus this helix is straighter than in class A GPCRs (Fig. 2e). Similarly, in helix VII that typically has the conserved NPxxY motif in class A GPCRs, the proline is also absent (Fig. 2f). Although these prolines are missing in the SMO receptor, we observed a large number of glycines in helices V, VI and VII (Supplementary Fig. 2). Conceivably, these glycines could facilitate both flexibility and bending of the helices, thereby enabling 7TM packing and conformational changes during the activation of the SMO receptor. In the current structure of the SMO receptor in complex with an antagonist, helix VI is found in an inactive-like, closed state (Supplementary Fig. 7), presumably precluding G-protein binding.
Binding site of LY2940680
LY2940680 is a SMO receptor antagonist designed for the treatment of solid tumors18. The SMO receptor binding pocket has a long and narrow shape and is connected to the extracellular aqueous environment via a small opening formed by the ECD linker domain, ECL2 and ECL3 (Fig. 3a). This orifice likely facilitates small molecule ligand entry into the 7TM core region. Residues from the extracellular tips of helices I, II, V, and VII interact with LY2940680, most notably R4005.39 of helix V, which hydrogen bonds with the phthalazine ring system of the ligand. Most of the other contact residues belong to the ECD linker domain and ECLs (Fig. 3b, c and Supplementary Fig. 8). Several structured water molecules are identified in the ligand pocket, including two waters mediating the hydrogen bonding network between R4005.39, H4706.52, D4736.55, E5187.38, N5217.41 side chains (Supplementary Fig. 9). Although these waters do not directly contact LY2940680, they may play an important role in the conformational properties and dynamics of the pocket. Mutation of D4736.55, which participates in this water-mediated hydrogen bonding network, to histidine results in resistance to the approved Genentech drug GDC-0449 (ref 25). Direct contact of this residue with LY2940680 is limited (distance between the carboxylate of D4736.55 and LY2940680 is 4.04 Å in molecule A and 4.31 Å in molecule B). Cyclopamine, a naturally occurring steroid and the first identified small molecule SMO receptor ligand, inhibits the Hh signaling pathway26. Radioligand assays revealed that LY2940680 and the SMO receptor agonist SAG compete with the binding of 3H-cyclopamine (Supplementary Fig. 3), indicating these ligands bind within the long and narrow cavity embedded in the 7TM domain of the SMO receptor.
Figure 3. Ligand binding pocket for LY2940680.

a, The ligand binding pocket surface is colored according to binding properties (green: hydrophobic; blue: hydrogen-bond donor; red: hydrogen-bond acceptor). LY2940680 is shown by sticks with magenta carbons. The membrane boundary is shown as a red line. b, |2FO|-|FC| map (blue mesh) is shown for LY2940680 (contoured at 1.0σ, 0.10 e/ Å). Binding pocket residues (4.0 Å cut-off distance from LY2940680) are shown in sticks with colors indicating the location (white: TM helices; cyan: ECD linker domain; yellow: ECL1; pink: ECL2; light blue: ECL3). The hydrogen bond interactions between R4005.39, N219 and LY2940680 are shown as black dashed lines. c, Diagram of ligand interactions in the binding pocket. The color scheme of the boxes for the residues is the same as b.
ECD linker domain and ECLs structures
The SMO receptor has a unique ECD linker domain and ECLs that are long compared to most class A GPCRs. These extracellular domains are organized into complex tertiary structures through covalent and non-covalent interactions forming a lid on the 7TM bundle (Fig. 4a, b). The unusually long ECL1 (Fig. 4c) is connected to the ECD linker domain through a disulfide bond between C217 and C295, which divides ECL1 into two distinctive segments. Preceding C295, there is a short α-helical structure (G288 to V294) stabilized by cation-π interactions between the guanidinium group of R290 and the imidazole group of H2311.36 from helix I. After C295, ECL1 is packed into a U-shaped loop that is stabilized by an ionic interaction between R302 and E305. In addition, this loop segment forms contacts with the ECD linker domain through a hydrogen bonding network involving residues N202, S205, D298 and T300. ECL2 forms a β-hairpin and connects to helix III by a disulfide bond between C3143.25 and C390. This loop is positioned deep within the cavity formed by the 7TM bundle and makes extensive contacts with LY2940680. ECL3 is the longest loop of the SMO receptor and forms a protrusion from the 7TM bundle into the extracellular space (Fig. 4d). The long extension of helix VI adopts a well ordered α-helical structure that is partially stabilized by an ionic interaction between E479 and R482. This α-helical extension connects to helix VI through a 45° non-proline kink that is stabilized by several water molecules (Supplementary Fig. 10). On top of the ECL3 helical structure there is C490, which forms a disulfide bond with C507. The loop between C490 and C507 is mostly disordered, while the segment between C507 and the extracellular tip of helix VII forms an extended strand. ECL3 also makes contact with the ECD linker domain: R485 interacts with E208 within the ECD linker domain through a salt bridge; the amide side chain of Q491 and the guanidinium group of R512 form hydrogen bonds with the main chain carbonyl groups of V195 and L221, respectively. The integrity of the ECL structures is essential for maintaining the SMO receptor in an inactive state since disruption of the extracellular structures by mutations of the extracellular cysteines increases SMO receptor activity27.
Figure 4. The structure of the ECD linker domain and ECLs.

a and b, Different views of the cartoon presentation of the structure of the ECD linker domain and ECLs. ECD linker domain is colored cyan. ECL1 is colored yellow. ECL2 is colored pink. ECL3 is colored light blue. Disulfide bonds are shown in orange sticks. c, Side chain interactions that stabilize the structure of ECL1. The hydrogen bond network among residues N202, S205, D298 and T300 and the ionic interaction between R302 and E305 are shown in dashed lines. d, Side chain interactions that stabilize the structure of ELC3. The ionic interactions between E208 and R485, E479 and R482, and hydrogen bond interaction between Q491 and V195, R512 and L221 are shown in dashed lines.
In the extracellular region, the only structural feature that the SMO receptor shares with class A GPCRs is the β-hairpin structure of ECL2 that is linked to the extracellular tip of helix III via a disulfide bond (Fig 5). The corresponding cysteine in position 3.25 (B&W numbering), is conserved in the vast majority of class A and other GPCRs. The β-hairpin structure of ECL2 appears to be a hallmark of class A peptide-binding GPCRs21,28–33, which has been shown by docking studies30 and peptide-receptor co-crystal structures21,31 to play an important role in the recognition of peptide ligands. The ECL2s of peptide binding receptors all point outwards from the 7TM core domain, leaving relatively open and spacious binding cavities for their cognate peptide ligands (Fig. 5c–f). In contrast, the β-hairpin structure of ECL2 in rhodopsin folds on top of its covalently attached ligand retinal, sealing the extracellular entrance of the pocket (Fig. 5b). Interestingly, the ECL2 structure of the SMO receptor is distinct from both rhodopsin and peptide-binding GPCRs. Though ECL2 sits much deeper in the SMO receptor than in class A peptide receptors and occupies a substantial space in the cavity of 7TM bundle (Fig. 5a), unlike in rhodopsin, the β-hairpin in the SMO receptor does not occlude the ligand entrance. Instead, the ECL2 of the SMO receptor is positioned laterally to LY2940680 (Supplementary Fig. 11), forming a large part of the binding pocket for this ligand.
Figure 5. Comparison of ECL2 of the SMO receptor with class A GPCRs.
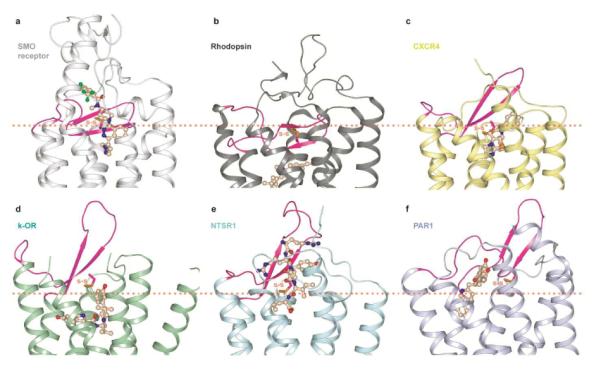
a–f, Cartoon presentation of the extracellular part of the structures for (a) the SMO receptor (white), (b) rhodopsin (PDB accession: 1U19) (dark grey), (c) CXCR4 chemokine receptor (3ODU) (yellow), (d) κ-opioid receptor (4DJH) (light green, representing the opioid receptor family), (e) neurotensin receptor NTSR1 (4GRV) (cyan), (f) protease activated receptor 1 (PAR1) (3VW7) (light blue). The ECL2 of each structure is shown in magenta. All ligands are shown in wheat. The approximate position of the extracellular membrane boundary is shown in orange dotted lines. The disulfide bonds connecting ECL2 and helix III are shown in orange sticks.
Homology with Frizzled family receptors
Within class F receptors, one can find a gapless alignment at TM helices (Supplementary Fig. 1), and 45 residues are fully conserved within the CRD linker and 7TM domains (Fig. 6a, b). The cysteines that form disulfide bonds maintaining the structures of the ECD linker domain and the ECLs in the SMO receptor are highly preserved in the FZD receptors (the only exception is FZD4, of which the disulfide bond in ECL3 is missing probably due to a very short loop), implying the importance of disulfide bonds in maintaining the ECL structures for the FZD receptors. In the extracellular half of the 7TM bundle, the conserved residues form a cluster of hydrophobic side chains buried between helices III (F3183.29, Y3223.33, M3263.37), V (F4035.42, V4045.43, P4075.46), and ECL2 (V392), which is apparently important for the structural integrity of these receptors. Closer to the intracellular membrane boundary, there is an unusually high number of conserved tryptophans: W3313.42 and W3393.50 in helix III, W3654.50 in helix IV, and W5357.55 in helix VII. The latter tryptophan, conserved among class F and superimposable with the location of the NPxxY motif of class A, is shown to play an important role in receptor activation, as mutation of W5357.55 leads to a constitutively active SMO receptor34. In the FZD receptors, the KTxxxW motif in helix VIII is highly conserved and has been shown to be critical for the activation of the WNT/β-catenin signaling pathway by interacting directly with Disheveled35,36. Sequence alignment shows that the SMO receptor has an extra alanine between lysine and threonine, but that the rest of the motif is conserved. In the SMO receptor structure, this motif stabilizes the α-helical structure of helix VIII which packs parallel to the membrane (Fig. 6c). The hydroxyl group of the conserved T541 of this motif forms a hydrogen bond with the main chain carbonyl group of V536 at the intracellular end of helix VII; while the indole nitrogen of W545 forms a hydrogen bond interaction with the hydroxyl group of T2511.56, which is conserved in the FZD receptors. The α-helical structure of helix VIII therefore likely plays a critical role for the interaction of activated FZD receptors with downstream signaling proteins, such as Disheveled.
Figure 6. Structural insight for the Frizzled (FZD) receptors based on their sequence homology with the SMO receptor.

a and b, Fully conserved residues in class F members are highlighted on the SMO structure. The side chains of all these conserved residues are shown in sticks with green carbons. c, Alignment of the KTxxxW motif in the FZD receptors to the helix VIII of the SMO receptor. The similar α-helical structure could exist for the KTxxxW motif of the FZD receptors which is stabilized by the hydrogen bond interactions between the hydroxyl group of the conserved residue T541 and the main chain carbonyl group of V536, the indole nitrogen of the conserved residue W545 and the hydroxyl group of the conserved residue T2511.56, shown in dashed lines. The non-conserved residues are colored yellow.
Conclusion
Evolutionarily, class F receptors precede class A receptors — as revealed by extensive phylogenetic analysis37. Despite the earlier emergence, the diversity within class F is significantly less than that of class A. This remarkable conservation likely reflects the pivotal function of the SMO and FZD receptors in the regulation of cell proliferation polarity and differentiation along with tissue formation, some of the most fundamental physiological processes for multicellular metazoan. The SMO receptor structure highlights the incredible use of the 7TM structural fold, with little sequence similarity to other GPCR classes, and is a great example of structural convergence in protein space. As we learn more about different “GPCR” classes and their structure-function, including the expansion of many different intracellular interacting partners beyond G-proteins, it is likely the term “GPCR” may be of limited value in describing the incredible power of the 7TM fold38 in biology.
Methods
Generation of BRIL-ΔCRD-SMO-ΔC fusion construct for structural studies
Human SMO gene was obtained from Origene (Cat#SC122724). A thermally stabilized apocytochrome b562 RIL from E. coli (M7W, H102I, R106L), referred to as BRIL, was fused to the truncated N-terminus at S190 of human SMO receptor, using overlapping PCR. The C-terminus of SMO receptor was truncated at Q555. The resulting receptor chimera sequence was subcloned into a modified pFastBac1 vector (Invitrogen), designated as pFastBac1-833100, which contained an expression cassette with a haemagglutinin (HA) signal sequence followed by a Flag tag, a 10× His tag, and a TEV protease recognition site at the N-terminus before the receptor sequence. Subcloning into the pFastBac1-833100 was achieved using PCR with primer pairs encoding restriction sites KpnI at the 5′ and HindIII at the 3′ termini with subsequent ligation into the corresponding restriction sites found in the vector.
Expression and purification of BRIL-ΔCRD-SMO-ΔC protein for crystallization
The resulting BRIL-ΔCRD-SMO-ΔC construct was expressed in Spodoptera frugiperda (Sf9) insect cells using the Bac-to-Bac Baculovirus Expression System (Invitrogen). Sf9 cells at cell density of 2–3 × 106 cells/ml were infected with baculovirus at 27 °C. Cells were harvested by centrifugation at 48 h post infection and stored at −80 °C until use.
Insect cell membranes were lysed by thawing frozen cell pellets in a hypotonic buffer containing 10 mM HEPES, pH 7.5, 10 mM MgCl2, 20 mM KCl and EDTA-free complete protease inhibitor cocktail tablets (Roche). Extensive washing of the raw membranes was performed by repeated centrifugation (two-three times) in a high osmotic buffer comprised of 1.0 M NaCl in the hypotonic buffer described above.
The washed membranes were resuspended into buffer containing 30 μM LY2940680 (Active Biochemicals Co. Limited), 2 mg/ml iodoacetamide (Sigma), and EDTA-free complete protease inhibitor cocktail tablets, and incubated at 4 °C for 1 h prior to solubilization. The membranes were then solubilized in buffer containing 50 mM HEPES, pH 7.5, 200 mM NaCl, 1% (w/v) n-dodecyl-β-D-maltopyranoside (DDM, Anatrace), 0.2% (w/v) cholesteryl hemisuccinate (CHS, Sigma), for 3–4 h at 4 °C. The supernatant containing solubilized SMO protein was isolated from the cell debris by high-speed centrifugation, and subsequently incubated with TALON IMAC resin (Clontech) overnight at 4 °C in the presence of 20 mM imidazole and 1 M NaCl. After binding, the resin was washed with 10 column volumes of Wash I Buffer comprised of 50 mM HEPES, pH 7.5, 800 mM NaCl, 10% (v/v) glycerol, 0.1% (w/v) DDM, 0.02% (w/v) CHS, 8 mM ATP, 20 mM imidazole, 10 mM MgCl2 and 15 μM LY2940680, followed by 6 column volumes of Wash II Buffer comprised of 50 mM HEPES, pH 7.5, 500 mM NaCl, 10% (v/v) glycerol, 0.05% (w/v) DDM, 0.01% (w/v) CHS, 50 mM imidazole and 20 μM LY2940680. The protein was then eluted by 3 column volumes of Elution Buffer containing 50 mM HEPES, pH 7.5, 300 mM NaCl, 10% (v/v) glycerol, 0.03% (w/v) DDM, 0.006% (w/v) CHS, 250 mM imidazole and 50 μM LY2940680. PD MiniTrap G-25 column (GE Healthcare) was used to remove imidazole. The protein was then treated overnight with TEV protease (His-tagged) to cleave the N-terminal His-tag and FLAG-tag. TEV protease and cleaved N-terminal fragment were removed by TALON IMAC resin incubation at 4 °C for 2 h. The tag-less protein was collected as the TALON IMAC column flow-through. The protein was then concentrated to 50–60 mg/ml with a 100 kDa cut-off Vivaspin concentrator. Protein monodispersity was tested by analytical size-exclusion chromatography (aSEC). Typically, the aSEC profile showed a monodisperse peak.
Lipidic cubic phase crystallization
Protein samples of the SMO receptor in a complex with LY2940680 were reconstituted into lipidic cubic phase (LCP) by mixing with molten lipid (10%w/w cholesterol, 90%w/w monoolein) in a mechanical syringe mixer39 at a ratio of 2/3 v/v protein solution/lipid. LCP crystallization trials were performed using an NT8-LCP crystallization robot (Formulatrix) as previously described40. 96-well glass sandwich plates (Marienfeld) were incubated and imaged at 20 °C using an automated incubator/imager (RockImager 1000, Formulatrix). Initial crystal hits were found from a precipitant condition containing 100 mM HEPES, pH 7.4, 30% (v/v) PEG400, 100 mM ammonium fluoride. After optimization, crystals grew in 100 mM HEPES, pH 7.8, 70 mM ammonium fluoride, 32% (v/v) PEG400, 4%–8% (v/v) Polypropylene glycol P 400 to the size of 100×20×20 μm for 2–3 d, and were harvested using MiTeGen micromounts and flash frozen in liquid nitrogen for data collection.
Crystallographic data collection and processing
X-ray data were collected at the 23ID-D beamline (GM/CA CAT) at the Advanced Photon Source, Argonne, IL using a 20 μm minibeam at a wavelength of 1.0330 Å and a MarMosaic 300 CCD detector. Crystals were aligned and data were collected using strategy similar to other GPCR structures41. Typically 20 frames at 1° oscillation and 1 s exposure with non-attenuated beam were collected following by a translation of the crystal to a non-exposed position or changing the crystal to minimize the effect of radiation damage. A complete data set was obtained by indexing, integrating, scaling, and merging data from 5 crystals using HKL2000 (ref 42) (Supplementary Table 1).
Experimental phasing
The attempts to find a molecular replacement solution using all previous class A GPCR structures as a search model did not generate any reliable solutions due to the low sequence similarity. Therefore, experimental phasing was performed by soaking the crystals in the presence of 5 mM tantalum bromide ([Ta6Br12]2+•2Br−, Jena Bioscience) for 24 h. The data were collected at 23ID-D beamline (GM/CA CAT) at the Advanced Photon Source using the peak wavelength of the tantalum L3 edge (9.880 keV). A complete 360° data set was acquired from a single crystal by using a 20 μm minibeam at 50 × attenuation with 1° oscillation and 1 s exposure per frame and collecting 30° wedges with direct and inverse beam. The SAD data set was integrated and scaled at 3.5 Å resolution using HKL2000 (ref 42), and PHENIX.AutoSol43 was used to search for the heavy atom sites.
Structure determination and refinement
The structure was initial solved using 3.5 Å Ta SAD data with PHENIX.AutoSol, the map clearly showed transmembrane helices. Further heavy atom refinement and phasing, combining the high resolution native data and SAD (SIRAS) was carried out using SHARP44 based on two heavy atom sites identified from the anomalous difference map. Density modification and automatic tracing were then performed using PHENIX.AutoBuild45. Refinement was performed by rounds of REFMAC5 (ref 46) and autoBUSTER47 using the 2.5 Å resolution native dataset followed by manual examination and rebuilding of the refined coordinates in the program COOT 48 using both |2Fo| - |Fc| and |Fo| - |Fc| maps, as well as omit maps. Data collection and refinement statistics are shown in Supplementary Table 1.
Radioligand binding assays
Radioligand binding assays used Sf9 pellets expressing the crystallization construct BRIL-ΔCRD-SMO-ΔC (described in expression section) and crude HEK 293T membrane preparations expressing wild type human SMO receptor in 96-well plates at a final volume of 125 μl. To obtain crude HEK 239T membrane preparations, HEK 293T cells were transfected with a human SMO receptor expression plasmid for 24 h and scraped into conical centrifuge tubes. Collected cells were centrifuged at 1,000 × g for 10 min and the cell pellet was hypotonically lysed by cold lysis buffer (50 mM Tris-HCl, pH 7.4). Crude membrane fractions were isolated by centrifugation at 21,000 × g for 20 min at 4 °C. The membrane pellets were resuspended with lysis buffer at 3× volume of pellet size, subjected to protein concentration determination, and stored in aliquots at −80 °C if not used immediately. To determine equilibrium dissociation constant (Kd) for 3H-cyclopamine, a serial of 8 concentrations of 3H-cyclopamine (0.2 – 36 nM in triplicate) were incubated with 6 μg of SMO Sf9 membranes or 20 μg of above SMO HEK 293T membranes in binding buffer (50 mM HEPES, 3 mM MgCl2, EDTA-free protease inhibitor cocktail, 0.5 mg/ml BSA, pH 7.2, modified from ref 49) for 2.5 h in dark at room temperature. Nonspecific binding was defined by 10 μM SMO receptor antagonist LY2940680. To determine equilibrium dissociation constant (Ki) for cyclopamine, LY2940680, and SAG (Cayman Chemical #11914), a serial of 11 concentrations of test compound (0.1 nM to 10 μM in triplicate sets) were incubated with a fixed concentration of 3H-cyclopamine (~Kd of 3H-cyclopamine) and SMO Sf9 membranes or SMO HEK293 T membranes for 2.5 h in the dark and at the room temperature. At the end of the incubation period, the reactions were stopped by rapid filtration onto 0.3% PEI-soaked GF/A filters and washed three times with cold PBS (pH 7.2). The filters were then microwave dried and scintillant was melted on the filters on a hot plate. The filters were wrapped in plastic wrap and counted for radioactivity. Results (Supplementary Fig. 3) were analyzed using GraphPad Prism 5.0.
Supplementary Material
Acknowledgements
This work was supported by NIH Common Fund grant P50 GM073197 for technology development (V.C. and R.C.S.), PSI:Biology grant U54 GM094618 for biological studies and structure production (target GPCR-131) (V.K., V.C. and R.C.S.); F32 DK088392 (F.Y.S.); R01 MH61887, U19 MH82441, R01 DA27170 and the NIMH Psychoactive Drug Screening Program (X.-P.H. and B.L.R.) and the Michael Hooker Chair of Pharmacology (B.L.R.). We thank J. Velasquez for help on molecular biology; T. Trinh and M. Chu for help on baculovirus expression; K. Kadyshevskaya for assistance with figure preparation; A. Walker for assistance with manuscript preparation; D. Wacker for assistance with SAD data collection and processing; J. Smith, R. Fischetti, and N. Sanishvili for assistance in development and use of the minibeam and beamtime at GM/CA-CAT beamline 23-ID at the Advanced Photon Source, which is supported by National Cancer Institute grant Y1-CO-1020 and National Institute of General Medical Sciences grant Y1-GM-1104.
Footnotes
Supplementary Information is linked to the online version of the paper at (web).
Author Contributions C.W. designed and made the constructs, purified and crystallized the receptor in LCP, optimized crystallization conditions, grew crystals for data collection, assisted with crystal harvesting and heavy atom soaking experiment, collected the diffraction data and prepared the manuscript. H.W. performed baculovirus expression of the receptor, purified the receptor, optimized crystallization conditions and prepared the manuscript. V.K. prepared the manuscript. G.W.H. solved and refined the structure and assisted with preparing the manuscript. X.-P.H. performed radioligand binding assay. W.L. assisted with data collection. F.Y.S. provided material and conditions for heavy atom soaking experiment. B.L.R. supervised the radioligand binding experiment and assisted with preparing the manuscript. V.C. harvested crystals, performed heavy atom soaking experiment, collected and processed diffraction data, assisted with preparation of the manuscript. R.C.S. was responsible for the overall project strategy and management and wrote the manuscript.
Author Information The coordinates and the structure factors have been deposited in the Protein Data Bank under the accession code (4JKV). Reprints and permissions information is available at (web). The authors declare no competing financial interests. Readers are welcome to comment on the online version of this article at (web).
References
- 1.Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001;15:3059–3087. doi: 10.1101/gad.938601. doi:10.1101/gad.938601. [DOI] [PubMed] [Google Scholar]
- 2.Robbins DJ, Fei DL, Riobo NA. The Hedgehog signal transduction network. Sci Signal. 2012;5:re6. doi: 10.1126/scisignal.2002906. doi:10.1126/scisignal.2002906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marigo V, Davey RA, Zuo Y, Cunningham JM, Tabin CJ. Biochemical evidence that patched is the Hedgehog receptor. Nature. 1996;384:176–179. doi: 10.1038/384176a0. doi:10.1038/384176a0. [DOI] [PubMed] [Google Scholar]
- 4.Stone DM, et al. The tumour-suppressor gene patched encodes a candidate receptor for Sonic hedgehog. Nature. 1996;384:129–134. doi: 10.1038/384129a0. doi:10.1038/384129a0. [DOI] [PubMed] [Google Scholar]
- 5.Taipale J, Cooper MK, Maiti T, Beachy PA. Patched acts catalytically to suppress the activity of Smoothened. Nature. 2002;418:892–897. doi: 10.1038/nature00989. doi:10.1038/nature00989. [DOI] [PubMed] [Google Scholar]
- 6.Corbit KC, et al. Vertebrate Smoothened functions at the primary cilium. Nature. 2005;437:1018–1021. doi: 10.1038/nature04117. doi:10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- 7.Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer. 2008;8:387–398. doi: 10.1038/nrc2389. doi:10.1038/nrc2389. [DOI] [PubMed] [Google Scholar]
- 8.Huang HC, Klein PS. The Frizzled family: receptors for multiple signal transduction pathways. Genome Biol. 2004;5:234. doi: 10.1186/gb-2004-5-7-234. doi:10.1186/gb-2004-5-7-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schulte G. International Union of Basic and Clinical Pharmacology. LXXX. The class Frizzled receptors. Pharmacol Rev. 2010;62:632–667. doi: 10.1124/pr.110.002931. doi:10.1124/pr.110.002931. [DOI] [PubMed] [Google Scholar]
- 10.Bhanot P, et al. A new member of the frizzled family from Drosophila functions as a Wingless receptor. Nature. 1996;382:225–230. doi: 10.1038/382225a0. doi:10.1038/382225a0. [DOI] [PubMed] [Google Scholar]
- 11.Janda CY, Waghray D, Levin AM, Thomas C, Garcia KC. Structural basis of Wnt recognition by Frizzled. Science. 2012;337:59–64. doi: 10.1126/science.1222879. doi:10.1126/science.1222879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Foord SM, et al. International Union of Pharmacology. XLVI. G protein-coupled receptor list. Pharmacol Rev. 2005;57:279–288. doi: 10.1124/pr.57.2.5. doi:10.1124/pr.57.2.5. [DOI] [PubMed] [Google Scholar]
- 13.Ayers KL, Therond PP. Evaluating Smoothened as a G-protein-coupled receptor for Hedgehog signalling. Trends Cell Biol. 2010;20:287–298. doi: 10.1016/j.tcb.2010.02.002. doi:10.1016/j.tcb.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 14.Chen W, et al. Activity-dependent internalization of smoothened mediated by beta-arrestin 2 and GRK2. Science. 2004;306:2257–2260. doi: 10.1126/science.1104135. doi:10.1126/science.1104135. [DOI] [PubMed] [Google Scholar]
- 15.Riobo NA, Saucy B, Dilizio C, Manning DR. Activation of heterotrimeric G proteins by Smoothened. Proc Natl Acad Sci U S A. 2006;103:12607–12612. doi: 10.1073/pnas.0600880103. doi:10.1073/pnas.0600880103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Polizio AH, et al. Heterotrimeric Gi proteins link Hedgehog signaling to activation of Rho small GTPases to promote fibroblast migration. J Biol Chem. 2011;286:19589–19596. doi: 10.1074/jbc.M110.197111. doi:10.1074/jbc.M110.197111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heretsch P, Tzagkaroulaki L, Giannis A. Modulators of the hedgehog signaling pathway. Bioorg Med Chem. 2010;18:6613–6624. doi: 10.1016/j.bmc.2010.07.038. doi:10.1016/j.bmc.2010.07.038. [DOI] [PubMed] [Google Scholar]
- 18.Bender MH, et al. Identification and characterization of a novel smoothened antagonist for the treatment of cancer with deregulated hedgehog signaling. Cancer Res. 2011;71:2819. [Google Scholar]
- 19.Hipskind PA, Patel BK, Wilson T. Preparation of disubstituted phthalazine derivatives for use as hedgehog pathway antagonists and useful in treatment of cancer. 2010 WO2010147917A1. [Google Scholar]
- 20.Cherezov V, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. doi:10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu B, et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330:1066–1071. doi: 10.1126/science.1194396. doi:10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao Y, Tong C, Jiang J. Hedgehog regulates smoothened activity by inducing a conformational switch. Nature. 2007;450:252–258. doi: 10.1038/nature06225. doi:10.1038/nature06225. [DOI] [PubMed] [Google Scholar]
- 23.Ballesteros JA, Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- 24.Rasmussen SG, et al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature. 2011;469:175–180. doi: 10.1038/nature09648. doi:10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yauch RL, et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science. 2009;326:572–574. doi: 10.1126/science.1179386. doi:10.1126/science.1179386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taipale J, et al. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature. 2000;406:1005–1009. doi: 10.1038/35023008. doi:10.1038/35023008. [DOI] [PubMed] [Google Scholar]
- 27.Carroll CE, Marada S, Stewart DP, Ouyang JX, Ogden SK. The extracellular loops of Smoothened play a regulatory role in control of Hedgehog pathway activation. Development. 2012;139:612–621. doi: 10.1242/dev.075614. doi:10.1242/dev.075614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Granier S, et al. Structure of the delta-opioid receptor bound to naltrindole. Nature. 2012;485:400–404. doi: 10.1038/nature11111. doi:10.1038/nature11111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manglik A, et al. Crystal structure of the micro-opioid receptor bound to a morphinan antagonist. Nature. 2012;485:321–326. doi: 10.1038/nature10954. doi:10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thompson AA, et al. Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic. Nature. 2012;485:395–399. doi: 10.1038/nature11085. doi:10.1038/nature11085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.White JF, et al. Structure of the agonist-bound neurotensin receptor. Nature. 2012;490:508–513. doi: 10.1038/nature11558. doi:10.1038/nature11558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu H, et al. Structure of the human kappa-opioid receptor in complex with JDTic. Nature. 2012;485:327–332. doi: 10.1038/nature10939. doi:10.1038/nature10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang C, et al. High-resolution crystal structure of human protease-activated receptor 1. Nature. 2012;492:387–392. doi: 10.1038/nature11701. doi:10.1038/nature11701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xie J, et al. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature. 1998;391:90–92. doi: 10.1038/34201. doi:10.1038/34201. [DOI] [PubMed] [Google Scholar]
- 35.Umbhauer M, et al. The C-terminal cytoplasmic Lys-thr-X-X-X-Trp motif in frizzled receptors mediates Wnt/beta-catenin signalling. EMBO J. 2000;19:4944–4954. doi: 10.1093/emboj/19.18.4944. doi:10.1093/emboj/19.18.4944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wong HC, et al. Direct binding of the PDZ domain of Dishevelled to a conserved internal sequence in the C-terminal region of Frizzled. Mol Cell. 2003;12:1251–1260. doi: 10.1016/s1097-2765(03)00427-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krishnan A, Almen MS, Fredriksson R, Schioth HB. The origin of GPCRs: identification of mammalian like Rhodopsin, Adhesion, Glutamate and Frizzled GPCRs in fungi. PLoS One. 2012;7:e29817. doi: 10.1371/journal.pone.0029817. doi:10.1371/journal.pone.0029817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schwartz TW, Rosenkilde MM. Is there a `lock' for all agonist `keys' in 7TM receptors? Trends Pharmacol Sci. 1996;17:213–216. doi: 10.1016/0165-6147(96)10017-1. [DOI] [PubMed] [Google Scholar]
- 39.Caffrey M, Cherezov V. Crystallizing membrane proteins using lipidic mesophases. Nat Protoc. 2009;4:706–731. doi: 10.1038/nprot.2009.31. doi:10.1038/nprot.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cherezov V, Peddi A, Muthusubramaniam L, Zheng YF, Caffrey M. A robotic system for crystallizing membrane and soluble proteins in lipidic mesophases. Acta Crystallogr D Biol Crystallogr. 2004;60:1795–1807. doi: 10.1107/S0907444904019109. doi:10.1107/S0907444904019109. [DOI] [PubMed] [Google Scholar]
- 41.Cherezov V, et al. Rastering strategy for screening and centring of microcrystal samples of human membrane proteins with a sub-10 microm size X-ray synchrotron beam. J R Soc Interface. 2009;6(Suppl 5):S587–597. doi: 10.1098/rsif.2009.0142.focus. doi:10.1098/rsif.2009.0142.focus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. doi:Doi 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 43.Terwilliger TC, et al. Decision-making in structure solution using Bayesian estimates of map quality: the PHENIX AutoSol wizard. Acta Crystallogr D. 2009;65:582–601. doi: 10.1107/S0907444909012098. doi:10.1107/S0907444909012098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bricogne G, Vonrhein C, Flensburg C, Schiltz M, Paciorek W. Generation, representation and flow of phase information in structure determination: recent developments in and around SHARP 2.0. Acta Crystallogr D. 2003;59:2023–2030. doi: 10.1107/s0907444903017694. doi:10.1107/S0907444903017694. [DOI] [PubMed] [Google Scholar]
- 45.Terwilliger TC, et al. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr D. 2008;64:61–69. doi: 10.1107/S090744490705024X. doi:10.1107/S090744490705024x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D. 1997;53:240–255. doi: 10.1107/S0907444996012255. doi:10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 47.BUSTER v. 2.8.0. Global Phasing Ltd.; Cambridge, U.K.: 2009. [Google Scholar]
- 48.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D. 2010;66:486–501. doi: 10.1107/S0907444910007493. doi:10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rominger CM, et al. Evidence for allosteric interactions of antagonist binding to the smoothened receptor. J Pharmacol Exp Ther. 2009;329:995–1005. doi: 10.1124/jpet.109.152090. doi:10.1124/jpet.109.152090. [DOI] [PubMed] [Google Scholar]
- 50.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. doi:10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dann CE, et al. Insights into Wnt binding and signalling from the structures of two Frizzled cysteine-rich domains. Nature. 2001;412:86–90. doi: 10.1038/35083601. doi:10.1038/35083601. [DOI] [PubMed] [Google Scholar]
- 52.Chun E, et al. Fusion partner toolchest for the stabilization and crystallization of G protein-coupled receptors. Structure. 2012;20:967–976. doi: 10.1016/j.str.2012.04.010. doi:10.1016/j.str.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.