

Abstract
Inflammatory and anti-inflammatory cytokines play an important role in the generation of effector and memory CD8+ T cells. We used two different models, transgenic expression of truncated (dominant negative) form of TGF-β receptor II (dnTGFβRII) and Cre-mediated deletion of the floxed TGFβRII to examine the role of TGF-β signaling in the formation, function, and homeostatic proliferation of memory CD8+ T cells. Blocking TGF-β signaling in effector CD8+ T cells using both of these models demonstrated a role for TGF-β in regulating the number of short-lived effector cells, but did not alter memory CD8+ T cell formation and their function upon Listeria monocytogenes infection in mice. Interestingly however, a massive lymphoproliferative disorder and cellular transformation were observed in antigen-experienced and homeostatically generated memory CD8+ T cells only in cells that express the dnTGFβRII, but not in cells with a complete deletion of TGFβRII. Furthermore, the development of transformed memory CD8+ T cells expressing dnTGFβRII was IL-7- and IL-15-independent, and MHC class I was not required for their proliferation. We show that transgenic expression of the dnTGFβRII, rather than the absence of TGFβRII-mediated signaling, is responsible for dysregulated expansion of memory CD8+ T cells. This study uncovers a previously unrecognized dominant function of the dnTGFβRII in CD8+ T cell proliferation and cellular transformation, which is caused by a mechanism that is different than the absence of TGF-β signaling. These results should be considered during both basic and translational studies where there is a desire to block TGF-β signaling in CD8+ T cells.
The formation of immunological memory begins as naive T cells come in contact with their cognate antigen on activated antigen-presenting cells, undergo clonal expansion, and differentiate into effector T cells, most of which soon die by apoptosis during the “contraction” phase (1–4). During clonal expansion in response to most viral and bacterial infections, multiple subpopulations of effector CD8+ T cells exist, whose survival is regulated by inflammatory and anti-inflammatory cytokines. The subset that survives and becomes memory cells are referred to as memory precursor effector cells (MPECs), which are enriched in CD127 (IL-7Rα)hi KLRG1lo populations (1, 4). The fraction that dies during contraction is referred to as short-lived effector cells (SLECs), which are enriched in IL-7RαloKLRG1hi populations. Although both subsets have similar functional abilities at the peak of the immune response, they greatly differ in their memory potential and survivability (1, 4).
Functional memory CD8+ T cells can also be differentiated from naive cells by homeostatic proliferation, or when naive CD8+ T cells undergo a proliferative response in a lymphopenic environment (5, 6). It has been suggested that these so-called “memory-like” CD8+ T cells are the progeny of cells that have responded to either self or environmental antigens in the presence of common γ chain cytokines IL-7 and IL-15 (5–7). Indeed, mice deficient in IL-15 signaling have a marked decrease in the number of CD8+CD44+ T cells (8, 9), whereas transgenic (Tg) mice expressing IL-7 or IL-15 have an increase number of cells in this population (10, 11). Dysregulated IL-7 or IL-15 signaling is linked to lymphoproliferative disorders and cellular transformation (10, 12, 13).
Transforming growth factor-β (TGF-β) is a pleiotropic cytokine that activates a broad range of cellular responses (14, 15). Active TGF-β binds to TGF-β receptor II (TGFβRII), triggering the kinase activity of the cytoplasmic domain that in turn activates TGFβRI (16, 17). The activated receptor complex leads to activation of both smad-dependent and -independent signaling pathways (16, 17). TGF-β signaling in T cells is essential to restrain self-reactivity and immune homeostasis, as demonstrated by the fatal, rapidly progressing autoimmune lesions observed in T cell-specific TGFβRII conditional knockout (KO) mice (CD4Cre-Tgfbr2f/f) (18, 19). Autoimmunity, in the form of inflammatory bowel disease (IBD) is also observed in mice in which a truncated form of TGFβRII that is missing the kinase domain, referred to here as dominant negative form of TGFβRII (dnTGFβRII), is expressed under the control of Cd4 promoter (20). However, autoimmune pathology of dnTGFβRII mice is much weaker than that of T cell-specific TGFβRII conditional knockout mice or Tgfb1−/− mice (18, 20), suggesting that dnTGFβRII mice perhaps still express a functional TGFβRII that can induce some level of TGF-β signaling. Due to the early-onset lethal autoimmune disease development in CD4Cre-Tgfbr2f/f mice, the dnTGFβRII mice have been used to study the importance of TGF-β signaling in the regulation of effector CD8+ T cell function during autoimmunity and tumor development (20, 21). In fact, these promising results have prompted the idea of over expressing the dnTGFβRII in CD8+ T cells as a means to enhance the activity of tumor-specific CD8+ T cells toward a feasible strategy for T cell-mediated immunotherapy (22, 23). Furthermore, TGF-β controls the number of CD8+ effector cells that are generated in response to bacterial and viral infections (24, 25). Blockade of TGF-β signaling in this model increased the number of SLECs by regulating Bcl2 expression induced by IL-15 (24). In addition, homeostatic proliferation of CD8+ memory T cells is also thought to be regulated by TGF-β under steady state conditions (26, 27). Mice expressing dnTGFβRII under the control of the Cd2 promoter do not develop autoimmunity, but instead develop polyclonal memory-like CD8+ T cell hyperproliferation that eventually leads to cellular transformation (26, 28, 29). However, Cd4 promoter-driven dnTGFβRII mice on a polyclonal background do not exhibit dysregulated expansion of the CD8+ T cells prior to developing autoimmunity (20). Thus, it is still not clear how TGF-β signaling regulates CD8+ T cell homeostasis as well as peripheral CD8+ T cell lymphoma development. Furthermore, since dnTGFβRII cells can still respond to TGF-β signaling, the exact role of TGF-β signaling in CD8+ T cell function, especially its contribution to memory CD8+ T cell formation and homeostatic maintenance remains unknown.
In this study we utilized two different mouse models, Tg mice expressing dnTGFβRII under the control of Cd4 promoter and Tgfbr2 conditional KO mice crossed to either granzyme B (GzmB)- or CD4-Cre mice, to address the role of TGF-β signaling in CD8+ T cell development during pathogen-induced memory formation, function, and homeostatic proliferation. We demonstrate that antigen-induced expansion, cytokine production, and cytolytic activity of memory CD8+ T cells is not impacted by the blockade of TGF-β signaling in either model. Interestingly however, we observed a massive lymphoproliferative disorder in both antigen-experienced and homeostatically produced memory phenotype CD8+ T cells only in the dnTGFβRII model, suggesting that this previously observed phenotype may be due to the expression of the truncated form of TGFβRII, rather than the absence of TGFβRII-mediated signaling, in memory CD8+ T cells.
Materials and Methods
Mice
CD4-dnTGFβRII, CD11c-dnTGFβRII, and CD4Cre mice on the C57BL/6 background were previously described (20, 30, 31). Rag1−/−, b2m−/−, OTI, OTII, and P14 mice were obtained from Jackson Laboratory. C57BL/6 and C57BL/6-Ly5.2/Cr (CD45.1) congenic mice were obtained from National Cancer Institute. Il15−/− mice were purchased from Taconic. Tgfbr2f/f, Il7−/− and GzmB-Cre mice were kindly provided by H. Moses (Vanderbilt University), Schering-Plough (Palo Alto, CA), and J. Jacobs (Emory University), respectively (32–34). All animal experimentation was conducted in accordance with institutional guidelines.
Adoptive transfer and LM-OVA infection
Naive CD44loCD62LhiCD8+ OTI-Rag1−/− cells were purified from spleen and peripheral lymph nodes (LNs) by FACS sorting. Every co-transfer experiment consisted of 0.5~1.0 × 105 sorted naive OTI-control-Rag1−/−, OTI-dnTGFβRII-Rag1−/−, OTI-CD4Cre-Tgfbr2f/f-Rag1−/−, or OTI-GzmBCre-Tgfbr2f/f-Rag1−/− cells each on a different congenic background mixed at a 1:1 ratio. One day after adoptive transfer, mice were infected intravenously (i.v.) with 1 × 105 recombinant Listeria monocytogenes expressing OVA (LM-OVA), which was a generous gift from H. Shen (University of Pennsylvania). Memory OTI-WT-Rag1−/− or OTI-dnTGFβRII-Rag1−/− cells were isolated from LNs and spleen of LM-OVA infected mice based on staining with CD8α and the corresponding congenic markers. For recall experiments, mice were infected i.v. with 5 × 105 of the LM-OVA.
Flow cytometry
Fluorescent dye-labeled Abs against CD8α, TCRβ, CD45.1, CD45.2, CD43 (1B11), CD62L, CD69, CD25 were purchased from BioLegend, CD127, KLRG1, and CD122 were from eBioscience, IFN-γ, IL-2, and TNF were from BD Biosciences, and GzmB was from Caltag Laboratories. For intracellular cytokine staining, splenocytes from infected mice were stimulated with SIINFEKL peptide in the presence of Golgi-stop (BD Biosciences) for 4 hr, followed by fixing and permeabilization according to the manufacturer’s instructions (BD Biosciences). For cell division assay, purified memory OTI cells were stained with CFSE (Molecular Probes), and cultured in the presence of anti-CD3, PMA/ionomycin, IL-2, or IL-15 for 2–4 days. The cells were analyzed on a LSRII or FACSCariber (Becton Dickinson) and data were analyzed with FlowJo software (Tree Star).
In vivo CTL assay
OTI cells of each genotype were FACS sorted based on their congenic markers, and mixed with CFSE-labeled target cells at 1:5 ratio. The mixture of effector and target cells were injected i.v. into naive C57BL/6 hosts. Spleens from the recipient mice were harvested 14–16 hr later, and the ratio of CFSEhi and CFSElo cells was measured by FACS analysis, and in vivo CTL activity was determined as described (35).
Isolation of lymphocytes from non-lymphoid organs
Peripheral blood lymphocytes (PBLs) were isolated from tail or retro-orbital vein of infected mice. Liver and lung cell homogenates were digested for 1 hour at 37°C with digestion buffer [RPMI + 5% FBS + 100 U/ml DNase I (Sigma) + 0.2mg/ml Collagenase IV (Sigma)]. Liver homogenates were centrifuged at 300 rpm for 3 min to remove hepatocytes, and lung homogenates were run through a 70μm filter mesh. Non-hepatic supernatant and lung lymphocytes were centrifuged at 1500 rpm for 10 min. The cell pellet was resuspended in 1 ml of RPMI + 5% FBS and mixed with 4 ml of 27.5% OptiPrep (Axis-Shield). To make a gradient, 1ml of RPMI was layered on top of the cells, and centrifuged at 2000 rpm for 20 min. Lymphocytes were removed from the interface of the gradient.
Real-time RT-PCR analysis
Total RNA was extracted with Qiagen mRNAEasy kit (Qiagen) according to the manufacturer’s instructions. RNA was reverse transcribed with the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). The cDNA was subsequently used for quantitative PCR on an ABI Prism instrument using commercial primer-probe sets.
Karyotype analysis
Memory OTI-dnTGFβRII cells were isolated from LNs and spleen of LM-OVA infected mice, and stimulated with PMA and ionomysin for 2 days. Twenty mitoses were subjected to G-banding analysis (the Laboratory of Genome Biology and Cytogenesis, Van Andel Institute).
Statistical analysis
Unpaired Student’s t test was used to calculate the statistical significance of data comparison. p value of < 0.05 was considered to be significant.
Results
Conditional deletion of Tgfbr2 in OTI cells results in increased number of SLECs, but does not lead to increased accumulation of total effector cells during acute Listeria infection
Using OTI cells expressing dnTGFβRII driven by the Cd4 promoter, we have shown that TGF-β signaling in CD8+ T cells controls the number of SLECs during acute Listeria monocytogenes expressing OVA (LM-OVA) infection (24). In this model, OTI cells isolated from dnTGFβRII-Rag1−/− mice cannot be adoptively transferred into wild-type (WT) C57BL/6 recipients due to immunological rejection (24, 36). This problem is overcome by using CD11c-dnTGFβRII mice as hosts, as they also express human dnTGFβRII under the control of CD11c promoter (30). As reported previously (24), effector OT-dnTGFβRII cells expanded 2–3 fold more than OTI-WT cells by day 7 and 14 post infection (p.i.), and contracted normally by day 21 p.i. (Fig. 1A–B). An increased percentage of SLECs (KLRGhiCD127lo) in OTI-dnTGFβRII cells was also observed during the effector phase of LM-OVA infection (Fig. 1C).
FIGURE 1.

Conditional deletion of Tgfbr2 in OTI cells results in increased number of SLECs, but does not lead to increased accumulation of total effector cells during acute Listeria infection (A) Naive OTI-WT and OTI-dnTGFβRII cells on different CD45 congenic backgrounds were adoptively co-transferred into naive CD11c-dnTGFβRII hosts, followed by infection with LM-OVA 1 day later. The percentage of OTI-WT and OTI-dnTGFβRII cells was monitored among the PBLs of infected mice over time. (B) The ratio of OTI-dnTGFβRII over OTI-WT cells in PBLs. (C) The percentage of OTI-WT and OTI-dnTGFβRII SLECs in the blood after LM-OVA infection at the indicated time point (left). Representative plots of the expression of CD127 and KLRG1 on blood OTI-WT and OTI-dnTGFβRII cells at day 7 p.i. (right). (D) The percentage of OTI-control (CD45.1.1) and OTI-CD4Cre-Tgfbr2f/f (CD45.1.2) cells among PBLs was monitored over time. (E) The ratio of OTI-CD4Cre-Tgfbr2f/f over OTI-control cells in PBLs. (F) The percentage of SLECs in the blood OTI-control and OTI-CD4Cre-Tgfbr2f/f cells after LM-OVA infection at the indicated time point. Representative plots of the expression of CD127 and KLRG1 in blood OTI-WT or OTI-CD4Cre-Tgfbr2f/f cells at day 5 p.i. are shown on the right. Each dot represents 1 mouse. NS, not significant. *** p<0.001, ** p<0.01, * p<0.05 versus control. Data are representative of at least 3 independent experiments.
Because of partial blockade of TGF-β signaling and the problem of immunological rejection in dnTGFβRII cells (18, 20), we used the TGFβRII conditional KO system as an alternate method to block TGF-β signaling in CD8+ T cells to study the effect of TGF-β on formation and function of memory CD8+ T cells. To delete TGFβRII in CD8+ T cells, we crossed Tgfbr2f/f mice to OTI-Rag1−/− as well as to mice expressing Cre recombinase driven by the Cd4 promoter (CD4Cre-Tgfbr2f/f), and we used their littermates (CD4Cre-Tgfbr2f/+ or CD4Cre-negative Tgfbr2f/f) as controls. We adoptively co-transferred OTI-control or OTI-CD4Cre-Tgfbr2f/f cells into naive C57BL/6 mice, and infected them with LM-OVA. The percentage of transferred cells was measured over the course of infection. We expected that the phenotype of CD4Cre-Tgfbr2f/f cells would be much stronger than that of dnTGFβRII cells. However, in contrast to OTI-dnTGFβRII cells (Fig. 1A–B), the expansion of effector OTI-CD4Cre- Tgfbr2f/f cells was comparable to control OTI cells during effector phase of acute LM-OVA infection (Fig, 1D–E). By contrast, similar to what was observed in OTI-dnTGFβRII cells (Fig. 1C), the OTI-CD4Cre-Tgfbr2f/f cells had a higher percentage of SLECs and lower percentage of MPECs (Fig. 1F). These data suggest that dnTGFβRII cells and conditional TGFβRII KO cells have a different phenotype with regard to total effector T cell expansion, but show similarly increased percent of SLECs.
Late memory OTI-dnTGFβRII cells show enhanced accumulation after acute Listeria infection
Using the dnTGFβRII model, we next wanted to study the role of TGF-β signaling in the formation and function of memory CD8+ T cells. OTI-WT cells were either transferred alone into C57BL/6 mice or co-transferred together with OTI-dnTGFβRII cells into CD11c-dnTGFβRII hosts. At early time points, days 25–101 p.i., the frequency of transferred OTI-WT or OTI-dnTGFβRII cells among PBLs ranged from 0.01 to 8.73% in C57BL/6 or CD11c-dnTGFβRII hosts (Fig. 2A). Interestingly, by about day 300 p.i., CD11c-dnTGFβRII hosts that still retained OTI cells showed a dramatically elevated frequency of OTI-dnTGFβRII cells (Fig. 2A and 2B). The time at which this expansion could first be detected varied among the animals and for each experiment, and ranged from day 180 to 250 p.i. (data not shown). OTI-WT cells transferred into the CD11c-dnTGFβRII hosts behaved similarly to those transferred into C57BL/6 hosts, and the percentage of OTI-WT cells in PBLs was only slightly increased in either host (Fig. 2A). However, we observed that the engraftment efficiency of transferred OTI-WT cells in CD11c-dnTGFβRII hosts was significantly lower when compared to their engraftment into C57BL/6 hosts at 28–40 days p.i. (Fig. S1A). Although the exact reason for this lower engraftment of the OTI-WT cells in the CD11c-dnTGFβRII hosts is currently unclear, we hypothesize that this may be due to enhanced innate immunity in CD11c-dnTGFβRII hosts (30, 37), and a potential NK-mediated rejection of the OTI-WT cells over time. This long-term rejection of engrafted OTI-WT memory cells in CD11c-dnTGFβRII hosts is different than the early rejection of the OT-dnTGFβRII cells in C57BL/6 hosts, which occurs within days as previously reported (24, 36) (Fig. S1B–C). Since the long-term engraftment of OTI-WT cells was considerably reduced in CD11c-dnTGFβRII hosts, we had to use C57BL/6 hosts that received OTI-WT cells as our age-matched controls in most of the subsequent long-term experiments.
FIGURE 2.

Transgenic expression of dnTGFβRII, but not conditional deletion of Tgfbr2, results in massive accumulation of late memory OTI cells after Listeria infection. (A) Naive OTI-WT or OTI-dnTGFβRII cells were adoptively transferred into CD11c-dnTGFβRII hosts, followed by infection with LM-OVA 1 day later. C57BL/6 mice receiving naive OTI-WT were also used as controls. The percentage of OTI-WT and OTI-dnTGFβRII after contraction was monitored in PBLs over time. (B) Representative dot plots of transferred OTI-WT (top) and OTI-dnTGFβRII (bottom) cells in the PBLs at day 307 p.i. (C) Gross morphology of spleen and LNs from infected hosts at 11 months p.i. (D–E) The percentage (D) and absolute number (E) of donor OTI cells in the indicated tissues at day 374–448 p.i. Results are combined from 4 independent experiments. (F) The percentage of host CD8+ T cells in the spleen of the same mice as in (D). (G) Representative dot plots of donor OTI (CD45.1+) and host CD8+ T cells (CD45.1−) in the spleen at day 374 p.i (top). The expression of CD44 and CD62L in the spleen of host CD8+ T cells (bottom). (H) Expression pattern of indicated surface cell markers was analyzed on OTI-WT and OTI-dnTGFβRII cells isolated from spleen of LM-OVA-infected mice at day 340 p.i. (I) Naive OTI-control and OTI-GzmBCre-Tgfbr2f/f cells on different congenic backgrounds were adoptively co-transferred into naive C57BL/6 hosts, followed by infection with LM-OVA 1 day later. The percentage of OTI-control and OTI-GzmBCre-Tgfbr2f/f cells in PBLs was monitored over time at indicated time points. (J) The percentage of SLECs, MPECs and KLRG1+CD127+ (DP) cells among the OTI-control or OTI-GzmBCre-Tgfbr2f/f cells in the PBLs at 280 days p.i. Representative plots of the expression of CD127 and KLRG1 at day 280 p.i. are shown on the right. (K) The percentage of OTI-control and OTI-GzmBCre-Tgfbr2f/f cells in the spleen at day 438 p.i. NS, not significant versus OTI-WT. * p<0.05, ** p<0.01 *** p<0.001 versus OTI-WT. (A, B, G–K) Data are representative of 3 independent experiments. Each dot represents 1 mouse.
To measure the number of late memory OTI cells in different tissues, recipients were sacrificed at day 374–448 p.i. The CD11c-dnTGFβRII hosts that had received OTI-dnTGFβRII cells showed splenomegaly and lymphoadenopathy (Fig. 2C). The percentage of OTI-dnTGFβRII cells was significantly higher than that of OTI-WT cells in both lymphoid and non-lymphoid tissues (Fig. 2D), and greater than 20-fold more OTI-dnTGFβRII cells was recovered from each organ than OTI-WT cells (Fig. 2E). Unlike the transferred OTI-dnTGFβRII cells, the frequency of host CD8+ T cells from the spleen of CD11c-dnTGFβRII host did not increase over time (day 374–448 p.i.) (Fig. 2F), even though they had the CD44hiCD62Llo activated phenotype (Fig. 2G), suggesting that the OTI-dnTGFβRII phenotype in the CD11c-dnTGFβRII hosts is unique to the donor OTI cells that express the dnTGFβRII transgene.
Analyses of cell surface markers revealed that OTI-dnTGFβRII cells were phenotypically identical to memory OTI-WT cells before they exhibited lymphoproliferation (data not shown). However, although the expression of CD25, CD122 (IL-2Rβ/IL-15Rβ) and CD127, KLRG1, CD27, CD69, and CD44 was not different, the expanded memory OTI-dnTGFβRII cells showed reduced CD43, CD8α, and CD62L expression when compared with OTI-WT cells at about 300 days p.i. (Fig. 2H). As previously reported (29), these results demonstrate that over-expression of dnTGFβRII results in accumulation of dysregulated CD8+ T cells that phenotypically resemble memory CD8+ cells.
Deletion of Tgfbr2 does not lead to dysregulated expansion of late memory OTI cells upon Listeria infection
To eliminate any potential effect of TGF-β during T cell development and priming and to further focus on the role of TGF-β signaling on activated and memory CD8+ T cells, we crossed OTI-Tgfbr2f/f-Rag1−/− mice to GzmB-Cre animals (OTI-GzmBCre-Tgfbr2f/f). Interestingly, although the OTI-GzmBCre-Tgfbr2f/f cells had an increased percentage of SLECs as was seen in OTI-dnTGFβRII and OTI-Cd4Cre-Tgfbr2f/f cells during effector phase of Listeria infection (Fig. 1C and 1F, and data not shown), the frequency of OTI-control and OTI-GzmBCre-Tgfbr2f/f cells in PBLs was similar at day 7, 14 and 280 p.i. (Fig. 2I). At this late memory time point, almost all of the blood OTI-control cells were KLRG1 negative as expected; however, OTI-GzmBCre-Tgfbr2f/f cells still showed a SLEC and double positive phenotype, as ~35% of the cells were still KLRG1+ (Fig. 2J), again confirming the role of TGF-β signaling in controlling the survival of the SLEC population. Furthermore, no significant difference was observed in the percentage of OTI-control and OTI-GzmBCre-Tgfbr2f/f cells in the spleen even at day 438 p.i. (Fig. 2K). These results demonstrate that the accumulation of late memory OTI cells is unique to the dnTGFβRII model, and it is not recapitulated in the conditional KO system.
Development and proliferation of late memory OTI-dnTGFβRII cells is not dependent on IL-7 or IL-15
Given that IL-7 and IL-15 are important cytokines for memory CD8+ T cell homeostasis, we next examined whether dysregulated expansion of OTI-dnTGFβRII cells is dependent on IL-7 or IL-15 signaling. We previously reported that survival of effector OTI-dnTGFβRII cells is largely dependent on IL-15 signaling (24). However, we found that a small fraction of effector OTI-dnTGFβRII cells could differentiate into memory cells in Il15+/+ or Il15−/− CD11c-dnTGFbRII hosts, and they eventually massively expanded at 225 days p.i. (Fig. 3A and Fig. 3B). OTI-WT cells were hardly detected in the CD11c-dnTGFbRII co-transfer setting (Fig. 3A–3B), possibly as a result of out-competition of OTI-dnTGFβRII cells and reduced engraftment of OTI-WT cells (Fig. S1D). To further examine the role of IL-15 in OTI-dnTGFβRII cell expansion, we isolated these dysregulated memory OTI-dnTGFβRII cells 370 days post LM-OVA immunization, labeled them with CFSE, and re-transferred them into Il15+/+ CD11c-dnTGFbRII or Il15−/− CD11c-dnTGFbRII hosts. The proliferation of donor cells was assessed by cell division-induced loss of CFSE fluorescence. Memory OTI-dnTGFβRII cells proliferated similarly in IL-15 deficient and IL-15 sufficient hosts over a two-week period (Fig. 3C). These results suggest that in contrast to previous reports, IL-15 is not required for the development, survival, and dysregulated proliferation of late memory OTI-dnTGFβRII cells (29).
FIGURE 3.
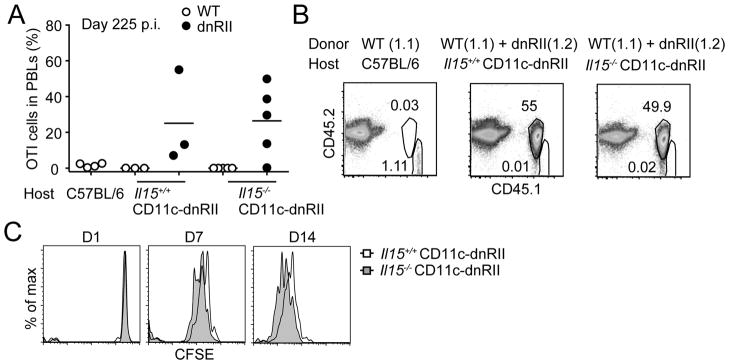
IL-15 is not required for accumulation of late memory OTI-dnTGFβRII cells. (A) C57BL/6 hosts (CD45.2.2) receiving OTI-WT cells (CD45.1.1) (left), Il15+/+ CD11c-dnTGFβRII (middle) or Il15−/− CD11c-dnTGFβRII (right) hosts (CD45.2.2) receiving both OTI-WT (CD45.1.1) and OTI-dnTGFβRII (CD45.1.2) cells, were infected with LM-OVA, and the percentage of donor cells in the PBLs was analyzed at day 225 p.i. Each dot represents 1 mouse. (B) Representative plots of transferred OTI cells in the PBLs shown in (A). (C) Naive OTI-dnTGFβRII cells were transferred into CD11c-dnTGFβRII mice, followed by LM-OVA infection 1 day later. On day 370 p.i., dysregulated memory OTI-dnTGFβRII cells were isolated and CFSE-labeled. CFSE-labeled late memory cells (2 × 106) were transferred into naive Il15+/+ CD11c-dnTGFβRII or Il15−/− CD11c-dnTGFβRII mice. Animals were bled at indicated time points and CFSE dilution was measured. Data are representative of 2 independent experiments with similar results.
To determine whether IL-7 plays a critical role in memory OTI-dnTGFβRII cell dysregulated expansion, we co-transferred OTI-WT and OTI-dnTGFβRII cells into Il7+/+ CD11c-dnTGFβRII or Il7−/− CD11c-dnTGFβRII hosts. Interestingly, effector OTI-dnTGFβRII cells expanded at a much higher magnitude compared to OTI-WT cells in the IL-7 deficient environment than in the IL-7 sufficient environment (Fig. 4A). An increased percentage of SLECs in OTI-dnTGFβRII cells was also observed in IL-7 deficient hosts at day 7 and 12 p.i. (Fig. 4B). Furthermore, similar to the results with Il15−/− CD11c-dnTGFβRII hosts (Fig. 3A), effector OTI-dnTGFβRII cells differentiated into memory T cells and eventually exhibited dysregulated expansion in Il7−/− CD11c-dnTGFβRII mice, and this expansion occurred sooner than in Il7+/+ hosts (Fig. 4C and 4D; compare day 146 p.i.). In addition, when CFSE-labeled dysregulated memory OTI-dnTGFβRII cells were transferred into Il7+/+ CD11c-dnTGFβRII or Il7−/− CD11c-dnTGFβRII host as performed in Fig. 3C, the proliferation of donor cells was accelerated in IL-7 deficient hosts, most likely due to the lymphopenic environment of the Il7−/− hosts (Fig. 4E) (32). Overall, these results suggest that IL-7 is also not essential for the development and dysregulated proliferation of late memory OTI-dnTGFβRII cells.
FIGURE 4.

IL-7 is not required for accumulation of late memory OTI-dnTGFβRII cells. (A) Naive OTI-WT and OTI-dnTGFβRII cells were adoptively co-transferred into naive Il7+/+ CD11c-dnTGFβRII (left) or Il7−/− CD11c-dnTGFβRII (right) hosts, followed by infection with LM-OVA 1 day later. The percentage of OTI-WT and OTI-dnTGFβRII in PBLs was monitored over time. Each dot represents 1 mouse. (B) The percentage of OTI-WT and OTI-dnTGFβRII SLECs in the blood of the same mice as (A) after LM-OVA infection at the indicated time points. (C–D) C57BL/6 hosts (CD45.2.2) receiving OTI-WT (CD45.1.1) cells, Il7+/+ CD11c-dnTGFβRII or Il7−/− CD11c-dnTGFβRII hosts (CD45.2.2) receiving both OTI-WT (CD45.1.1) and OTI-dnTGFβRII (CD45.1.2) cells, were infected with LM-OVA, and the percentage of donor cells in the PBLs was monitored over time. (D) Representative dot plots of transferred OTI-WT and OTI-dnRII cells in the PBLs at day 223 p.i. from (C). (E) Naive OTI-dnTGFβRII cells were transferred into CD11c-dnTGFβRII mice, followed by LM-OVA infection 1 day later. On day 448 p.i., dysregulated memory OTI-dnTGFβRII cells were isolated and CFSE-labeled. CFSE-labeled late memory cells (5 × 105) were transferred into naive Il7+/+ CD11c-dnTGFβRII or Il7−/− CD11c-dnTGFβRII mice. Animals were bled at indicated time points and CFSE dilution was measured. Each dot represents 1 mouse. * p<0.05, ** p<0.01, NS, not significant. Data are representative of 2 independent experiments.
Dysregulated memory OTI-dnTGFβRII cells can proliferate independent of MHC class I
Memory CD8+ T cells can persist in the absence of antigen (1, 3, 4). To examine whether proliferation of dysregulated memory OTI-dnTGFβRII cells is dependent on MHC class I, we isolated massively accumulated memory OTI-dnTGFβRII cells from infected CD11c-dnTGFβRII hosts as performed in Fig. 3C, and transferred (2 × 106 cells) into sublethally irradiated C57BL/6 or b2m−/− mice, which are deficient in the expression of functional MHC class I. At day 30 after transfer, the frequency of transferred OTI-dnTGFβRII cells in both spleen and LNs was greater in b2m−/− mice most likely due to the lymphopenia caused by the absence of host CD8+ T cells (Fig. 5A and 5B). In addition, dysregulated memory OTI-dnTGFβRII cells proliferated extensively in both irradiated C57BL/6 and b2m−/− mice (Fig. 5C), indicating that dysregulated memory OTI-dnTGFβRII do not need MHC class I to survive and proliferate.
FIGURE 5.

Proliferation of dysregulated memory OTI-dnTGFβRII cells is MHC class I-independent. Naive OTI-dnTGFβRII cells were adoptively transferred into CD11c-dnTGFβRII hosts, followed by LM-OVA infection 1 day later. Dysregulated memory OTI-dnTGFβRII cells were isolated and 2 × 106 cells were transferred into sublethally irradiated C57BL/6 and b2m−/− mice. (A) Representative plots of transferred OTI-dnTGFβRII cells in the LNs and spleen 30 days post transfer. Plot represents cells in the live lymphocyte gate. (B–C) The percentage (B) and absolute number (C) of host CD8+ T cells and transferred OTI-dnTGFβRII cell in the LNs and spleen 30 days post transfer. Each dot represents 1 mouse. Data are representative of 2 independent experiments.
OTI-dnTGFβRII cells, but not OTI-CD4Cre-Tgfbr2f/f cells, show dysregulated expansion under steady state condition
C57BL/6 mice expressing dnTGFβRII driven by the Cd2 promoter developed polyclonal memory-like CD8+ T cell hyperproliferation under steady state condition, without developing autoimmunity (29). However, our dnTGFβRII mice on the Rag1+/+ background did not show this phenotype as they develop colitis so rapidly it was not possible to assess this (20). However, we did observe the hyperproliferation phenotype in pathogen-induced memory OTI-dnTGFβRII-Rag1−/− cells (Fig. 2). To eliminate the autoimmune phenotype in dnTGFβRII mice as well as the complications associated with the CD11c-dnTGFβRII hosts, we analysed the phenotype of OTI-dnTGFβRII cells on the Rag1−/− background. The expression of CD44 on OTI cells was analyzed in OTI-dnTGFβRII-Rag1−/− and OTI-CD4Cre-Tgfbr2f/f-Rag1−/− mice under steady state conditions, in which endogenous T cell receptors are excluded and autoimmunity was eliminated. Despite the fact that OTI-dnTGFβRII-Rag1−/− mice do not express the cognate OVA antigen, a large proportion (35~95%) of OTI-dnTGFβRII cells isolated from LNs and spleen were CD44hi memory phenotype by 13 weeks of age, whereas OTI-WT cells retained the CD44lo naive phenotype even at 32 weeks of age (Fig. 6A and data not shown). In contrast to OTI-dnTGFβRII cells, more than half of OTI-CD4Cre-Tgfbr2f/f cells, similar to control cells, still retained CD44lo naive phenotype at 11–25 weeks of age (Fig. 6B). Interestingly, OTI-dnTGFβRII-Rag1−/− mice displayed a considerable increase in the absolute number of OTI cells in both LNs and spleen by 12–32 weeks of age (Fig. 6C). Most of the expanded OTI cells were CD44hi memory-like cells but CD44lo naive phenotype cells were also expanded in some of the animals (Fig. 6A and 6C). Furthermore, the dysregulated dnTGFβRII cell expansion is not OTI-Tg line specific, as it was also seen in lymphocytic choriomeningitis virus-specific CD8+ P14-dnTGFβRII cells (Fig. S2A–D). As expected, consistent with the late memory OTI cell phenotype, this dysregulated expansion of OTI cells was detected only when TGF-β signaling was blocked by over-expression of dnTGFβRII, and not when the whole receptor was deleted (Fig. 6D). Indeed, no significant difference in the absolute number of OTI cells in the LNs and spleen was observed between control and OTI-CD4Cre-Tgfbr2f/f-Rag1−/− mice (Fig. 6D), further demonstrating that the deletion of the TGFβRII itself, at the same time as when the dnTGFβRII would be expressed (thymic double positive stage) does not lead to dysregulated expansion of memory-like OTI cells, even though a fraction of naive OTI-CD4Cre-Tgfbr2f/f cells do become CD44hi over time (Fig. 6B).
FIGURE 6.

OTI-dnTGFβRII cells, but not OTI-CD4Cre-Tgfbr2f/f cells, show dysregulated expansion under steady state condition. (A) The percentage of CD44hi memory-like OTI cells in the LNs of OTI-dnTGFβRII-Rag1−/− or OTI-WT-Rag1−/− mice under steady-state conditions are plotted against age. Representative plots at 20 weeks of age are shown at the bottom. Plots show cells gated on CD8α+TCRβ+ cells. (B) The percentage of CD44hi memory-like OTI cells in the LNs of OTI-control (CD4Cre-Tgfbr2f/+ or Tgfbr2f/f)-Rag1−/− or OTI-CD4Cre-Tgfbr2f/f-Rag1−/− mice under steady-state conditions are plotted against age. Representative plots at 22 weeks of age are shown at the bottom. Plots show cells gated on CD8α+TCRβ+ cells. (C) The absolute number of CD44hi memory-like OTI cells in the LNs and spleen of OTI-dnTGFβRII-Rag1−/− or OTI-WT-Rag1−/− mice under steady-state conditions are plotted against age. (D) The absolute number of CD44hi memory-like OTI cells in the LNs and spleen of OTI-control (CD4Cre-Tgfbr2f/+ or Tgfbr2f/f)-Rag1−/− or OTI-CD4Cre-Tgfbr2f/f-Rag1−/− mice under steady-state conditions are plotted against age. (E) Cell surface makers on the CD8α+TCRβ+ OTI cells from the LNs of indicated 7-week old Rag1−/− mice under steady-state conditions. Data are representative of 2 independent experiments. Each dot represents 1 mouse.
Rapid differentiation of naive OTI-dnTGFβRII cells into CD44hi memory-like cells occurs even in the absence of endogenous Tgfbr2 gene
Because the effects of TGF-β signaling in T cell differentiation is context dependent (38), it is possible that a low level of leaked TGF-β signaling in OTI-dnTGFβRII cells promotes CD44hi memory CD8+ T cell differentiation, which then somehow causes progressive expansion. To examine this possibility, we crossed OTI-dnTGFβRII-Rag1−/− mice with CD4Cre-Tgfbr2f/f-Rag1−/− mice to completely delete TGF-β signaling in naive OTI cells. We still found an increased percentage of CD44hiCD122hi memory-like cells in the LNs of OTI-dnTGFβRII-Rag1−/− × CD4Cre-Tgfbr2f/f-Rag1−/− mice when compared with OTI-CD4Cre-Tgfbr2f/f cells (Fig. 6E), suggesting a dominant effect of truncated form of TGFβRII on CD8+ T cell expansion that is independent of blocking TGF-β signaling in these cells.
Memory OTI-dnTGFβRII cells develop lymphoma
As memory-like CD8+ T cells that express dnTGFβRII under the control of the Cd2 promoter have been shown to develop lymphoma (28), we analyzed whether over-expression of dnTGFβRII by Cd4 promoter also causes cellular transformation. Dysregulated memory-like OTI-dnTGFβRII cells or Listeria-induced memory OTI-dnTGFβRII cells were isolated from 6 months old OTI-dnTGFβRII-Rag1−/− mice or CD11c-dnTGFβRII hosts at day 420 p.i., respectively, and karyotype analysis was performed. These cells showed chromosomal abnormality, including trisomy of chromosome 15 and reciprocal translocation between chromosome 4 and 15. (Table 1 and Fig. 7A). Gain of chromosome 15 is frequently present in several mouse models of lymphoma (39–41), and it has been suggested that Myc protooncogene located on mouse chromosome 15 contribute to trisomy 15 and leukemic transformation (42). We therefore analysed Myc expression in memory-like OTI-dnTGFβRII cells from 2–3 months old OTI-dnTGFβRII-Rag1−/− mice before they exhibited dysregulated expansion. We found that although the expression level of Myc target genes, such as Id2 (inhibitor of DNA binding 2) (43) and Cdkn1a (cyclin-dependent kinase inhibitor 1A; also called p21) (44) were increased in CD44hi memory-like OTI-dnTGFβRII cells, Myc expression did not differ between these groups (Fig. 7B). However, when we analyzed dysregulated memory-like OTI-dnTGFβRII cells isolated from OTI-dnTGFβRII-Rag1−/− mice at 6 months of age, Myc expression was increased in memory-like OTI-dnTGFβRII cells compared with that of OT-I WT cells (Fig. 7C). These results suggest that over-expression of dnTGFβRII in OTI cells by Cd4 promoter results in cellular transformation most likely through a Myc-mediated mechanism.
Table 1.
Karyotype analysis of OTI-dnTGFβRII cells.
| ID | Cell types | Sampling | Karyotype |
|---|---|---|---|
| 1 | Memory-like OTI | 6 months old | 41,XY +15 [17/30]* 40,XY [13/30] |
| 2 | Memory-like OTI | 6 months old | 41,XY +15 [8/30] 40,XY [22/30] |
| 3 | Listeria-induced memory OTI | 420 days p.i. | 40,XX [3/20] 41,XX +15 [15/20] 40,XX Der(4)T(4E;15A2) [2/20] |
Number of chromosomes with aberration/total number of chromosomes studied.
FIGURE 7.
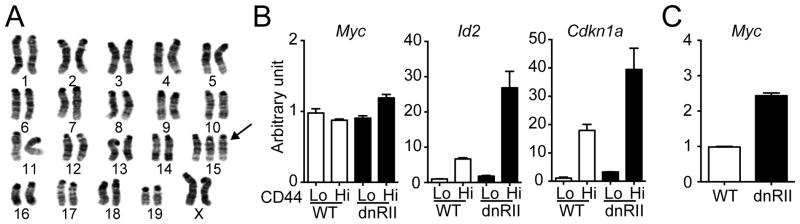
Memory OTI-dnTGFβRII cells develop lymphoma. (A) Karyotype analysis of late memory OTI-dnTGFβRII cells isolated form LM-OVA-infected mice at day 420 p.i. as described in Fig. 1. Arrow indicates trisomy of chromosome 15. (B) CD44lo naive or CD44hi memory-like OTI cells were isolated from OTI-dnTGFβRII-Rag1−/− or OTI-WT-Rag1−/− mice at 2–3 months of age (prior to cellular transformation), and the mRNA expression of Myc, Id2 and Cdkn1a was determined with real-time PCR. The RNA sample was pooled from 3–4 mice for each group. The mRNA expression level in the CD44lo OTI-WT cells is defined as 1. Data represent the means +/− SEM. (C) CD44hi memory-like OTI cells were isolated from OTI-dnTGFβRII-Rag1−/− or OTI-WT-Rag1−/− mice at 6 months of age (after cellular transformation), and the mRNA expression of Myc was determined with real-time PCR. The RNA sample was pooled from 3–4 mice for each group. The mRNA expression level in the OTI-WT cells is defined as 1. Data represent the means +/− SEM. (B–C) Data are representative of 2 independent experiments.
Transformed memory OTI-dnTGFβRII cells produce effector cytokines and have CTL activity, but can not proliferate after recall response
To determine whether transformed memory OTI-dnTGFβRII cells retain their functionality as memory cells, we assessed the capacity of these cells to produce effector cytokines, and to kill antigen-sensitized target cells in vivo. We isolated memory OTI-WT cells or transformed memory OTI-dnTGFβRII cells from infected C57BL/6 or CD11c-dnTGFbRII mice at day 380 p.i,, respectively, and re-stimulated them with SIINFEKL peptide ex vivo. Interestingly, the transformed memory OTI-dnTGFβRII cells were functional in terms of IFN-γ, TNF and GzmB production, when compared to memory OTI-WT cells (Fig 8A). Furthermore, when the same number of each memory OTI population was re-transferred into naive hosts along with SIINFEKL-pulsed target cells, no difference was observed in the in vivo ability of OTI-dnTGFβRII cells to kill target cells when compared to memory OTI-WT cells (Fig. 8B), suggesting that they also retain their cytolytic activity. Similar results were also obtained when we used early memory OTI-WT and OTI-dnTGFβRII cells that had not yet exhibited dysregulated expansion (Fig. S3A and S3B). Similarly, early memory OTI-CD4Cre-Tgfbr2f/f cells exhibited a normal ability to produce IL-2 and IFN-γ after SIINFEKL peptide stimulation ex vivo (Fig. S4A). Interestingly, however, transformed memory OTI-dnTGFβRII cells proliferated less than memory OTI-WT cells in response to SIINFEKL peptide or anti-CD3 ex vivo, while they responded similar to OTI-WT cells to PMA/ionomycin stimulation (Fig. 8C). Furthermore, transformed memory OTI-dnTGFβRII cells did not respond to IL-2 or IL-15 stimulation (Fig. 8D), whereas early memory OTI-dnTGFβRII cells isolated from the hosts at day 70 p.i. proliferated similarly to OT-WT cells in response to these cytokines (Fig. S3C). To determine the ability of transformed memory OTI-dnTGFβRII cells to respond to a secondary infection in vivo, we isolated transformed memory OTI-dnTGFβRII cells and memory OTI-WT cells at day 461 p.i., and re-transferred the same number of cells from each into naive hosts followed by LM-OVA infection. Consistent with results from ex vivo experiments, OTI-dnTGFβRII cells responded poorly to a secondary infection, whereas memory OTI-WT cells mounted a proliferative response (Fig. 8E). In contrast, no difference in proliferative response was observed when we transferred memory OTI-WT and OTI-dnTGFβRII cells isolated 180 days p.i. that had not exhibited dysregulated expansion (Fig. S3D). The early memory OTI-control and OTI-GzmBCre-Tgfbr2f/f cells also expanded similarly in vivo upon re-challenge (Fig. S4B). These results indicate that blocking TGF-β signaling on memory CD8+ T cells does not impact their function and recall response. In addition, although transformed late memory OTI-dnTGFβRII cells are capable of producing effector cytokines and killing target cells, they have defects in antigen- or cytokine-induced proliferative responses. The consequence of long-term over expression of dnTGFβRII should be significantly considered for studies where the over expression of this construct is used for enhancing T cell function during T cell mediated immunotherapy.
FIGURE 8.

Transformed late memory OTI-dnTGFβRII cells have effector and CTL activity, but do not proliferate after recall response. Adoptive transfer and LM-OVA infection was performed as described in Fig. 2. (A) Intracellular cytokine staining of splenocytes isolated at day 380 p.i. and stimulated ex vivo with SIINFEKL peptide. Memory OTI-WT and transformed memory OTI-dnTGFβRII cells were identified based on staining with CD8α and the corresponding congenic markers. (B) In vivo CTL assay performed with memory OTI-WT and transformed memory OTI-dnTGFβRII cells isolated from the spleen of infected mice at day 380 p.i. Representative histograms are shown on the left. NS, not significant compared to OTI-WT. (C–D) Memory OTI-WT or transformed memory OTI-dnTGFβRII cells isolated from LM-OVA-infected mice at day 375 p.i. were CFSE-labeled, and stimulated with indicated mitogens (C) or cytokines (D). CFSE dilution was analyzed 2 days (C) or 4 days (D) after stimulation. (E) Memory OTI-WT or transformed memory OTI-dnTGFβRII cells isolated from LM-OVA-infected mice at day 461 p.i. (5 × 104 cells) were transferred into naive CD11c-dnTGFβRII recipients that were subsequently infected with LM-OVA. The percentage of OTI-WT and OTI-dnTGFβRII in PBLs was monitored over time. NS, not significant versus OTI-WT. *** p<0.001 versus OTI-WT. Data are representative of 2 independent experiments.
Discussion
In this study, we utilized two different mouse models, Tg mice expressing truncated form of TGFβRII under the Cd4 promoter and TGFβRII conditional knockout mice crossed to GzmB- or CD4-Cre mice to address the role of TGF-β signaling in memory CD8+ T cell development, function, and homeostatic proliferation. We demonstrate that antigen-induced expansion and cytokine production of memory CD8+ T cells is not effected by the blockade of TGF-β signaling in both genetic models. Interestingly however, we observe a massive lymphoproliferative disorder in both antigen-experienced and homeostatically generated memory CD8+ T cells in the Tg model only, suggesting that the truncated form of TGFβRII has functions that are independent of endogenous TGFβRII signaling. This study has therefore revealed a previously unrecognized function of truncated TGFβRII in CD8+ T cell differentiation and proliferation, which should be considered during the potential therapeutic use of the dnTGFβRII as a means to enhance antigen-specific CD8+ T cell function.
Blockade of TGF-β signaling by over-expressing dnTGFβRII using a tissue-specific promoter has been widely used to study the role of TGF-β signaling in vivo. We expected that conditional deletion of Tgfbr2 in CD8+ T cells would result in a much stronger phenotype compared to CD8+ dnTGFβRII cells, because the latter still express a functional TGFβRII that can induce some level of TGF-β signaling (18, 20). However, we found that memory OTI-dnTGFβRII cells expanded massively and developed a lymphoproliferative disorder both in Listeria infected hosts and in unimmunized OTI-dnTGFβRII-Rag1−/− mice, whereas the OTI-conditional TGFβRII KO cells lacked this phenotype. These results indicate that dnTGFβRII itself has functions independent of the endogenous TGFβRII in CD8+ T cells. In support of this, increased differentiation of naive OTI cells into CD44hi memory-like T cells was still observed in OTI-dnTGFβRII × CD4Cre-Tgfbr2f/f mice on the Rag1−/− background. Previous reports showed that immunocompetent mice expressing dnTGFβRII driven by Cd2 promoter exhibited dysregulated memory CD8+ T cell homeostasis and cellular transformation without any sign of autoimmune disease (26), whereas the dnTGFβRII-Rag1+/+ strain driven by Cd4 promoter developed autoimmune disease prior to any obvious signs of memory CD8+ T cell expansion (20). By contrast, a recent study demonstrated that lymphoproliferative disorder can also be seen in the dnTGFβRII-Rag1+/+ strain driven by Cd4 promoter, in which dnTGFβRII are homozygously expressed and IL-6 or IL-12p40 were also deleted to prevent autoimmune disease development (45). The reason for the differences observed between the two dnTGFβRII strains is unknown, but it is suggested that differential expression of the dnTGFβRII transgene may affect the efficiency of TGF-β signaling blockade. However, we showed here that dnTGFβRII phenotype is not recapitulated in the conditional KO system, suggesting that lymphoproliferative disorder seen by increasing copy number of dnTGFβRII is not simply due to increased suppression of TGF-β signaling. In fact, dysregulated memory OTI-dnTGFβRII cell homeostasis was observed despite the use of hemygygous OTI-dnTGFβRII-Rag1−/− mice in this study. In this regard, over-expression of dnTGFβRII in a tumor cell line has been shown to impair both TGF-β1 and BMP2 signaling (46). Thus, it is possible that dnTGFβRII interacts with receptors other than TGFβRI and also block TGF-β-independent signaling pathway such as BMP signaling, which may have synergistic effect on proliferation and transformation of memory CD8+ T cells. Further studies, outside the scope of the current study, are required to identify such a dnTGFβRII-binding ligand, the composition of the receptor complex as well as the downstream signaling pathway(s) in CD8+ T cells.
As the human origin of the dnTGFβRII, rendered this molecule immunogenic in mice, we had to adoptively transfer the OTI-dnTGFβRII cells into the CD11c-dnTGFβRII hosts, which also express the same transgene in the NK and dendritic cell compartments and thus are tolerant to the transgene (24, 36). The naive OTI-dnTGFβRII and OTI-WT cells highly expanded after immunization, and differentiated memory cells successfully engrafted in the CD11c-dnTGFβRII hosts; however, the OT-WT cell engraftment of memory cells diminished over time in these hosts. On the rare occasions that we were able to find engrafted OTI-WT late memory cells in the CD11c-dnTGFβRII hosts, they did not display the same expansion phenotype as the OTI-dnTGFβRII cells, and the host CD8+ T cells did not expand in this setting either. Furthermore, we also found that OTI-dnTGFβRII-Rag1−/− or P14-dnTGFβRII-Rag1−/− mice, where the dnTGFβRII transgene is only expressed in the CD8+ T cells, also showed accumulation of memory-like OTI or P14 cells under steady state condition. Therefore, we believe that the expansion and transformation of late memory OTI-dnTGFβRII cells is cell intrinsic, although we cannot completely exclude the contributions from the CD11c-dnTGFβRII transgene on our findings.
We found that the kinetics of proliferation between OTI-dnTGFβRII cells isolated from Listeria-infected CD11c-dnTGFβRII host and those from uninfected OTI-dnTGFβRII-Rag1−/− mice is clearly different; in the former case, cells start expanding around 180–220 days (25–32 wks) p.i., whereas homeostatic proliferation begins around 10–12 weeks of age in the latter hosts. After Listeria infection in the CD11c-dnTGFβRII hosts, the sorted naive OTI-dnTGFbRII cells clonally expand, contract, form proper memory cells and remain quiescent until about 180 days p.i., after which they form lymphomas and begin proliferating again. Several studies have shown that CD8+ T cell clonal expansion (TCE), which is nonmalignant expansion of monoclonal populations of CD8+ T cells, can appear with increasing frequency as individuals and mice age (47, 48). In fact, we did observe this phenomenon in our aged mice as the frequency of OTI-WT cells in the PBLs of C57BL/6 hosts was slightly increased over time. However, unlike the CD11c-dnTGFβRII host that received OTI-dnTGFβRII cells, mice having TEC do not show increased number of total CD8+ T cells, but rather an expansion of a sub-clone of the CD8+ T cells (47, 48). It has been reported that transfer of oncogene-expressing TCR-Tg T cell (OTI or P14) into Rag1−/− hosts induces lymphoma, and transformation of these cells is suppressed by co-transferring polyclonal T cells (49). This clonal competition model may explain the difference between these two experimental models; presence of polyclonal T cells in CD11c-dnTGFβRII hosts may delay proliferation and transformation of memory OTI-dnTGFbRII cells after Listeria infection.
Given the essential role of IL-15 and IL-7 in regulating memory CD8+ T cell proliferation and survival, and the critical role of these cytokine signaling pathways in lymphoma development (10, 12), we examined the involvement of these cytokines in expansion of memory OTI-dnTGFβRII cells. However, we found that neither IL-15 nor IL-7 was required for the development and expansion of memory OTI-dnTGFβRII cells, as the OTI-dnTGFβRII cells showed enhanced accumulation even in the Il7−/− and Il15−/− mice. Furthermore, we also observed that dysregulated memory-like OTI-dnTGFβRII cells transferred into Il7−/−Il15−/− mice massively proliferated (data not shown). Although transformed memory OTI-dnTGFβRII cells displayed normal CD122 and CD127 expression, cytokine production and cytolytic activity, they had a poor proliferative response to IL-2 or IL-15, and exhibited a reduced capacity to participate in recall proliferative response both in vivo and in vitro, whereas early memory OTI-dnTGFβRII cells that had not yet exhibited dysregulated expansion did proliferate and responded to cytokines. These results indicate that the normal homeostatic regulation of memory CD8+ T cells becomes lost once they become lymphomas. However, naive OTI-CD2-dnTGFβRII cells proliferated extensively in Il7−/−Il15−/− hosts (27), suggesting that additional cytokines or mediators may regulate the proliferation and maintenance of CD8+ T cells expressing dnTGFβRII.
TCR signaling has been shown to play an important role in lymphoma development in several murine lymphoma models (50, 51). Furthermore, it has been reported that the TCR-MHC interaction is required for the development of memory-like CD8+ CD2-dnTGFβRII T cells (27, 29). Indeed, male, but not female, HY-TCR-Tg CD2-dnTGFβRII mice on the Rag1−/− background showed an increased accumulation of CD44hi CD8+ T cells (29), and naive OTI-CD2-dnTGFβRII cells failed to expand in MHC class I deficient host (27). However, we found that transformed memory OTI-dnTGFβRII cells could expand in an MHC class I deficient environment, although the molecular mechanisms by which these cells can proliferate so extensively are still under investigation. It is likely that presence of dnTGFβRII in CD8+ T cells may induce cell intrinstic alterations, such as acquisition of mutations or epigenetic alteration that affect a common signaling pathway that leads to cell proliferation. Indeed, we found chromosomal abnormalities in infection-induced late memory OTI- or memory-like OTI-dnTGFβRII cells. Up-regulation of proto-oncogenes, such as Myc, and epigenetic changes may provide a mechanism by which the growth properties of memory CD8+ dnTGFβRII T cells could be altered, ultimately resulting in a CD8+ T cell expansion and transformation (45). Thus, we suggest that TCR signaling is most likely required for initial proliferation and transformation of CD8+ dnTGFβRII cells; however, it does not drive the proliferation of the lymphoma after transformation.
We previously reported that TGF-β selectively controls the number of SLECs during effector phase of Listeria infection (24). However, in contrast to OTI-dnTGFβRII cells, the present study, and a recent published paper showed that the total number of effector OT-I cells was not affected by depletion of TGF-β signaling using a conditional KO system (52). We do, however show here that TGF-β signaling regulates the ratio between SLECs and MPECs; nonetheless, TGF-β signaling deficient early memory CD8+ T cells produce normal level of effector cytokines, and mount recall responses, suggesting that TGF-β signaling is not essential for memory CD8+ T cell function during Listeria infection. The role of TGF-β signaling in CD8+ T cells in infectious diseases is still unclear. Previous studies showed that dnTGFβRII mice showed rapid viral eradication, and more functional effector and memory CD8+ T cells after chronic viral infection (25, 53), whereas therapeutic blockade of TGF-β signaling by a neutralizing Ab or small inhibitor did not recapitulate the phenotype of dnTGFβRII mice (53, 54). This discrepancy might be due to the effects of spontaneous accumulation of memory-like CD8+ T cells in dnTGFβRII mice (53). As blockade of TGF-β signaling by dnTGFβRII results in unexpected effects in CD8+ T cells, future studies need to address the mechanism of regulation of SLEC formation by TGF-β signaling, and its contribution to host protection against pathogens using conditional KO system.
In summary, we have provided evidence showing that TGF-β signaling in CD8+ T cells controls the formation of SLECs during the effector phase of Listeria infection. However, TGF-β signaling does not regulate the homeostatic proliferation of memory CD8+ T cells and transgenic over-expression of dnTGFβRII leads to dysregulated expansion and ultimately transformation of memory CD8+ T cells. Thus, investigators should be cautious concerning the use of these different models to study the role of TGF-β signaling in CD8+ T cells and the potential use of the dnTGFβRII for therapeutic purposes. Nonetheless, identification of the dnTGFβRII-dependent alternative signaling pathway might provide a new mechanism for the regulation of memory CD8+ T cell expansion and lymphoma development.
Supplementary Material
Acknowledgments
We thank Caroline Lieber for assistance with manuscript preparation, and the Laboratory of Genome Biology and Cytogenesis (Van Andel Institute) for karyotype analysis.
This work was supported by the Juvenile Diabetes Research Foundation (4-2007-1059) (R.A.F.) and Uehara Memorial Foundation Fellowship (H.I.). R.A. Flavell is an investigator of the Howard Hughes Medical Institute.
Footnotes
Disclosures
The authors declare no competing financial interests.
References
- 1.Cui W, Kaech SM. Generation of effector CD8+ T cells and their conversion to memory T cells. Immunol Rev. 2010;236:151–166. doi: 10.1111/j.1600-065X.2010.00926.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lefrancois L, Obar JJ. Once a killer, always a killer: from cytotoxic T cell to memory cell. Immunol Rev. 2010;235:206–218. doi: 10.1111/j.0105-2896.2010.00895.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sallusto F, Lanzavecchia A, Araki K, Ahmed R. From vaccines to memory and back. Immunity. 2010;33:451–463. doi: 10.1016/j.immuni.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang N, Bevan MJ. CD8+ T cells: foot soldiers of the immune system. Immunity. 2011;35:161–168. doi: 10.1016/j.immuni.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sprent J, Surh CD. Normal T cell homeostasis: the conversion of naive cells into memory-phenotype cells. Nat Immunol. 2011;131:478–484. doi: 10.1038/ni.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hamilton SE, Jameson SC. CD8 T cell quiescence revisited. Trends Immunol. 2012;33:224–230. doi: 10.1016/j.it.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marrack P, Kappler J. Control of T cell viability. Annu Rev Immunol. 2004;22:765–787. doi: 10.1146/annurev.immunol.22.012703.104554. [DOI] [PubMed] [Google Scholar]
- 8.Lodolce JP, Boone DL, Chai S, Swain RE, Dassopoulos T, Trettin S, Ma A. IL-15 receptor maintains lymphoid homeostasis by supporting lymphocyte homing and proliferation. Immunity. 1998;9:669–676. doi: 10.1016/s1074-7613(00)80664-0. [DOI] [PubMed] [Google Scholar]
- 9.Kennedy MK, Glaccum M, Brown SN, Butz EA, Viney JL, Embers M, Matsuki N, Charrier K, Sedger L, Willis CR, Brasel K, Morrissey PJ, Stocking K, Schuh JC, Joyce S, Peschon JJ. Reversible defects in natural killer and memory CD8 T cell lineages in interleukin 15-deficient mice. J Exp Med. 2000;191:771–780. doi: 10.1084/jem.191.5.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fehniger TA, Suzuki K, Ponnappan A, VanDeusen JB, Cooper MA, Florea SM, Freud AG, Robinson ML, Durbin J, Caligiuri MA. Fatal leukemia in interleukin 15 transgenic mice follows early expansions in natural killer and memory phenotype CD8+ T cells. J Exp Med. 2001;193:219–231. doi: 10.1084/jem.193.2.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kieper WC, Tan JT, Bondi-Boyd B, Gapin L, Sprent J, Ceredig R, Surh CD. Overexpression of interleukin (IL)-7 leads to IL-15-independent generation of memory phenotype CD8+ T cells. J Exp Med. 2002;195:1533–1539. doi: 10.1084/jem.20020067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rich BE, Campos-Torres J, Tepper RI, Moreadith RW, Leder P. Cutaneous lymphoproliferation and lymphomas in interleukin 7 transgenic mice. J Exp Med. 1993;177:305–316. doi: 10.1084/jem.177.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sato N, Sabzevari H, Fu S, Ju W, Petrus MN, Bamford RN, Waldmann TA, Tagaya Y. Development of an IL-15-autocrine CD8 T-cell leukemia in IL-15-transgenic mice requires the cis expression of IL-15Ralpha. Blood. 2011;117:4032–4040. doi: 10.1182/blood-2010-09-307504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol. 2006;24:99–146. doi: 10.1146/annurev.immunol.24.021605.090737. [DOI] [PubMed] [Google Scholar]
- 15.Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limon P. The polarization of immune cells in the tumour environment by TGFbeta. Nat Rev Immunol. 2010;10:554–567. doi: 10.1038/nri2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kang JS, Liu C, Derynck R. New regulatory mechanisms of TGF-beta receptor function. Trends Cell Biol. 2009;19:385–394. doi: 10.1016/j.tcb.2009.05.008. [DOI] [PubMed] [Google Scholar]
- 17.Massague J. TGFbeta in Cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li MO, Sanjabi S, Flavell RA. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity. 2006;25:455–471. doi: 10.1016/j.immuni.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 19.Marie JC, Liggitt D, Rudensky AY. Cellular mechanisms of fatal early-onset autoimmunity in mice with the T cell-specific targeting of transforming growth factor-beta receptor. Immunity. 2006;25:441–454. doi: 10.1016/j.immuni.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 20.Gorelik L, Flavell RA. Abrogation of TGFbeta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity. 2000;12:171–181. doi: 10.1016/s1074-7613(00)80170-3. [DOI] [PubMed] [Google Scholar]
- 21.Gorelik L, Flavell RA. Immune-mediated eradication of tumors through the blockade of transforming growth factor-beta signaling in T cells. Nat Med. 2001;7:1118–1122. doi: 10.1038/nm1001-1118. [DOI] [PubMed] [Google Scholar]
- 22.Wang L, Wen W, Yuan J, Helfand B, Li Y, Shi C, Tian F, Zheng J, Wang F, Chen L, Liang L, Zhou L, Lee C, Chen Z, Guo Y, Wang H, Zhang Q, Qin W. Immunotherapy for human renal cell carcinoma by adoptive transfer of autologous transforming growth factor beta-insensitive CD8+ T cells. Clin Cancer Res. 2010;16:164–173. doi: 10.1158/1078-0432.CCR-09-1758. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Q, Yang X, Pins M, Javonovic B, Kuzel T, Kim SJ, Parijs LV, Greenberg NM, Liu V, Guo Y, Lee C. Adoptive transfer of tumor-reactive transforming growth factor-beta-insensitive CD8+ T cells: eradication of autologous mouse prostate cancer. Cancer Res. 2005;65:1761–1769. doi: 10.1158/0008-5472.CAN-04-3169. [DOI] [PubMed] [Google Scholar]
- 24.Sanjabi S, Mosaheb MM, Flavell RA. Opposing effects of TGF-beta and IL-15 cytokines control the number of short-lived effector CD8+ T cells. Immunity. 2009;31:131–144. doi: 10.1016/j.immuni.2009.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tinoco R, Alcalde V, Yang Y, Sauer K, Zuniga EI. Cell-intrinsic transforming growth factor-beta signaling mediates virus-specific CD8+ T cell deletion and viral persistence in vivo. Immunity. 2009;31:145–157. doi: 10.1016/j.immuni.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lucas PJ, Kim SJ, Melby SJ, Gress RE. Disruption of T cell homeostasis in mice expressing a T cell-specific dominant negative transforming growth factor beta II receptor. J Exp Med. 2000;191:1187–1196. doi: 10.1084/jem.191.7.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson LD, Jameson SC. TGF-beta sensitivity restrains CD8+ T cell homeostatic proliferation by enforcing sensitivity to IL-7 and IL-15. PLoS One. 2012;7:e42268. doi: 10.1371/journal.pone.0042268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lucas PJ, McNeil N, Hilgenfeld E, Choudhury B, Kim SJ, Eckhaus MA, Ried T, Gress RE. Transforming growth factor-beta pathway serves as a primary tumor suppressor in CD8+ T cell tumorigenesis. Cancer Res. 2004;64:6524–6529. doi: 10.1158/0008-5472.CAN-04-0896. [DOI] [PubMed] [Google Scholar]
- 29.Lucas PJ, Kim SJ, Mackall CL, Telford WG, Chu YW, Hakim FT, Gress RE. Dysregulation of IL-15-mediated T-cell homeostasis in TGF-beta dominant-negative receptor transgenic mice. Blood. 2006;108:2789–2795. doi: 10.1182/blood-2006-05-025676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laouar Y, Sutterwala FS, Gorelik L, Flavell RA. Transforming growth factor-beta controls T helper type 1 cell development through regulation of natural killer cell interferon-gamma. Nat Immunol. 2005;6:600–607. doi: 10.1038/ni1197. [DOI] [PubMed] [Google Scholar]
- 31.Lee PP, Fitzpatrick DR, Beard C, Jessup HK, Lehar S, Makar KW, Perez-Melgosa M, Sweetser MT, Schlissel MS, Nguyen S, Cherry SR, Tsai JH, Tucker SM, Weaver WM, Kelso A, Jaenisch R, Wilson CB. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity. 2001;15:763–774. doi: 10.1016/s1074-7613(01)00227-8. [DOI] [PubMed] [Google Scholar]
- 32.von Freeden-Jeffry U, Vieira P, Lucian LA, McNeil T, Burdach SE, Murray R. Lymphopenia in interleukin (IL)-7 gene-deleted mice identifies IL-7 as a nonredundant cytokine. J Exp Med. 1995;181:1519–1526. doi: 10.1084/jem.181.4.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chytil A, Magnuson MA, Wright CV, Moses HL. Conditional inactivation of the TGF-beta type II receptor using Cre:Lox. Genesis. 2002;32:73–75. doi: 10.1002/gene.10046. [DOI] [PubMed] [Google Scholar]
- 34.Jacob J, Baltimore D. Modelling T-cell memory by genetic marking of memory T cells in vivo. Nature. 1999;399:593–597. doi: 10.1038/21208. [DOI] [PubMed] [Google Scholar]
- 35.Ingulli E. Tracing tolerance and immunity in vivo by CFSE-labeling of administered cells. Methods Mol Biol. 2007;380:365–376. doi: 10.1007/978-1-59745-395-0_23. [DOI] [PubMed] [Google Scholar]
- 36.Sanjabi S, Flavell RA. Overcoming the hurdles in using mouse genetic models that block TGF-beta signaling. J Immunol Methods. 2010;353:111–114. doi: 10.1016/j.jim.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Laouar Y, Town T, Jeng D, Tran E, Wan Y, Kuchroo VK, Flavell RA. TGF-beta signaling in dendritic cells is a prerequisite for the control of autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2008;105:10865–10870. doi: 10.1073/pnas.0805058105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li MO, Flavell RA. TGF-beta: a master of all T cell trades. Cell. 2008;134:392–404. doi: 10.1016/j.cell.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dofuku R, Biedler JL, Spengler BA, Old LJ. Trisomy of chromosome 15 in spontaneous leukemia of AKR mice. Proc Natl Acad Sci U S A. 1975;72:1515–1517. doi: 10.1073/pnas.72.4.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gibbons DL, MacDonald D, McCarthy KP, Cleary HJ, Plumb M, Wright EG, Greaves MF. An E mu-BCL-2 transgene facilitates leukaemogenesis by ionizing radiation. Oncogene. 1999;18:3870–3877. doi: 10.1038/sj.onc.1202721. [DOI] [PubMed] [Google Scholar]
- 41.Wiener F, Babonits M, Spira J, Bregula U, Klein G, Merwin RM, Asofsky R, Lynes M, Haughton G. Chromosome 15 trisomy in spontaneous and carcinogen-induced murine lymphomas of B-cell origin. Int J Cancer. 1981;27:51–58. doi: 10.1002/ijc.2910270109. [DOI] [PubMed] [Google Scholar]
- 42.Jones L, Wei G, Sevcikova S, Phan V, Jain S, Shieh A, Wong JC, Li M, Dubansky J, Maunakea ML, Ochoa R, Zhu G, Tennant TR, Shannon KM, Lowe SW, Le Beau MM, Kogan SC. Gain of MYC underlies recurrent trisomy of the MYC chromosome in acute promyelocytic leukemia. J Exp Med. 2010;207:2581–2594. doi: 10.1084/jem.20091071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perk J, Iavarone A, Benezra R. Id family of helix-loop-helix proteins in cancer. Nat Rev Cancer. 2005;5:603–614. doi: 10.1038/nrc1673. [DOI] [PubMed] [Google Scholar]
- 44.Dang CV, O’Donnell KA, Zeller KI, Nguyen T, Osthus RC, Li F. The c-Myc target gene network. Semin Cancer Biol. 2006;16:253–264. doi: 10.1016/j.semcancer.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 45.Zhang W, Tsuda M, Yang GX, Tsuneyama K, He XS, Ansari AA, Ridgway WM, Coppel RL, Lian ZX, Leung PS, Gershwin ME. Lymphoma-like T cell infiltration in liver is associated with increased copy number of dominant negative form of TGFbeta receptor II. PLoS One. 2012;7:e49413. doi: 10.1371/journal.pone.0049413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dumont N, Arteaga CL. A kinase-inactive type II TGFbeta receptor impairs BMP signaling in human breast cancer cells. Biochem Biophys Res Commun. 2003;301:108–112. doi: 10.1016/s0006-291x(02)02977-7. [DOI] [PubMed] [Google Scholar]
- 47.Clambey ET, White J, Kappler JW, Marrack P. Identification of two major types of age-associated CD8 clonal expansions with highly divergent properties. Proc Natl Acad Sci U S A. 2008;105:12997–13002. doi: 10.1073/pnas.0805465105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kohlmeier JE, Connor LM, Roberts AD, Cookenham T, Martin K, Woodland DL. Nonmalignant clonal expansions of memory CD8+ T cells that arise with age vary in their capacity to mount recall responses to infection. J Immunol. 2010;185:3456–3462. doi: 10.4049/jimmunol.1001745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Newrzela S, Al-Ghaili N, Heinrich T, Petkova M, Hartmann S, Rengstl B, Kumar A, Jack HM, Gerdes S, Roeder I, Hansmann ML, von Laer D. T-cell receptor diversity prevents T-cell lymphoma development. Leukemia. 2012;26:2499–2507. doi: 10.1038/leu.2012.142. [DOI] [PubMed] [Google Scholar]
- 50.Wang X, Werneck MB, Wilson BG, Kim HJ, Kluk MJ, Thom CS, Wischhusen JW, Evans JA, Jesneck JL, Nguyen P, Sansam CG, Cantor H, Roberts CW. TCR-dependent transformation of mature memory phenotype T cells in mice. J Clin Invest. 2011;121:3834–3845. doi: 10.1172/JCI37210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Piccaluga PP, Agostinelli C, Califano A, Rossi M, Basso K, Zupo S, Went P, Klein U, Zinzani PL, Baccarani M, Dalla Favera R, Pileri SA. Gene expression analysis of peripheral T cell lymphoma, unspecified, reveals distinct profiles and new potential therapeutic targets. J Clin Invest. 2007;117:823–834. doi: 10.1172/JCI26833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang N, Bevan MJ. TGF-beta signaling to T cells inhibits autoimmunity during lymphopenia-driven proliferation. Nat Immunol. 2012;13:667–673. doi: 10.1038/ni.2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Garidou L, Heydari S, Gossa S, McGavern DB. Therapeutic blockade of transforming growth factor Beta fails to promote clearance of a persistent viral infection. J Virol. 2012;86:7060–7071. doi: 10.1128/JVI.00164-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boettler T, Cheng Y, Ehrhardt K, von Herrath M. TGF-beta Blockade Does Not Improve Control of an Established Persistent Viral Infection. Viral Immunol. 2012;25:232–238. doi: 10.1089/vim.2011.0079. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.