

Abstract
An increasing number of studies involve integrative analysis of gene and protein expression data, taking advantage of new technologies such as next-generation transcriptome sequencing and highly sensitive mass spectrometry (MS) instrumentation. Recently, a strategy, termed ribosome profiling (or RIBO-seq), based on deep sequencing of ribosome-protected mRNA fragments, indirectly monitoring protein synthesis, has been described. We devised a proteogenomic approach constructing a custom protein sequence search space, built from both Swiss-Prot- and RIBO-seq-derived translation products, applicable for MS/MS spectrum identification. To record the impact of using the constructed deep proteome database, we performed two alternative MS-based proteomic strategies as follows: (i) a regular shotgun proteomic and (ii) an N-terminal combined fractional diagonal chromatography (COFRADIC) approach. Although the former technique gives an overall assessment on the protein and peptide level, the latter technique, specifically enabling the isolation of N-terminal peptides, is very appropriate in validating the RIBO-seq-derived (alternative) translation initiation site profile. We demonstrate that this proteogenomic approach increases the overall protein identification rate 2.5% (e.g. new protein products, new protein splice variants, single nucleotide polymorphism variant proteins, and N-terminally extended forms of known proteins) as compared with only searching UniProtKB-SwissProt. Furthermore, using this custom database, identification of N-terminal COFRADIC data resulted in detection of 16 alternative start sites giving rise to N-terminally extended protein variants besides the identification of four translated upstream ORFs. Notably, the characterization of these new translation products revealed the use of multiple near-cognate (non-AUG) start codons. As deep sequencing techniques are becoming more standard, less expensive, and widespread, we anticipate that mRNA sequencing and especially custom-tailored RIBO-seq will become indispensable in the MS-based protein or peptide identification process. The underlying mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium with the dataset identifier PXD000124.
The integrative analysis of gene and protein expression is getting more widespread. Integration of new technologies such as next-generation transcriptome sequencing (RNA-seq)1 and highly sensitive mass spectrometry (MS) emerges as a very powerful method for fast and comprehensive profiling of mammalian proteomes (1, 2). Generally, after MS/MS spectra acquisition, protein sequence database searching (Mascot (3), X!Tandem (4), and OMSSA (5), among others) is used for peptide identification. When performing these searches, publicly available protein databases, such as UniProtKB (6) or Ensembl (7), are commonly used. Although convenient for routine use, these public databases only serve as reference proteomes of experimentally verified and/or predicted protein sequences and thus are not likely to represent the real protein pool of a specific sample or even be all-inclusive. In contrast, translation products predicted based on mRNA-seq data give a more representative expression state of the sample under investigation by viewing the fact that unexpressed gene products are (largely) eliminated according to transcript quantification (8). Hence, the search database would only contain expressed gene products, thus reducing the database size. This is beneficial, because it has been demonstrated that larger databases yield more distraction, lower signal-to-noise ratio, and reduce sensitivity under the search criteria needed to maintain a low false discovery rate (9). Also, databases are usually incomplete with respect to sequence variation information, such as single nucleotide polymorphisms (SNPs) and RNA-splice and -editing variants (8, 10, 11). Without taking them into account, proteomic studies may fail to detect novel, unexplored protein forms (1, 12).
Recently, a new strategy, termed ribosome profiling (or RIBO-seq), based on deep sequencing of ribosome-protected mRNA fragments, monitoring protein synthesis, has been described (13, 14). RIBO-seq assembles a genome-wide snapshot of mRNA that enters the translation machinery thus allowing us to comprehensively determine the in vivo synthesis of true translation products measured at the mRNA level. Furthermore, exploiting the properties of harringtonine or lactimidomycin that cause ribosomes to stall at sites of translation initiation in RIBO-seq experiments enables the study of (alternative) translation initiation sites (a)TIS with sub-codon to single-nucleotide resolution (15–19), a strategy also referred to as global translation initiation sequencing or GTI-seq (19). As a consequence, ribosome profiling is thus more suitable than mRNA-seq to delineate the exact ORFs and thus derive protein sequences, which are highly informative, to create a custom sequence search space for MS/MS-based peptide identification. Although the RIBO-seq outcome by itself can readily be applied to detect coding transcripts, no direct in vivo evidence of the genuine and mature protein products is generated. Mass spectrometry assisted validation is in many cases indispensable (20), as some noncoding transcripts, showing an association with ribosomes, do not result in protein products (21, 22). Needless to say, RIBO-seq-predicted translation products compose the ideal search space for protein identification in MS experiments.
Next to canonical translation products, ribosome profiling (15, 18, 19) enabled the identification of a multitude of previously nonannotated N-terminally extended and truncated protein variants. Furthermore, it was demonstrated that the majority of un-annotated and mainly near-cognate start sites drives the translation of 5′UTR located upstream of the open reading frames (uORFs). Moreover, internal out-of-frame translation products and a small number of translations of polycistronic, ribosome-associated small open reading frames, were observed. In fact, for more than 65% of the annotated proteins, more than one translation initiation site was determined (15).
The aim of this study was to create an ideal search space for mass spectrometry-assisted proteome identification based on sample matching (Next Generation Sequencing-based) ribosome profiling results. We devised an MS/MS database search pipeline, taking full advantage of the RIBO-seq information (i.e. the ribosome-targeted mRNA), by merging the derived translation products with public protein sequence databases to construct the ideal search space. To the 16,570 mouse proteins listed in UniProtKB-SwissProt version 10_12, another 7,785 extra unique translation products, derived from the Ingolia et al. (15) ribosome profiling study, were added. We investigated the identification gain using sample-matched RIBO-seq, gel-free shotgun, and N-terminal combined fractional diagonal Chromatography (COFRADIC) proteomics data from an undifferentiated mouse embryonic stem cell line (i.e. an E14 mESC cell line as used in Ref. 15). We demonstrated that this customized database increases the identification rate in a standard shotgun experiment with an additional 2.5% on the protein level (e.g. new protein products, new protein splice variants, SNP variant proteins, and N-terminally extended forms of known proteins) and 4.9% on the peptide level as compared with only searching against UniProtKB-SwissProt. Furthermore, using this custom sequence database, identification of N-terminal COFRADIC data overall resulted in the identification of over 1,800 protein N termini, including the detection of 16 alternative start sites giving rise to N-terminally extended protein variants besides the identification of four translated uORFs. In addition, characterization of these new translation products revealed the use of near-cognate (non-AUG) start codons. As deep sequencing techniques are becoming more standard, less expensive, and widespread, we anticipate that mRNA-seq and especially custom-tailored RIBO-seq will become indispensable in the MS-based protein or peptide identification process.
EXPERIMENTAL PROCEDURES
mESC Cell Culture and Expansion in Feeder Free Medium
E14Tg2a mESC cells (a kind gift of Prof. I. Chambers, University of Edinburgh, Scotland, UK) were maintained at 21% O2, 5% CO2 and passaged every 2 days on 0.1% gelatin (Chemicon)-coated dishes using TripLETM Express (Invitrogen) as a cell-dissociating agent in 80% KO DMEM high glucose, 20% KO serum replacement, 4 mm l-glutamine, 0.1 mm penicillin/streptomycin, 1× nonessential amino acids, 1 mm sodium pyruvate, 110 mm 2-mercaptoethanol (all from Invitrogen), supplemented with 1 unit/ml murine leukaemia inhibitory factor (Chemicon), 1 mm MEK inhibitor PD0325901 (Axon MedChem), and 3 mm GSK3 inhibitor CHIR99021 (Axon MedChem) (23).
Shotgun Proteome Analysis
15 × 106 mESC E14 cells were lysed by three rounds of freeze-thaw lysis in 1 ml of 50 mm NH4HCO3 (pH 7.9). Lysates were cleared by centrifugation for 15 min at 16,000 × g. Protein concentrations were measured using the protein assay kit (Bio-Rad) according to the manufacturer's instructions. To partially denature proteins, guanidinium hydrochloride (final concentration 0.5 m) and acetonitrile (final concentration 2%) were added to the cleared protein extracts. 1 mg of the protein sample was digested overnight at 37°C using sequencing-grade modified trypsin (Promega, Madison, WI) (enzyme/substrate of 1:100 w/w). Samples were acidified with acidic acid to a final concentration of 0.5%. The digest was vacuum-dried, and the equivalent of 250 μg of the original protein material was loaded onto a reverse phase-HPLC column for fractionation as described previously (24). To prevent oxidation of methionines between reverse phase-HPLC runs, methionines were oxidized in the injector compartment by transferring 20 μl of a freshly prepared aqueous 3% H2O2 solution to a vial containing 90 μl of the acidified peptide mixture (final concentration of 0.54% H2O2). This reaction proceeded for 30 min at 30°C after which the sample was immediately injected onto the reverse phase-HPLC column and separated. Fractions of 0.5 min were collected from 20 to 80 min after sample injection (120 fractions). These peptide fractions were vacuum-dried, and fractions eluting 12 min apart were pooled by re-dissolving these in a final volume of 40 μl of 2 mm tris(2-carboxyethyl)phosphine and 2% acetonitrile, similar to a pooling strategy described previously (24). In total, 24 samples were analyzed by LC-MS/MS.
N-terminal COFRADIC Analysis
Cell lysis, N-terminal COFRADIC analyses, and sample analyses were performed as described previously (25). To enable the assignment of in vivo N-terminal acetylation events, all primary protein amines were blocked making use of an (stable isotopically encoded) N-hydroxysuccinimide ester at the protein level (i.e. N-hydroxysuccinimide-[13C2D3]acetate). In total, 45 samples were analyzed by LC-MS/MS.
LC-MS/MS Analysis Using an Ultimate 3000 RSLC Nano-LTQ Orbitrap Velos System
LC-MS/MS analysis was performed using an Ultimate 3000 RSLC nano-LC-MS/MS system (Dionex, Amsterdam, The Netherlands) in-line connected to an LTQ Orbitrap Velos (Thermo Fisher Scientific, Bremen, Germany). 2 μl of the sample mixture was first loaded on a trapping column (made in-house, 100 μm internal diameter × 20 mm long, 5-μm Reprosil-Pur Basic-C18-HD beads, Dr. Maisch, Ammerbuch-Entringen, Germany). After back-flushing from the trapping column, the sample was loaded on a reverse-phase column (made in-house, 75 μm internal diameter × 150 mm long, 3-μm C18 Reprosil-Pur Basic-C18-HD beads). Peptides were loaded with solvent A′ (0.1% trifluoroacetic acid in 2% acetonitrile) and were separated with a linear gradient from 98% of solvent A″ (0.1% formic acid in 2% acetonitrile) to 50% of solvent B′ (0.1% formic acid in 80% acetonitrile) with a linear gradient of 1.8% of solvent B′ increase per min at a flow rate of 300 nl/min, followed by a steep increase to 100% of solvent B′. The Orbitrap Velos mass spectrometer was operated in a data-dependent mode, automatically switching between MS and MS/MS acquisition for the 10 most abundant peaks in an MS spectrum. Full scan MS spectra were acquired in the Orbitrap at a target value of 1E6 with a resolution of 60,000. The 10 most intense ions were then isolated for fragmentation in the linear ion trap, with a dynamic exclusion of 20 s. Peptides were fragmented after filling the ion trap at a target value of 1E4 ion counts. From the MS/MS data in each LC run, Mascot Generic Files were created using the Mascot Distiller software (version 2.3.2.0, Matrix Science). While generating these peak lists, a grouping of spectra was allowed with a maximum intermediate retention time of 30 s and maximum intermediate scan count of 5. Grouping was done with a 0.005-Da precursor tolerance. A peak list was only generated when the MS/MS spectrum contained more than 10 peaks. There was no de-isotoping, and the relative signal-to-noise limit was set at 2.
Peptide/Protein Identification and Interpretation
The obtained fragmentation spectra were searched against the custom database (combination of UniProtKB-SwissProt and RIBO-seq-derived translation sequences) using three different search engines as follows: OMSSA (version 2.1.9), X!Tandem (TORNADO, version 2010.01.01.04) and Mascot (version 2.3). The first two were run from the SearchGUI graphical user interface, version 1.10.4 (26). A combination of X!Tandem and Mascot was used for the N-terminal COFRADIC analysis, a combination of all three search engines for the shotgun proteome analysis. Note that OMMSA cannot cope with the protease setting semi-ArgC/P needed to analyze the N-terminal COFRADIC data (see below).
For the shotgun proteome data, trypsin was set as a cleavage enzyme allowing for one missed cleavage, and singly to triply charged precursors or singly to quadruply charged precursors were taken into account, respectively, for the Mascot or X!Tandem/OMSSA search engines, and the precursor and fragment mass tolerance were set to 10 ppm and 0.5 Da, respectively. Methionine oxidation to methionine sulfoxide, pyroglutamate formation of N-terminal glutamine, and acetylation (protein N terminus) were set as variable modifications. For the N-terminal COFRADIC analysis, the protease setting semi-ArgC/P (Arg-C specificity with arginine-proline cleavage allowed) was used. No missed cleavages were allowed, and the precursor and fragment mass tolerance were also set to 10 ppm and 0.5 Da, respectively. Carbamidomethylation of cysteine and methionine oxidation to methionine sulfoxide and 13C3D2 acetylation of lysines were set as fixed modifications. Peptide N-terminal acetylation or 13C3D2 acetylation and pyroglutamate formation of N-terminal glutamine were set as variable modifications, and instrument setting was put on ESI-TRAP.
Protein and peptide identifications in addition to data interpretations were done using the PeptideShaker algorithm, setting the false discovery rate to 1% at all levels (protein, peptide, and peptide to spectrum matching). Aforementioned tools and algorithms (SearchGui, X!Tandem, OMSSA, and PeptideShaker) are freely available as open source.
Ribosome Profiling
Raw sequencing reads of the mESC ribosome profiling data (15) were downloaded from the Gene Expression Omnibus (dataset GSE30839). All reads from the control (cycloheximide-treated sample GSM765292) and harringtonine-treated (sample GSM765295) were remapped using bowtie (version 0.12.7) on the mouse genome (assembly version 37) using the protocol described (16).
Genome-wide Visualization
Genome-wide visualization of the experimental data, in combination with publicly available data, was accomplished using an in-house-developed genome browser (H2G2). Information tracks containing the ribosome profile mappings of the cycloheximide-treated sample (generating a translation profile all over the coding mRNA) and harringtonine-treated sample (translation profile accumulation at the TIS) are available (see Ref. 15 for more information). Furthermore, an information track was constructed showing the predicted translation products track, based on the TIS predictions from Ingolia et al. (15) and the UCSC transcript annotation. The genomic locations of the N-terminal peptides identified with the COFRADIC experiment are also visualized in the H2G2 browser. In addition, several other information tracks are available for visualizing public data as follows: genomic information from a local Ensembl (7) instance (NCBIM37.66) and PhastCons conservation scores (27), among others. More information on how to use the H2G2 browser and what login credentials to use can be found in the supplemental File S1.
Swiss-Prot/RIBO-seq Integrated Database Construction
The combined protein database was constructed using translation products derived from the mouse RIBO-seq data presented by Ingolia et al. (15) and all mouse UniProtKB-SwissProt (6) protein sequences (downloaded version 2012_10). Fig. 1 gives an overall representation of the identification strategy. The RIBO-seq-derived translation products were reconstructed based on both the predicted (a)TIS genomic locations (15) and the corresponding mRNA sequences obtained from the UCSC Genome Browser resource (table browser: assembly = NCBI37/mm9, track = Old UCSC Genes). After reconstructing the amino acid sequences, the UCSC identifiers were mapped to UniProtKB-SwissProt identifiers (to safeguard uniformity) using the UCSC gene annotation information (mm9.knownGene table using the Table browser). For several UCSC IDs, mapping to a RefSeq and thus not to a UniProtKB-SwissProt identifier, the Uniprot Mapping Service was applied to retrieve the corresponding UniProtKB-TrEMBL identifiers. Finally, to remove redundancy, introduced by the combination of the RIBO-seq-derived translation products and the set of known UniProtKB-SwissProt protein sequences, duplicated sequences were removed, retaining the UniProtKB-SwissProt sequence where possible. Moreover, only the longest form of a series of gene translation products (N-terminal extended or canonical) is withheld in the combined database. The custom database contains 24,355 sequences as compared with UniProtKB-SwissProt version 2012_10 holding 16,570 proteins. We opted to merge the RIBO-seq-derived translation products to the UniProtKB-SwissProt protein repository for several reasons as follows: (i) Swiss-Prot contains only reviewed, maintained, and well annotated sequences and thus serves well as a species-specific base-line protein repository; (ii) opting for Swiss-Prot instead of trEMBL (containing the UniProtKB unreviewed entries and hence also partial sequences) makes the downstream protein inference task less complex, and (iii) sample specific translation products that are missing in the Swiss-Prot reference will be added to the custom database because they are contained in the RIBO-seq-derived translation products sequence set.
Fig. 1.

Overview of the custom database construction and protein/peptide identification pipeline. Proteome samples were prepared from an mESC cell lysate. Following Velos Orbitrap LC-MS/MS analyses, fragmentation spectra are searched against a custom combined database (DB), merging sequences from a public protein database (e.g. UniProtKB-SwissProt) and sequences derived from RIBO-seq data. The latter sequences are constructed based on the predicted TIS and the known gene models of the corresponding genes. In this study, three different search engines were applied (X!Tandem, OMSSA, and Mascot), and the peptide identification and interpretation were performed using the PeptideShaker algorithm (version 0.18.3).
RESULTS
To record the impact of using the constructed deep proteome database (combining well annotated UniProtKB-SwissProt proteins and RIBO-seq derived translation product sequences), we performed two types of proteome analysis as follows: (i) a regular shotgun proteomic and (ii) an N-terminal COFRADIC approach. Although the former gives an overall assessment on the protein and peptide level, the latter, by enriching for N-terminal peptides, is highly suited for validating the RIBO-seq translation initiation site observations (15).
Shotgun Proteomics
Using the custom combined database as search space, the number of protein identifications increases with 2.64% (from 3,166 to 3,252 protein identifications) as compared with searching the UniProtKB-SwissProt reference set only (see Fig. 2). Along the same line, the number of peptide identifications, using the same 1% false discovery rate threshold, increases with 4.93% (from 29,343 to 30,865 peptide identifications). The majority of these newly identified proteins are recorded in UniProtKB/trEMBL. Only a few originate from peptide identifications that overlap (part of) an N-terminal extension, an exonic region of an alternative spliced isoform, or an SNP mutation site. Similarly and due to the increased protein coverage, these phenomena account for the improved protein identification and score significance for another 1.5% of proteins. Twenty seven protein identifications have an increased coverage because peptide(s) were identified that coincide with a mutation site, another 11 proteins because peptide(s) are partly contained in the N-terminal extension of the RIBO-seq-derived translation product, and another 11 proteins because peptides are located within an exonic region of an alternative translation product (see Fig. 3 exemplifying these three categories). All protein identifications, grouped into the different categories (Fig. 2), are provided as supplemental Table S1. Also, the different search engine performances, measured as validated peptide-to-spectrum matches, are listed in supplemental Table S2.
Fig. 2.

Pie chart representing the different groups of protein identifications obtained from the shotgun proteomics experiment based on searching the custom combined database. The majority of protein identifications (i.e. 3,117 identifications or 95.8% of total) map to canonical, annotated UniProtKB-SwissProt protein entries (highlighted in gray). Slightly more than 2.6% or 135 identifications (green) map to new protein variants (almost all are recorded in UniProt-trEMBL). Around 1.5% of the canonical protein identifications show an improved score searching the custom combined database as compared with a standard searching against UniProtKB-SwissProt. This is because extra tryptic peptides are identified coinciding with an SNP mutation or residing in an N-terminal extension or an exonic region from an alternative splice isoform.
Fig. 3.
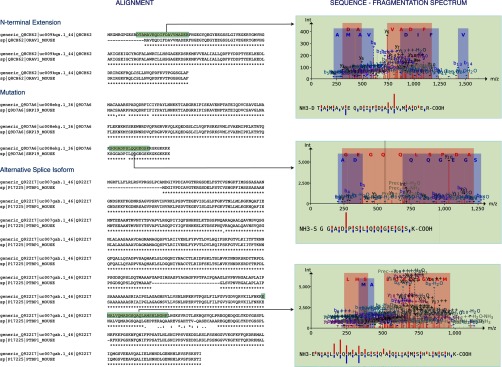
Illustrations of improved/new mouse protein identifications using a shotgun proteomics analysis. Three examples are depicted as follows: (i) a new N-terminally extended protein identification (Swiss-Prot accession Q8CH62); (ii) a new identification because of an SNP mutation (Swiss-Prot accession Q9D7A6), and (iii) a better scoring, improved identification due to the identification of an alternative splicing event (Swiss-Prot accession Q922I7). For all three examples, the Clustal Omega alignment (59) is presented in the left panels, aligning both the annotated UniProtKB-SwissProt and the RIBO-seq-derived amino acid sequence. The tryptic isoform-indicative peptide identification is highlighted in green. In the right panels, the corresponding annotated fragmentation spectra and delineated sequence fragmentation are depicted, indicative of the confidence/quality of the peptide identification.
N-terminal COFRADIC
Protein inference is responsible for the fact that specific ORFs (i.e. truncated protein forms) are difficult to monitor using conventional LC-MS/MS-based analyses of total protein digests. Because the protein N termini are highly indicative for the N-terminal protein isoforms expressed and thus indicative for the TIS identified by means of GTI-seq, we applied the N-terminal COFRADIC positional proteomics approach, strongly enriching for protein N-terminal peptides (28). Prior to tryptic digestion, all primary amines are modified by 13C2D3 acetylation, which allows us to differentiate between in vivo-acetylated and free N termini (in vitro 13C2D3-acetylated) by introducing a spacing of 5 Da between these types of N-terminal peptides. If needed, this labeling strategy allows for a straightforward calculation of the extent of N-terminal acetylation (29). After tryptic digestion, all protein N-terminal peptides will thus be blocked, whereas internal peptides acquired a newly generated primary α-amine, a property exploited to isolate N-terminal peptides from internal peptides in a diagonal chromatography setup that follows strong cation exchange enrichment at low pH (25).
To select for TIS-indicative N termini, we relied on the co-translational nature of N-terminal acetylation of protein N termini (30) by N-terminal acetyltransferases, the near universal requirement of a Met-encoding (or near-cognate) initiator codon (iMet) and the co-translational processing of iMet by methionine aminopeptidases (rules reported in Ref. 31). Following LC-MS/MS analyses and combined database searching, we identified 1,835 TIS-indicative N termini; an overview of the different categories of these N termini is presented in Fig. 4. The majority of the identified N termini map to canonical translation start sites (1,556 peptides, 84.8%). Another 259 peptides (14.1%) start beyond protein position 2 and are indicative for alternative or wrongly annotated protein translation initiation sites. The peptides positioned in RIBO-seq-annotated N-terminal extensions (16 occurrences, 0.9%) and uORFs (completely within the 5′UTR or out-of-frame and overlapping with canonical CDS; 4 occurrences, 0.2%) could only be identified using the custom combined database strategy. These extended translation products point to upstream TIS (uTIS). Compared with the RIBO-seq sequencing results (see supplemental Fig. S1) (15), it is clear that the N-terminal COFRADIC technique only validated a small number of translated uORFs and N-terminal extensions (relative to the annotated CDS), although in sharp contrast, nearly half of the identified translation products from the RIBO-seq results consist of translated uORFs (supplemental Fig. S1). Although N-terminal COFRADIC appears to be more sensitive as compared with a regular shotgun proteomics for identifying some uORF translation products, it is clear that RIBO-seq outranks both proteomics technologies.
Fig. 4.

Pie chart representing the different classes of selected N-terminal mouse peptide identifications identified using N-terminal COFRADIC and searching the custom combined database. The majority of N-terminal peptide identifications map to canonical, annotated UniProtKB-SwissProt protein N termini (highlighted in gray, 84.8%). 14.1% of the identified N termini are indicative of N-terminally truncated protein forms (light green), whereas nearly 1% (light blue) of the N termini map to new N-terminally extended protein forms. Besides, four (in red and orange) translated uORFs were identified. Identification of the N-terminal extended proteins and translated uORFs would not have been possible using merely UniProtKB-SwissProt as search space.
A listing of all identified N-terminal peptides, categorized into the different classes (canonical, extension, truncation, uORF, and overlapping uORF) is provided as supplemental Table S3. The different search engine performances, measured as validated peptide-to-spectrum matches, can be found in supplemental Table S2. Furthermore, the N-terminal peptides have also been made available as s visualization track in our in-house developed genome-browser. Next to Ensembl Gene and Transcript visualization tracks (Ensembl annotation version 66 (7)), experimental RIBO-seq data (15) are also presented as custom tracks, allowing manual inspection of the co-occurrence of N-terminal COFRADIC and RIBO-seq experimental evidence. The N-terminal COFRADIC information is also provided in BED-format (32) for upload in a genome browser of choice (supplemental File S2).
Next to annotation of nonregular translation products as (i) N-terminal protein extensions or truncations pointing to aTIS, uTIS, or downstream TIS, or (ii) translation of uORFs, the N-terminal COFRADIC technology provides us with evidence of translation initiation at near-cognate start sites (noninitiator methionine). This was also a remarkable observation within the RIBO-seq studies (13, 15, 18, 19), and notably, we here observe the outcome of this phenomenon at the protein level instead of the mRNA level. Weblogos (33) were created based on the sequence contexts flanking the newly identified uTIS (from the extended protein products) and the TIS of the translated uORFs (see Fig. 6). The weblogos clearly show the presence of translation initiation at near-cognate start codons. Next to the four N termini indicative for N-terminal protein extension starting at the canonical AUG start codon, others were formed by translation initiation at CUG (five N termini), GUG (four N termini), ACG (two N termini), and UUG (one N terminus) (also see supplemental Table S3). These near-cognate start codons respectively encode for leucine, valine, threonine, and again leucine but are recoded to the regular methionine as, for example, exemplified by the N-terminally acetylated iMet-retaining N-terminal MDPPTSEKAVAQGAGR originating from translation initiation of the thyroid receptor-interacting protein 13 transcript at an upstream near-cognate GUG codon. In parallel, next to the newly translated uORF identification starting from the canonical AUG (one N terminus), others were also found starting from near-cognate codons GUG (two N termini), ACG (one N terminus), and UUG (one N terminus) (see supplemental Table S3). In line with these results, the majority of discovered uORFs in RIBO-seq studies was found to start from near-cognate start sites (15, 18, 19). The recoding of the alternative amino acids, resulting from the near-cognate start sites, seems to take place in all identified candidates, pointing to the fact that no non-iMet translation products are formed.
Fig. 6.

Weblogos depicting the sequence context (three bases upstream and four bases downstream) of the newly identified translation initiation sites from the N-terminal extended protein forms (16) and the translated uORFs (4). Near-cognate start site initiation is observed from the logos. The most crucial Kozak elements are present as follows: a purine at position −3 and a guanine at position +4 within the Kozak motif (A/G)ccAUGG, further corroborating the observation of upstream translation initiation and/or translation of the uORFs.
It is also apparent from the sequence context of the start sites that the most crucial Kozak elements (34, 35) are present as follows: a purine at position −3 and a guanine at position +4 within the Kozak motif (A/G)ccAUGG, further corroborating the upstream translation initiation and/or translation of the uORFs.
DISCUSSION
Our results show that our strategy performs better than previously reported efforts mapping the mouse embryonic stem cell proteome (36) by identifying 3,252 translation products (supplemental Table S1) instead of 1,895, while combining three different proteomic approaches. This is in part attributable to the increase in the total number of peptides correctly identified by using a state-of-the-art mass spectrometer capable of making measurements with high resolution and improved mass accuracy and a robust identification pipeline. Subtle changes in database searching algorithms are known to provide different identification results, thus combining multiple search engines in the identification pipeline results in increased rate of peptide assignments and consequently an increased number of protein identifications (37). Moreover, building an MS-based strategy, integrating ribosome profiling information to provide an optimized search space, clearly aids the identification process. In fact, both optimal experimental MS design (shotgun and/or N-terminal COFRADIC) and a robust identification strategy in combination with the construction of an optimal sequence search space are equally important. Searching the constructed deep proteome sequence database with the publicly available MS/MS data described by Gundry et al. (36) (PRIDE accessions 11364–79) did not result in the identification of any N-terminally extended protein isoforms, although we identified 11 N-terminal extended forms in our shotgun analysis (and up to 16 in the N-terminal COFRADIC study). This again stresses the importance of MS sensitivity, accuracy, and optimal design, next to optimization of the search space.
This study confirms three notable observations made in recent RIBO-seq studies (15, 18, 19). Whereas the RIBO-seq strategy represents ribosomal targeting of mRNA, thereby revealing protein-coding regions, mass spectrometry is highly complementary in revealing true translation products. First, it is clear from the large number of annotated N-terminal truncated (259 N termini) and extended forms (16 N termini) within the N-terminomics results that alternative TIS selection, next to already acknowledged alternative splicing, contributes substantially to protein diversification, leading to an even greater proteome complexity. Integration of the RIBO-seq data into the MS-based proteomics pipeline, and thus optimizing the search space, clearly aids the identification of these N-terminal protein variants (Fig. 5), especially using the N-terminal COFRADIC approach. The results presented (see Fig. 6 and supplemental Table S2) contribute to the scarce reports on alternative TIS usage available at the proteome-wide level (31, 38–40), focusing on translation initiation and its influence on protein structure and function and the translation process regulation thereof. As an example and in line with previous Western blot observations (41) and GTI-seq data (15, 19), an N-terminal extended protein isoform for the hdgf gene was identified with the N-terminal COFRADIC technique (depicted in Fig. 7).
Fig. 5.

Illustrations of the different classes of identified N termini using N-terminal COFRADIC. Three examples are depicted as follows: (i) an N terminus indicative of a new N-terminally extended protein identification (Swiss-Prot accession P51859); (ii) a new N-terminally truncated protein identification (Swiss-Prot accession Q9ES46), and (iii) a translation of an uORF overlap (Swiss-Prot accession Q80UY2). For the first two examples, the Clustal Omega alignment is presented in the left panels, aligning both the annotated UniProtKB-SwissProt and the RIBO-seq-derived amino acid sequence. (Note that only a truncated alignment is given focusing on the N terminus.) For the uORF-overlap example, an Ensembl Genome Browser screenshot is presented, depicting the out-of-frame starting uORF-overlap peptide at near-cognate start site ACG, where the regular TIS site is also annotated. The N-terminal peptide identification is highlighted in green or red, respectively, for examples 1–2 and example 3. In the right panels the corresponding annotated fragmentation spectra and delineated sequence fragmentation are depicted, indicative of the confidence/quality of the peptide identification.
Fig. 7.

Representation of the mouse HDGF protein in the H2G2 genome browser. Several information tracks are presented. From top to bottom: (i) Ensembl Gene annotation; (ii) Ensembl Transcript annotation; (iii) predicted translation products derived from the RIBO-seq data; (iv) ribosome profile data of the control mESC cell line sample showing profiles alongside the CDS; (v) ribosome profile data of the harringtonine-treated cell line sample showing profiles at the TIS; (vi) UniProtKB-SwissProt annotated TIS; (vii) RIBO-seq predicted TIS, and (viii) Ensembl annotated TIS. Two zoomed figures (in blue boxes) are also depicted, representing a more detailed view of the genomic region around the translation initiation site of the N-terminal extended HDGF protein isoform that has been identified using the N-terminal COFRADIC study, clearly demonstrating the near-cognate initiation site (GTG) and accumulated RIBO-seq signal at the start site. Furthermore, an alignment with the homologues human peptide sequence is presented (brown box).
Second, to a lower extent, we were also successful in demonstrating the translation of upstream ORFs using our MS-based approach. Ingolia et al. (15) (supplemental Fig. S1) and Lee et al. (19) presented that the majority of initiation sites result from RIBO-seq drives translation of uORFs. The fact that we cannot validate this vast amount of uORF translation events can be attributed to several reasons. It is possible that the translated uORFs are rapidly degraded and are thus not functional, but nevertheless they exert a regulatory function by influencing translation of downstream canonical translation product(s) (20, 42, 43). Also, the sensitivity and dynamic range of state-of-the-art mass spectrometers might be a limiting factor in the identification of uORF translations, mainly because of the small size and possibly also low abundance of the members of this new gene class. Moreover, technical aspects in the N-terminal COFRADIC protocol, more specifically the strong cation exchange chromatographic enrichment step, only allows for less than 65% identifiable N-terminal peptides, dependent on peptide sequence and peptide charge state (44). RIBO-seq, however, is generally unbiased toward this peptide “sequence-identity” issue. Notable is also that other high throughput proteomics endeavors were not very successful in identifying short translation products of long intergenic noncoding RNAs, for example (22). Upstream ORFs are typically very short. In combination with their “nontryptic” nature, this makes these very difficult to be identified, because the available algorithms are typically not suited for identification of such MS/MS spectra. In other words, experimental validation of RIBO-seq-derived small translation products still represents a challenge, and further RIBO-seq might also yield false positives caused by ribosomal drug treatment, for example.
Third, usage of the RIBO-seq-derived sequence database allowed us to confirm translation initiation from near-cognate start sites (Figs. 6 and 7 and supplemental Table S2), which is notable because this seems to be severely overlooked in the past, and only limited evidence is available for noncognate translation initiation (40, 45, 46).
Furthermore, the size of the protein product plays a role for its successful MS-based identification. Current proteomics technologies, in contrast to the RIBO-seq technique, are problematic in isolating molecular species below 45–50 amino acids (20). To capture a larger fraction of the small proteins or (oligo-) peptides, specific pipelines need to be elaborated and applied, ensuring the enrichment of short (47, 48) and, very frequently, less abundant (49) translation products, for example encoded within small ORFs (50–53)2 or more specifically uORFs (54). Combining these specific MS pipelines with the custom DB approach described in this study should aid for more comprehensive identification of this class of small translation products in the future.
This study focuses on the advantages of searching against a translation product database derived from RIBO-seq sequencing data. Some of the remarkable findings, like N-terminal protein extension through 5′ uTIS selection, have here been further validated at the level of the proteome. Next to sequence conservation analysis based on PhastCons (27, 55), showing high sequence context conservation around aTIS, in vitro mutagenesis studies corroborate our mass spectrometry evidence of aTIS.3
CONCLUSION
As deep sequencing techniques are becoming more routine, less expensive, and widespread, ribosome profiling will in the future probably serve as an alternative to MS-based protein and peptide identification, certainly seen its large dynamic range, sensitivity, and comprehensive nature. On the other hand, enrichment techniques preceding MS experiments (such as for example N-terminal COFRADIC (24) or enrichment for small (48) or low abundant (49) protein products) will still be indispensable to lower the complexity of the peptide mixture and/or enrich for selected peptide classes. Furthermore, MS techniques in general, provide us with true in vivo evidence of proteins/peptides existence, and concurrently their co- and post-translational modification status. Moreover, MS experiments are still indispensable in protein or peptide localization (56) and/or stability studies (57) as are antibody staining immunohistochemistry approaches. In any case, a combination of deep sequencing and mass spectrometry will definitely become more mainstream in revealing protein translation levels. As combined, these technologies are capable of capturing regulated degradation events. Ultimately, building an automated pipeline and intuitive user interface, converting RIBO-seq data into a custom searchable database comprising both SNP mutation, ORF delineation, and thus also TIS information, alongside performing quality control of the data and genome-wide visualization, proves to be very beneficial.
Supplementary Material
Acknowledgments
We acknowledge the Nucleotide 2 Networks Multidisciplinary Research Partnership (Special Research Fund Ghent University).
Footnotes
 This article contains supplemental material.
This article contains supplemental material.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium, via the PRIDE partner repository (58) with the dataset identifier PXD000124 and DOI 10.6019/PXD000124.
2 Jeroen Crappé, Wim Van Criekinge, Geert Trooskens, Eisuke Hayakawa, Walter Luyten, Geert Baggerman and Gerben Menschaert. Combining in silico prediction and ribosome profiling in a genome-wide search for novel micropeptides. In review BMC Genomics.
3 Petra Van Damme*, Gerben Menschaert*, Daria Gawron, Kimberly Demeyer, Wim Van Criekinge and Kris Gevaert N-terminal proteomics provides ample evidence of the alternative translation initiation landscape in higher eukaryotes. In preparation to submit to Molecular System Biology. *: joined first authors
1 The abbreviations used are:
- RNA-seq
- RNA sequencing
- aTIS
- alternative translation initiation site
- GTI-seq
- global translation initiation sequencing
- H2G2
- HitchHikers Guide to the Genome
- mESC
- mouse embryonic stem cell
- OMSSA
- open mass spectrometry search algorithm
- RIBO-seq
- ribosome profiling sequencing
- SNP
- single nucleotide polymorphism
- uORF
- upstream open reading frame
- uTIS
- upstream translation initiation site
- COFRADIC
- combined fractional diagonal chromatography
- iMet
- initiator codon Met
- CDS
- coding sequence.
REFERENCES
- 1. Beck M., Schmidt A., Malmstroem J., Claassen M., Ori A., Szymborska A., Herzog F., Rinner O., Ellenberg J., Aebersold R. (2011) The quantitative proteome of a human cell line. Mol. Syst. Biol. 7, 549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nagaraj N., Wisniewski J. R., Geiger T., Cox J., Kircher M., Kelso J., Pääbo S., Mann M. (2011) Deep proteome and transcriptome mapping of a human cancer cell line. Mol. Syst. Biol. 7, 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Perkins D. N., Pappin D. J., Creasy D. M., Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 4. Craig R., Beavis R. C. (2004) TANDEM: matching proteins with tandem mass spectra. Bioinformatics 20, 1466–1467 [DOI] [PubMed] [Google Scholar]
- 5. Geer L. Y., Markey S. P., Kowalak J. A., Wagner L., Xu M., Maynard D. M., Yang X., Shi W., Bryant S. H. (2004) Open mass spectrometry search algorithm. J. Proteome Res. 3, 958–964 [DOI] [PubMed] [Google Scholar]
- 6. UniProt Consortium (2012) Reorganizing the protein space at the Universal Protein Resource (UniProt). Nucleic Acids Research 40, D71–D75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Flicek P., Amode M. R., Barrell D., Beal K., Brent S., Carvalho-Silva D., Clapham P., Coates G., Fairley S., Fitzgerald S., Gil L., Gordon L., Hendrix M., Hourlier T., Johnson N., Kähäri A. K., Keefe D., Keenan S., Kinsella R., Komorowska M., Koscielny G., Kulesha E., Larsson P., Longden I., McLaren W., Muffato M., Overduin B., Pignatelli M., Pritchard B., Riat H. S., Ritchie G. R., Ruffier M., Schuster M., Sobral D., Tang Y. A., Taylor K., Trevanion S., Vandrovcova J., White S., Wilson M., Wilder S. P., Aken B. L., Birney E., Cunningham F., Dunham I., Durbin R., Fernández-Suarez X. M., Harrow J., Herrero J., Hubbard T. J., Parker A., Proctor G., Spudich G., Vogel J., Yates A., Zadissa A., Searle S. M. (2012) Ensembl 2012. Nucleic Acids Res. 40, D84–D90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang X., Slebos R. J., Wang D., Halvey P. J., Tabb D. L., Liebler D. C., Zhang B. (2012) Protein identification using customized protein sequence databases derived from RNA-Seq data. J. Proteome Res. 11, 1009–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nesvizhskii A. I. (2010) A survey of computational methods and error rate estimation procedures for peptide and protein identification in shotgun proteomics. J. Proteomics 73, 2092–2123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ning K., Nesvizhskii A. I. (2010) The utility of mass spectrometry-based proteomic data for validation of novel alternative splice forms reconstructed from RNA-Seq data: a preliminary assessment. BMC Bioinformatics 11, S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ning K., Fermin D., Nesvizhskii A. I. (2012) Comparative analysis of different label-free mass spectrometry based protein abundance estimates and their correlation with RNA-Seq gene expression data. J. Proteome Res. 11, 2261–2271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Djebali S., Davis C. A., Merkel A., Dobin A., Lassmann T., Mortazavi A., Tanzer A., Lagarde J., Lin W., Schlesinger F., Xue C., Marinov G. K., Khatun J., Williams B. A., Zaleski C., Rozowsky J., Röder M., Kokocinski F., Abdelhamid R. F., Alioto T., Antoshechkin I., Baer M. T., Bar N. S., Batut P., Bell K., Bell I., Chakrabortty S., Chen X., Chrast J., Curado J., Derrien T., Drenkow J., Dumais E., Dumais J., Duttagupta R., Falconnet E., Fastuca M., Fejes-Toth K., Ferreira P., Foissac S., Fullwood M. J., Gao H., Gonzalez D., Gordon A., Gunawardena H., Howald C., Jha S., Johnson R., Kapranov P., King B., Kingswood C., Luo O. J., Park E., Persaud K., Preall J. B., Ribeca P., Risk B., Robyr D., Sammeth M., Schaffer L., See L. H., Shahab A., Skancke J., Suzuki A. M., Takahashi H., Tilgner H., Trout D., Walters N., Wang H., Wrobel J., Yu Y., Ruan X., Hayashizaki Y., Harrow J., Gerstein M., Hubbard T., Reymond A., Antonarakis S. E., Hannon G., Giddings M. C., Ruan Y., Wold B., Carninci P., Guigo R., Gingeras T. R. (2012) Landscape of transcription in human cells. Nature 489, 101–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ingolia N. T. (2010) Genome-wide translational profiling by ribosome footprinting. Methods Enzymol. 470, 119–142 [DOI] [PubMed] [Google Scholar]
- 14. Guo H., Ingolia N. T., Weissman J. S., Bartel D. P. (2010) Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466, 835–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ingolia N. T., Lareau L. F., Weissman J. S. (2011) Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147, 789–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ingolia N. T., Brar G. A., Rouskin S., McGeachy A. M., Weissman J. S. (2012) The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat. Protoc. 7, 1534–1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Brar G. A., Yassour M., Friedman N., Regev A., Ingolia N. T., Weissman J. S. (2012) High-resolution view of the yeast meiotic program revealed by ribosome profiling. Science 335, 552–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fritsch C., Herrmann A., Nothnagel M., Szafranski K., Huse K., Schumann F., Schreiber S., Platzer M., Krawczak M., Hampe J., Brosch M. (2012) Genome-wide search for novel human uORFs and N-terminal protein extensions using ribosomal footprinting. Genome Res. 22, 2208–2218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee S., Liu B., Lee S., Huang S. X., Shen B., Qian S. B. (2012) Global mapping of translation initiation sites in mammalian cells at single-nucleotide resolution. Proc. Natl. Acad. Sci. U.S.A. 109, E2424–E2432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stern-Ginossar N., Weisburd B., Michalski A., Le V. T., Hein M. Y., Huang S. X., Ma M., Shen B., Qian S. B., Hengel H., Mann M., Ingolia N. T., Weissman J. S. (2012) Decoding human cytomegalovirus. Science 338, 1088–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guttman M., Rinn J. L. (2012) Modular regulatory principles of large noncoding RNAs. Nature 482, 339–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Volders P. J., Helsens K., Wang X., Menten B., Martens L., Gevaert K., Vandesompele J., Mestdagh P. (2013) LNCipedia: a database for annotated human lncRNA transcript sequences and structures. Nucleic Acids Res. 41, D246–D251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ying Q. L., Wray J., Nichols J., Batlle-Morera L., Doble B., Woodgett J., Cohen P., Smith A. (2008) The ground state of embryonic stem cell self-renewal. Nature 453, 519–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Staes A., Impens F., Van Damme P., Ruttens B., Goethals M., Demol H., Timmerman E., Vandekerckhove J., Gevaert K. (2011) Selecting protein N-terminal peptides by combined fractional diagonal chromatography. Nat. Protoc. 6, 1130–1141 [DOI] [PubMed] [Google Scholar]
- 25. Staes A., Van Damme P., Helsens K., Demol H., Vandekerckhove J., Gevaert K. (2008) Improved recovery of proteome-informative, protein N-terminal peptides by combined fractional diagonal chromatography (COFRADIC). Proteomics 8, 1362–1370 [DOI] [PubMed] [Google Scholar]
- 26. Vaudel M., Barsnes H., Berven F. S., Sickmann A., Martens L. (2011) SearchGUI: An open-source graphical user interface for simultaneous OMSSA and X!Tandem searches. Proteomics 11, 996–999 [DOI] [PubMed] [Google Scholar]
- 27. Siepel A., Bejerano G., Pedersen J. S., Hinrichs A. S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L. W., Richards S., Weinstock G. M., Wilson R. K., Gibbs R. A., Kent W. J., Miller W., Haussler D. (2005) Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 15, 1034–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gevaert K., Impens F., Van Damme P., Ghesquière B., Hanoulle X., Vandekerckhove J. (2007) Applications of diagonal chromatography for proteome-wide characterization of protein modifications and activity-based analyses. FEBS J. 274, 6277–6289 [DOI] [PubMed] [Google Scholar]
- 29. Van Damme P., Van Damme J., Demol H., Staes A., Vandekerckhove J., Gevaert K. (2009) A review of COFRADIC techniques targeting protein N-terminal acetylation. BMC Proc. 3 Suppl. 6, S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Arnesen T., Van Damme P., Polevoda B., Helsens K., Evjenth R., Colaert N., Varhaug J. E., Vandekerckhove J., Lillehaug J. R., Sherman F., Gevaert K. (2009) Proteomics analyses reveal the evolutionary conservation and divergence of N-terminal acetyltransferases from yeast and humans. Proc. Natl. Acad. Sci. U.S.A. 106, 8157–8162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Helsens K., Van Damme P., Degroeve S., Martens L., Arnesen T., Vandekerckhove J., Gevaert K. (2011) Bioinformatics analysis of a Saccharomyces cerevisiae N-terminal proteome provides evidence of alternative translation initiation and post-translational N-terminal acetylation. J. Proteome Res. 10, 3578–3589 [DOI] [PubMed] [Google Scholar]
- 32. Meyer L. R., Zweig A. S., Hinrichs A. S., Karolchik D., Kuhn R. M., Wong M., Sloan C. A., Rosenbloom K. R., Roe G., Rhead B., Raney B. J., Pohl A., Malladi V. S., Li C. H., Lee B. T., Learned K., Kirkup V., Hsu F., Heitner S., Harte R. A., Haeussler M., Guruvadoo L., Goldman M., Giardine B. M., Fujita P. A., Dreszer T. R., Diekhans M., Cline M. S., Clawson H., Barber G. P., Haussler D., Kent W. J. (2013) The UCSC Genome Browser database: extensions and updates 2013. Nucleic Acids Res. 41, D64–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Crooks G. E., Hon G., Chandonia J. M., Brenner S. E. (2004) WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kozak M. (1987) An analysis of 5′-noncoding sequences from 699 vertebrate messenger RNAs. Nuclic Acids Res. 15, 8125–8148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kozak M. (1991) Structural features in eukaryotic mRNAs that modulate the initiation of translation. J. Biol. Chem. 266, 19867–19870 [PubMed] [Google Scholar]
- 36. Gundry R. L., Tchernyshyov I., Sheng S., Tarasova Y., Raginski K., Boheler K. R., Van Eyk J. E. (2010) Expanding the mouse embryonic stem cell proteome: combining three proteomic approaches. Proteomics 10, 2728–2732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Searle B. C., Turner M., Nesvizhskii A. I. (2008) Improving sensitivity by probabilistically combining results from multiple MS/MS search methodologies. J. Proteome Res. 7, 245–253 [DOI] [PubMed] [Google Scholar]
- 38. Kochetov A. V., Sarai A., Rogozin I. B., Shumny V. K., Kolchanov N. A. (2005) The role of alternative translation start sites in the generation of human protein diversity. Mol. Genet. Genomics 273, 491–496 [DOI] [PubMed] [Google Scholar]
- 39. Thomas D., Plant L. D., Wilkens C. M., McCrossan Z. A., Goldstein S. A. (2008) Alternative translation initiation in rat brain yields K2P2.1 potassium channels permeable to sodium. Neuron 58, 859–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Touriol C., Bornes S., Bonnal S., Audigier S., Prats H., Prats A. C., Vagner S. (2003) Generation of protein isoform diversity by alternative initiation of translation at non-AUG codons. Biol. Cell 95, 169–178 [DOI] [PubMed] [Google Scholar]
- 41. Abouzied M. M., Baader S. L., Dietz F., Kappler J., Gieselmann V., Franken S. (2004) Expression patterns and different subcellular localization of the growth factors HDGF (hepatoma-derived growth factor) and HRP-3 (HDGF-related protein-3) suggest functions in addition to their mitogenic activity. Biochem. J. 378, 169–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Medenbach J., Seiler M., Hentze M. W. (2011) Translational control via protein-regulated upstream open reading frames. Cell 145, 902–913 [DOI] [PubMed] [Google Scholar]
- 43. Wethmar K., Bégay V., Smink J. J., Zaragoza K., Wiesenthal V., Dörken B., Calkhoven C. F., Leutz A. (2010) C/EBPβδuORF mice–a genetic model for uORF-mediated translational control in mammals. Genes Dev. 24, 15–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Van Damme P., Staes A., Bronsoms S., Helsens K., Colaert N., Timmerman E., Aviles F. X., Vandekerckhove J., Gevaert K. (2010) Complementary positional proteomics for screening substrates of endo- and exoproteases. Nat. Methods 7, 512–515 [DOI] [PubMed] [Google Scholar]
- 45. Zhang F., Hinnebusch A. G. (2011) An upstream ORF with non-AUG start codon is translated in vivo but dispensable for translational control of GCN4 mRNA. Nucleic Acids Res. 39, 3128–3140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Németh A. L., Medveczky P., Tóth J., Siklódi E., Schlett K., Patthy A., Palkovits M., Ovádi J., Tõkési N., Németh P., Szilágyi L., Gráf L. (2007) Unconventional translation initiation of human trypsinogen 4 at a CUG codon with an N-terminal leucine. A possible means to regulate gene expression. FEBS J. 274, 1610–1620 [DOI] [PubMed] [Google Scholar]
- 47. Slavoff S. A., Mitchell A. J., Schwaid A. G., Cabili M. N., Ma J., Levin J. Z., Karger A. D., Budnik B. A., Rinn J. L., Saghatelian A. (2013) Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nat. Chem. Biol. 9, 59–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tinoco A. D., Tagore D. M., Saghatelian A. (2010) Expanding the dipeptidyl peptidase 4-regulated peptidome via an optimized peptidomics platform. J. Am. Chem. Soc. 132, 3819–3830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fonslow B. R., Carvalho P. C., Academia K., Freeby S., Xu T., Nakorchevsky A., Paulus A., Yates J. R., 3rd (2011) Improvements in proteomic metrics of low abundance proteins through proteome equalization using ProteoMiner prior to MudPIT. J. Proteome Res. 10, 3690–3700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Galindo M. I., Pueyo J. I., Fouix S., Bishop S. A., Couso J. P. (2007) Peptides encoded by short ORFs control development and define a new eukaryotic gene family. PLoS Biol. 5, e106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hashimoto Y., Niikura T., Tajima H., Yasukawa T., Sudo H., Ito Y., Kita Y., Kawasumi M., Kouyama K., Doyu M., Sobue G., Koide T., Tsuji S., Lang J., Kurokawa K., Nishimoto I. (2001) A rescue factor abolishing neuronal cell death by a wide spectrum of familial Alzheimer's disease genes and Aβ. Proc. Natl. Acad. Sci. U.S.A. 98, 6336–6341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kondo T., Plaza S., Zanet J., Benrabah E., Valenti P., Hashimoto Y., Kobayashi S., Payre F., Kageyama Y. (2010) Small peptides switch the transcriptional activity of Shavenbaby during Drosophila embryogenesis. Science 329, 336–339 [DOI] [PubMed] [Google Scholar]
- 53. Ladoukakis E., Pereira V., Magny E. G., Eyre-Walker A., Couso J. P. (2011) Hundreds of putatively functional small open reading frames in Drosophila. Genome Biol. 12, R118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Calvo S. E., Pagliarini D. J., Mootha V. K. (2009) Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proc. Natl. Acad. Sci. U.S.A. 106, 7507–7512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fan X., Zhu J., Schadt E. E., Liu J. S. (2007) Statistical power of phylo-HMM for evolutionarily conserved element detection. BMC Bioinformatics 8, 374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sadowski P. G., Dunkley T. P., Shadforth I. P., Dupree P., Bessant C., Griffin J. L., Lilley K. S. (2006) Quantitative proteomic approach to study subcellular localization of membrane proteins. Nat. Protoc. 1, 1778–1789 [DOI] [PubMed] [Google Scholar]
- 57. Boisvert F. M., Ahmad Y., Gierlinski M., Charriere F., Lamont D., Scott M., Barton G., Lamond A. I. (2012) A quantitative spatial proteomics analysis of proteome turnover in human cells. Mol. Cell. Proteomics 11, M111.011429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Vizcaíno J. A., Côté R., Reisinger F., Barsnes H., Foster J. M., Rameseder J., Hermjakob H., Martens L. (2010) The Proteomics Identifications database: 2010 update. Nucleic Acids Res. 38, D736–D742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., Higgins D. G. (2011) Fast, scalable generation of high quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}