

Abstract
Arsenic is an environmental toxicant and carcinogen. Exposure to arsenic is associated with development of liver fibrosis and portal hypertension through ill defined mechanisms. We evaluated hepatic fibrogenesis after long term arsenic exposure in a murine model. BALB/c mice were exposed to arsenic by daily gavages of 6 μg/ gm body weight for 1 year and were evaluated for markers of hepatic oxidative stress and fibrosis, as well as pro-inflammatory, pro-apoptotic and pro-fibrogenic factors at 9 and 12 months. Hepatic NADPH oxidase activity progressively increased in arsenic exposure with concomitant development of hepatic oxidative stress. Hepatic steatosis with occasional collection of mononuclear inflammatory cells and mild portal fibrosis were the predominant liver lesion observed after 9 months of arsenic exposure, while at 12 months, the changes included mild hepatic steatosis, inflammation, necrosis and significant fibrosis in periportal areas. The pathologic changes in the liver were associated with markers of hepatic stellate cells (HSCs) activation, matrix reorganization and fibrosis including α-smooth muscle actin, transforming growth factor-β1, PDGF-Rβ, pro-inflammatory cytokines and enhanced expression of tissue inhibitor of metalloproteinase-1 and pro (α) collagen type I. Moreover, pro-apoptotic protein Bax was dominantly expressed and Bcl-2 was down-regulated along with increased number of TUNEL positive hepatocytes in liver of arsenic exposed mice. Furthermore, HSCs activation due to increased hepatic oxidative stress observed after in vivo arsenic exposure was recapitulated in co-culture model of isolated HSCs and hepatocytes exposed to arsenic. These findings have implications not only for the understanding of the pathology of arsenic related liver fibrosis but also for the design of preventive strategies in chronic arsenicosis.
Keywords: Arsenic, Oxidative stress, Reactive oxygen species, Apoptosis, Liver
Introduction
Environmental exposure to inorganic arsenic, primarily due to geophysiochemical contamination of drinking water, is a major public health hazard in both the developing and developed world (Harvey, 2008; Fendorf et al., 2010). An estimated 13 million people in the United States live in areas where arsenic concentration in the drinking water exceeds the US Environmental Protection Agency’s safe cut-off value of 10 μg/L (EPA, 2001). However, the greatest catastrophe in the history of arsenic poisoning of a population is attributed to the thickly populated deltas of the great rivers flowing through South and South-East Asia, where more than 100 million people are being exposed to unsafe level of arsenic by drinking contaminated groundwater (Fendorf et al., 2010).
The toxicity of inorganic arsenic usually occurs after a considerable latent period. It is responsible for the genesis of a wide range of health problems involving skin, nervous system, pancreas, lung, cardiovascular and peripheral vascular system and is also implicated as an etiological agent of cancers of the skin, lung and urinary bladder (National Research Council, 1999; Smith et al., 2000; Guha Mazumdar et al., 1998; Goddard et al., 1992; Rahman et al., 1998; Guha Mazumder et al., 2000; Navas-Acien et al., 2005; Chi and Blackwell, 1968; Chen et al., 1988; IARC, 2004; Navas-Aciens et al., 2008). Previous works, from the Gangetic delta of West Bengal, India, where arsenic levels in drinking water can be as high as 1500 to 3400 μg/L (World Health Organization, 1999; National Research Council, 1999; Guha Mazumdar et al., 1998; Bagla and Kaiser, 1996), highlighted such toxicity and were the first to define the hepatotoxic effects of chronic arsenic ingestion (Guha Mazumder et al., 1988; Santra et al., 1999; Guha Mazumder, 2005). Non cirrhotic portal hypertension associated with portal fibrosis has also been a major consequence of chronic arsenic toxicity in human clinicopathological studies from India and the West (Guha Mazumder et al., 1988; Santra et al., 1999; Guha Mazumder, 2005; Dutta et al., 1979; Morris et al., 1974; Huet et al., 1975; Nevens et al., 1990). Our preliminary studies on chronic arsenic toxicity in mice had revealed that it induces oxidative stress, mitochondrial injury and apoptosis of hepatocytes (Santra et al., 2000a, 2007). Interestingly, low levels of arsenic exposure cause liver sinusoidal endothelial cells (SECs) injury that includes SECs defenestration, capillarization, and decreased liver clearance in experimental animal models (Straub et al., 2007, 2008). However, these animal models did not develop features of portal fibrosis which is an important feature of arsenic related hepatotoxicity in humans (Guha Mazumder et al., 1988; Santra et al., 1999; Guha Mazumder, 2005; Dutta et al., 1979).
Based on clinical and morphological studies in this region, it is well established that hepatic fibrosis is an important morphological lesion in chronic arsenic toxicity (Guha Mazumder, 2008). Activation of hepatic fibrogenesis is therefore a likely consequence of this arsenic induced liver injury. We hypothesized that sustained arsenic induced oxidative stress in the liver leads to increased apoptosis of hepatocytes and liberation of some mediators which help in the activation of hepatic stellate cells (HSCs), the predominant fibrogenic cell type producing collagen type I in the liver (Friedman et al., 1985; Friedman, 2008). HSCs also produce tissue inhibitor of matrix metalloproteinases (TIMP), which inhibits collagen-degrading matrix metalloproteinase (MMPs) and shifts the balance between extra cellular matrix (ECM) synthesis and degradation towards fibrogenesis (Arthur, 2000; Elsharkawy et al., 2005).
The overall goal of this study is to evaluate the impact of chronic arsenic exposure on the progression of hepatic fibrogenesis in mice. Our experimental data suggest that arsenic induces fibrosis after prolonged exposure that involves an intricate crosstalk between apoptotic hepatocytes, pro-inflammatory cytokines and HSCs.
Materials and methods
Animals
Adult (age 6–8 week) male wild type BALB/c mice were purchased from the National Center for Laboratory Animal Sciences (Hyderabad, India). Treatment of animals and procedures performed were done in accordance with the guidelines stipulated by the animal ethics committee of Institute of Post Graduate Medical Education & Research, Kolkata, India.
Animal treatment
Mice (n=40) were exposed to sodium arsenite (6 μg/g body weight in bottled water/day by gavages) 6 days a week for one year. Control mice received an equal volume of bottled water by gavages as per schedule of the arsenic exposed mice. Details of animal termination procedure and biochemical assays are provided in the supplemental material and methods.
Serum aminotransferases
Serum ALT was measured with a commercial kit according to the manufacturer’s instruction.
Hepatic triglyceride and collagen estimation
A 10% liver homogenate was used for determination of liver triglyceride concentration using a spectrophotometric kit from Sigma Diagnostics (St Louis, MO). Collagen content of the liver tissue was measured as described previously (Das et al., 2005).
Histology, immunohistochemistry, Western blot and TUNEL assay
Liver tissues embedded in paraffin were cut in sections (5 μm) and stained with hematoxylin–eosin (H&E) and Sirius red for collagen I detection using standard procedures. To detect fat deposition, frozen sections of the livers were processed for Oil Red O staining and evaluated. Immunohistochemistry and Western blots were performed using antibodies against 4 hydroxy-2 non-enal (4HNE) [R&D System clone 198960], α-smooth muscle actin (α-SMA) [Sigma, clone 1A4], cytochrome c (R&D System, clone 7H8.2C12), Bax (Sigma, clone 5B7), Bcl2 (Abcam), and β-actin (Abcam). Details of the immunohistochemistry procedure are provided in the supplemental materials and methods. TUNEL assays were performed using the in situ cell death detection kit (Roche) according to the manufacture’s instruction. The extent of injury, apoptosis and fibrosis was evaluated by an investigator (GKD), who was blinded to the experimental protocol.
Reverse-transcription PCR (RT-PCR) and real-time quantitative PCR (qRT-PCR)
Six mice from each of the groups were studied. RNA was extracted from liver tissues using TRIzol® Reagent (Invitrogen). A high-capacity cDNA reverse transcription kit (Applied Biosystems) was used to generate cDNA from extracted RNA. Conditions and primers for reverse transcriptase polymerase chain reaction (RT-PCR) and quantitative real time polymerase chain reaction (qRT-PCR) are available on request. qRT-PCR was carried out on cDNA using primer sets and SYBR® green PCR master mix (Applied Biosystems) according to the manufacturer’s instructions. Data were normalized against the expression of β-actin.
Caspase activity
The activity of caspase 3 was determined in liver homogenates by measuring proteolytic cleavage of the specific fluorogenic substrates DEVD-AFC (Asp-Glu-Val-Asp) (AFC: 7-amido-4-trifluoromethyl coumarin, respectively; Sigma). The results were expressed as percentage of control.
Cytokine quantification
Hepatic tissue necrosis factor-α (TNF-α) and transforming growth factor β-1 (TGF-β1) levels were evaluated by enzyme-linked immunosorbant assay (ELISA) using Quantikine kits of R & D systems.
Gelatin zymography
Gelatinase activity was determined using SDS-PAGE zymography as described previously (Nakamura et al., 2007). Gels were scanned using a Kodak Gel scanning system and analyzed using Adobe Photoshop 6 (Adobe System Inc., San Jose, CA) in gray scale at 450 dpi.
Estimation of hepatic and urinary arsenic contents
Liver tissues and urine samples were digested with 2 M NaOH as described previously (Yamauchi and Yamamura, 1984). Arsenic content in the digested samples were analyzed by hydride generation atomic absorption spectroscopy (HGAA) using Perkin Elmer AA100 atomic absorption spectrophotometer equipped with a flow injection atomic spectroscopy system (FIAS-100) as we have described previously (Santra et al., 2007).
Isolation of mouse hepatocytes and stellate cells
Hepatocytes were isolated from 25 to 30 g overnight-fasted male BALB/c mice by collagenase perfusion as described previously (Pertoft and Smedsrød, 1987). Hepatocytes were resuspended in DMEM containing 2 mM L-glutamine, 10% fetal bovine serum, 100 nM insulin, 100 nM dexamethasone, 100 U/mL penicillin, and 100 μg/mL streptomycin. Cell viability was greater than 97%, as determined by trypan blue exclusion test. After primary culture, the viable cells were seeded onto the cell inserts (3 μm pore size) at a density of 1×106 cells in DMEM without serum.
Primary HSCs were isolated from BALB/c mice (25 to 30 g) by in situ liver perfusion with collagenase and pronase, followed by density gradient centrifugation with Nycodenz according to published protocol (Bataller et al., 2003). Cell viability (95%) was assessed by trypan blue exclusion test. Purity of HSC (96%) was determined as described previously (Nieto et al., 2001). HSC were incubated overnight in DMEM supplemented with 10% fetal bovine serum.
Co-culture of hepatocytes and HSCs
Cells were cultured using cell culture inserts (3 μm pore size) to separate both the cell population; the HSCs were plated on the bottom of the collagen I coated plate and the hepatocytes were plated on the insert to make a gravity gradient of the released mediators (Nieto et al., 2002). A ratio of hepatocytes/ HSC of 5:1 was chosen, because it is representative of the ratio of parenchymal/nonparenchymal cells in liver (Fontana et al., 1997). Details of the co-culture model are shown in Fig. 6A. After overnight incubation, both the hepatocytes and the HSCs were washed 3 times in serum free DMEM, and the cell-culture inserts containing the hepatocytes were transferred onto the HSCs. Fresh medium (3 mL without serum) was added to HSCs cultured with hepatocytes or with empty inserts, the latter considered as the co-culture control. At this time all additions were made (t=0 h). 2 μM of arsenic was added to the inserts and in some selected wells, 2000 U of catalase was added in the co-culture media (t=0 h). In another set of experiment, hepatocytes were pre-incubated for 1 h with 10 mM N-acetyl cysteine (NAC) (Sigma) before incubation in the presence and absence of 2 μM arsenic.
Fig. 6.

A) Scheme of the co-culture model. HSCs were seeded on the bottom plate at a density of 2×105 cells in 3 mL of culture medium. Primary cultured isolated hepatocytes were plated at a density of 1×106 on the insert in 1.5 mL of culture medium. After overnight incubation, the medium from the HSCs was discarded, the cells were washed 3 times in PBS, and the inserts were transferred together with the incubation medium from the hepatocytes onto the HSCs. New medium was added to HSCs plated with empty inserts; these were considered as non-co cultured controls. Various additions were made at this time (t=0 h), and samples of HSC, Hepatocytes, or medium were collected at selected time points. B) Arsenic induced formation of ROS was observed in the hepatocytes and HSCs after 1, 3 and 5 days of treatment. The result was plotted graphically as the representative figure of 4 sets of independent experiments. The formation of ROS was expressed in arbitrary fluorescence units (AFU). A: arsenic treated; B: blank insert; C: control; N: NAC pretreated arsenic exposed groups. Hep and HSC represent hepatocytes and hepatic stellate cells respectively. The cells from where the ROS was quantified were underlined in the figure.
The two cells were co-cultured for 5 days. Aliquots of both the cells were sampled for analysis of ROS generation (Ding et al., 2004) and expression of α-SMA after 1, 3 and 5 days of culture. After 5 days of co-culture, the media from the inserts and the culture plates were collected for quantification of H2O2 and TBARs. The ROS generation in HSCs and immunohistochemistry of α-SMA were studied by Confocal Microscopy. The HSCs were also harvested for the study of mRNA expression of Pro-(α) collagen I, PDGF-Rβ, and β-actin.
Statistical analysis
Results are expressed as mean±SD. Statistical analysis was performed with Student’s t test and analysis of variance. Values of p<0.05 were considered statistically significant. Correlation was calculated and drawn using SPSS version 10.1 and Microsoft excel 2003.
Results
Hepatic oxidative stress and arsenic exposure
Our previous studies suggested that oxidative stress might be involved in the pathogenesis of arsenic induced liver injury (Santra et al., 2000a, 2000b, 2007). In the present study, we evaluated hepatic accumulation of 4-HNE adduct, a reliable index of oxidative stress, by immunohistochemistry. Although deposition of 4-HNE adduct was observed at 9 month in the liver of mice exposed to arsenic, arsenic exposure for 12 months resulted in a more pronounced accumulation of 4-HNE protein adduct that was most prominent in the mid zonal and periportal regions of the lobule (Fig. 1A).
Fig. 1.

Arsenic and hepatic oxidative stress: (A) Immunohistochemistry for 4-HNE adducts in control and arsenic exposed mouse liver. Magnification: 40×. The 4-HNE adducts are shown with black arrows. (B) Hepatic NADPH oxidase activity. (C) Arsenic accumulation in the liver due to prolonged arsenic exposure. (D) Urinary excretion of arsenic during different study periods. Data (B to D) are means±SD of 10 mice per group (**p<0.001).
The mechanism underlying the arsenic induced oxidative stress response remains unclear. One potential candidate for the generation of ROS in chronic arsenic exposure may be NADPH oxidase. We therefore, assessed hepatic NADPH oxidase after prolonged arsenic exposure. Hepatic NADPH oxidase progressively increased from 9 months to 12 months of arsenic exposure (Fig. 1B). To confirm that arsenic activates NADPH oxidase in murine liver, real time mRNA expression study of the NADPH subunits was also performed and revealed a significant increase in the expression of p67phox and gp91phox subunits (Table 1).
Table 1.
Increased expression of NADPH subunits in liver and activation of Kupffer cells during chronic arsenic exposure.
| Gene | Fold increase
|
|||
|---|---|---|---|---|
| Cont 9 | As9 | Cont 12 | As12 | |
| gp91 phox | 1.01±0.01 | 3.25*±0.06 | 1.00±0.02 | 14.42*,**±1.05 |
| p67phox | 1.00±0.01 | 9.19*±1.04 | 1.01±0.02 | 48.50*,**±2.04 |
| TNF-α | 1.02±0.04 | 3.78*±0.41 | 1.04±0.02 | 10.86*,**±0.39 |
| CD-14 | 1.05±0.03 | 3.31*±0.05 | 1.01±0.02 | 8.80*,**±0.19 |
| TLR-4 | 1.01±0.01 | 1.72*±0.51 | 1.05±0.04 | 10.83*,**±0.62 |
| Pro-(α) collagen I | 1.01±0.02 | 1.52*±0.55 | 1.02±0.03 | 5.83*,**±0.41 |
Mice were treated with 6 μg arsenic/g body weight by gastric intubations for 6 days a week for 9 and 12 months respectively and were subsequently terminated. Abbreviations: C9 and C12 are respective control of 9 and 12 months while As9 and As12 are arsenic treated animals for the 9 and 12 months respectively. The results are expressed as mean±SD of 6 mice of each group.
Significantly different from control at p<0.05 and
significantly different from As9 at p < 0.05.
To evaluate whether progression of hepatic oxidative stress was associated with accumulation of arsenic in the liver, atomic absorption spectroscopic analysis of liver tissues were conducted. Excretion of arsenic through urine in the mice was also evaluated. Mice that received arsenic for 12 months showed significant increases in total arsenic accumulation (p<0.001) (Fig. 1C) as well as less urinary excretion of arsenic (p<0.001) (Fig. 1D) compared to mice that received arsenic for 9 months.
Characterization of liver injury and hepatic fibrosis
Morphological changes caused by prolonged arsenic exposure were visualized in liver sections stained with H&E and Sirius red. The changes included fatty infiltration, necrosis, inflammation, and fibrosis. No abnormal hepatic morphology was observed by light microscopy during the first six months of arsenic exposure. Nine months after arsenic exposure, there were accumulation of cytoplasmic macrovesicular lipid droplets and occasional foci of inflammatory cells in the liver of mice (Fig. 2A). Sirius red-stained sections also revealed mild portal fibrosis (Fig. 2B). Steatosis was further confirmed by Oil Red O staining (Fig. 2C) and significant increase in hepatic triglyceride content (Fig. 2D). After 12 months of arsenic exposure, fatty infiltration was minimal but inflammatory cell infiltration within the liver lobules was also increased (Fig. 2A) and portal fibrosis became more marked and pericellular fibrosis accumulated near the portal tract (Fig. 2B). Increased deposition of collagen in the liver was confirmed biochemically (Fig. 2E) and quantification of mRNA expression of Pro-(α) collagen I gene by real time PCR (Table 1).
Fig. 2.

Liver histology of arsenic exposed and control mice A) Hematoxylin and eosin staining of control and arsenic exposed mice for 9 and 12 months respectively (magnification: 40×). The infiltrations of inflammatory cells are shown with black arrows. B) Histology with Sirius red collagen staining of liver section of control and arsenic exposed mice for 9 and 12 months respectively (magnification: 20×). The fibrosis in the liver sections are shown with red arrows. C) Frozen sections were stained with Oil Red O; lipid droplets stained red (magnification 10×). D) Hepatic triglyceride content. E) Hepatic collagen content after 9 and 12 months of arsenic exposure. Results of D & E are expressed as mean±SD of 10 mice per group (**p<0.001).
Activated HSCs are the major source of hepatic collagen, therefore we examined their number by immunohistochemistry of α-SMA using confocal microscopy (Fig. 3A). By 12 months of arsenic exposure, the number of activated HSCs had increased significantly (Fig. 3B). A positive correlation between the hepatic TBARs and activated HSCs was also observed, r=0.886; p<0.001 (Fig. 3C).
Fig. 3.
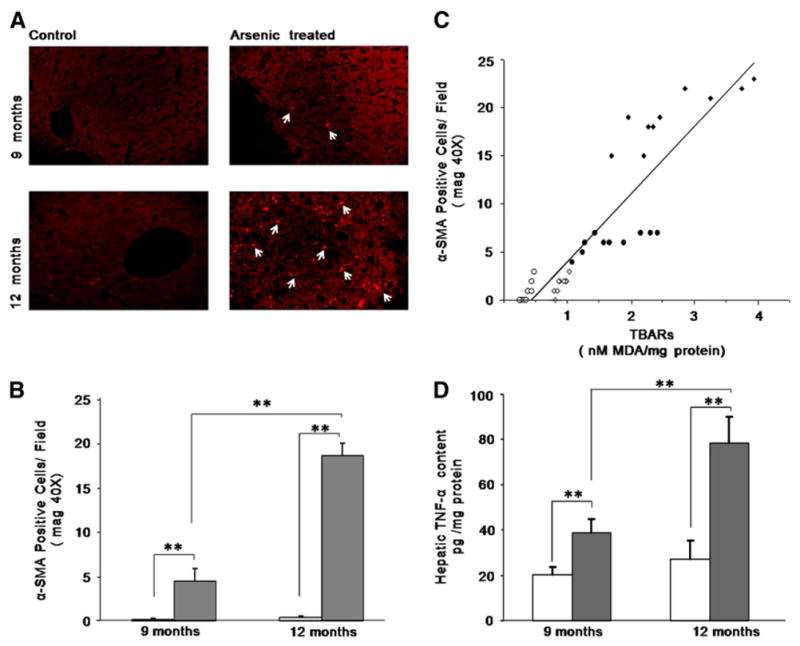
Detection of immunofluorescence of activated stellate cells at different time periods after arsenic treatment. A) Laser scanning confocal microscopy of α-SMA demonstrates the localization of activated HSCs in mouse liver (magnification: 40×). The white arrows indicate activated HSCs. B) The numbers of activated HSCs as identified by the immunohistochemistry of α-SMA from the paraffin sections of the liver tissues are shown graphically. Eight different fields from each section were observed and the mean value was taken. The result was expressed as mean±SD of 10 mice per group (**p<0.001). C) Data showing positive correlation between hepatic TBARs level and α-SMA positive HSCs of control (○: 9 months; □: 12 months) and arsenic (●: 9 months; ■: 12 months) exposed mice (r=0.886; p<0.001). D) Hepatic TNF-α level in control and arsenic exposed mice in different study periods. The TNF-α level was assayed using ELISA kit. The result was expressed as mean±SD of 10 mice per group. (**p<0.001).
Proinflammatory factors mediating arsenic induced liver injury
Oxidative stress in the liver and the resultant liberation of cytokines during liver injury are the two interrelated process that may induce liver fibrosis. TNF-α, a proinflammatory cytokine, plays an important role in the initiation and maintenance of the inflammatory response and is involved in the activation of HSCs. Moreover, TNF-α has an important role in the development of hepatic steatosis. Therefore, we quantified hepatic TNF-α levels (Fig. 3D) by ELISA and its mRNA level by real time PCR (Table 1). Activation of Kupffer cells are also important initiating event that leads to the production of TNF-α. Therefore we quantified mRNA expression of cluster of differentiation 14 (CD14) and Toll like receptor-4 (TLR-4) which are involved in Kupffer cell activation. As shown in Table 1, CD14 and TLR-4 mRNA levels were found to be higher than that of control mice after 12 months of arsenic exposure (p<0.001).
Apoptosis
Liver cell apoptosis is an important mechanism for hepatocytes elimination and is a useful biomarker to monitor the severity of injury. The number of apoptotic cells was gradually increased with arsenic exposure from 9 to 12 months as evident by an increase in percentage of TUNEL positive cells (Fig. 4A). Increased TNF-α and oxidative stress can induce hepatocytes apoptosis. TNF-α may induce the release of cytochrome c by mitochondria and the activation of caspases (Kim et al., 1997), the final common pathway in the apoptotic cascade. Moreover, steatosis is considered to be the manifestation of mitochondrial dysfunction (Day and James, 1998). To examine these possibilities, we measured the expression of the specific anti-apoptotic protein Bcl-2, which has been shown to act on mitochondria and prevent the release of cytochrome c and subsequent caspase activation, as well as the pro-apoptotic protein Bax, which has been shown to accelerate these processes (Oltvai et al., 1993). As illustrated in Fig. 4B, the western blot showed a progressive increase in translocation of cytochrome c from 9 to 12 months of exposure, consistent with the findings from the TUNEL assay. To confirm these findings caspase 3 activity was estimated in the cytosolic fraction of mouse liver and revealed a progressive increase in arsenic exposed mice from 9 to 12 months (Fig. 4C). In contrast the anti-apoptotic factor, Bcl-2 mRNA was only weakly expressed, whereas pro-apoptotic Bax expression was enhanced (Fig. 4D). These results strongly suggest that arsenic exposure causes cell death mediated by the mitochondrial apoptotic pathway.
Fig. 4.

Influence of long term arsenic exposure (6 μg/g body weight/day) in the induction of apoptosis in mice liver. A) Apoptosis in the liver sections was assessed by Tunnel Assay. Eight different fields from each liver section were observed and the mean value was plotted graphically. B) Western blot analysis for Bcl2, Bax and Cytochrome c from liver extracts from control and arsenic exposed mice at the end of 9 and 12 months of arsenic exposure. The protein bands were quantified and expressed as fold induction compared with that of controls from the respective months. Data were expressed under the blotting bands as mean±SD (n=6) for the different study periods. C) Caspase 3 activity of liver extract was determined using a fluorometric assay with Ac-DEVD AFC, in control and arsenic treated mice after 9 and 12 months of exposure. Results are presented as the percentage increase from control values. The results of A and C were expressed as mean±SD of 10 mice per group. (**p<0.001). D) Reverse transcript PCR demonstrate mRNA expression of anti-apoptotic protein Bcl2, pro-apoptotic protein Bax and Caspase 3 at the end of 9 months and 12 months respectively. β-actin was used as in house control. Representative mRNA expression of control mice of 9 months (C9 in Lane 1); 12 months (C12 in Lane 4); arsenic exposed mice for 9 months (As9 in Lanes 2 and 3) and arsenic exposed mice for 12 months (As12 in Lanes 5 and 6) respectively. The quantification of the signal corrected by β-actin was referred to that of their respective control liver, which was assigned a value of 1. The results were expressed as fold induction compared with that of controls. Data are expressed as mean±SD (n=6).
Profibrogenic factors responsible for arsenic induced liver fibrosis
Liver fibrosis results from the deposition and accumulation of type I collagen with simultaneous inhibition of collagen degradation by interstitial collagenase. TIMP-1 plays a key role in the regulation of ECM accumulation through its ability to inhibit many other MMPs (Arthur, 2000). Therefore, we measured TIMP-1 expression in liver of mice exposed to arsenic for 9 and 12 months and found that TIMP-1 expression was markedly increased in liver of the arsenic exposed mice after 12 months of exposure (Fig. 5A).
Fig. 5.

Arsenic exposure and liver fibrosis in mice: (A) Reverse transcriptase PCR demonstrates enhanced expression of TIMP-1 during long term arsenic exposure. Lane 1 and lane 4 represent 9 months and 12 months control values while lane 2, 3 and lane 5, 6 were from mice treated with arsenic for 9 months and 12 months respectively. The quantification of the signal corrected by β-actin was referred to that of the respective control (B) Upper panel (i) shows hepatic TGF-β protein level of mice exposed to arsenic for different time periods. Hepatic TGF-β content was quantified by ELISA. The results were expressed as mean±SD of 10 mice per group. **p<0.001. Lower panel (ii) shows reverse transcriptase PCR that demonstrates enhanced expression of TGF-β. C) mRNA expression of α-SMA was observed after 12 months of arsenic treatment. Lane 1 and lane 4 represent 9 months and 12 months control values while lane 2, 3 and lane 5, 6 were from mice treated with arsenic for 9 months and 12 months respectively. β-actin was used as in house control. D) Treatment with arsenic for a prolonged period causes HSCs activation and remodeling of the hepatic matrix. Upper panel (i) shows mRNA expression of PDGF-Rβ, MMP-2 and MMP-9 by RT-PCR at the end of 9 months and 12 months respectively. Lane 1 and lane 4 represent 9 months and 12 months control values while lane 2, 3 and lane 5, 6 were from mice treated with arsenic for 9 months and 12 months respectively. β-actin was used as in house control. Lower panel (ii) shows enhanced MMP-2 activity in the liver of mice exposed for 12 months by gelatin zymography (lane 4). All the mRNA expression data and zymography was performed with 6 mice per group.
TIMP-1 is synthesized and secreted by activated HSCs in response to fibrogenic cytokines, in particular TGF-β1 (Cao et al., 2002). Increased levels of TGF-β1 have been found in experimental models of liver fibrosis. When hepatic TGF-β levels were measured by ELISA in arsenic exposed and control mice, a significant increase in hepatic TGF-β level was also seen after 12 months of arsenic exposure compared to the control mice (Fig. 5Bi), a finding that was confirmed by RT-PCR mRNA expression for TGF-β (Fig. 5Bii). Because α-SMA is a marker of HSCs activation, we also studied the mRNA for α-SMA. Normally α-SMA mRNA expression is not found in control mice. In the current study we observed increase in α-SMA mRNA expression in the liver tissue only after 12 months of arsenic exposure (Fig. 5C). As previously noted, the number of stellate cells, identified by immunofluorescent staining of α-SMA by confocal microscopy was also increased, per 40× field at 12 months compared to 9 months of arsenic exposure (Fig. 3B).
We also determined the effect of chronic arsenic exposure on stellate cell proliferation, a crucial step in the development of liver fibrosis. Platelet derived growth factor (PDGF) is thought to be a fibrogenic cytokine that enhances proliferation of HSCs in the fibrotic liver. As seen in Fig. 5Di, PDGF-Rβ mRNA expression was also increased in liver tissue after 9 and 12 months of arsenic exposure, consistent with the activation of stellate cells.
Finally since TIMP production was detected at 12 months of arsenic exposure we also assessed the levels of MMPs. As noted in Fig. 5Di, marked upregulation of MMP-2 and 9 mRNA expressions was observed between 9 and 12 months of arsenic exposure. This finding was confirmed by increased MMP-2 synthesis as suggested by gelatin zymography (Fig. 5Dii). Together, these findings suggest significant matrix remodeling, a pre-requisite criteria of fibrosis (Mandal et al., 2003).
In vitro activation of HSCs
To understand the activation of HSCs due to oxidative stress in hepatocytes caused by arsenic, we co-cultured mouse primary hepatocytes with HSCs. We evaluated ROS generation in both hepatocytes and HSCs after 1, 3 and 5 days of co-culture by a fluorescence multi-plate reader using the 2′, 7′-dichlorofluorescein diacetate (DCF-DA). There was a time dependent increase of ROS generation in both the cells of the co-culture in presence of arsenic, while ROS level remained the same in these cells without arsenic exposure (Fig. 6B). The increase of ROS generation by hepatocytes was not altered when co-cultured with HSCs (data not shown). We visualized intracellular ROS such as H2O2 of HSCs by confocal microscopy (Fig. 7Ai). We also examined whether increased ROS caused a phenotypic change of HSCs to myofibroblast like state accompanied by the appearance of α-SMA filaments, a marker of HSCs activation. A time course experiment revealed the presence of α-SMA filaments in a significant number of HSCs after 5 days co-culture with hepatocytes in presence of arsenic (Fig. 7Aii). Moreover RT-PCR revealed an elevated mRNA expression of PDGF-Rβ and pro-(α) collagen I in HSCs after 5 days co-culture with hepatocytes in presence of arsenic (Fig. 7B). Addition of NAC in the cell insert containing hepatocytes 1 h before treatment of arsenic not only caused significant reduction of ROS generation in HSCs and immunofluorescence of α-SMA filaments (Fig. 7A) but also inhibited mRNA expression of PDGF-Rβ and pro-(α) collagen I (Fig. 7B).
Fig. 7.

Arsenic exposed hepatocytes cause HSCs activation. A)(i) Intracellular ROS, mainly H2O2, levels were observed in each cell type (HSCs and hepatocytes) individually, as well as in the co-cultures, using DCF-DA fluorescent probe and (ii) Immunofluorescence for α-SMA in HSCs cultured alone or with arsenic exposed hepatocytes pretreated with or without NAC were evaluated by confocal microscopy. HSCs were maintained in culture or co-incubated with hepatocytes for 5 days. Original magnification 40×; zoom 3.2 in case of (i). The nucleus was stained with DAPI. B) mRNA expression of Pro-(α) collagen I and PDGF-Rβ in HSCs by RT-PCR at the end of 5 days of co-culture. Lane 1 and 2 represents HSCs co-cultured without or with hepatocytes without arsenic treatment. Lane 3 and 4 represents HSCs co-cultured with arsenic exposed hepatocytes without or with NAC pretreatment. β-actin was used as in house control. C) Accumulation of ROS mainly H2O2 in the culture medium was evaluated by measuring the DCF fluorescence in a fluorescence multi-plate reader. Results were expressed as micro molar amount of H2O2 released to the incubation medium in the presence or absence of 2000 U of catalase. D) The amount of lipid peroxidation end-products released to the culture medium due to arsenic treatment was evaluated. Results are expressed in picomole (pM) of TBARs. All results refer to mean±SD of 4 sets of individual experiments. **p<0.001.
Because H2O2 can be easily permeable to the plasma membrane, it is possible that oxidative stress derived products from hepatocytes can diffuse and produce a pro-oxidant stress in HSCs. We measured the levels of H2O2 in the culture media to determine whether ROS could diffuse from the hepatocytes through the cell insert filter to HSCs. H2O2 was elevated by more than 4 fold in the culture medium in the HSCs/hepatocytes (exposed to arsenic) co-culture compared with HSCs incubated alone or with hepatocytes without arsenic exposure (p<0.001) (Fig. 7C). Further, this effect was blocked by the addition of NAC in cell inserts and catalase in the incubating medium (Fig. 7C). These data suggest that the arsenic exposure to hepatocytes produces ROS, such as H2O2, which can diffuse to the HSCs culture medium.
Lipid peroxidation end-products in the co-cultures
Collagen production in HSCs is stimulated by end-products of lipid peroxidation. We therefore, assessed the lipid peroxidation end-products secreted to the culture medium by the thiobarbituric acid reactive substances (TBARS) method. Similar to the DCF fluorescence, the amount of lipid peroxidation end-products present in the culture medium was estimated and an increase of about 8 fold was found in HSCs co-incubated with the hepatocytes exposed with arsenic when compared with the other two co-cultures (p<0.001) (Fig. 7D). This effect was prevented by the addition of 10 mmol/L NAC in the cell inserts.
Discussion
In this study we present evidence, for the first time, of the pathophysiological mechanism by which arsenic induced oxidative stress and hepatocyte apoptosis leads to progressive portal fibrosis, the pathological lesion seen in human liver, in a murine model of chronic arsenic toxicity. Our study demonstrates significant increase in fibrosis on liver histology along with evidence for up-regulation of profibrogenic cytokines and markers of HSCs activation in mice, after exposure to high doses of arsenic for 1 year. This was preceded by accumulation of arsenic in the liver, development of oxidative stress, liver cell injury, and increase in hepatocyte apoptosis. Together, this data provides support for a sequence whereby chronic exposure to arsenic produces oxidative stress with generation of ROS via the NADPH oxidase pathway as well as increased Kupffer cell activation and TNF-α production. TNF-α production leads to hepatocyte apoptosis via the mitochondrial cytochrome c pathway as well as activation of HSCs. Activated HSCs produce collagen I and the pro-fibrogenic PDGF and TGF-β, resulting in up-regulation of TIMPs and MMPs, leading to remodeling of the ECM and histological hepatic fibrosis. These intertwined networks of inflammation, cell death and HSCs activation were morphologically represented by initial hepatic steatosis leading to ultimate portal fibrosis with continued prolonged exposure to arsenic in drinking water.
The rationale for the current experiments came from our earlier studies that demonstrated increased lipid peroxidation, steatosis, and portal fibrosis after prolonged exposure to arsenic in high doses (Santra et al., 2000b), where the mice were allowed to drink arsenic contaminated water 3.4 mg/L ad libitum and their food was prepared with this contaminated water. Neither the amount of arsenic consumed by the mice was properly documented nor were the pathophysiological mechanisms contributing to hepatic fibrogenesis adequately investigated in our previous study.
We speculate that oxidative stress may be an important initiating factor in the pathogenesis of the liver injury. We found a persistent and progressive increase in oxidant stress in the liver, after exposure to arsenic for 9 to 12 months as demonstrated by increased deposition of 4-HNE adduct in the liver section over this time period. 4-HNE is a prototypic toxic end product of lipid peroxidation that results from oxidative stress (Esterbauer et al., 1991). The source of oxidant production in liver due to arsenic exposure is still unknown, although we have previously reported that the early phase of acute arsenic exposure results in oxidative stress due to generation of ROS both from mitochondrial as well as from the non-mitochondrial pathways (Santra et al., 2007). Similar to our previous report, in the present study also, we noted increases in SOD, TBARs and GSSG as well as decrease in GSH peroxidase at 9 months of arsenic exposure (see Table S1), all markers of increased oxidative stress (Santra et al., 2000a, 2000b; Choi and Ou, 2006). Although there was evidence of hepatic oxidative stress (Table S1) associated with increased accumulation of arsenic in the liver after 6 months of arsenic exposure (Table S2), the hepatic morphology was normal.
NADPH oxidase is a potential non-mitochondrial source of ROS. In the current study we tried to further elucidate the role of NADPH oxidase in arsenic induced liver injury and subsequent development of fibrosis, since it is well established that ROS activate HSCs and stimulate fibrogenesis (Nieto et al., 1999). We found that enhanced accumulation of 4-HNE adduct in the liver was associated with increased NADPH oxidase activity and increased expression of gp91phox and p67phox, the subunits of NADPH oxidase complex. At the same time collagen deposition and quantum of activated HSCs were also increased in the tissue. This supports a role for NADPH oxidase in the development of liver fibrosis following chronic exposure to high levels of arsenic.
To further pursue a functional link between increased oxidant stress and liver fibrosis, we evaluated the role of TNF-α signaling and Kupffer cell activation in arsenic exposed mice liver. TNF-α plays an important role in the activation of HSCs, the predominant cells involved in the production of type I collagen (Bachem et al., 1993; Arthur, 2000; Friedman, 2008). We observed enhanced hepatic TNF-α production, as we previously reported (Das et al., 2005), accompanied by up regulation of CD14 and TLR-4 mRNA transcripts after prolonged arsenic exposure. Although the process by which arsenic activates Kupffer cells is not known, Kupffer cell activation seems to be mediated by CD14 and TLR-4 (Su, 2002). Arsenic induced oxidative stress in liver may up regulate CD14 expression and thus increase the susceptibility of Kupffer cells to arsenic or its metabolites. It seems likely that the Kupffer cells, in turn, up regulate TNF-α production and it is well known that cytokines released from activated Kupffer cells stimulate collagen synthesis in HSCs (Friedman, 1999). Thus, although the precise mechanism remains to be determined, arsenic induced ROS in the liver appears to play an important role in development of liver injury.
We observed an increase in caspase activity and an increase in TUNEL positive hepatocytes in the liver exposed to arsenic for prolonged periods, indicative of apoptosis. The pro-apoptotic proteins Bax and cytosolic cytochrome c, which are released from mitochondria during apoptosis were also increased after arsenic exposure, whereas the anti-apoptotic protein Bcl2 was reduced (Figs. 4B and D). These findings suggest that the mechanism of hepatocyte apoptosis after arsenic exposure in mice is most likely attributable to the mitochondrial pathway for apoptosis.
Several lines of evidence strongly suggest that arsenic exposure in this study resulted in activation of HSCs. First, both immunohistochemical staining (Figs. 3A and B) as well as mRNA levels for α-SMA (Fig. 5C), a hallmark for stellate cell activation were detected in the livers of mice treated with arsenic after 12 months as compared to control animals and mice treated for 9 months. In addition, both the growth factors, TGF-β and PDGF-Rβ were detected in hepatic tissues after arsenic exposure compared to control livers (Figs. 5B and D). Both proteins are important in induction of stellate cell activation and proliferation and the development of fibrosis (Bissell et al., 2001).
Finally our study provides conclusive evidence for the development of hepatic fibrosis after 9 to 12 months of exposure to high levels of arsenic. This is visually demonstrated by Sirius red stained liver tissues (Fig. 2B) which show progressive accumulation of Sirius red stained fibers that develop in periportal regions of the hepatocytes. Quantitative assessment of fibrosis is provided by measurements of hepatic collagen (Fig. 2D) and pro-(α) collagen I mRNA expression (Table 1) that confirms the histological findings. Hepatic levels of TIMP-1 mRNA were modestly increased at 12 months while MMP-2 and 9 levels were also increased. Heightened expression of MMPs likely contributes to the pathogenesis of arsenic induced liver injury, as several studies also report that MMP-2 and MMP-9 were up-regulated in the fibrotic liver (Bansal et al., 2005). The normal ECM is essential for maintaining the homeostasis of all resident liver cells. Therefore, it is likely that degradation of the ECM by MMPs alters cell matrix as well as cell–cell interactions and enhances hepatocytes susceptibility to necrosis and/or apoptosis caused by prolonged arsenic exposure. The ECM also serves as a binding reservoir for several key cytokines such as TNF-α and growth factors such as TGF-β, and PDGF (Taipale and Keskioja, 1997; Davis et al., 2000). MMPs release soluble bioactive factors through ECM degradation and regulate macrophage chemoattractants and leukocyte infiltration during injury (Uchinami et al., 2006). Collectively, these data strongly suggest that arsenic mediated liver injury is associated with HSCs activation and hepatic fibrogenesis.
To validate our in vivo findings and to investigate whether sustained oxidative stress in the hepatocytes due to arsenic exposure were responsible for activation of HSCs that may transdifferentiate into myofibroblasts, we used a co-culture model containing HSC with hepatocytes (Fig. 6A) with or without arsenic exposure. The dose of arsenic (2 μM) was selected in this in-vitro study because it may mimic chronic environmental exposure as described previously (Peng et al., 2007). This system displayed several lines of evidence to provide role of oxidative stress in the hepatocytes in arsenic toxicity. First, we have identified excessive ROS generation in both hepatocytes as well as HSCs. ROS are known to play a key role in HSC activation (Nieto et al., 2002). Second, we have detected H2O2 and TBARS accumulation, derived from hepatocytes, in the incubating medium of HSCs and third, higher levels of α-SMA, PDGF-Rβ and Pro-(α) collagen I expression in HSCs. H2O2 derived from hepatocytes may induce collagen transcription in HSCs (Nieto et al., 2002). Further pretreatment of hepatocytes with antioxidant, NAC, 1 h before they were exposed to arsenic abrogates both generation of ROS in the cells, H2O2 and TBARS accumulation in the medium and expression of α-SMA along with decreased expression of PDGF-Rβ and collagen I. Thus, results of the present study clearly support a role of oxidative stress in the hepatocytes due to arsenic exposure in the activation of HSCs.
Our results differ from another study which used a comparatively low to moderate exposure to arsenic (250 ppb in drinking water) for a shorter duration (5 weeks) (Straub et al., 2007). Such exposure caused increased SECs capillarization, vascularisation of the peribiliary vascular plexus, and constriction of hepatic arterioles in mice. However, this lower level of arsenic exposure, equivalent to human dose of 32 μg/day for 3.75 years, did not cause arsenic deposition in the liver or result in portal fibrosis, an important feature in liver histological lesion in chronic arsenic induced hepatotoxicity of humans (Guha Mazumder et al., 1988; Santra et al., 1999; Guha Mazumder, 2005; Dutta et al., 1979; Morris et al., 1974; Huet et al., 1975; Nevens et al., 1990). The rationale for the relatively high dose of arsenic used in our current experimental study comes from epidemiological studies of arsenicosis in this region (Guha Mazumder, 2005). These studies suggest that clinical manifestations of arsenic induced liver injury develops only after a long latency with the intake of large volumes of arsenic contaminated water, usually since childhood (Guha Mazumder et al., 1988; Santra et al., 1999; Guha Mazumder, 2005; Dutta et al., 1979). The dose we used in the present study was simulated to be equivalent to a human exposure of 6 mg of arsenic/day for more than 20 years, a time when liver toxicity might be expected to occur. However, the precise relationship between duration of arsenic exposure and the onset of liver lesions is uncertain and remains to be clarified.
In conclusion, the present study contributes substantially to the understanding of the arsenic induced liver fibrosis. On the basis of the present findings, we propose that chronic arsenic exposure results in oxidative stress in the liver via NADPH oxidase, leading to Kupffer cell activation and subsequent TNF-α signaling mechanism that result in hepatic cell apoptosis, activation of HSCs, and the development of hepatic fibrosis. Future studies will need to delineate the intertwined pathways of this fibrogenic process as well as to determine treatment strategies for patients chronically exposed to arsenic.
Supplementary Material
Acknowledgments
The authors would like to acknowledge the help of Dr. Kshaunish Das for his critical appraisal of the manuscript.
Grant support: This study was funded by Indian Council of Medical Research, New Delhi (grant no.: 5/8/4-17(Env) 04-NCD-I to A.S and grant no.: 68/13/2004-NCD-I to A.C).
Abbreviations
- 4HNE
4 hydroxy-2 non-enal
- α-SMA
α-smooth muscle actin
- ΔCT
difference in threshold cycle
- CD14
cluster of differentiation 14
- DCF-DA
2′,7′-dichlorofluorescein diacetate
- DMEM
Dulbecco’s modified eagle’s medium
- ECM
extra cellular matrix
- ALT
alanine aminotransferases
- ELISA
enzyme-linked immunosorbant assay
- GSH
reduced glutathione
- GSSG
oxidized glutathione
- H&E
Hematoxylin & eosin
- HSCs
hepatic stellate cells
- MMP
matrix metalloproteinase
- NAC
N-acetyl cysteine
- NOX2
NADPH oxidase 2
- PDGF-Rβ
platelet derived growth factor receptor β
- ROS
reactive oxygen species
- RT-PCR
reverse transcriptase polymerase chain reaction
- qRT-PCR
quantitative real time polymerase chain reaction
- SECs
sinusoidal endothelial cells
- SOD
superoxide dismutase
- TBARs
thiobarbituric acid reactive substances
- TGF-β1
transforming growth factor β-1
- TIMP-1
tissue inhibitor of matrix metalloprotei-nases-1
- TLR-4
toll like receptor-4
- TNF-α
tissue necrosis factor-α
- TUNEL
terminal deoxynucleodidyl transferase-mediated deoxyuridine triphosphate nick-end labeling
Appendix A. Supplementary data
Supplementary data to this article can be found online at doi:10.1016/j.taap.2010.11.016.
Footnotes
Disclosures: The authors have nothing to disclose financially and there is no conflict of interest.
References
- Arthur MJ. Fibrogenesis II (Metalloproteinases and their inhibitors in liver fibrosis) Am J Physiol Gastrointest Liver Physiol. 2000;279:245–249. doi: 10.1152/ajpgi.2000.279.2.G245. [DOI] [PubMed] [Google Scholar]
- Bachem MG, Shell KM, Melchior R, Kroof J, Gressner AM. TNF-alpha and TGF-beta1 stimulate fibronectin synthesis and the transdifferentiation of fat storing cells in the rat liver into myofibroblast. Virchows Arch B Cell Pathol. 1993;63:123–130. doi: 10.1007/BF02899251. [DOI] [PubMed] [Google Scholar]
- Bagla P, Kaiser J. India’s spreading health crisis draws global arsenic experts. Science. 1996;274:174–175. doi: 10.1126/science.274.5285.174. [DOI] [PubMed] [Google Scholar]
- Bansal MB, Kovalovich K, Gupta R, Li W, Agarwal A, Radbill B, et al. Interleukin-6 protects hepatocytes from CCl4-mediated necrosis and apoptosis in mice by reducing MMP-2 expression. J Hepatol. 2005;42:548–556. doi: 10.1016/j.jhep.2004.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bataller R, Schwabe RF, Choi YH, Yang L, Paik YH, Lindquist J, et al. NADPH oxidase signal transduces angiotensin II in hepatic stellate cells and is critical in hepatic fibrosis. J Clin Invest. 2003;112:1383–1394. doi: 10.1172/JCI18212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissell DM, Roulot D, George J. Transforming growth factor beta and the liver. Hepatology. 2001;34:859–867. doi: 10.1053/jhep.2001.28457. [DOI] [PubMed] [Google Scholar]
- Cao Q, Mak KM, Lieber CS. Dilinoleoylphosphatidylcholine prevents transforming growth factor-β1-mediated collagen accumulation in cultured rat hepatic stellate cells. J Lab Clin Med. 2002;139:202–210. doi: 10.1067/mlc.2002.121853. [DOI] [PubMed] [Google Scholar]
- Chen CJ, Chen CW, Wu MM. Arsenic and cancer. Lancet. 1988;1:414–415. doi: 10.1016/s0140-6736(88)91207-x. [DOI] [PubMed] [Google Scholar]
- Chi IC, Blackwell RQ. A controlled retrospective study of blackfoot disease, an endemic peripheral gangrene disease in Taiwan. Am J Epidemiol. 1968;88:7–24. doi: 10.1093/oxfordjournals.aje.a120869. [DOI] [PubMed] [Google Scholar]
- Choi J, Ou JH. Mechanisms of Liver Injury. III Oxidative stress in the pathogenesis of hepatitis C virus. Am J Physiol Gastrointest Liver Physiol. 2006;290:G847–G851. doi: 10.1152/ajpgi.00522.2005. [DOI] [PubMed] [Google Scholar]
- Das S, Santra A, Lahiri S, Guha Mazumder DN. Implications of oxidative stress and hepatic cytokine (TNF-α and IL-6) response in the pathogenesis of hepatic collagenesis in chronic arsenic toxicity. Toxicol Appl Pharmacol. 2005;204:18–26. doi: 10.1016/j.taap.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Davis G, Bayless KJ, Davis M, Meininger GA J. Regulation of tissue injury responses by the exposure of matricryptic sites within extracellular matrix molecules. Am J Pathol. 2000;156:1489–1498. doi: 10.1016/S0002-9440(10)65020-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day CP, James OF. Hepatic steatosis: innocent bystander or guilty party? Hepatology. 1998;27:1463–1466. doi: 10.1002/hep.510270601. [DOI] [PubMed] [Google Scholar]
- Ding WX, Ni HM, Francesca DD, Stolz DB, Yin XM. Bid-dependent generation of oxygen radicals promotes death receptor activation-induced apoptosis in murine hepatocytes. Hepatology. 2004;40:403–413. doi: 10.1002/hep.20310. [DOI] [PubMed] [Google Scholar]
- Dutta DV, Mitra SK, Chhuttani PN, Chakravarti RN. Chronic oral arsenic intoxication as a possible etiological factor in idiopathic portal hypertension (non-cirrhotic portal fibrosis) in India. Gut. 1979;20:378–384. doi: 10.1136/gut.20.5.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsharkawy AM, Oakley F, Mann DA. The role and regulation of hepatic stellate cell apoptosis in reversal of liver fibrosis. Apoptosis. 2005;10:927–939. doi: 10.1007/s10495-005-1055-4. [DOI] [PubMed] [Google Scholar]
- Environmental Protection Agency. National primary drinking water regulations; arsenic and clarifications to compliance and new source contaminants monitoring; final rule. Fed Regist. 2001;66:6976–7066. [Google Scholar]
- Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- Fendorf S, Michael HA, Van Geen A. Spatial and temporal variations of groundwater arsenic in south and Southeast Asia. Science. 2010;328:1123–1127. doi: 10.1126/science.1172974. [DOI] [PubMed] [Google Scholar]
- Fontana L, Jerez D, Rojas-Valencia L, Solís-Herruzo JA, Greenwel P, Rojkind M. Ethanol induces the expression of alpha 1(I) procollagen mRNA in a co-culture system containing a liver stellate cell-line and freshly isolated hepatocytes. Biochim Biophys Acta. 1997;1362:135–144. doi: 10.1016/s0925-4439(97)00056-2. [DOI] [PubMed] [Google Scholar]
- Friedman SL. Cytokines and fibrogenesis. Semin Liver Dis. 1999;19:129–140. doi: 10.1055/s-2007-1007105. [DOI] [PubMed] [Google Scholar]
- Friedman SL. Mechanism of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–1669. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman SL, Roll FJ, Boyles J, Bissell DM. Hepatic lipocytes: the principal collagen-producing cells of normal rat liver. Proc Nat Acad Sci USA. 1985;82:8681–8685. doi: 10.1073/pnas.82.24.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard MJ, Tanhehco JL, Dau PC. Chronic arsenic poisoning masquerading as Landry–Guillian–Barré syndrome. Electromyogr Clin Neurophysiol. 1992;32:419–423. [PubMed] [Google Scholar]
- Guha Mazumdar DN, Haque R, Ghosh N, De BK, Santra A, Chakraborty D, et al. Arsenic levels in drinking water and the prevalence of skin lesions in West Bengal, India. Int J Epidemiol. 1998;27:871–877. doi: 10.1093/ije/27.5.871. [DOI] [PubMed] [Google Scholar]
- Guha Mazumder DN. Effect of chronic intake of arsenic-contaminated water on liver. Toxicol Appl Pharmacol. 2005;206:169–175. doi: 10.1016/j.taap.2004.08.025. [DOI] [PubMed] [Google Scholar]
- Guha Mazumder DN. Chronic Arsenic toxicity and human health. Indian J Med Res. 2008;128:436–447. [PubMed] [Google Scholar]
- Guha Mazumder DN, Chakraborty AK, Ghose A, Gupta JD, Chakraborty DP, Dey SB, et al. Chronic arsenic toxicity from drinking tubewell water in rural West Bengal. Bull World Health Organ. 1988;66:499–506. [PMC free article] [PubMed] [Google Scholar]
- Guha Mazumder DN, Haque R, Ghosh N, De BK, Santra A, Chakraborty D, et al. Arsenic in drinking water and the prevalence of respiratory effects in West Bengal, India. Int J Epidemiol. 2000;29:1047–1052. doi: 10.1093/ije/29.6.1047. [DOI] [PubMed] [Google Scholar]
- Harvey CF. Poisoned waters traced to source. Nature. 2008;454:415–416. doi: 10.1038/454415a. [DOI] [PubMed] [Google Scholar]
- Huet PM, Guillaime E, Cote J, Légaré A, Lavoie P, Viallet A. Non-cirrhotic perisinusoidal portal hypertension—association with chronic arsenical intoxication. Gastroenterology. 1975;68:1270–1277. [PubMed] [Google Scholar]
- IARC. IARC Monographs. Vol. 84. IARC; Lyon: 2004. Some drinking water disinfectants and contaminants, including arsenic. Monographs on the evaluation of carcinogenic risk to humans. [PMC free article] [PubMed] [Google Scholar]
- Kim YM, Talanian RV, Billiar TR. Nitric oxide inhibits apoptosis by preventing increases in caspase-3-like activity via two distinct mechanisms. J Biol Chem. 1997;272:31138–31148. doi: 10.1074/jbc.272.49.31138. [DOI] [PubMed] [Google Scholar]
- Mandal M, Mandal A, Das S, Chakraborti T, Sajal C. Clinical implications of matrix metalloproteinases. Mol Cell Biochem. 2003;252:305–329. doi: 10.1023/a:1025526424637. [DOI] [PubMed] [Google Scholar]
- Morris JS, Schmid M, Newman S, Scheuer PJ, Sherlock S. Arsenic and noncirrhotic portal hypertension. Gastroenterology. 1974;66:86–94. [PubMed] [Google Scholar]
- Nakamura T, Torimura T, Sakamoto M, Hashimoto O, Taniguchi E, Inoue K, et al. Significance and therapeutic potential of endothelial progenitor cell transplantation in a cirrhotic liver rat model. Gastroenterology. 2007;133:91–107. doi: 10.1053/j.gastro.2007.03.110. [DOI] [PubMed] [Google Scholar]
- National Research Council. Arsenic in Drinking Water. National Academy Press; Washington, D.C: 1999. [Google Scholar]
- Navas-Acien A, Sharrett AR, Silbergeld EK, Schwartz BS, Nachman KE, Burke TA, et al. Arsenic exposure and cardiovascular disease: a systematic review of the epidemiologic evidence. Am J Epidemiol. 2005;162:1037–1049. doi: 10.1093/aje/kwi330. [DOI] [PubMed] [Google Scholar]
- Navas-Aciens A, Silbergeld EK, Pastor-Barriuso R, Guallar E. Arsenic exposure and prevalence of Type 2 diabetes in US adults. JAMA. 2008;300:814–822. doi: 10.1001/jama.300.7.814. [DOI] [PubMed] [Google Scholar]
- Nevens F, Fevery J, Van Steenbergen W, Sciot R, Desmet V, De Groote J. Arsenic and non-cirrhotic portal hypertension. A report of eight cases. J Hepatol. 1990;11:80–85. doi: 10.1016/0168-8278(90)90276-w. [DOI] [PubMed] [Google Scholar]
- Nieto N, Friedman SL, Greenwel P, Cederbaum AI. CYP2E1 mediated oxidative stress induces collagen type I expression in rat hepatic stellate cells. Hepatology. 1999;30:987–995. doi: 10.1002/hep.510300433. [DOI] [PubMed] [Google Scholar]
- Nieto N, Dominguez-Rosales JA, Fontana L, Salazar A, Armendariz-Borunda J, Greenwel P, et al. Rat hepatic stellate cells contribute to the acute-phase response with increased expression of alpha1(I) and alpha1(IV) collagens, tissue inhibitor of metalloproteinase-1, and matrix-metalloproteinase-2 messenger RNAs. Hepatology. 2001;33:597–607. doi: 10.1053/jhep.2001.22520. [DOI] [PubMed] [Google Scholar]
- Nieto N, Friedman SL, Cederbaum AI. Stimulation and proliferation of primary rat hepatic stellate cells by cytochrome p450 2e1-derived reactive oxygen species. Hepatology. 2002;35:62–73. doi: 10.1053/jhep.2002.30362. [DOI] [PubMed] [Google Scholar]
- Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- Peng Z, Peng L, Yunxia F, Zandi E, Shertzer HG, Xia Y. A critical role of I B kinase β in Metallothionein-1 expression and protection against arsenic toxicity. J Biol Chem. 2007;282:21487–21496. doi: 10.1074/jbc.M702510200. [DOI] [PubMed] [Google Scholar]
- Pertoft H, Smedsrød B. In: Cell Separation. Pretlow TG, Pretlow TB, editors. Vol. 4 Academic Press; New York: 1987. pp. 1–24. [Google Scholar]
- Rahman M, Tondel M, Ahmad SA, Axelson O. Diabetes mellitus associated with arsenic exposure in Bangladesh. Am J Epidemiol. 1998;148:198–203. doi: 10.1093/oxfordjournals.aje.a009624. [DOI] [PubMed] [Google Scholar]
- Santra A, DasGupta J, De BK, Guha Mazumder DN. Hepatic manifestations in chronic arsenic toxicity. Indian J Gastroenterol. 1999;18:152–155. [PubMed] [Google Scholar]
- Santra A, Maiti A, Chowdhury A, Guha Mazumder DN. Oxidative stress in liver of mice exposed to arsenic contaminated water. Indian J Gastroenterol. 2000a;19:112–115. [PubMed] [Google Scholar]
- Santra A, Maiti A, Das S, Lahiri S, Charkaborty SK, Guha Mazumder DN. Hepatic damage caused by arsenic (As) toxicity in experimental animals. Clin Toxicol. 2000b;38:395–405. doi: 10.1081/clt-100100949. [DOI] [PubMed] [Google Scholar]
- Santra A, Chowdhury A, Ghatak S, Biswas A, Dhali GK. Arsenic induces apoptosis in mouse liver is mitochondria dependent and is abrogated by N-acetylcysteine. Toxicol Appl Pharmacol. 2007;220:146–155. doi: 10.1016/j.taap.2006.12.029. [DOI] [PubMed] [Google Scholar]
- Smith AH, Lingas EO, Rahman M. Contamination of drinking-water by arsenic in Bangladesh: a public health emergency. Bull World Health Organ. 2000;78:1093–1103. [PMC free article] [PubMed] [Google Scholar]
- Straub AC, Stolz DB, Ross MA, Hernández-Zavala A, Soucy NV, Klei LR, et al. Arsenic Stimulates Sinusoidal Endothelial Cell Capillarization and Vessel Remodeling in Mouse Liver. Hepatology. 2007;45:205–212. doi: 10.1002/hep.21444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub AC, Clark KA, Ross MA, Chandra AG, Li S, Gao X, et al. Arsenic-stimulated liver sinusoidal capillarization in mice requires NADPH oxidase-generated superoxide. J Clin Invest. 2008;118:3980–3989. doi: 10.1172/JCI35092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su GL. Lipopolysaccharides in liver injury: molecular mechanisms of Kupffer cell activation. Am J Physiol Gastrointest Liver Physiol. 2002;283:256–265. doi: 10.1152/ajpgi.00550.2001. [DOI] [PubMed] [Google Scholar]
- Taipale J, Keski-Oja J. Growth factors in the extracellular matrix. FASEB J. 1997;11:51–59. doi: 10.1096/fasebj.11.1.9034166. [DOI] [PubMed] [Google Scholar]
- Uchinami H, Seki E, Brenner DA, D’Armiento J. Loss of MMP 13 attenuates murine hepatic injury and fibrosis during cholestasis. Hepatology. 2006;44:420–429. doi: 10.1002/hep.21268. [DOI] [PubMed] [Google Scholar]
- World Health Organization. Arsenic in Drinking Water. Fact Sheet No. 210. WHO Press; Geneva: 1999. pp. 1–6. [Google Scholar]
- Yamauchi H, Yamamura Y. Metabolism and excretion of orally administered dimethylarsinic acid in the hamster. Toxicol Appl Pharmacol. 1984;74:134–140. doi: 10.1016/0041-008x(84)90279-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.