

Abstract
There is growing evidence that cerebrovascular reactivity to carbon dioxide (CVRCO2) is impaired in Alzheimer’s disease (AD). Preclinical and animal studies suggest chronic hypercontractility in brain vessels in AD. We review (a) preclinical studies of mechanisms for impaired CVRCO2 in AD; (b) clinical studies of cerebrovascular function in subjects with AD dementia, mild cognitive impairment (MCI), and normal cognition. Although results of clinical studies are inconclusive, an increasing number of reports reveal an impairment of vascular reactivity to carbon dioxide in subjects with AD, and possibly also in MCI. Thus, CVRCO2 may be an attractive means to detect an early vascular dysfunction in subjects at risk.
Vasoreactivity is a vasodilatory or constrictor reaction of a blood vessel to a stimulus. In neuroscience, the often-studied measure of cerebrovascular reactivity is the response of brain vessels to changing arterial tension of carbon dioxide: the CVRCO2 which is a topic of this review. Carbon dioxide elevation causes vasodilatation and an increase in cerebral blood flow (CBF), while CO2 reduction results in vasoconstriction and CBF decrease. This process is crucial for maintaining pH and respiratory drive [1]. The response of the cerebral circulation to a changing arterial CO2 concentration is not linear: the circulatory response to hypercapnia is greater than the reaction to hypocapnia [2–4]. The percentage of CBF increase in the human brain is estimated at about 6% per 1 mm Hg rise in arterial tension of CO2 (PaCO2); hypocapnia decreases CBF by approximately 3% per 1 mm Hg change in PaCO2 [5]. With end-tidal CO2 (PetCO2), the effects are 5% CBF increase per 1 mmHg rise in PetCO2, and 2% CBF reduction per 1 mm Hg PetCO2 decrease [6]. PetCO2 measurements are often used instead of PaCO2 to avoid invasive sampling. However, despite the tight relationship between the two, relying on PetCO2 leads to overestimation of PaCO2 in hypercapnic conditions [7] and underestimation of CVRCO2 in hypercapnia [8].
Although commonly studied, the mechanisms behind CVRCO2 are not completely understood (Figure 1). The effect of increased CO2 on vessels is most likely mediated through the increase in perivascular pH, activation of potassium ATP [9] and non-ATP channels [10], influx of potassium, hyperpolarization of the endothelial cell [1], and subsequent decrease in intracellular calcium levels in vascular smooth muscles leading to their relaxation (for review [10]). There is growing evidence that nitric oxide (NO) is an important mediator of CO2-related vessel dilatation [1,10–12], and that it is released by the brain during hypercapnic challenge [2]. The sources of this NO increase are still debated: some argue that neurons are possible NO suppliers (reviewed in [10]), while others suggest that the endothelium may play an important role, and that endothelial dysfunction compromises CVRCO2 [12]. The response to hypocapnia likely involves a pH –related rise in inositol triphosphate leading to increased calcium concentration in vascular smooth muscle cells and vasoconstriction [13]. Altogether, the mechanisms involved in hypercapnic vasodilatation are much better described than the pathways of hypocapnic vasoconstriction. It has been suggested that vessels react more strongly to the vasodilators released during hypercapnia than to the constrictors acting during hypocapnia [14].
Figure 1.

Schematic representation of proposed mechanisms involved in hypercapnic vasodilatation and hypocapnic vasoconstriction. Increase in CO2 results in increased pH and activation of potassium channels. Endothelial cells become hyperpolarized due to the rise in intracellular potassium levels. The hyperpolarizing current is transmitted to VSMC, causing closure of Ca++ channels, decrease in intracellular calcium and muscle relaxation [1]. Increase in CO2 through changes in pH also activates nitric oxide synthase [11]. Increased NO concentration in VSMC leads to activation of guanyl cyclase, increase of cGMP, phosphorylation of calcium channels, decreased Ca++ and myorelaxation of VSM [10]. During hypocapnia, reduced CO2 and the resulting increased pH stimulate production of inositol triphosphate [113], which increases intracellular Ca++ concentration in VSM, leading to muscle constriction [13].
CO2 – carbon dioxide, Ca++ - calcium, K+ - potassium, NO – nitric oxide, NOS - nitric oxide synthase, cGMP – cyclic guanosine monophosphate, VSMC- vascular smooth muscle cells, VSM- vascular smooth muscle.
Vasoreactivity is also referred to as cerebrovascular reserve [15], emphasizing the capability of the vascular system to increase its performance above resting blood flow in response to a challenge. Thus CVRCO2 can be considered a measure of vascular health. There is indeed a body of data showing that CVRCO2 is impaired in conditions affecting cerebral vasculature [16–20]. Interestingly, there is also growing evidence that CVRCO2 may be impaired in neurodegenerative diseases like Alzheimer’s disease (AD). It should be noted that CVRCO2 is a different phenomenon from the vascular response to changes in blood pressure (autoregulation) [4,11]. There is also evidence that CVRCO2 is distinct from neurovascular coupling (the increase in blood flow in response to increased brain activation) [21].
In this review, we will first outline data from preclinical experiments that may illuminate possible causes of impaired CVRCO2 in AD, even though it will not be possible to encompass all the mechanisms at play. Second, we will present clinical studies that examined CVRCO2 in subjects with normal cognition, mild cognitive impairment, (MCI), and AD dementia. They are presented by method and, when possible, in chronological order.
Mechanisms of CVRCO2 reduction in AD
Vascular risk factors and atherosclerosis
The prevalence of both neurodegeneration and vascular diseases rises with age. The coexistence of these two conditions, even without considering a possible causal relationship between them, can explain the vessel dysfunction observed in AD. Hypertension, hypercholesterolemia, obesity, and diabetes all contribute to small and large vessel damage [22–25], leading to impairment of CBF regulation and vessels’ contractility [26]. Furthermore, vascular disease does not only coexist with the neurodegenerative process, but also contributes to it [27–29] and, as recently suggested, may even initiate it [30]. Epidemiological evidence indicates that conditions leading to atherosclerosis are associated with higher incidence of cognitive impairment, dementia and AD [31–35]. Irrespective of the initiating trigger, the AD process, once underway, brings into play a number of mechanisms influencing vessels’ contractility and consequently CBF and CVRCO2.
Amyloid
Together with neurofibrillary tangles, β-amyloid deposition is a key feature of the AD process [36]. Amyloid deposits form extracellular plaques [37], and also accumulate in vessel walls [30]. In addition to its neurotoxic effects, β-amyloid has also vasoactive properties. It has been shown that β-amyloid increased vasoconstriction of rat aorta pretreated with the vasoconstrictor phenylepinephrine [38] or endothelin-1 [39]. Furthermore, in guinea pigs, both Aβ42 and Aβ40 augmented contraction of isolated aorta induced by application of norepinephrine [40]. Vasoactive properties of β-amyloid are not limited to the aorta; the amino acid sequence 25–35 constricted skin microvasculature in rats [41], and Aβ42 and Aβ40 also exerted a constrictive effect on the pulmonary artery [40]. Niwa et al. showed that similar effects can be observed directly in the cerebral vasculature and that pharmacological pretreatment is not necessary. Superfusion of mouse neocortex with both Aβ40 and Aβ42 reduced CBF and further potentiated CBF reduction caused by a tromboxane analogue [42]. It was also subsequently reported that mice overexpressing amyloid precursor protein (APP) exhibit an impairment of cerebral autoregulation. Niwa et al. found a linear relationship between brain Aβ40 and Aβ42 levels and the disruption of the vessels’ response to changes in blood pressure. Interestingly, the effects occurred with no parenchymal or vessel amyloid deposition [43]. These findings were later extended to show that resting CBF and glucose utilization were also decreased, by 20–45% and 30–35% respectively, in mice expressing mutated human APP and high brain amyloid levels [44]. Like in the preceding report [43], no brain or vessel deposits were necessary to cause the functional impairment. This stands in contrast to a later study by Shin et al. [45]. They found that the response of the cerebral vasculature to CO2 challenge or to whisker stimulation in young transgenic mice overexpressing a mutant human APP, who have increased soluble amyloid but no amyloid deposition, did not differ from the response of wild type animals. The CVRCO2 differences were apparent only in mice with developed amyloid deposits. Even though it is uncertain whether parenchymal or vascular deposits are a prerequisite for the impairment of vessels’ contractility, it seems that the most abundant β-amyloid peptides act as vasoconstrictors both in systemic and cerebral circulation.
Endothelin
Endothelin-1 (ET-1) is the most potent vessel constrictor in humans [46]. It is produced by enzymatic cleavage of the precursor protein big-ET-1 by endothelin-converting enzymes (ECE) [47]. Their role in AD has been brought to light with the observation that both ECE-1 (produced by endothelial cells) and ECE-2 (a product of neural tissue) are able to catabolize β-amyloid [46,48]. It was suggested that an increase in ET-1 production, possibly leading to CBF disturbances [47], may be a side effect of ECE - β-amyloid interactions [49]. Palmer at al. showed that both ECE-2 mRNA and ECE-2 protein are increased in AD brain tissue as compared to control brains. Furthermore, exposure to Aβ42 caused an increase in ECE-2 mRNA in cell culture, suggesting that higher ECE-2 levels in the AD brain may be an adaptive response to a higher amyloid load [49]. In a subsequent study, the same group reported a nearly 6-fold increase in the expression of the ET-1 precursor gene mRNA in the AD brain. ET-1 and ECE-2 levels were positively correlated and the addition of Aβ42 was shown to release ET-1 in cell culture [46]. Others also observed increased ET-1 expression in microvessels of AD brains as compared to controls [50]. In this study, ET-1 had a neuroprotective effect: in cell culture, preconditioning with a low dose of ET-1 attenuated the deleterious effects of a subsequent administration of thrombin. The authors argued that the expression of other substances, like cytokines, may determine whether ET-1 acts in a protective or harmful way. Overall, it seems that in AD, both ECEs and endothelin are upregulated in response to increased amyloid concentration, and possibly contribute to the impairment in CBF and CVRCO2.
Serum response factor and myocardin
Myocardin and serum response factor are nuclear transcription factors which act in concert to regulate the expression of cardiomyogenic and smooth muscle genes [51]. It has been shown that both of these factors are upregulated in vascular smooth muscle cells (VSMC) isolated from the cortical pial arteries of AD patients [52]. This upregulation was accompanied by an increase in proteins orchestrating smooth muscle contraction in the vascular wall. Furthermore, experimental overexpression of myocardin in culture of cerebral VSMC from age- matched controls resulted in the hypercontractile phenotype. These effects did not seem to result from β-amyloid deposition. Subsequently, the same group observed that the capacity of VSMC from AD cerebral arteries to clear β-amyloid was reduced by 70%, and showed that serum response factor/myocardin reduced β-amyloid clearance by interfering with the function of lipoprotein receptor-related protein 1 [53]. Altogether, changes in transcription factors are yet another putative path leading to a pro-contractile state and vascular dysfunction in AD.
Thrombin
Thrombin is a product of the proteolytic cleavage of prothrombin. In addition to its role in the coagulation cascade, it also acts though proteinase activated receptors (PAR 1 and PAR 4) [54]. Activation of PAR affects expression of multiple genes for cytokines, possibly promoting a pro-inflammatory phenotype. PAR stimulation can also lead to vasoconstriction or vasodilatation, depending on the artery. Procontractile properties of thrombin in healthy systemic or cerebral circulation are rather weak [55]. However, it has been shown that in some cases thrombin can act as a significant vasoconstrictor. For example, it has the capacity to induce contraction of the porcine pulmonary artery though generation of reactive oxygen species [56]. Most interestingly, thrombin plays an important role in cerebral vasospasm after subarachnoid hemorrhage, and its levels in CSF correlate with the vasospasm severity [55]. Furthermore, in many vascular diseases, expression of PAR receptors is increased, and their activation by thrombin may contribute to the hypercontractile state and thickening of the vessel wall (for review [54]). Although the primary source of prothrombin is the liver, this protein is also expressed in neural cells and astrocytes [57]. The relevance of thrombin in the AD process was highlighted by observations that a) upon thrombin activation platelets secrete amyloid precursor protein and β-amyloid [58], b) thrombin is neurotoxic and its administration causes cognitive deficits in rats [59], c) it is found both in neurofibrillary tangles and senile plaques [57], d) its levels are elevated in microvessels isolated from AD brains [60], and finally e) it is synthesized in endothelial cells in AD but not in controls [61]. The reasons for this increased concentration are not fully understood, but overall, it is likely that thrombin may be an additional factor contributing to the procontractile state.
Cholinergic dysfunction
The cholinergic contribution to vascular impairment in AD has been comprehensively reviewed [29,62–64]. As summarized by Claassen and Jansen [62], the cholinergic deficit in AD not only reduces cholinergic innervation of cortical neurons, but also results in reduction of cholinergic input to cortical blood vessels. Animal studies revealed that cholinergic neurons originating in the basal forebrain and the substantia innominata project to cortical microvessels directly [65] or through interneurons secreting nitric oxide [66]. The densest projections to cortical vessels were found in the fronto-parietal cortex [65]. Moreover, stimulation of cholinergic neurons in the basal forebrain causes CBF increases in the animal neocortex, while inhibition or destruction of the same neurons results in CBF decrease [29,62]. The vasodilatation is mediated directly by muscarinic receptors [62,63] and most likely indirectly by nicotinic receptors [63,67]. The observations presented above, coupled with the fact that extensive loss of cholinergic fibers projecting to cortical areas is a long known hallmark of AD [68], and that in humans, cholinergic projections to the cortex begin almost exclusively in the basal forebrain (in [65]), clearly support the notion that dysfunction of the acetylcholine system can contribute to the impairment of CBF and CVRCO2. A revised form of the cholinergic hypothesis suggests that cholinergic dysfunction is not a causative factor for AD. Instead, progressive age-related decline of acetylcholine system compromises compensatory mechanisms for multiple types of injuries, thus facilitating cognitive impairment [69]. Although the authors do not list CVRCO2 among the many mechanisms influenced by the cholinergic system, one could argue that it should be included. As supporting evidence, we can cite reports of CBF increases or preservation after treatment with acetylcholinesterase inhibitors in dementia patients [70–74], or the CBF reduction in healthy volunteers after scopolamine administration [75–77]. Furthermore, galantamine normalized CVRCO2 both in patients with AD and patients with vascular dementia [78]. As reviewed by Claassen et al. [62], increases in CBF induced by cholinomimetics do not seem to result from augmentation of metabolism but rather from direct effects of these drugs on brain vessels. Altogether, there is a substantial body of evidence to support the hypothesis of cholinergic involvement in the regulation of CBF and vasoreactivity in AD.
Inflammatory factors
The role of the inflammatory process in AD is widely acknowledged [79,80] and it is beyond the scope of this review. For the sake of completeness, however, we would like to mention that changes in inflammation- related signaling proteins found in the blood of AD patients [81] most likely affect vascular tone and contractility, either directly or indirectly by changing the expression of other molecules.
Anatomical changes in microvasculature
Specific structural changes occur in microvasculature in AD. In up to 95% of AD patients, amyloid is deposited in medium and small vessels of the cortex and leptomeninges, causing cerebral amyloid angiopathy. In these vessels amyloid accumulates in the tunica media adjacent to smooth muscle cells, in some cases inside the smooth muscle cells, and in the adventitia [82], forming transverse bands [83]. In the capillaries Scheibel at al. observed swelling and vessel distortion. The capillary surface was encrusted with rounded nodules filled with amyloid. Other vessels appeared perforated with cavities most likely created by the detachment of amyloid nodules. Lesions spanned the width of the basement membrane, while the endothelial cells were spared. In addition, the perivascular plexus was lost where pathology was the most severe [84]. Interestingly, capillary amyloid angiopathy seem to better correlate with clinical severity and neuropathological grading of AD than with angiopathy of bigger vessels [85].
Other distinctive features of microcirculation in AD were also described [for review [29]]. Some authors found swelling of astrocytic end-feet with deposition of glycogen granules, distorted and degenerated pericytes, irregularity and hypertrophy of the basement membrane [86], irregular and degenerated layers of smooth muscles, or endothelial cells with irregular nuclei [87]. Large vessels were not affected; lesions were prominent in arterioles and capillaries, where the wall was uneven with focal constrictions [86,87]. These findings were further confirmed and extended by Buee et al. [88], who reported thickening of the basement membrane, thinning of the microvessels also known as string vessels, increased vessel tortuosity, decreased number of long microvessels, and reduced number of branches. Vessel architecture seemed to be relatively preserved in the occipital cortex, where AD pathology was also less pronounced. Microvascular changes were found mostly in the regions with more AD lesions: highly metabolic areas and the hippocampus.
In conclusion, both functional and anatomical alterations in AD may contribute to the dysfunction of the vascular system through different mechanisms compromising flow and contractility. It is unclear whether structural lesions in microvessels are the result or the cause of biochemical and molecular changes. The relationship is most likely bidirectional. Since presented data concerning possible mechanisms of vessels dysfunction comes largely from animal or postmortem studies, the importance of these pathways in humans still remains to be determined.
CVRCO2 studies in AD
Xenon 133 and HMPAO (99mTc-hexamethylpropyleneaminoxime) imaging
One of the first reports regarding CVRCO2 in dementia came from a study using hypocapnic challenge [89]. Simard et al. observed that voluntary hyperventilation produced declines in CBF, and concluded that vasoreactivity was not impaired. The subjects under study consisted of a mixed group with “presenile” and senile dementia (presumed AD n=15), vascular dementia (n=3), Korsakoff psychosis (n=4) and other dementing conditions (n=2). CBF was measured with multi-probe scintillation detectors after intracarotid Xenon 133 (Xe133) injection. Even though no reactivity impairment was reported, the authors found that most AD cases had reduced baseline flow based on a predefined threshold of 50 ml/100g/ min. No control group was available for comparisons. Using the same Xe133 technique, Hachinski et al. reported foci of diminished reactivity in response to decreased CO2 levels among AD subjects [90]. Abnormal areas were located in the temporal lobes where baseline perfusion was also low. However, when average hemispheric flow was compared between baseline and challenge conditions, patients with AD (n=8) and vascular dementia (n=9) both showed significant reduction in CBF. The overall reactivity was deemed to be close to normal. The AD group did not differ from the control group (n=5) in baseline average hemispheric CBF. A later study used the Xe133 inhalation method in conjunction with bilateral scintillation detectors [91]. The authors examined CVRCO2 in 5 AD patients, 10 subjects with other types of dementia, and 21 normal volunteers. Hypercapnic challenge was used: participants breathed air mixed with with 5% CO2. Surprisingly, 2 out of 5 AD patients showed higher vasoreactivity as compared to both the control and multi-infarct dementia groups. A low resting flow was given as an explanation for this finding. Reduced CVRCO2 was found in subjects with multi-infarct dementia.
Bonte et al. examined 35 AD patients, 16 stroke patients and 15 normal controls. To measure blood flow, the authors used single photon emission computed tomography (SPECT) after Xe133 inhalation. Acetazolamide was used to increased blood concentration of CO2. In AD patients, acetazolamide increased CBF in over half of the regions of interest (ROI) that displayed low CBF at rest (below 2 standard deviations [SD] relative to the normal group), and in 3 out of 7 ROIs where baseline CBF was below 4 SD. This effect was absent in stroke patients who, in addition to a lack of CBF increases, showed additional areas of CBF reduction after acetazolamide injection [92]. No direct comparisons of percentage of CBF increase were performed between normal and AD subjects. The authors concluded that vasoreactivity was preserved in AD.
In contrast to these results, Stoppe et al. [93] found a significant CVRCO2 impairment in AD. Twelve patients with AD and 9 controls were studied. Here, blood flow was measured with a gamma camera after intracubital injection of 99mTc-hexamethylpropyleneaminoxime (HMPAO). Hypercapnic challenge was achieved with acetazolamide injection. In this study, the percentage increase in CBF between baseline and challenge condition was calculated in each ROI, and directly compared between groups. Not only was the resting CBF lower in the AD group, but the percentage increase in CBF in response to acetazolamide was also uniformly reduced in dementia patients across almost all ROIs. Vasoreactivity correlated with MMSE score in AD subjects, despite high variability of the data. Knapp et al. employed the same imaging technique and the same activation method (although acetazolamide dose was reduced by 50%, to 500 mg). The conclusions, however, were different. Baseline flow was compared among 30 AD patients, 50 subjects with MCI, and 14 healthy controls. The MCI group, as compared to controls, showed decreased CBF in the temporal and parietal regions. In the AD group, flow reduction was also seen in the prefrontal areas. Hypercapnic challenge was performed in 11 MCI and 12 AD subjects. They were analyzed together. In the entire group, acetazolamide injection resulted in significant CBF increase in the prefrontal, frontal and temporal regions; CBF increased to some degree in all areas. The authors concluded that there were no vasoreactivity deficits [94]. Unfortunately, there were no direct comparisons between patients and the control group.
The same HMPAO – SPET (single photon emission tomography) technique was used by Pavics et al., who investigated the value of acetazolamide challenge in discrimination between AD (n=33) and vascular dementia (n=18). Both visual examination and quantitative assessments of means within predefined ROIs were performed. Means were compared to the means obtained from the group of 20 healthy controls. Impaired vasoreactivity was defined as an increase in the number of abnormal regions or an increase in the severity of the abnormality after drug injection. Altogether, by visual comparisons of pre- and post- acetazolamide scans, 73% of AD patients showed preserved vasoreactivity as compared with only 22% in the vascular dementia group. In quantitative assessment these numbers were 76% and 29%, respectively [95]. Again, no data on the magnitude of the CBF increase in the control group were provided.
Direct comparisons between 10 controls and 10 AD patients were given by Oishi et al. They studied response to acetazolamide using Xenon inhalation and subsequent CT scanning. As in the study by Stoppe et al. [93], contrasting rates of CBF increase between patients and controls revealed significant CVRCO2 differences in the frontal, parietal and temporal cortices. Baseline flow in these regions was also lower than in controls. Neither resting CBF nor CVRCO2 differed in the thalamus, caudate or putamen [96]. Overall, these studies provided confusing results, with no convincing evidence for diminished CVRCO2 in AD.
PET (positron emission tomography) studies
15O-labeled water is one of the most commonly used tracers for the measurement of CBF by PET [97,98]. With this method, it is necessary to measure the arterial input function in addition to the radioactivity distribution in the brain. Probably due to the complexity of dual injection experiments, there are very few PET studies of CVRCO2. Kuwabara et al. used PET to compare CVRCO2 among 5 AD patients, 5 subjects with Binswanger’s disease and 5 age-matched normal controls. CVRCO2 was defined as the percent change in CBF per 1 mm Hg change in PaCO2 in response to inhalation of air with 5% CO2. Although resting CBF was significantly lower in the AD group than in healthy age-matched controls in the frontal, temporal, and parietal cortices, as well as in the periventricular white matter, the CVRCO2 did not differ between the two groups. Both resting flow and vasoreactivity were diffusely reduced in the vascular dementia group [99]. This lack of difference between AD and controls was also observed in another PET study by Jagust et al. Here the tracer was N,N,N′-trimethyl-N′-(2-hydroxy -3- methyl -5-iodobenzyl) -1,3- propane-diamine (HIPDM) labeled with a positron-emitting isotope of iodine. Again, though resting CBF levels were lower in AD than in controls, reactivity to hypercapnia did not differ [100]. In a recent work by Rodell et al. it is implied that CVRCO2 in AD is preserved, or at least not completely abolished. The authors attempted to factor out the effects of CO2 on flow, and showed that after accounting for CO2 levels, CBF variability in AD is reduced, which in itself may be pathology [4].
In summary, the gamma camera (Xe133), SPECT and PET studies did not show much evidence for impaired CVRCO2 in AD. While interpreting these findings, several factors should be considered: First, in the earlier publications, the definition of AD was not clearly presented or not fully comparable to the present criteria for probable AD. Terms like senile dementia, presenile dementia and degenerative dementia were used. Second, both hypo- and hypercapnic challenges were employed, and it is known that the associated CBF changes are not fully comparable [2,3]. Patients’ comorbidities may have been different. Furthermore, these early techniques of CBF assessment have poor precision and accuracy; they may involve dual injection, facing potential problems related to the need to correct for residual activity from the first acquisition. Methods of radiation detection also varied across studies. Many of the Xe133 and HMPAO studies are difficult to interpret due to the fact that no group comparisons of CBF change in response to the challenge were presented. The authors reported only the changes from pre- to post- challenge within diagnostic groups, and drew conclusions based on this somewhat limited view. When rates of CBF change are directly compared between AD and control groups, the results are equivocal. Two out of 4 reports utilizing molecular imaging methods (Table 1) showed reduced CVRCO2 in AD when compared directly with the control group.
Table 1.
Studies comparing CBF and CVRCO2 between healthy controls, MCI and AD patients.
| Study group | Technique | CO2 challenge | Resting CBF | CVRCO2 | |
|---|---|---|---|---|---|
| Kuwabara et al., 1992 | 5 CTL/ 5 AD | Water [15O] PET | Gas mixture enriched with CO2 | NL>AD | No difference |
| Stoppe et al., 1995 | 9 CTL/ 12 AD | HMPAO – SPET | Acetazolamide | NL>AD | NL>AD |
| Jagust et al., 1997 | 16 CTL/ 5 AD | HIPDM- PET | Gas mixture enriched with CO2 | NL>AD | No difference |
| Oishi et al., 1999 | 10 CTL/ 10 AD | Xenon CT | Acetazolamide | NL>AD | NL>AD |
| Bar et al., 2007 | 20 CTL/ 17 AD | TCD | Gas mixture enriched with CO2 | Not reported | NL>AD |
| Vincenzini et al., 2007 | 62 CTL/ 60 AD | TCD | Gas mixture enriched with CO2 | NL>AD | NL>AD |
| Glodzik et al., 2010 | 17 CTL/ 7 MCI | ASL | Rebreathing | No difference | NL> MCI |
| Yezhuvath et al., 2010 | 17 CTL / 17 AD | ASL (CBF), BOLD fMRI (CVRCO2) | Gas mixture enriched with CO2 | NL>AD | NL>AD |
| Cantin et al., 2011 | 11 CTL/ 7 MCI/ 9 AD | ASL (CBF), BOLD fMRI (CVRCO2) | Gas mixture enriched with CO2 | No difference | Nl>MCI=AD |
CTL: controls, AD: patients with Alzheimer’s disease, MCI: patients with mild cognitive impairment, HMPAO – SPET: single photon emission tomography with 99mTc-hexamethylpropyleneaminoxime, HIPDM- PET: positron emission tomography with N,N,N′-trimethyl-N′-(2-hydroxy -3- methyl -5-iodobenzyl) -1,3- propane-diamine, CT: computed tomography, TCD: transcranial Doppler, ASL: arterial spin labeling, CBF: cerebral blood flow, BOLD fMRI: blood oxygenation level dependent functional MRI, CVRCO2: cerebral vasoreactivity to carbon dioxide
Transcranial Doppler studies
While not strictly a CBF technique, transcranial Doppler (TCD) studies lend more consistent support to the notion that CVRCO2 is impaired in AD patients. TCD is an inexpensive and easy to use modality. A major advantage of TCD is its very high temporal resolution, which makes it suited to study rapid changes in cerebral haemodynamics. This has led to its use in the measurement of CVRCO2. One potential problem in this setting is that TCD measures blood flow velocity and not absolute blood flow. Early angiography studies demonstrated that the change in the middle cerebral artery diameter during routine carbon dioxide inhalation experiments is negligible [101]. However, this observation has been contested [102], and others have shown that large cerebral arteries dilate in response to increased PaCO2 [7]. Unfortunately, due to significant pulsation artifacts, there is currently no reliable method (achieving precision of 1%) to measure the diameter of cerebral arteries. There remains a concern that arterial stiffness may vary across individuals and groups. Given this potential bias, the TCD method to estimate cerebral blood flow and microvascular reactivity should be interpreted with caution unless changes in the vessels’ diameter during the intervention can be precisely monitored on an individual basis. Using TCD, Vicenzini et al. [103] examined a large group of 60 AD, 58 vascular dementia and 62 healthy control subjects, comparable for age and gender distribution. The authors assessed reactions to both hypercapnia (inhalation of gas mixture enriched with 6% CO2) and hypocapnia (hyperventilation). In both dementia groups MFV (mean flow velocity) in the middle cerebral artery at rest was lower than in controls. Moreover, vasoreactivity was significantly different during the hypercapnic challenge, even after controlling for risk factors. Both AD and vascular dementia patients had lower responses as compared with healthy peers. There was no difference between the 2 impaired groups. A similar trend was observed during the hyperventilation. This observation was confirmed by another experiment [78] of 17 AD patients, 17 vascular dementia patients, and a group of age- matched and younger controls (n=20). CVRCO2 differed significantly between age- matched controls and either dementia group, again with no differences between AD and vascular dementia patients. The authors also tested whether galantamine (an acetylcholinesterase inhibitor) could improve CVRCO2 in both dementia groups. After 5 weeks of galantamine treatment CVRCO2 improved significantly in both groups, with no differences between the two diagnostic categories [78]. The lack of differences in CVRCO2 between AD (n=9) and vascular dementia (n=9) was also reported in another recent TCD study, where challenge was elicited by acetazolamide injection. Increases in MFV were observed in both groups, and they were not statistically different. No control group was available for comparisons [104].
Vasoreactivity seems to correlate with cognitive decline. In a group of 53 AD subjects, breath holding index (BHI) was the best predictor of a 2-year change in MMSE and ADAS-Cog scores. BHI was defined as the ratio ((MFVpost−MFVpre)/ MFVpre)/DT where MFVpre, MFVpost, are MFV values before and during breath-holding, and DT is the length of time subjects held their breath. When BHI fell below 1, a steep decline in MMSE was observed. For values > 1, there was a weak correlation between cognitive decline and CVRCO2 [105]. Altogether, TCD experiments reveal noticeable hypoperfusion at challenge in AD, when compared with age-matched healthy elderly. It remains to be seen whether these changes represent large vessel or microvascular disease.
Magnetic resonance imaging
Arterial spin labeling (ASL) provided a means of non-invasive investigation of global and regional cerebral blood flow. Similarly, blood oxygenation level dependent (BOLD) functional MRI (fMRI) allows for regional examination of changes in the magnetic properties of the blood occurring with flow changes. BOLD response is primarily driven by CBF, but it is also strongly modulated by the amount of deoxyhemoglobin present. Surprisingly, there are few reports of vasoreactivity in AD subjects using MRI methods. In a recently published study, Yezhuvath et al. [106] studied 17 AD patients and 17 age-matched healthy controls. Baseline flow was determined with EPI ASL, reactivity to hypercapnia was examined with fMRI, and CVRCO2 was calculated in units of percent BOLD signal change per mmHg of end-tidal CO2 change. The challenge was induced with the inhalation of air containing 5% CO2. The whole brain vasoreactivity maps were created after normalization of CVRCO2 to the cerebellar value. Patients’ CVRCO2 differed significantly from controls in the frontal cortex, anterior cingulate and insula. Group differences in resting flow were observed mostly in the parietal and temporal cortices. CVRCO2 in the frontal and insular cortices correlated with Boston Naming Test scores [106]. A similar methodology was employed in another study that used Q2TIPS ASL to examine cerebral perfusion in 9 AD, 7 MCI and 11 normal controls. In contrast to the Yezhuvath et al. no baseline CBF differences were found. This was most likely due to limited statistical power, as there was a trend for resting flow to be lower in the AD group. However, CVRCO2 of the global gray matter was significantly lower in AD and MCI subjects than in healthy peers and did not differ between impaired groups. Comparisons of vasoreactivity maps showed widespread reduction in the AD group, comprising mostly posterior areas (parietal and occipital cortex, posterior cingulate). In the entire group, CVRCO2 correlated positively with MMSE scores and negatively with hippocampal atrophy [107]. Our study [108] investigated perfusion and CVRCO2 in an axial slice comprising both hippocampi. Unlike the 2 previously described reports, we used true fast imaging in steady-precession (TrueFISP) ASL sequence. While the EPI and Q2TIPS techniques afford greater brain coverage at a shorter acquisition times, the key advantage of TrueFISP ASL is the spatial resolution (1.2 mm in-plane, Figure 2). The resolution is important to separate the contribution of the arterial vessels passing near the medial aspect of the hippocampus that confound PET and SPECT measurements (Figure 3). Our technique allows these vessels to be separated from hippocampal region. In addition to spatial resolution, the sequence is not affected by susceptibility artifacts found in the vicinity of the temporal bone and hippocampus [109]. Our rationale for using this technique was to examine vasoreactivity and CBF in the hippocampus, a structure affected early in the course of the disease [110–112]. CVRCO2 and CBF were determined in 7 subjects with MCI and 17 healthy controls. Re-breathing through a mouthpiece and a tube was used to induce hypercapnia (Figure 4 presents baseline hippocampal CBF and the CBF during a re-breathing session in subjects studied at our Center; Glodzik, Rusinek, de Leon, unpublished data). Vasoreactivity was defined as a percentage increase in CBF in a given ROI, per 1 mm Hg increase in end-tidal CO2. Although resting hippocampal and neocortical CBF did not distinguish the groups, averaged hippocampal CVRCO2 was significantly lower in the MCI patients. Across the control and MCI groups, CVRCO2 correlated with vascular risk as measured with Framingham Cardiovascular Risk Score. There were no differences in this study between MCI and control participants in their Framingham Risk ratings [108]. Overall, the evidence from MRI studies is still limited but the existing reports point towards vasoreactivity impairment in AD and MCI. However, at present there is no agreement on the regional distribution of these deficits.
Figure 2.

Perfusion image (a), with corresponding tag (b) and untag (c) images obtained with TrueFISP ASL.
Figure 3.

Four subjects imaged using TrueFISP ASL (tag images presented). Subjects (a through d) show various degrees of hippocampal atrophy. Note large arteries around the pons and near the medial border of the hippocampi.
Figure 4.
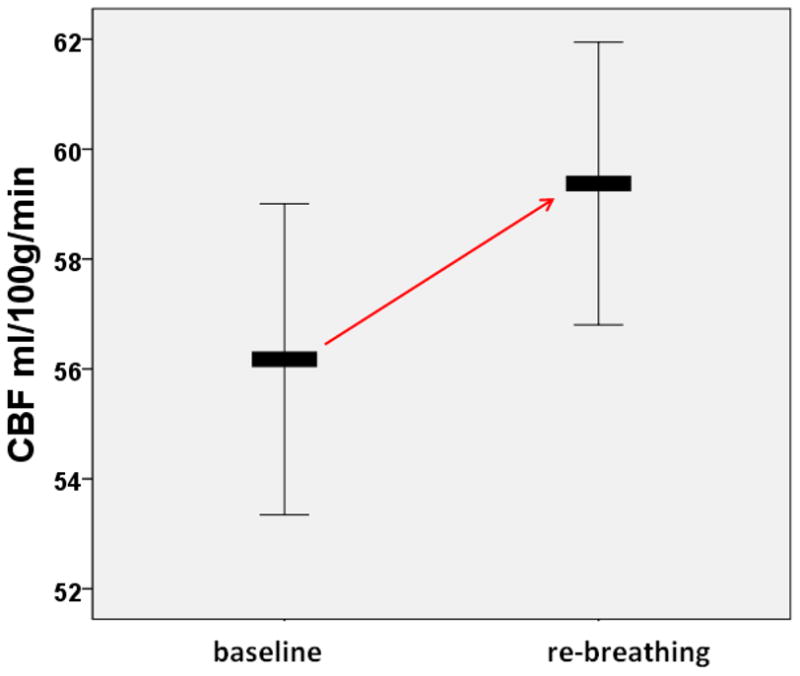
Hippocampal cerebral blood flow at baseline and during a re-breathing session in 84 elderly subjects (mean age 70±8 years). The graph represents means; the error bars indicate a 95% confidence interval (Glodzik, Rusinek, de Leon, unpublished data).
CVRCO2 or CBF impairment: Which comes earlier?
One of the most interesting questions arising when one examines the issue of CBF and vasoreactivity in AD is whether employing challenge techniques affords additional benefits and provides supplementary information beyond what can be provided by resting CBF. As pointed out earlier, vasoreactivity, or cerebrovascular reserve [15], is the capacity of brain vasculature to enhance flow above basal levels in response to a challenge. Thus, it is possible that CVRCO2 measurement could reveal dysfunction earlier than CBF in still relatively healthy tissue and could serve as a sort of “stress test” for brain circulation. In Table 1 we summarized 9 studies, where the authors directly compared CBF and CVRCO2 between healthy controls, MCI and AD patients. In all but two, CVRCO2 was significantly lower in the AD group. In most of them, resting flow deficits were also already present. However, this does not rule out the possibility that vasoreactivity is affected earlier. When a diagnosis of AD is given, both flow and response to the challenge may be already compromised. Interestingly, in two studies where MCI were examined, a pattern emerged: while CBF seemed to be preserved, CVRCO2 was impaired. Although more research is needed to confirm this hypothesis, CVRCO2 measurement may detect an earlier dysfunction of the vascular system in subjects at risk for AD.
Conclusions
Preclinical and animal experiments provide evidence for hypercontractility of cerebral (and possibly systemic) vessels in AD. Even though results of clinical studies are inconsistent, an increasing number of recent reports with larger sample sizes and improved brain sampling, directly comparing CVRCO2 in control and impaired groups, reveals an impairment of vascular reactivity to carbon dioxide in subjects with AD, and possibly also in MCI. Thus, CVRCO2 may be able to detect an early dysfunction of the vascular system in subjects at risk. As such, it may facilitate further research on vascular mechanisms in AD. Due to the lack of large studies in AD and populations at risk, and the non-specific nature of CVRCO2 impairment, it is too early to judge its utility as a diagnostic test. Future studies should focus on elucidating the regional distribution of CVRCO2 reductions in AD. In addition, when effective treatments become available it will be crucial to determine whether they can modify vascular reserve.
Acknowledgments
This study was supported by the following grants: HL111724-01, NIH-NIA AG12101, AG13616 and AG022374. We thank Ms. Elizabeth Pirraglia for her help with Figures.
Reference List
- 1.Anslie PN, Duffin J. Integration of cerebrovascular CO2 reactivity and chemoreflex control of breathing: mechanisms of regulation, measurement, and interpretation. American Journal of Physiology- Regulatory, Integrative and Comparative Physiolog. 2009;296:R1473–R1495. doi: 10.1152/ajpregu.91008.2008. [DOI] [PubMed] [Google Scholar]
- 2.Peebles KC, Richards AM, Celi M, McGrattan K, Murrell CJ, Anslie PN. Human cerebral arteriovenous vasoactive exchange during alterations in arterial blood gases. Journal of Applied Physiology. 2008;105:1060–1068. doi: 10.1152/japplphysiol.90613.2008. [DOI] [PubMed] [Google Scholar]
- 3.Ogoh S, Hayashi N, Inagaki M, Anslie PN, Miyamoto T. Interaction between the ventilatory and cerebrovascular responses to hypo- and hypercapnia at rest and during exercise. The Journal Of Physiology. 2008;586:4327–4338. doi: 10.1113/jphysiol.2008.157073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rodell AB, Aanerud J, Braendgaard H, Gjedde A. Low Residual CBF Variability in Alzheimer’s Disease after Correction for CO(2) Effect. Front Neuroenergetics. 2012;4:8. doi: 10.3389/fnene.2012.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ito H, Kanno I, Fukuda H. Human cerebral circulation: positron emission tomography studies. Annals Of Nuclear Medicine. 2005;19:65–74. doi: 10.1007/BF03027383. [DOI] [PubMed] [Google Scholar]
- 6.Claassen JA, Zhang R, Fu Q, Witkowski SL, Levine BD. Transcranial Doppler estimation of cerebral blood flow and cerebrovascular conductance during modified rebreathing. Journal of Applied Physiology. 2007;102:870–877. doi: 10.1152/japplphysiol.00906.2006. [DOI] [PubMed] [Google Scholar]
- 7.Willie CK, Macleod DB, Shaw AD, Smith KJ, Tzeng YC, Eves ND, Ikeda K, Graham J, Lewis NC, Day TA, Anslie PN. Regional brain blood flow in man during acute changes in arterial blood gases. Journal of Physiology. 2012;590:3261–3275. doi: 10.1113/jphysiol.2012.228551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peebles K, Celi L, McGrattan K, Murrell C, Thomas K, Ainslie PN. Human cerebrovascular and ventilatory CO2 reactivity to end-tidal, arterial and internal jugular vein PCO2. J Physiol. 2007;584:347–357. doi: 10.1113/jphysiol.2007.137075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang X, Wu J, Li L, Chen F, Wang R, Jiang C. Hypercapnic Acidosis Activates KATP Channels in Vascular Smooth Muscles. Circulation research. 2003;92:1225–1232. doi: 10.1161/01.RES.0000075601.95738.6D. [DOI] [PubMed] [Google Scholar]
- 10.Brian JE. Carbon Dioxide and the cerebral circulation. Anesthesiology. 1998;88:1365–1386. doi: 10.1097/00000542-199805000-00029. [DOI] [PubMed] [Google Scholar]
- 11.Lavi S, Egbarya R, Lavi R, Jacob G. Role of Nitric Oxide in the Regulation of Cerebral Blood Flow in Humans: Chemoregulation Versus Mechanoregulation. Circulation. 2003;107:1901–1905. doi: 10.1161/01.CIR.0000057973.99140.5A. [DOI] [PubMed] [Google Scholar]
- 12.Lavi S, Gaitini D, Milloul V, Jacob G. Impaired cerebral CO2 vasoreactivity: association with endothelial dysfunction. American Journal of Physiology - Heart & Circulatory Physiology. 2006;291(4):H1856–61. doi: 10.1152/ajpheart.00014.2006. [DOI] [PubMed] [Google Scholar]
- 13.Mirro R, Lowery-Smith R, Armstead WR, Shibata M, Zuckerman LS, Leffler CW. Cerebral vasoconstriction in response to hypocapnia is maintained after ischemia/reperfusion injury in newborn pigs. Stroke. 1992;23:1613–1616. doi: 10.1161/01.str.23.11.1613. [DOI] [PubMed] [Google Scholar]
- 14.Ogoh S, Anslie PN. Cerebral blood flow during exercise: mechanisms of regulation. Journal of Applied Physiology. 2009;107:1370–1380. doi: 10.1152/japplphysiol.00573.2009. [DOI] [PubMed] [Google Scholar]
- 15.Boles Ponto LL, Magnotta VA, Moser DJ, Duff KM, Schultz SK. Global cerebral blood flow in relation to cognitive performance and reserve in subjects with mild memory deficits. Molecular Imaging and Biology. 2006;8:372. doi: 10.1007/s11307-006-0066-z. [DOI] [PubMed] [Google Scholar]
- 16.Chollet F, Celsis P, Clanet M, Guirard-Chaumeil B, Rascol A, Marc-Vergnes JP. SPECT study of cerebral blood flow reactivity after acetazolamide in patients with transient ischemic attacks. Stroke. 1989;20:458–464. doi: 10.1161/01.str.20.4.458. [DOI] [PubMed] [Google Scholar]
- 17.De Reuck J, Decoo D, Hasenbroeckx MC, Lamont B, Santens P, Goethals P, Strijckmans K, Lemahieu I. Acetazolamide vasoreactivity in vascular dementia: a positron emission tomographic study. European Neurology. 1999;41:31–36. doi: 10.1159/000007995. [DOI] [PubMed] [Google Scholar]
- 18.Groschel K, Terborg C, Riecker A, Witte OW, Kastrup A. Effects of physiological aging and cerebrovascular risk factors on the hemodynamic response to brain activation: A functional transcranial doppler study. Clinical Neurophysiology. 2007;118:e35–e36. doi: 10.1111/j.1468-1331.2006.01563.x. [DOI] [PubMed] [Google Scholar]
- 19.Settakis G, Pall D, Molnar C, Bereczki D, Csiba L, Fulesdi B. Cerebrovascular Reactivity in Hypertensive and Healthy Adolescents: TCD With Vasodilatory Challenge. J Neuroimaging. 2003;13:106–112. [PubMed] [Google Scholar]
- 20.Fujiwara Y, Mizuno T, Okuyama C, Nagakane Y, Watanabe-Hosomi A, Kondo M, Kuriyama N, Tokuda T, Matsushima S, Nishimura T, Nakagawa M. Simultaneous impairment of intracranial and peripheral artery vasoreactivity in CADASIL patients. Cerebrovascular Diseases. 2012;33:128–134. doi: 10.1159/000334185. [DOI] [PubMed] [Google Scholar]
- 21.Claassen JA, Zhang R. Cerebral autoregulation in Alzheimer’s disease. Journal of Cerbral Blood Flow and Metabolism. 2011;31:1572–1577. doi: 10.1038/jcbfm.2011.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kennelly SA, Lawlor B, Kenny RA. Blood pressure and the risk for dementia—A double edged sword. Aging Reaserch Reviews. 2009;8:61–70. doi: 10.1016/j.arr.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 23.Nagai M, Hoshide S, Kario K. Hypertension and Dementia. American Journal of Hypertension. 2010;23:116–124. doi: 10.1038/ajh.2009.212. [DOI] [PubMed] [Google Scholar]
- 24.Murray AD, Staff RT, Shenkin SD, Deary IJ, Starr JM, Whalley LJ. Brain white matter hyperintensities: relative importance of vascular risk factors in nondemented elderly people. Radiology. 2005;237:251–257. doi: 10.1148/radiol.2371041496. [DOI] [PubMed] [Google Scholar]
- 25.Usman K, Porteous L, Hassan A, Markus HS. Risk factor profile of cerebral small vessel disease and its subtypes. Journal of Neurology, Neurosurgery & Psychiatry. 2007;78:702–706. doi: 10.1136/jnnp.2006.103549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pase MP, Grima NA, Sarris J. The effects of dietary and nutrient interventions on arterial stiffness: a systematic review. Am J Clin Nutr. 2011;93:446–454. doi: 10.3945/ajcn.110.002725. [DOI] [PubMed] [Google Scholar]
- 27.Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nature Reviews Neuroscience. 2004;5(5):347–60. doi: 10.1038/nrn1387. [Review] [183 refs] [DOI] [PubMed] [Google Scholar]
- 28.De La Torre JC. Carotid artery ultrasound and echocardiography testing to lower the prevalence of Alzheimer’s disease. Journal of Stroke and Cerebrovascular Diseases. 2009;18:319–328. doi: 10.1016/j.jstrokecerebrovasdis.2008.11.014. [DOI] [PubMed] [Google Scholar]
- 29.Farkas E, Luiten PGM. Cerebral microvascular pathology in aging and Alzheimer’s disease. Progress in Neurobiology. 2001;64:575–611. doi: 10.1016/s0301-0082(00)00068-x. [DOI] [PubMed] [Google Scholar]
- 30.Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci. 2011;12:723–738. doi: 10.1038/nrn3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.kivipelto M, Helkala EL, Hanninen T, Laakso MP, Hallikainen M, Alhainen K, Soininen H, Tuomilehto J, Nissinen A. Midlife vascular risk factors and late-life mild cognitive impairment: A population-based study. Neurol. 2001;56:1683–1689. doi: 10.1212/wnl.56.12.1683. [DOI] [PubMed] [Google Scholar]
- 32.Moore PM, Baker GA. Psychometric properties and factor structure of the Wechsler Memory Scale-Revised in a sample of persons with intractable epilepsy. J Clin Exp Neuropsychol. 1997;19:897–905. doi: 10.1080/01688639708403770. [DOI] [PubMed] [Google Scholar]
- 33.Kilander L, Nyman H, Boberg M, Hansson L, Lithell H. Hypertension is related to cognitive impairment: a 20-year follow-up of 999 men. Hypertension. 1998;31:780–786. doi: 10.1161/01.hyp.31.3.780. [DOI] [PubMed] [Google Scholar]
- 34.kivipelto M, Ngandu T, Fratiglioni L, Viitanen M, Kareholt I, Winblad B, Helkala E-L, Tuomilehto J, Soininen H, Nissinen A. Obesity and vascular risk factors at midlife and the risk of dementia and Alzheimer disease. Arch Neurol. 2005;62:1556–1560. doi: 10.1001/archneur.62.10.1556. [DOI] [PubMed] [Google Scholar]
- 35.Craft S, Watson GS. Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol. 2004;3:169–178. doi: 10.1016/S1474-4422(04)00681-7. [DOI] [PubMed] [Google Scholar]
- 36.Blennow K, de Leon MJ, Zetterberg H. Alzheimer’s disease. Lancet Neurology. 2006;368:387–403. doi: 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- 37.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathologica. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 38.Thomas T, Thomas G, McLendon C, Sutton T, Mullan M. [beta]-Amyloid-mediated vasoactivity and vascular endothelial damage. Nature. 1996;380:168–171. doi: 10.1038/380168a0. [DOI] [PubMed] [Google Scholar]
- 39.Crawford F, Suo Z, Fang C, Mullan M. Characteristics of the in vitro vasoactivity of beta-amyloid peptides. Exp Neurol. 1998;150:159–168. doi: 10.1006/exnr.1997.6743. [DOI] [PubMed] [Google Scholar]
- 40.Said SI, Raza S, Berisha HI. Enhancement of systemic and pulmonary vasoconstriction by beta-amyloid peptides and its suppression by vasoactive intestinal peptide. Ann N Y Acad Sci. 1998;865:582–585. doi: 10.1111/j.1749-6632.1998.tb11240.x. [DOI] [PubMed] [Google Scholar]
- 41.Khalil Z, Chen Hb, Helme RD. Mechanisms underlying the vascular activity of [beta]-amyloid protein fragment ([beta]A425-35) at the level of skin microvasculature. Brain Res. 1996;736:206–216. doi: 10.1016/0006-8993(96)00685-3. [DOI] [PubMed] [Google Scholar]
- 42.Niwa K, Porter VA, Kazama K, Cornfield D, Carlson GA, Iadecola C. A beta-peptides enhance vasoconstriction in cerebral circulation. American Journal of Physiology - Heart & Circulatory Physiology. 2001;281(6):H2417–24. doi: 10.1152/ajpheart.2001.281.6.H2417. [DOI] [PubMed] [Google Scholar]
- 43.Niwa K, Kazama K, Younkin L, Younkin SG, Carlson GA, Iadecola C. Cerebrovascular autoregulation is profoundly impaired in mice overexpressing amyloid precursor protein. American Journal of Physiology - Heart & Circulatory Physiology. 2002;283:H315–23. doi: 10.1152/ajpheart.00022.2002. [DOI] [PubMed] [Google Scholar]
- 44.Niwa K, Kazama K, Younkin SG, Carlson GA, Idecola C. Alterations in Cerebral Blood Flow and Glucose Utilization in Mice Overexpressing the Amyloid Precursor Protein. Neurobiology of Disease. 2002;9:61–68. doi: 10.1006/nbdi.2001.0460. [DOI] [PubMed] [Google Scholar]
- 45.Shin HK, Jones PB, Garcia-Alloza M, Borrelli L, Greenberg SM, Bacskai BJ, Frosch MP, Hyman BT, Moskowitz MA, Ayata C. Age-dependent cerebrovascular dysfunction in a transgenic mouse model of cerebral amyloid angiopathy. Brain. 2007;130(Pt 9):2310–9. doi: 10.1093/brain/awm156. [DOI] [PubMed] [Google Scholar]
- 46.Palmer JC, Barker R, Kehoe PG, Love S. Endothelin-1 is elevated in Alzheimer’s disease and upregulated by amyloid-beta. J Alzheimers Dis. 2012;29:853–861. doi: 10.3233/JAD-2012-111760. [DOI] [PubMed] [Google Scholar]
- 47.Kawanabe Y, Nauli SM. Endothelin. Cell Mol Life Sci. 2011;68:195–203. doi: 10.1007/s00018-010-0518-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eckman EA, Reed DK, Eckman CB. Degradation of the Alzheimer’s amyloid beta peptide by endothelin-converting enzyme. J Biol Chem. 2001;276:24540–24548. doi: 10.1074/jbc.M007579200. [DOI] [PubMed] [Google Scholar]
- 49.Palmer JC, Baig S, Kehoe PG, Love S. Endothelin-converting enzyme-2 is increased in Alzheimer’s disease and up-regulated by Abeta. Am J Pathol. 2009;175:262–270. doi: 10.2353/ajpath.2009.081054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Luo J, Grammas P. Endothelin1 is elevated in Alzheimer’s disease brain microvessels and is neuroprotective. Journal of Alzheimer’s Disease. 2010;21:887–896. doi: 10.3233/JAD-2010-091486. [DOI] [PubMed] [Google Scholar]
- 51.Madonna R, De CR, Willerson JT, Geng YJ. Biologic function and clinical potential of telomerase and associated proteins in cardiovascular tissue repair and regeneration. Eur Heart J. 2011;32:1190–1196. doi: 10.1093/eurheartj/ehq450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chow N, Bell RD, Deane R, Streb JW, Chen J, Brooks A, Van Nostrand W, Miano JM, Zlokovic BV. Serum response factor and myocardin mediate arterial hypercontractility and cerebral blood flow dysregulation in Alzheimer’s phenotype. PNAS. 2007;104:823–828. doi: 10.1073/pnas.0608251104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bell RD, Deane R, Chow N, Long X, Sagare A, Singh I, Streb JW, Guo H, Rubio A, Van NW, Miano JM, Zlokovic BV. SRF and myocardin regulate LRP-mediated amyloid-beta clearance in brain vascular cells. Nat Cell Biol. 2009;11:143–153. doi: 10.1038/ncb1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hirano K. The roles of proteinase-activated receptors in the vascular physiology and pathophysiology. Arterioscler Thromb Vasc Biol. 2007;27:27–36. doi: 10.1161/01.ATV.0000251995.73307.2d. [DOI] [PubMed] [Google Scholar]
- 55.Hirano K, Hirano M. Current perspective on the role of the thrombin receptor in cerebral vasospasm after subarachnoid hemorrhage. J Pharmacol Sci. 2010;114:127–133. doi: 10.1254/jphs.10r03cp. [DOI] [PubMed] [Google Scholar]
- 56.Maki J, Hirano M, Hoka S, Kanaide H, Hirano K. Involvement of reactive oxygen species in thrombin-induced pulmonary vasoconstriction. Am J Respir Crit Care Med. 2010;182:1435–1444. doi: 10.1164/rccm.201002-0255OC. [DOI] [PubMed] [Google Scholar]
- 57.Arai T, Miklossy J, Klegeris A, Guo JP, McGeer PL. Thrombin and prothrombin are expressed by neurons and glial cells and accumulate in neurofibrillary tangles in Alzheimer disease brain. J Neuropathol Exp Neurol. 2006;65:19–25. doi: 10.1097/01.jnen.0000196133.74087.cb. [DOI] [PubMed] [Google Scholar]
- 58.Engelberg H. Pathogenic factors in vascular dementia and Alzheimer’s disease. Multiple actions of heparin that probably are beneficial. Dement Geriatr Cogn Disord. 2004;18:278–298. doi: 10.1159/000080034. [DOI] [PubMed] [Google Scholar]
- 59.Mhatre M, Nguyen A, Kashani S, Pham T, Adesina A, Grammas P. Thrombin, a mediator of neurotoxicity and memory impairment. Neurobiol Aging. 2004;25:783–793. doi: 10.1016/j.neurobiolaging.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 60.Grammas P, Samany PG, Thirumangalakudi L. Thrombin and inflammatory proteins are elevated in Alzheimer’s disease microvessels: implications for disease pathogenesis. J Alzheimers Dis. 2006;9:51–58. doi: 10.3233/jad-2006-9105. [DOI] [PubMed] [Google Scholar]
- 61.Yin X, right J, all T, rammas P. Brain endothelial cells synthesize neurotoxic thrombin in Alzheimer’s disease. The American Journal of Pathology. 2010;176:1600–1606. doi: 10.2353/ajpath.2010.090406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Claassen JA, Jansen RW. Cholinergically mediated augmentation of cerebral perfusion in Alzheimer’s disease and related cognitive disorders: the cholinergic-vascular hypothesis. J Gerontol A Biol Sci Med Sci. 2006;61:267–271. doi: 10.1093/gerona/61.3.267. [DOI] [PubMed] [Google Scholar]
- 63.van Beek AH, Claassen JA. The cerebrovascular role of the cholinergic neural system in Alzheimer’s disease. Behav Brain Res. 2011;221:537–542. doi: 10.1016/j.bbr.2009.12.047. [DOI] [PubMed] [Google Scholar]
- 64.Sato A, Sato Y, Uchida S. Regulation of cerebral cortical blood flow by the basal forebrain cholinergic fibers and aging. Auton Neurosci. 2002;96:13–19. doi: 10.1016/s1566-0702(01)00367-8. [DOI] [PubMed] [Google Scholar]
- 65.Vaucher E, Hamel E. Cholinergic basal forebrain neurons project to cortical microvessels in the rat: electron microscopic study with anterogradely transported Phaseolus vulgaris leucoagglutinin and choline acetyltransferase immunocytochemistry. J Neurosci. 1995;15:7427–7441. doi: 10.1523/JNEUROSCI.15-11-07427.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tong XK, Hamel E. Basal forebrain nitric oxide synthase (NOS)-containing neurons project to microvessels and NOS neurons in the rat neocortex: cellular basis for cortical blood flow regulation. Eur J Neurosci. 2000;12:2769–2780. doi: 10.1046/j.1460-9568.2000.00158.x. [DOI] [PubMed] [Google Scholar]
- 67.Uchida S, Hotta H. Cerebral cortical vasodilatation mediated by nicotinic cholinergic receptors: effects of old age and of chronic nicotine exposure. Biol Pharm Bull. 2009;32:341–344. doi: 10.1248/bpb.32.341. [DOI] [PubMed] [Google Scholar]
- 68.Davies P, Maloney AJF. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet. 1976;2:1403. doi: 10.1016/s0140-6736(76)91936-x. [DOI] [PubMed] [Google Scholar]
- 69.Craig LA, Hong NS, McDonald RJ. Revisiting the cholinergic hypothesis in the development of Alzheimer’s disease. Neurosci Biobehav Rev. 2011;35:1397–1409. doi: 10.1016/j.neubiorev.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 70.Lojkowska W, Ryglewicz D, Jedrzejczak T, Minc S, Jakubowska T, Jarosz H, Bochynska A. The effect of cholinesterase inhibitors on the regional blood flow in patients with Alzheimer’s disease and vascular dementia. J Neurol Sci. 2003;216:119–126. doi: 10.1016/s0022-510x(03)00229-6. [DOI] [PubMed] [Google Scholar]
- 71.Li W, Antuono PG, Xie C, Chen G, Jones JL, Ward BD, Franczak MB, Goveas JS, Li SJ. Changes in regional cerebral blood flow and functional connectivity in the cholinergic pathway associated with cognitive performance in subjects with mild Alzheimer’s disease after 12-week donepezil treatment. Neuroimage. 2012;60:1083–1091. doi: 10.1016/j.neuroimage.2011.12.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Imamura K, Wada-Isoe K, Kowa H, Tanabe Y, Nakashima K. The effect of donepezil on increased regional cerebral blood flow in the posterior cingulate cortex of a patient with Parkinson’s disease dementia. Neurocase. 2008;14:271–275. doi: 10.1080/13554790802269984. [DOI] [PubMed] [Google Scholar]
- 73.Nobili F, Vitali P, Canfora M, Girtler N, De LC, Mariani G, Pupi A, Rodriguez G. Effects of long-term Donepezil therapy on rCBF of Alzheimer’s patients. Clin Neurophysiol. 2002;113:1241–1248. doi: 10.1016/s1388-2457(02)00110-4. [DOI] [PubMed] [Google Scholar]
- 74.Venneri A, Shanks MF, Staff RT, Pestell SJ, Forbes KE, Gemmell HG, Murray AD. Cerebral blood flow and cognitive responses to rivastigmine treatment in Alzheimer’s disease. NeuroReport. 2002;13:83–87. doi: 10.1097/00001756-200201210-00020. [DOI] [PubMed] [Google Scholar]
- 75.Honer WG, Prohovnik I, Smith G, Lucas LR. Scopolamine reduces frontal cortex perfusion. J Cereb Blood Flow Metab. 1988;8:635–641. doi: 10.1038/jcbfm.1988.110. [DOI] [PubMed] [Google Scholar]
- 76.Prohovnik I, Arnold SE, Smith G, Lucas LR. Physostigmine reversal of scopolamine-induced hypofrontality. J Cereb Blood Flow Metab. 1997;17:220–228. doi: 10.1097/00004647-199702000-00012. [DOI] [PubMed] [Google Scholar]
- 77.Bahro M, Molchan SE, Sunderland T, Herscovitch P, Schreurs BG. The effects of scopolamine on changes in regional cerebral blood flow during classical conditioning of the human eyeblink response. Neuropsychobiology. 1999;39:187–195. doi: 10.1159/000026582. [DOI] [PubMed] [Google Scholar]
- 78.Bar KJ, Boettger MK, Seidler N, Mentzel HJ, Terborg C, Sauer H. Influence of galantamine on vasomotor reactivity in Alzheimer’s disease and vascular dementia due to cerebral microangiopathy. Stroke. 2007;38:3186–3192. doi: 10.1161/STROKEAHA.107.492033. [DOI] [PubMed] [Google Scholar]
- 79.Wyss-Coray T, Rogers J. Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harb Perspect Med. 2012;2:a006346. doi: 10.1101/cshperspect.a006346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kamer AR, Dasanayake AP, Craig RG, Glodzik-Sobanska L, Brys M, de Leon MJ. Alzheimer’s disease and peripheral infections: the possible contribution from periodontal infections, model and hypothesis. [Review] Journal of Alzheimer’s Disease. 2008;13:437–449. doi: 10.3233/jad-2008-13408. [DOI] [PubMed] [Google Scholar]
- 81.Ray S, Britschgi M, Herbert C, Takeda-Uchimura Y, Boxer A, Blennow K, Friedman LF, Galasko DR, Jutel M, Karydas A, Kaye JA, Leszek J, Miller BL, Minthon L, Quinn JF, Rabinovici GD, Robinson WH, Sabbagh MN, So YT, Sparks DL, Tabaton M, Tinklenberg J, Yesavage JA, Tibshirani R, Wyss-Coray T. Classification and prediction of clinical Alzheimer’s diagnosis based on plasma signaling proteins. Nat Med. 2007;13:1359–1362. doi: 10.1038/nm1653. [DOI] [PubMed] [Google Scholar]
- 82.Pezzzini A, Del Zotto E, Volonghi I, Giossi A, Costa P, Padovani A. Cerebral amyloid angiopathy: a common cause of cerebral hemorrhage. Current Medicinal Chemistry. 2009;16:2498–2513. doi: 10.2174/092986709788682047. [DOI] [PubMed] [Google Scholar]
- 83.Vonsattel J-P, Myers RH, Hedley-Whyte ET, Ropper AH, Bird ED, Richardson EP., Jr Cerebral amyloid angiopathy without and with cerebral hemorrhages: a comparative histological study. Ann Neurol. 1991;30:637–649. doi: 10.1002/ana.410300503. [DOI] [PubMed] [Google Scholar]
- 84.Scheibel AB, Duong T, Jacobs R. Alzheimer’s disease as a capillary dementia. Annals of Medicine. 1989;21:103–107. doi: 10.3109/07853898909149194. [DOI] [PubMed] [Google Scholar]
- 85.Jellinger KA, Attems J. Prevalence and impact of cerebrovascular pathology in Alzheimer’s disease and parkinsonism. Acta Neurologica Scandinavica. 2006;114:38–46. doi: 10.1111/j.1600-0404.2006.00665.x. [DOI] [PubMed] [Google Scholar]
- 86.Miyakawa T, Katsuragi S, Higuchi Y, Yamashita K, Kimura T, Teraoka K, Ono T, Ishizuka K. Changes of microvessels in the brain with Alzheimer’s disease. Annals of the New York Academy of Science. 1997;826:428–432. doi: 10.1111/j.1749-6632.1997.tb48497.x. [DOI] [PubMed] [Google Scholar]
- 87.Hashimura T, Kimura T, Miyakawa T. Morphological changes of blood vessels in the brain with Alzheimer’s disease. Japanese Journal of Psychiatry and Neurology. 1991;45:661–665. doi: 10.1111/j.1440-1819.1991.tb01187.x. [DOI] [PubMed] [Google Scholar]
- 88.Buee L, Hof PR, Delacourte A. Brain microvascular changes in Alzheimer’s disease and other dementias. Ann N Y Acad Sci. 1997;826:7–24. doi: 10.1111/j.1749-6632.1997.tb48457.x. [DOI] [PubMed] [Google Scholar]
- 89.Simard D, Olesen J, Paulson OB, Lassen NA, Skinhoj E. Regional cerebral blood flow and its regulation in dementia. Brain. 1971;94:273–288. doi: 10.1093/brain/94.2.273. [DOI] [PubMed] [Google Scholar]
- 90.Hachinski VC, Iliff LD, Zilhka E, Du Boulay GH, McAllister VL, Marshall J, Russell RWR, Symon L. Cerebral blood flow in dementia. Arch Neurol. 1975;32:632–637. doi: 10.1001/archneur.1975.00490510088009. [DOI] [PubMed] [Google Scholar]
- 91.Yamaguchi F, Meyer JS, Yamamoto M, Sakai F, Shaw T. Noninvasive regional cerebral blood flow measurements in dementia. Arch Neurol. 1980;37:410–418. doi: 10.1001/archneur.1980.00500560040003. [DOI] [PubMed] [Google Scholar]
- 92.Bonte FJ, Devous MD, Sr, Reisch JS, Ajmani AK, Weiner MF, Hom J, Tintner R. The effect of acetazolamide on regional cerebral blood flow in patients with Alzheimer’s disease or stroke as measured by single-photon emission computed tomography. Invest Radiol. 1989;24:99–103. doi: 10.1097/00004424-198902000-00002. [DOI] [PubMed] [Google Scholar]
- 93.Stoppe G, Schultze R, Kogler A, Staedt J, Munz DL, Emrich D, Ruther E. Cerebrovascular Reactivity to Acetazolamide in (Senile) Dementia of Alzheimer’s Type: Relationship to Disease Severity. Dementia. 1995;6:73–82. doi: 10.1159/000106925. [DOI] [PubMed] [Google Scholar]
- 94.Knapp WH, Dannenberg C, Marschall B, Zedlick D, Loschmann K, Bettin S, Barthel H, Seese A. Changes in local cerebral blood flow by neuroactivation and vasoactivation in patients with impaired cognitive function. European Journal of Nuclear Medicine. 1996;23:878–888. doi: 10.1007/BF01084360. [DOI] [PubMed] [Google Scholar]
- 95.Pavics L, Grunwald F, Reichmann K, Sera T, Ambrus E, Horn R, Hartmann A, Menzel C, Csernay L, Biersack HJ. rCBF SPECT and the acetazolamide test in the evaluation of dementia. Nucl Med Rev Cent East Eur. 1998;1:13–19. [PubMed] [Google Scholar]
- 96.Oishi M, Mochizuki Y, Takasu T. Regional differences in cerebrovascular reactivity to acetazolamide in Alzheimer’s disease. J Clin Neurosci. 1999;6:380–381. doi: 10.1054/jocn.1997.0085. [DOI] [PubMed] [Google Scholar]
- 97.Raichle ME, Martin WRW, Herscovitch P, Mintum MA, Markham J. Brain blood flow measured with intravenous 0–15 H2O. 2. Implementation and validation. J Nuc Med. 1983;24:790–798. [PubMed] [Google Scholar]
- 98.Herscovitch P, Markham J, Raichle ME. Brain blood flow measured with intravenous H2O15. I. Theory and error analysis. J Nucl Med. 2013;24:782–789. [PubMed] [Google Scholar]
- 99.Kuwabara Y, Ichiya Y, Otsuka M, Masuda K, Ichimiya A, Fujishima M. Cerebrovascular responsiveness to hypercapnia in Alzheimer’s dementia and vascular dementia of the Binswanger type. Stroke. 1992;23:594–598. doi: 10.1161/01.str.23.4.594. [DOI] [PubMed] [Google Scholar]
- 100.Jagust WJ, Eberling JL, Reed BR, Mathis CA, Budinger TF. Clinical studies of cerebral blood flow in Alzheimer’s disease. Ann NY Acad Sci. 1997;826:254–262. doi: 10.1111/j.1749-6632.1997.tb48477.x. [DOI] [PubMed] [Google Scholar]
- 101.Huber P, Handa J. Effect of contrast material, hypercapnia, hyperventilation, hypertonic glucose and papaverine on the diameter of the cerebral arteries. Angiographic determination in man. Investigative Radiology. 1967;2:17–32. doi: 10.1097/00004424-196701000-00016. [DOI] [PubMed] [Google Scholar]
- 102.Giller CA. The emperor has no clothes: velocity, flow, and the use of TCD. Journal of Neuroimaging. 2003;13:97–98. [PubMed] [Google Scholar]
- 103.Vincenzini E, Ricciardi MC, Altieri M, Puccinelli F, Bonaffini N, Di Piero V, Lenzi GL. Cerebrovascular Reactivity in Degenerative and Vascular Dementia: A Transcranial Doppler Study. European Neurology. 2007;58:84–89. doi: 10.1159/000103642. [DOI] [PubMed] [Google Scholar]
- 104.Likitjaroen Y, Suwanwela NC, Phanthumchinda K. Vasoreactivity induced by acetazolamide in patients with vascular dementia versus Alzheimer’s disease. J Neurol Sci. 2009;283:32–35. doi: 10.1016/j.jns.2009.02.363. [DOI] [PubMed] [Google Scholar]
- 105.Silvestrini M, Pasqualetti P, Baruffaldi R, Bartolini M, Handouk Y, Matteis M, Moffa F, Provinciali L, Vernieri F. Cerebrovascular reactivity and cognitive decline in patients with Alzheimer disease. Stroke. 2006;37:1010–1015. doi: 10.1161/01.STR.0000206439.62025.97. [DOI] [PubMed] [Google Scholar]
- 106.Yezhuvath US, Uh J, Cheng Y, Martin-Cook K, Weiner M, Diaz-Arrastia R, van OM, Lu H. Forebrain-dominant deficit in cerebrovascular reactivity in Alzheimer’s disease. Neurobiol Aging. 2012;33:75–82. doi: 10.1016/j.neurobiolaging.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cantin S, Villien M, Moreaud O, Tropres I, Keignart S, Chipon E, Le Bas JF, Warnking J, Krainik A. Impaired cerebral vasoreactivity to CO2 in Alzheimer’s disease using BOLD fMRI. Neuroimage. 2011;58:579–587. doi: 10.1016/j.neuroimage.2011.06.070. [DOI] [PubMed] [Google Scholar]
- 108.Glodzik L, Rusinek H, Brys M, Tsui WH, Switalski R, Mosconi L, Mistur R, Pirraglia E, De Santi S, Li Y, Goldowsky A, de Leon M. Framingham cardiovascular risk profile correlates with impaired hippocampal and cortical vasoreactivity to hypercapnia. J Cereb Blood Flow Metab. 2011;15:671–679. doi: 10.1038/jcbfm.2010.145. epub ahead of print, 2010 Sep. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rusinek H, Brys M, Glodzik L, Switalski R, Tsui WH, Haas F, McGorty K, Chen Q, de Leon MJ. Hippocampal blood flow in normal aging measured with arterial spin labeling at 3T. Magnetic Resonance in Medicine. 2011;65:128–137. doi: 10.1002/mrm.22611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.de Leon MJ, George AE, Stylopoulos LA, Smith G, Miller DC. Early marker for Alzheimer’s disease: The atrophic hippocampus. Lancet. 1989;2:672–673. doi: 10.1016/s0140-6736(89)90911-2. [DOI] [PubMed] [Google Scholar]
- 111.de Leon MJ, Golomb J, George AE, Convit A, Tarshish CY, McRae T, De Santi S, Smith G, Ferris SH, Noz M, Rusinek H. The radiologic prediction of Alzheimer’s disease: The atrophic hippocampal formation. American Journal of Neuroradiology. 1993;14:897–906. [PMC free article] [PubMed] [Google Scholar]
- 112.Rusinek H, De Santi S, Frid D, Tsui W, Tarshish C, Convit A, de Leon MJ. Regional brain atrophy rate predicts future cognitive decline: 6-year longitudinal MR imaging study of normal aging. Radiology. 2003;229:691–696. doi: 10.1148/radiol.2293021299. [DOI] [PubMed] [Google Scholar]
- 113.Albuquerque ML, Lowery-Smith L, Hsu P, Parfenova H, Leffler CW. Low CO2 stimulates inositol phosphate turnover and increased inositol 1,4,5-trisphosphate levels in piglet cerebral microvascular smooth muscle cells. Proceedings of the Society for Experimental Biology and Medicine. 1995;209:14–19. doi: 10.3181/00379727-209-43871. [DOI] [PubMed] [Google Scholar]