

Abstract
Aberrant androgen receptor (AR) activation is the major driver of castrate resistant prostate cancer (CRPC). CRPC is ultimately fatal and more therapeutic agents are needed to treat this disease. Compounds that target the androgen axis by inhibiting androgen biosynthesis and or AR signaling are potential candidates for use in CRPC treatment and are currently being pursued aggressively. Aldo-keto reductase 1C3 (AKR1C3) plays a pivotal role in androgen biosynthesis within the prostate. It catalyzes the 17-ketoreduction of weak androgen precursors to give testosterone and 5α-dihydrotestosterone. AKR1C3 expression and activity has been implicated in the development of CRPC, making it a rational target. Selective inhibition of AKR1C3 will be important however, due to the presence of closely related isoforms, AKR1C1 and AKR1C2 that are also involved in androgen inactivation. We examine the evidence that supports the vital role of AKR1C3 in CRPC and recent developments in the discovery of potent and selective AKR1C3 inhibitors.
Keywords: Type 5 17β-hydroxysteroid dehydrogenase, prostaglandin F synthase, prostate cancer, androgens, nonsteroidal anti-inflammatory drugs
1. Introduction
Androgen receptor (AR) activation by androgens is critical for the development of male secondary sexual characteristics as well as the growth and function of the normal prostate. However, AR signaling has also been implicated in the development and progression of prostate cancer (PC). PC is the second most common malignancy in American men and accounts for about 10% of all cancer related mortality [1, 2]. About 16% of men will develop PC in their lifetime resulting in an estimated 242000 new cases and 28000 deaths from PC in 2012 in the US alone [3]. The central role played by androgen receptor signaling in PC is underscored by the clinical utility of androgen deprivation therapy (ADT) in advanced prostate cancer patients. ADT targets the production of androgens, primarily testosterone by the Leydig cells of the testis. This is accomplished by chemical (using a gonadotrophic releasing hormone (GnRH) agonist, such as leuprolide, goserelin or busetelin) or surgical castration often in combination with anti-androgens like bicalutamide. ADT usually leads to an initial clinical improvement with a concomitant reduction in prostate specific antigen (PSA) levels. However, it is invariably followed by a period of therapeutic failure marked by resurgent PSA levels and consequently the development of a fatal and aggressive form of prostate cancer.
Recurrent prostate cancer was previously referred to as androgen independent prostate cancer due to the observation that the tumor growth continued in the presence of castrate levels of circulating androgens. However, studies over the last decade have established that the tumor proliferation occurs as a result of the reactivation of the AR within the tumor despite the low levels of circulating androgens, hence the more accurate classification, castrate resistant prostate cancer (CRPC) [4–6]. CRPC can arise by increased intratumoral androgen biosynthesis driven primarily by elevated expression of enzymes responsible for androgen production. This results in higher levels of intratumoral androgens that can bind and activate the AR within the tumor. CRPC could also be due to adaptive changes in the AR, usually amplification, mutation or the development of AR splice variants. The net result of these changes is that AR activation and signaling can occur in the presence of little or no circulating androgens. Considerable effort has been invested into the discovery of compounds targeting the androgen axis by inhibiting androgen biosynthesis and/or AR signaling as a means of treating CRPC. Two compounds, abiraterone (Abi; Zytiga®) and MDV3100 (Enzalutamide) have shown remarkable efficacy in clinical trials for CRPC [7–14].
Abi inhibits 17α-hydroxylase/17,20-lyase (P450 17A1), an enzyme involved in androgen biosynthesis by catalyzing the two step conversion of pregnenolone and progesterone to yield dehydroepiandrosterone (DHEA) and androstenedione, respectively [7–10]. The structure of P450 17A1 bound to Abi has been reported[15]. Abi was recently approved by the FDA for the treatment of CRPC and is marketed under the brand name ZYTIGA®. However, Abi also prevents glucocorticoid metabolism and leads to the accumulation of desoxycorticosterone (DOC), a potent mineralocorticoid that can cause life threatening hypertension. To prevent this, Abi is usually co-administered with prednisone to suppress the hypothalamic-pituitary-adrenal axis [7, 8, 16]. Despite this, 55% of patients on an Abi/prednisone combination will go on to develop a syndrome of secondary mineralocorticoid excess which necessitates the addition of an aldosterone antagonist to the treatment regimen [10]. Also, activation of the AR by prednisone is now thought to contribute to the development of resistance to Abi therapy in CRPC [17]. Enzalutamide is a super AR antagonist that blocks AR signaling and leads to AR degradation [11, 12]. Enzalutamide was recently approved (XTANDI ®) for the treatment of CRPC in patients that failed docetaxel therapy[18]. Enzalutamide has also been associated with dose-limiting CNS seizures, which may limit its potential therapeutic usage [13] [19]. Drug resistance has likewise been observed with both Abi and Enzalutamide. These factors indicate the need for additional compounds targeting the androgen axis, either at the level of androgen biosynthesis/AR signaling or both.
Aldo-keto reductase 1C3 (AKR1C3) also known as type 5, 17β-hydroxysteroid dehydrogenase (17β-HSD) is involved in the pre-receptor regulation of androgen action and represents an appealing alternative target. This review will focus on the evidence that AKR1C3 is a therapeutic target in CRPC. It will cover the role of AKR1C3 in CRPC and highlight recent advances in the development of potent inhibitors for this enzyme.
2. Properties of AKR1C3
AKR1C3 is a member of the aldo-keto reductase protein superfamily. AKR1C3 as well as other closely related AKR1C isoforms are soluble monomeric NAD(P)(H) dependent oxidoreductases that catalyze the stereospecific interconversion of carbonyl groups and alcohols. While AKR1C3 can catalyze both oxidation and reduction reactions in vitro, it is primarily a reductive enzyme in vivo due to its nanomolar affinity for NADPH, the major cellular co-reductant. AKR1C3 is highly expressed in the prostate where it catalyzes the formation of the potent androgens, testosterone (T) and 5α-dihydrotestosterone (5α-DHT) [20]. It catalyzes the NADPH dependent reduction of the weak androgen, Δ4-androstene-3, 17-dione (Δ4-AD) to give T, which can then be converted to DHT by 5α-reductases type 1 and type 2. AKR1C3 also catalyzes the reduction of 5α-androstane-3, 17-dione (5α-Adione) to yield DHT (Figure 1) [21]. Three pathways to DHT have been proposed in the prostate and AKR1C3 plays a role in each. The classical pathway involves the sequence DHEA→Δ4-AD→T→DHT, where AKR1C3 catalyzes the conversion of Δ4-AD→T. The alternative pathway bypasses T altogether and involves the sequence, DHEA→Δ4-AD→5α-Adione→DHT,[22] in which AKR1C3 catalyzes the conversion of 5α-Adione→DHT, and the backdoor pathway in which 5α-reduction occurs at the level of pregnanes and bypasses T[23]. This pathway involves the sequence, progesterone→5α-dihydroprogesterone→allopregnanolone→androsterone→3α-Diol→DHT,[23] where AKR1C3 converts androsterone into 3α-Diol. Which pathway predominates in prostate cancer is a matter of debate. However, irrespective of which pathway operates, AKR1C3 is essential for each.
Figure 1.

AKR1C3 and Androgen Metabolism in The Prostate (Δ5-Adiol, 5-Androstene-3β,17β-diol; Δ4-Adione, 4-Androstene-3,17-dione; 5α-Adione, 5α-Androstane-3,17-dione; AR, Androgen receptor; ARE, Androgen response element; DHEA, Dehydroepiandrosterone; 5α-DHT, 5α-Dihydrotestosterone; HSD3B, 3β-Hydroxysteroid dehydrogenase; PREG, Pregnenolone; SRD5A, 5α-Reductase); enzymes are also listed as their gene names.
AKR1C3 also catalyzes the formation of prostaglandin (PG) F2α and 11β-PGF2α from PGH2 and PGD2, respectively (Figure 2). These pro-proliferative signaling molecules can lead to proliferation of tumor cells [24–26]. PGF2α and 11β-PGF2 can bind to the prostanoid (FP) receptor, which activates MAPKinase pathways and leads to the phosphorylation and inactivation of the proliferator peroxisome activator receptor gamma (PPARγ) (a pro-proliferative response) [24, 27, 28]. By catalyzing the reduction of PGD2, AKR1C3 also prevents the non-enzymatic loss of two water molecules from PGD2 to form 15-deoxy-Δ12,14 PGJ2 (15d-PGJ2) [29, 30]. 15d-PGJ2 is a putative agonist for PPARγ, and displays anti-proliferative effects. 15d-PGJ2 also directly inhibits androgen receptor signaling [31]. AKR1C3 therefore has the potential to block the anti-proliferative effect of PPARγ by two mechanisms. Thus AKR1C3 inhibition could block both androgen dependent and independent prostate cancer cell growth.
Figure 2.

AKR1C3 and Prostaglandin Synthesis
With the exception of AKR1C3, all other known human 17β-HSDs belong to the short-chain dehydrogenase/reductase (SDR) superfamily of enzymes. Several of these enzymes play important roles in androgen biosynthesis and in the pre-receptor regulation of AR action. Type 2 17β-HSD (SDR9C2) plays an important role in the oxidation of testosterone to Δ4-AD and prevents testosterone binding to the androgen receptor[32]. Type 3 17β-HSD (SDR12C2) catalyzes the same reaction as AKR1C3 but is predominantly Leydig cell specific [33]. The importance of this enzyme in testosterone production is supported by male pseudohermaphroditism that occurs as a result of a Type 3 17β-HSD deficiency [32]. Type 3 17β-HSD is a target for prostate cancer and inhibition of this enzyme would be equivalent to a chemical castration. Type 6 17β-HSD (SDR9C6) is the predominant enzyme that catalyzes the conversion of 3α-Diol to DHT via the backdoor pathway in both normal prostate [34] and prostate cancer [35, 36]. Evidence exists that this pathway may operate in CRPC and could be an important therapeutic target [35, 36].
While SDRs are able to catalyze these reactions, important differences exist between the SDR and AKR family of enzymes. SDRs are mostly multimeric proteins, contain a Rossmann fold for cofactor binding, and catalyze pro-S hydride transfer from C4 position of the nicotinamide ring while AKRs are monomeric proteins, have a triosephosphate isomerase (TIM) barrel motif, and catalyze pro-R hydride transfer [37]. These differences might confer inhibitor selectivity for AKR1C3 over the other 17β-HSDs.
3. Involvement of AKR1C3 in Castrate Resistant Prostate Cancer
Studies conducted by us and other groups have underscored the involvement of AKR1C3 in the development of CRPC and the potential therapeutic usefulness of AKR1C3 inhibition in CRPC. First, Stanborough et al. showed that AKR1C3 is one of the most upregulated enzymes involved in androgen biosynthesis in CRPC patients at the RNA and protein level, both within the tumor and in soft-tissue metastasis [38]. They showed that compared to primary prostate cancer, AKR1C3 gene expression was increased 5.3 fold in CRPC, the highest fold change of all steroidogenic enzymes required for the formation of T and DHT starting from DHEA. Immunohistochemical staining of the tumor samples also showed that only 5.6 % of primary tumor stained strongly for AKR1C3 compared to 58 % of CRPC samples showing strong AKR1C3 expression. Other groups have since reported similar findings of AKR1C3 overexpression in CRPC [39–41]. Second, compared to other 17β-HSD isoforms, AKR1C3 was the most abundant isoform expressed in several prostate cancer cells. AKR1C3 transcript levels were 100–1000 fold higher than the others, pointing to a pivotal role for AKR1C3 [42]. Third, androgen treatment suppresses AKR1C3 expression in a variety of prostate cancer cell lines whereas growth in androgen depleted media (cell culture media supplemented with charcoal stripped fetal bovine serum) leads to a dramatic elevation of AKR1C3 expression [40, 41]. This observation has been recapitulated in murine xenograft models of CRPC [40]. These findings suggest that AKR1C3 upregulation in CRPC is an adaptive response to androgen deprivation. Fourth, measurement of intratumoral androgens by liquid chromatography-mass spectrometry in primary and metastatic disease showed an increase in T levels (0.23 to 0.74 ng/g) and a decrease in DHT levels (2.75 to 0.25 ng/g) as the tumor progresses to metastatic disease [39]. These changes result in a 30-fold increase in the ratio of T to DHT from primary to metastatic prostate cancer. This is consistent with a decline in type 2 5α-reductase (SRD5A2) expression, a modest increase in type 1 5α-reductase (SRD5A1) expression, and a robust increase in AKR1C3 expression [38]. Fifth, treatment of VCaP cells and their castrate resistant xenograft VSC2 cells with the AKR1C3 substrate, Δ4-AD led to robust PSA expression. The increase in PSA expression was blocked by indomethacin, a selective AKR1C3 inhibitor [42]. Knockdown of AKR1C3 using sh-AKR1C3-RNA resulted in a similar outcome. Likewise, treatment of a tumor xenograft with indomethacin resulted in decrease in T and DHT and inhibition of tumor cell proliferation. Sixth, siRNA knockdown of AKR1C3 led to increase in apoptosis and growth inhibition of prostate cancer cells while AKR1C3 overexpression increased prostate cancer cell proliferation [43, 44].
Collectively, these data suggest that AKR1C3 is the driver of steroidogenesis in CRPC, that it is necessary and sufficient to drive AR-regulated gene expression and that it is upregulated by ADT. Chang et al. [22] measured the major routes of Δ4-AD metabolism in six-different prostate cancer cell lines of differing AR status, and consistently found evidence for the “alternative pathway” of DHT formation described above. However, in those studies while the conversion of Δ4-AD to 5α-Adione was rapid, the formation of DHT was slow and indicates that AKR1C3 may catalyze the rate-determining step in reducing 5α-Adione to DHT. By targeting AKR1C3 for inhibitor development, all the pathways to potent androgens in the prostate may be blocked at a rate-determining step. AKR1C3 acts further downstream of 17-hydroxylase/17,20-lyase in the androgen biosynthetic pathway within the prostate and unlike Abi, AKR1C3 inhibitors will not interfere with glucocorticoid metabolism. The increase in testosterone levels seen with MDV3100 treatment may also involve AKR1C3 upregulation, which would be consistent with its upregulation in prostate cancer cells deprived of androgens [40]. Due to the pivotal role played by AKR1C3 and its intratumoral localization, AKR1C3 inhibitors are predicted to prevent cancer cell proliferation, have less side effects since they act further downstream than Abi and may be able to surmount drug resistance to MDV3100.
4. AKR1C3 Inhibitors
There has been remarkable interest in the development of inhibitors for AKR1C3 as therapeutic agents for malignancies. These efforts have led to the discovery of several structurally dissimilar, steroidal and non-steroidal AKR1C3 inhibitors. Steroidal AKR1C3 inhibitors include medroxyprogesterone acetate (MPA) and steroidal lactones [45] while benzodiazepines [46], jasmonates [47], cinnamic acids [48], flavonoids [49], and NSAIDs [50–53] have been reported as non-steroidal inhibitors of AKR1C3. These have been extensively reviewed along with the crystal structures of AKR1C3·NADP+·inhibitor complexes known at that time (See Byrns et. al, [54]). The present review will be limited to recent developments on AKR1C3 inhibitors reported within the last two years with insights from new crystal structures.
It is imperative that AKR1C3 be selectively inhibited in CRPC due to the presence of two closely related isoforms, AKR1C1 and AKR1C2 in the prostate. AKR1C1 and AKR1C2 share >86% sequence identity with AKR1C3 and are involved in DHT catabolism and deactivation within the prostate [20, 55]. AKR1C1 and AKR1C2 catalyze the NADPH dependent conversion of DHT to 5α-androstane-3β,17β-diol (3β-Adiol) and 5α-androstane-3α,17β-diol (3α-Diol), respectively [20, 56]. 3β-Adiol is a pro-apoptotic ligand for estrogen receptor β, while 3α-Diol is an inactive androgen [56, 57]. Lack of inhibitor selectivity for AKR1C3 will prevent DHT inactivation, increase the androgenic signal within the prostate and limit the efficacy of AKR1C3 inhibition.
4.1. N-Phenylanthranilate Based (N-PA) Inhibitors
These compounds are based on the non-steroidal anti-inflammatory drug (NSAID), flufenamic acid (FLU) (Figure 3). Our lab discovered that NSAIDs, which are used clinically for their ability to inhibit cyclooxygenases (COX) are non-selective AKR1C inhibitors at therapeutic concentrations required for COX inhibition [50]. The inhibitory activity of NSAIDs on AKR1C enzymes is believed to be due to the presence of carboxylic acid or ketone group which can interact with the conserved catalytic site/oxyanion hole of these enzymes [54]. Since, these compounds are relatively well tolerated and have been extensively studied; they can be used as lead compounds for the development of selective AKR1C3 inhibitors. Also, the relative simple structure of the N-PA scaffold is amenable to a large range of structural modification. However, to avoid adverse effects related to chronic COX inhibition, desirable compounds will have to be stripped of COX inhibitory activity and in addition lack inhibitory activity on AKR1C1 and AKR1C2. Using the known structure activity relationship (SAR) for COX, we were able to design compounds that were devoid of COX inhibitory activity; however, the compounds still had inhibitory activity for AKR1C1 and AKR1C2 at concentrations that will inhibit AKR1C3 [51].
Figure 3.

Structure and Potency of AKR1C3 Inhibitors. Rings are designated A and B for ease of discussion in the text. FLU = flufenamic acid.
Subsequent SAR studies on FLU that explored the effect of movement of the carboxylic acid group of the N-benzoic acid A-ring (Figure 3) and the introduction of various substituents on the phenylamino B-ring revealed key structural requirements for optimal AKR1C3 potency and selectivity [58, 59]. A fluorimetric assay based on the NADP+ dependent oxidation of S-(+)-1,2,3,4-tetrahydro-1-naphthol (S-tetralol) catalyzed by AKR1C3 at 37 °C was used to determine the inhibitory potencies of these compounds. Concentrations of S-tetralol used in the assays were equal to the KM (5 μM, 22.5 μM, 165 μM and 25 μM for AKR1C1-4, respectively) for the respective enzymes determined under the same experimental conditions enabling direct comparison of their IC50 values. The presence of the carboxylic acid group on the meta position of the benzoic acid ring and the introduction of an electron withdrawing group (EWG) at the para position of the phenylamino ring were critical for optimal AKR1C3 potency and selectivity. This led to the discovery of substituted 3-(phenylamino)benzoic acids as potent and selective AKR1C3 inhibitors [58]. These compounds represented the first reported compounds that inhibit AKR1C3 with nanomolar potency and display > 100 fold selectivity for AKR1C3 over AKR1C1-2. Unlike FLU, its isomer, compound 1 was 250 fold selective for AKR1C3. Of the 3-phenylaminobenzoic acids analogs synthesized and tested, the p-NO2 analog compound 2, with an IC50 value of 36 nM was the most potent AKR1C3 inhibitor, while the acetyl analog compound 3, with 360 fold selectivity for AKR1C3 over AKR1C1-2 was the most selective, Table 1. The AKR1C3 potency of these compounds displayed a statistically significant correlation with the electronic properties of the substituent on the phenylamino ring [58]. With the exception of 2, all the other compounds were devoid of COX 1 and 2 inhibitory activities at 100 μM concentrations. The compounds also did not display inhibitory activities on AKR1C4, AKR1B1 and AKR1B10 [59]. Importantly, lead compounds (10 μM) were effective in a LNCaP-AKR1C3 prostate cancer cell line where they produced a near complete inhibition of testosterone formation when the cells were treated with the AKR1C3 substrate Δ4-AD.
Table 1.
Inhibitory Potency of Compounds on AKR1C1-4
| Compound | AKR1C3 IC50 (μM) | AKR1C1 IC50 (μM) | AKR1C2 IC50 (μM) | AKR1C4 IC50 (μM) | Ref |
|---|---|---|---|---|---|
| 1 | 0.06 | 22.7 | 15.4 | 62.7 | [58, 59] |
| 2 | 0.03 | 6.74 | 3.38 | 32.7 | [58, 59] |
| 3 | 0.05 | 15.6 | 19.5 | 25.7 | [58, 59] |
| 4 | 0.31 | 50.8 | 50.2 | 20.1 | [60] |
| 5 | 0.35 | >100 | 63.0 | 30.3 | [60] |
| 6 | 0.013 | 20.3 | >30 | >30 | [61] |
| 7 | 0.006 | >30 | >30 | >30 | [61] |
| 8 | 0.05 | >30 | >30 | >30 | [61] |
| 9 | 0.03 | >30 | >30 | >30 | [61] |
| 10 | 0.08 | 11.7 | 8.17 | 8.17 | [64] |
| 11 | 0.21 | >300 | 206 | >300 | [65] |
| 12 | 0.29 | ND | ND | ND | [66] |
| Baccharin | 0.11 | >100 | >100 | >100 | [67] |
| Drupanin | 15.0 | >100 | 108 | >100 | [67] |
4.2. N-Benzoylanthranilic Acid Based Inhibitors
Due to the close structural similarity to N-phenylanthranilates, a series of N-benzoylanthranilic acids that were initially designed as inhibitors of the penicillin binding protein (PBP) were screened for AKR1C3 inhibitory activity and selectivity [60]. Of the 16 compounds that were tested, two compounds 4 and 5 inhibited AKR1C3 in a potent and selective manner. In the S-tetralol oxidation assay as described above, 4 inhibited AKR1C3 with an IC50 value of 310 nM and displayed 164-, 162- and 65- fold selectivity for AKR1C3 over AKR1C1, AKR1C2 and AKR1C4, respectively. Compound 5 likewise inhibited AKR1C3 with IC50 value of 350 nM and displayed 286-, 180- and 86- fold selectivity versus AKR1C1, AKR1C2 and AKR1C4, respectively (Table 1). SAR studies on these compounds indicate a need for a 3-OH group on the N- benzoylamino B- ring and a 5′-halogen on the benzoic acid A-ring. Movement of the carboxylic acid to other ring positions as well as substitution on other positions on the N-benzoylamino ring was not explored with these compounds. Hence, further modification of these compounds to increase potency as well as selectivity should be possible. It will be necessary to evaluate the efficacy of compounds 4 and 5 in cellular assays of AKR1C3 activity. Also, since the compounds are structurally similar to N-PA, it is essential to determine their effect on COX 1 and 2.
4.3. 3-(3,4-Dihydroisoquinolin-2(1H)-ylsulfonyl)benzoic acids
Using a high throughput screening (HTS) approach to identify putative AKR1C3 inhibitors, Jamieson et al [61] screened a total of 99000 compounds. Filtering of hits for undesirable properties and a potency cut off value of < 1 μM led to 187 compounds out of which 3-(3,4-dihydroisoquinolin-2(1H)-ylsulfonyl)benzoic acid (compound 6) was deemed to be the most promising lead AKR1C3 inhibitor. The authors used a fluorimetric assay that monitored the AKR1C catalyzed formation of a fluorescent alcohol from a non-fluorescent ketone substrate (probe 5) [62] at 37 °C and pH 7.2 with NADPH as the cofactor. The authors also utilized a cell-based assay that monitored product formation when HCT-116 cells stably transfected with AKR1C3 were treated with the AKR1C3 substrate, PR-104H in the presence of inhibitors [63]. PR-104A is a bioreductive prodrug that is activated by AKR1C3 into the DNA cross linking metabolites, PR-104H and PR-104M [63]. Compound 6 was a significantly more potent AKR1C3 inhibitor than FLU and other marketed NSAIDs. It inhibited AKR1C3 with an IC50 value of 13 nM and displayed > 1500 fold selectivity for AKR1C3 relative to the other AKR1C isoforms, Table 1. Compound 6 was likewise devoid of any inhibitory activity on COX 1 and 2 at 10 μM concentration. Subsequent SAR studies indicated that the orientation of the dihydroisoquinoline ring in compound 6 was optimal for AKR1C3 inhibition. AKR1C3 potency and selectivity was also found to be tolerant of small functional group substitutions on various positions of the dihydroisoquinoline ring. Relatively modest changes in AKR1C3 potency were observed as a result of the introduction of these substituents. These findings led to the identification of some of the most potent AKR1C3 inhibitors with several compounds having nanomolar potency and remarkable selectivity for AKR1C3 over other AKR1C isoforms. Compound 7, with a 6-Br group on the dihydroisoquinoline ring was the most potent and most selective inhibitor AKR1C3 inhibitor from the study. It inhibited AKR1C3 with an IC50 value of 6 nM and was > 5000 fold selective over other AKR1C isoforms. Compounds 6 – 9 displayed relatively similar inhibitory potency in the cell based assay. Table 1.
4.4. Indomethacin Analogs
New indomethacin based AKR1C3 inhibitors have been reported by us [68]. These include three classes of compounds which are (i) indomethacin analogs; (ii) 2′-des-methyl-indomethacin analogs in which the 2′-methyl group has been removed; and (iii) 3′alkyl indomethacin analogs in which the position of the 2′-methyl group and 3′ acetic side chain have been reversed, exemplified by compounds 13–15. Each of these compounds have mid-nanomolar potency for inhibition of AKR1C3, are selective for AKR1C3 over AKR1C isoforms, have weak inhibitory potency against COX isozymes, and inhibit AKR1C3 mediated testosterone production in LNCaP-AKR1C3 cells. A crystal structure of 2-des-methylindomethacin, 14 bound to AKR1C3 was also obtained to probe the binding modes of these compounds. A full description of these analogs has been recently published [68].
4.5. Natural Products
Phytochemicals such as flavonoids and cinnamic acids have long been known to display anticancer properties. Their anticancer properties is believed to be as a result of inhibition of one or more of the following processes including cell adhesion, matrix metalloproteinase (MMP) secretion, angiogenesis, protein kinase activities and tumor cell invasion (reviewed by Kanadaswami et al. 2005) [69]. However, some of these compounds also inhibit AKR1C3 at nanomolar to low micromolar concentrations suggesting that AKR1C3 inhibition is a potential mechanism for their antineoplastic effects [48, 49, 69–71]. Endo et al [67] recently studied the potential anticancer mechanism of two compounds, baccharin (3-prenyl-4-(dihydrocinnamoyloxy)cinnamic acid) and drupanin ((E)-3-(4-hydroxy-3-(3-methylbut-2-en-1-yl)phenyl)acrylic acid) which are components of Brazilian honey bee propolis, Figure 3. Both compounds are naturally occurring prenylated cinnamic acid derivatives that display growth inhibitory effects by inducing apoptosis in human cancer cell lines [72, 73]. Using a variation of the S-tetralol oxidation assay at 25 °C and pH 7.4 (S-tetralol concentrations were 100 μM for AKR1C1 and 1mM for AKR1C2-AKR1C4), baccharin inhibited AKR1C3 with an IC50 value of 110 nM (Ki = 56 nM) with no measurable inhibitory activity on AKR1C1 and AKR1C2 at 100 μM. This translates to > 1000 fold selectivity for AKR1C3. Drupanin on the other hand was about 150 fold less potent as a AKR1C3 inhibitor with little or no selectivity compared to baccharin. This disparity in AKR1C3 potency and selectivity was attributed to the importance of the dihydrocinnamoyloxy moiety of baccharin for AKR1C3 potency and selectivity. Baccharin was effective and selective for AKR1C3 inhibition in cellular assays where it inhibited the reduction of farnesal in MCF-7 cells and androsterone in A549 cells, with an IC50 of 30 μM.
4. 6. Bifunctional AKR1C3 Inhibitors and AR Antagonists
Due to the challenges encountered with Abiraterone and MDV3100 and the adaptive response that often results in development of resistance to these compounds, combinations of both drugs has been proposed for the treatment of CRPC [17, 74]. Concurrent inhibition of androgen biosynthesis and AR signaling-the two pathways implicated in the development of progression of CRPC should produce maximal blockade of the AR axis, increase therapeutic efficacy and reduce the incidence of resistance. Additionally, the use of a bifunctional compound may minimize the risk of adverse effects that may occur with using two separate drugs. The new P450 17A1 inhibitor TOK001 satisfies some of this need since it not only inhibits the enzyme but also causes degradation of the AR. Unfortunately, due to targeting P450 17A1 the issue of drug induced adrenal insufficiency will remain and will require the co-administration of prednisone.
Novel FLU analogs were recently described as AR antagonists [75]. This suggests that AR and AKR1C3 ligands share a common pharmacophore which could be exploited to develop bifunctional agents that target both proteins. We recently reported the discovery of one such compound, 3-((4-nitronaphthalen-1-yl)amino)benzoic acid (compound 10), a “first-in-class”, dual acting AKR1C3 inhibitor and AR antagonist [64]. In the S-tetralol oxidation assay, compound 10 inhibited AKR1C3 with IC50 value of 85 nM and was over 100 fold selective for AKR1C3 over other AKR1C isoforms. Unlike its phenyl analog 3, it did not display any significant effect on COX-1 and COX-2 with IC50 values > 100 μM. It also produced a robust reduction in testosterone formation in LNCaP-AKR1C3 cells at 10 μM concentrations. Notably, compound 10 competitively inhibited DHT induced AR transcriptional activity in an AR-luciferase reporter gene assay with an IC50 value of 4.7 μM and produced a concentration dependent reduction in cellular AR levels in the presence and absence of DHT. Dual inhibition of AKR1C3 and AR represents a novel concept with immense potential for the treatment of CRPC. It will be critical to understand the functional implication of using bifunctional agents like compound 10 in prostate cancer cell lines that express either one or both targets.
4.7. In- silico Screening
Using a fragment based virtual screening approach, a set of 143000 compounds were docked into the active site of AKR1C3 [65]. The highest ranked 33 compounds were then tested for AKR1C3 inhibitory activity. Of the new set of compounds, 11 was the most promising. In the S-tetralol oxidation assay, it inhibited AKR1C3 with an IC50 value of 200 nM, was about a 1000 fold selective versus AKR1C2 and was even more selective for AKR1C3 relative to AKR1C1/AKR1C4. This compound has yet to be evaluated in a cell based assay. Schuster et al also identified a novel AKR1C3 inhibitor from a pharmacophore –based virtual screening [66]. One of the most interesting compounds from their study is compound 12. Compound 12 displayed an IC50 value of 290 nM for AKR1C3 in a radiometric assay that measured the ability of the compound to inhibit the NADPH dependent reduction of [3H]-Δ4-AD by AKR1C3. The assay was conducted at pH 6.6 with a substrate concentration of 21 nM. The authors did not evaluate the compounds selectivity for AKR1C3 over other AKR1C enzymes but the compound was shown to preferentially inhibit AKR1C3 over 17β-HSD Types, 1, 2, 3, 4 and 7.
5. Structural Considerations/Crystal Structure Analysis
The critical role of AKR1C3 in CRPC and the potential therapeutic benefits of AKR1C3 inhibitors have generated immense interest in the structure of AKR1C3. These efforts are aimed at exploring the interaction of existing inhibitors with the enzyme, which can provide valuable insight that can be used in the design of next generation inhibitors. Sixteen new crystal structures of AKR1C3 in complex with different inhibitors have been reported since the last review published by Byrns et al. [54] (Table 2). These inhibitors are related to four major classes: N-phenylanthranilate based compounds (N-PA) [58, 59, 64, 76, 77], 3-(3,4-dihydroisoquinolin-2(1H)-ylsulfonyl)benzoic acids [61], indomethacin [77], and arylpropionic acids [77]. AKR1C3 contains the conical TIM barrel fold composed of eight alternating α-helices and β-strands. The active site is located at the C-terminal end of the β-strands and capped with three long flexible loops (Figure 5).
Table 2.
Recent Crystal Structures of AKR1C3 available in the Protein Data Bank (PDB)
| Inhibitor | PDB ID | Reference |
|---|---|---|
| N-phenylanthranilate (N-PA) | ||
| 3-phenoxybenzoic acid | 3UWE | [76] |
| Meclofenamic acid | 3R6I | [77] |
| Mefenamic acid | 3R43 | [77] |
| 1 | 4DBU | [64] |
| Arylpropionic acids | ||
| (R)-Flurbiprofen | 3R94 | [77] |
| (R)-Ibuprofen | 3R8G | [77] |
| (R)-Naproxen | 3UFY | [77] |
| (S)-Naproxen | 3R58 | [77] |
| Indomethacin analogs | ||
| Indomethacin pH 6.8 | 3UGR | [77] |
| Indomethacin pH 7.5 | 3UG8 | [77] |
| Zomepirac | 3R8H | [77] |
| Sulindac | 3R7M | [77] |
| 3-(3,4-dihydroisoquinolin-2(1H)-ylsulfonyl)benzoic acids | ||
| 6 | 4FAM | [61] |
| 8 | 4FAL | [61] |
| 9 | 4FA3 | [61] |
| Bifunctional inhibitor | ||
| 10 | 4DBS | [64] |
Figure 5.

Ligand binding pocket of AKR1C3 and AKR1C2. AKR1C3 and AKR1C2 share very similar overall structures. (A), sideview of the overall structure of AKR1C3 in complex with natural substrate prostaglandin D2 (PGD2, blue). (B), topview of the overall structure of AKR1C2 in complex with testosterone (green). Cofactor NADP+ is colored in red. β-strands are colored in yellow. The surface of the subpockets defined by Byrns et al. [54] are shown in the cross-section view of the ligand binding site of AKR1C3 (C) and AKR1C2 (D). AKR1C3 has much bigger subpockets compared to AKR1C2. The ligands and cofactors are not on the same plane as the protein structure/surface. The surface is colored according to protein secondary structure: yellow, β-stands; gray, α-helices and flexible loops. SC, steroid channel; OS, oxyanion site; SP, subpocket. All figures are prepared using The PyMOL Molecular Graphics System, Version 1.20 Schrödinger, LLC.
As proposed by Byrns et al. [54] the ligand binding pocket of AKR1C3 can be dissected into five compartments: oxyanion site, steroid channel, and three SubPockets, SP1, SP2, and SP3 (Figure 5C). The oxyanion site is the catalytic site and is formed by the cofactor and the conserved catalytic tetrad of Asp50, Tyr 55, Lys84, and His117. It is clear from the structural studies that the oxyanion site serves as an important anchorage point for the carboxylate/carbonyl group on most of the NSAID based inhibitors. The steroid channel is an open-ended cylindrical tunnel that is surrounded by Tyr24, Leu54, Ser129, and Trp227. The steroid channel guides the substrates down to the oxyanion site such that the steroid is roughly perpendicular to the cofactor. Even though this channel is a prerequisite for HSD activity, it is often not utilized by the inhibitors. Both the oxyanion site and the steroid channel are shared by all the human AKR1C enzymes and likely do not confer inhibitory selectivity. The difference among the AKR1C enzymes lies in the three subpockets (Figure 5C and D) [51, 54]. AKR1C3 has considerably larger and more flexible subpockets, which can accommodate bulky substrates like prostaglandins and allows special targeting by selective inhibitors. The SP1 pocket is lined by Ser118, Asn167, Phe306, Phe311, and Tyr319, the SP2 pocket is lined by Trp86, Leu122, Ser129, and Phe311 and the SP3 pocket is lined by Tyr24, Glu192, Ser221, and Tyr305. Formation of hydrogen bonding with the polar amino acids lining the SP1 pocket is expected to give more potent and selective AKR1C3 inhibitors.
5.1. N-PA based inhibitors and 3-(3,4-dihydroisoquinolin-2(1H)-ylsulfonyl)benzoic acids
Both N-PA based inhibitors and 3-(3,4-dihydroisoquinolin-2(1H)-ylsulfonyl)benzoic acids utilize the SP1 pocket and interact with the oxyanion site (Figure 6). This similarity in binding conformations is not too surprising since these compounds share a similar A-ring -linker- B-ring scaffold. The A-ring provides the basic anchorage of the inhibitor by hydrogen bonding between the carboxylate/carboxamide substituent and residues Tyr55 and His117. The B-ring optimizes inhibitor selectivity by targeting specific properties of the AKR1C3 SP1 pocket. The bulky bicyclic B-ring of the 3-(3,4-dihydroisoquinolin-2(1H)-ylsulfonyl)benzoic acids offers superior selectivity by taking advantage of the size of the AKR1C3 SP1 pocket,[61] whereas the smaller monocyclic B-ring of the N-PA compounds greatly benefits from strong electron withdrawing substitutions that may form dipole interactions or hydrogen bonding within the SP1 pocket [59, 77, 78]. Besides the B-ring, inhibitory selectivity is also affected by the linker region that directs B-ring into the SP1 pocket. Optimal selectivity is obtained when a single bridge atom connects the meta-carboxylate substituted A-ring and the substituted B ring [59, 61]. Choice of the bridge atom is not limited to nitrogen as in N-PA or sulfur as in the 3-(3,4-dihydroisoquinolin-2(1H)-ylsulfonyl)benzoic acids. 3-Phenoxybenzoic acid (3-PBA), which isosterically replaces the amine linker of the N-PA with an oxygen atom, adopts a similar binding pose in AKR1C3 as Compound 1 [76]. Not surprisingly, 3-PBA likewise displays similar AKR1C3 inhibitory potency and selectivity as the unsubstituted 3-phenylaminobenzoic acid ([58, 59] and Unpublished observation Adeniji.A.O). The rigidity of the linker is proposed to enhance selectivity once the B-ring is correctly positioned [61]. It should be noted that some of the 3-(3,4-dihydroisoquinolin-2(1H)-ylsulfonyl)benzoic acids carrying extraordinary large A-ring substitutions instead of the carboxylic acid are well accepted by AKR1C3. These substitutions apparently could not be accommodated in the oxyanion site, suggesting that the anchorage to the oxyanion site may be beneficial but not obligated for high binding affinity. These compounds likely adopt an alternative binding pose resembling the one used by indomethacin at lower pH as described below (Figure 7A). This conformation utilizes the SP3 pocket instead of the oxyanion site.
Figure 6.

Structures of the N-PA based analogs and 3-(3,4-dihydroisoquinolin-2(1H)-ylsulfonyl)benzoic acids bound to AKR1C3. Both classes of inhibitors utilize the oxyanion hole - SP1 cavity. Two slightly different binding poses exist for the N-PAs depending on the position of the carboxylate. A, for the o-CO2H-substituted N-PAs compounds, FLU (red), meclofenamic acid (blue), and mefenamic acid (green), the amine bridge forms hydrogen bonds with the cofactor nicotinamide head. B, for the m-CO2H-substituted N-PA based analogs, compound 1 (cyan) and 3-PBA (magenta), the bridge atom moves 2.4 Å away from the cofactor and no longer maintains the interaction. Compared to FLU, compound 1 projects 1 Å deeper into the SP1 pocket and induces significant movement of Phe306 and Phe311, which could be basis for its selectivity. C, Structures of the prototypical 3-(3,4-dihydroisoquinolin-2(1H)-ylsulfonyl)benzoic acids, compounds 6 (magenta), 7 (green), and 8 (gray). Even though there is only a slight change in the binding position between compounds 6 and 7, Trp227 is flipped and the C-terminal loop (Loop C: Asp300-end) is shifted to accommodate the two compounds.The presence of the methylamide in compound 8 causes significant movements of the benzyl ring and the sulfonyl linker. But the tetrahydroquinoline group is still positioned in the SP1 pocket. The linker regions of the N-PAs and the 3-(3,4-dihydroisoquinolin-2(1H)-ylsulfonyl)benzoic acids occupy the same location and the 3-(3,4-dihydroisoquinolin-2(1H)-ylsulfonyl)benzoic acids reach the same depth in the SP1 pocket as compound 1. But the plane of the bicyclic ring is roughly perpendicular to the phenyl ring in the N-PA compounds. Hydrogen bonds are shown as red dashes. Only the residues that are normally involved in significant movement are shown. Water molecules are shown as spheres. Distance is indicated by blue dashes.
Figure 7.
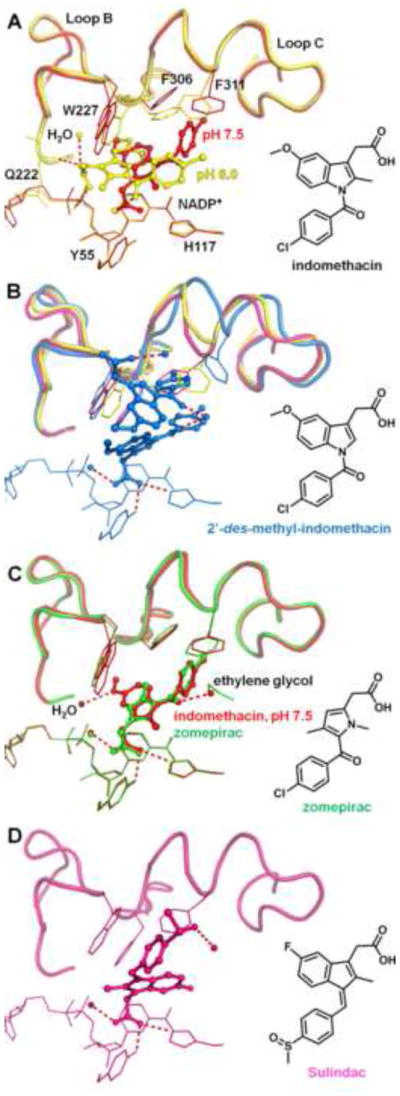
Structures of indomethacin analogs bound to AKR1C3. A, overlay of the two indomethacin conformations at different pH values. The pH 6.0 conformation (yellow) utilizes the SP3 pocket, whereas the pH 7.5 conformation (red) occupies the oxyanion site and the SP1 pocket. At intermediate pH (pH 6.8, PDB ID 3UGR)[77], the crystal structure shows presence of both conformations (70% occupancy by the low pH conformation, 25% by the high pH conformation, with the remaining 5% unaccounted for). Hydrogen bonds for the pH 6.0 conformation are shown as red dashes. B, two molecules of 2′-des-methyl-indomethacin (blue) are bound per active site. Only the lower molecule closer to the oxyanion site is contributing to inhibition. The accommodation of the second molecule forces significant loop movements of AKR1C3 compared to other indomethacin analogs. (indomethacin, yellow; sulindac, magenta) structures. C, overlay of indomethacin pH 7.5 conformation and zomepirac (green). Zomepirac adopts a very similar conformation to indomethacin. D, sulindac (magenta) is anchored to the oxyanion site but binds quite differently from the indomethacin conformation by extending towards the SP2 pocket.
5.2. Indomethacin Based Inhibitors
Indomethacin analogs are the class of AKR1C3 inhibitors that show the most diversity in binding positions (Figure 7). At pH 6.0 (PDB ID 1S2A) [78], indomethacin occupies the SP3 pocket with the indole ring and leaves its p-chorobenzoyl ring at the entrance to the SP1 pocket. The carboxylate group helps to anchor the indole ring by hydrogen bonding to the diphosphate bridge of the cofactor and Gln222. With a pH increase to 7.5 (PDB ID 3UG8), [77] the weakened interaction in the SP3 pocket allows the carboxylate group to occupy the oxyanion site, so that the p-chorobenzoyl ring extends into the SP1 pocket and the indole ring partially stays in the SP3 pocket (Figure 7A). The pH-dependency of the binding conformations is likely caused by the different protonation state of the phosphate bridge, O1N/O2N phosphate oxygens in particular, on the cofactor, have a pKa around 7 [77]. Since most of the inhibitor studies are performed around pH 7, it is likely that both binding poses contribute to the inhibitory potency of indomethacin analogs [53, 61]. Removing the methyl group on the indole ring gives 2′-des-methyl-indomethacin which binds differently. In this instance the indole ring enters the steroid binding channel while the carboxylate group remains tethered to the oxyanion site [79]. This binding pose also permits a second molecule of 2′-des-methyl-indomethacin to enter the active site. But kinetic data suggest the second molecule is unlikely to contribute to the inhibition potency. In contrast, replacing the indole ring with a pyrrole ring as in zomepirac elicits a minimal effect on the binding pose as zomepirac maintains the indomethacin binding pose at pH 7.5 (Figure 7C)(PDB ID 3R8H, pH undefined) [77]. Sulindac possesses minor structural changes compared to indomethacin. The inhibitor exhibits a novel binding pose not seen with the other NSAID based AKR1C3 inhibitors (PDB ID 3R7M, pH undefined) [77] (Figure 7D). The carboxylate group anchors sulindac to the oxyanion site while the p-chorobenzoyl ring extends to the entrance to the SP2 pocket, a location known to be occupied by substrate prostaglandin D2 (PDB ID 1RY0) and the inhibitor rutin (PDB ID 1RY8) [80]. Flanagan et al [77] stated that the sulindac conformation causes significant movement of the Ser129-Phe139 loop (part of Loop A), but the loop region was not modeled in the published structure. Sulindac has a bulkier sulfoxide substitution on the benzoic ring in place of the chorine in indomethacin, which likely precludes the compound from adopting the indomethacin binding poses. However, sulindac is a prodrug that is activated in the gastrointestinal tract by the reduction of the sulfoxide to a sulfide. The small size of the sulfide may allow the compound to act as a potent AKR1C3 inhibitor in vivo.
5.3. Arylpropionic acids
The arylpropionic acids cocrystallized with AKR1C3 ((R)-flurbiprofen, PDB ID 3R94; (R)-ibuprofen, PDB ID 3R8G; (R)-naproxen, PDB ID 3UFY; (S)-naproxen, PDB ID 3R58) are less potent and less selective Inhibitors. The inhibitors occupy the SP1 pocket and the oxyanion site and even extend into the SP1 pocket to a similar depth to that of potent N-PA compounds (Figure 8), however, it is not apparent from the structures why the compounds are not as selective or potent. Besides, different studies have reported discrepant IC50 values for these compounds using racemic mixtures and different assay methods, [61, 81] making it hard to extrapolate useful information.
Figure 8.

Structures of the arylpropionic acids bound to AKR1C3. A, overlay of (R)-flurbiprofen (blue) and (R)-ibuprofen (yellow) bound to AKR1C3. The two compounds adopt very similar binding poses despite their structural difference. B, overlay of (R)-naproxen (green) and (S)-naproxen (red). AKR1C3 shows only subtle preference for the (R)-configuration, which is reflected by the limited changes in binding conformation imposed by the chirality of the compounds. Hydrogen bonds are shown as red dashes.
5.4. Bifunctional inhibitors
In the course of developing AKR1C3 inhibitors, we identified a bifunctional compound 10 and recently determined its crystal structure in complex with AKR1C3 (PDB ID 4DBS) [64]. Compound 10 is designed based on the FLU scaffold and its binding pose showed significant resemblance to FLU and the other N-PA compounds. However, unlike the N-PA compounds, there are two molecules of inhibitor bound per enzyme monomer (Figure 9). Both molecules project their naphthyl rings into the SP1 pocket, forming a double decker arrangement and both molecules are deemed necessary for AKR1C3 inhibition as suggested by kinetic data [64]. The 3-(3,4-dihydroisoquinolin-2(1H)-ylsulfonyl)benzoic acids also possess bicyclic rings but bind only one molecule per AKR1C3 active site (Figure 6). The size and position of the sulfonyl linker probably prevents the binding of two molecules of the inhibitor per enzyme monomer. Future optimization of compound 10 may result in a single molecule of inhibitor bound at the active site. This may be accomplished by replacing the 1-naphthyl group of compound 10 with a 2-naphthyl group to mimic the structure of the 3-(3,4-dihydroisoquinolin-2(1H)-ylsulfonyl)benzoic acids. The mechanism by which compound 10 acts as an AR antagonist has not been fully elucidated. The compound could down regulate AR expression or directly interact with AR. N-PA compounds have been reported to bind to the AR surface and block coactivator recruitment [82]. Docking simulations demonstrate that compound 10 could bind to the AR ligand binding domain with high affinity (unpublished data).
Figure 9.

Structure of the bifunctional analog compound 10 (magenta) shown in overlay with compounds 1 (cyan) bound to AKR1C3. Two molecules of compound 10 are bound per AKR1C3 active site and the first molecule resembles the binding conformation of the N-PA compounds. The naphthyl rings of compound 10 are perpendicular to the B-ring of compound 1.
Despite the fruitful structural studies on AKR1C3-inhibitor interaction, molecular simulations to predict future inhibitor design have been hampered by the size and flexibility of the AKR1C3 subpockets. Inhibitors distinct in size, shape, and binding conformation could exhibit similar affinity for AKR1C3. Almost all the highly selective AKR1C3 inhibitors elicit significant residue/loop movement of the enzyme. The pH-dependency of the binding conformations could also be a setback for computer-based drug discovery. Docking studies have only succeeded in predicting the correct binding conformation on limited occasions [77]. For future in silico screening for AKR1C3 inhibitors, it will be necessary to allow highly flexible residues to assume multiple rotamer conformations and carefully examine the protonation state of the enzyme, cofactor, and ligand upon binding.
6. Preclinical Development
Despite the increase in the number of potent and selective AKR1C3 inhibitors and the immense potential therapeutic value, there are currently no AKR1C3 inhibitors in clinical trials for the treatment of CRPC or any other hormone dependent malignancy in which AKR1C3 has been implicated. Indeed, only indomethacin has been tested in an animal model and shown to block tumor cell proliferation in an AKR1C3 dependent manner [42].
While the AKR1C3 potency and selectivity of the inhibitors may vary due to the differences in assay conditions such as substrate concentration, pH, method of detection and cofactor used, it is clear that the new set of compounds reviewed here represent significant improvements in the development of candidate AKR1C3 inhibitors that can be funneled into preclinical development. Preclinical development of lead compounds involves a tiered screening approach and proof-of-principle that AKR1C3 inhibitors are effective in cell based and animal models of CRPC. Our tiered approach is shown in Fig. 10. This approach involves screening for nanomolar potency and selectivity for AKR1C3 versus its closely related isoform AKR1C2 (tier 1). In-vitro screening to determine that AKR1C3 inhibitors do not inhibit the other human AKR enzymes (9 total), the other human 17β-HSD isoforms and COX-1 and COX-2 (for those that are NSAID based (tier 2). Cell-based assays to show that they block AKR1C3 mediated testosterone production, endogenous gene expression (PSA, Transmembrane Protease, Serine 2-ETS Related gene fusion (TMPSS2ERG) and FK506 Binding Protein 5, FKBP5), and AKR1C3 mediated prostate cancer cell growth (tier 3). And, efficacy to block tumor growth in murine xenograft models of CRPC that over express AKR1C3 (tier 4).
Figure 10.

Tiered Approach to Preclinical Development of AKR1C3 Inhibitors
Compounds may fail in vivo screening due to unfavorable absorption, disposition, metabolism elimination and toxicological properties and thus a medicinal chemistry approach is required for lead optimization. Lead optimization involves removal of metabolic “soft-spots”, removal of structural alerts and re-screening and is by its very nature an iterative process.
7. Conclusions
Significant advances have been made in the discovery of therapeutic agents for use in CRPC. However, there is still a need for superior compounds that target the androgen axis. AKR1C3 plays a vital role in androgen biosynthesis and is critical for CRPC progression. It therefore represents a prime target and AKR1C3 inhibitors are expected to be more selective and have less side-effects than P450 17A1 inhibitors. Inhibitors with nanomolar potency and significant selectivity for AKR1C3 have been discovered and subsequent preclinical development of these drugs is required. These inhibitors have the potential to be efficacious in management of CRPC where they can be used alone or in combination with other compounds. They may also be useful in the neoadjuvant setting in combination with a LH-RH agonist. The discovery of a “first-in-class” bifunctional AKR1C3 inhibitor and AR antagonist is particularly exciting in this regard.
Figure 4.
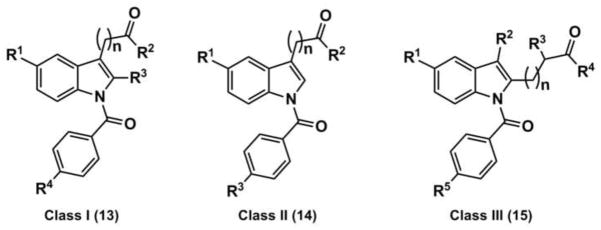
Representative Structures of Indomethacin based AKR1C3 Inhibitors. Compound 13 (n=2): R1 = OCH3, R2 = OH, R3 = CH3, R4 = Cl, AKR1C3 IC50 = 0.22 μM AKR1C2 IC50 = 56.5 μM; compound 14 (n=1): R1 = OCH3, R2 = OH and R3 = Cl, AKR1C3 IC50 = 0.96 μM, AKR1C2 IC50 = 100 μM; compound 15 (n=1): R1 = OCH3, R2 = CH2CH3, R3, = H, R4 = OH, R5 = Cl AKR1C3 IC50 = 0.09 μM AKR1C2 IC50 = 49.6 μM..
Highlights.
Superior therapeutic agents to Abiraterone and Enzalutamide are required to treat CRPC (103 characters).
AKR1C3 synthesizes potent ligands for the androgen receptor locally (68 characters)
AKR1C3 is upregulated by androgen deprivation in CRPC (53 characters)
Structurally diverse, potent and selective AKR1C3 inhibitors have been developed (80 characters)
AKR1C3 inhibitors need to be tested in xenograft models and taken into preclinical development.(94 characters)
Acknowledgments
This work was supported by P30-ES13508, R01-CA90744 and a Prostate Cancer Foundation Challenge Grant and TAPITMAT grant awarded to TMP. We thank Ms. Sonia D. Revello for her help with the figures used in the paper.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.SEER Cancer Statistics Review, 1975–2009. National Cancer Institute; Bethesda, MD: 2010. based on November 2009 SEER data submission, posted to the SEER web site, 2010. http://seer.cancer.gov/statfacts/html/prost.html. [Google Scholar]
- 2.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60(5):277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 3.http://www.cancer.org/Cancer/ProstateCancer/DetailedGuide/prostate-cancer-key-statistics.
- 4.Knudsen KE, Scher HI. Starving the addiction: new opportunities for durable suppression of AR signaling in prostate cancer. Clin Cancer Res. 2009;15(15):4792–4798. doi: 10.1158/1078-0432.CCR-08-2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Knudsen KE, Penning TM. Partners in crime: deregulation of AR activity and androgen synthesis in prostate cancer. Trends Endocrinol Metab. 2010;21(5):315–324. doi: 10.1016/j.tem.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Locke JA, Guns ES, Lubik AA, Adomat HH, Hendy SC, Wood CA, Ettinger SL, Gleave ME, Nelson CC. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Res. 2008;68(15):6407–6415. doi: 10.1158/0008-5472.CAN-07-5997. [DOI] [PubMed] [Google Scholar]
- 7.Attard G, Reid AH, Yap TA, Raynaud F, Dowsett M, Settatree S, Barrett M, Parker C, Martins V, Folkerd E, Clark J, Cooper CS, Kaye SB, Dearnaley D, Lee G, de Bono JS. Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J Clin Oncol. 2008;26(28):4563–4571. doi: 10.1200/JCO.2007.15.9749. [DOI] [PubMed] [Google Scholar]
- 8.Attard G, Reid AH, A’Hern R, Parker C, Oommen NB, Folkerd E, Messiou C, Molife LR, Maier G, Thompson E, Olmos D, Sinha R, Lee G, Dowsett M, Kaye SB, Dearnaley D, Kheoh T, Molina A, de Bono JS. Selective inhibition of CYP17 with abiraterone acetate is highly active in the treatment of castration-resistant prostate cancer. J Clin Oncol. 2009;27(23):3742–3748. doi: 10.1200/JCO.2008.20.0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reid AH, Attard G, Danila DC, Oommen NB, Olmos D, Fong PC, Molife LR, Hunt J, Messiou C, Parker C, Dearnaley D, Swennenhuis JF, Terstappen LW, Lee G, Kheoh T, Molina A, Ryan CJ, Small E, Scher HI, de Bono JS. Significant and sustained antitumor activity in post-docetaxel, castration-resistant prostate cancer with the CYP17 inhibitor abiraterone acetate. J Clin Oncol. 2010;28(9):1489–1495. doi: 10.1200/JCO.2009.24.6819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, Chi KN, Jones RJ, Goodman OB, Jr, Saad F, Staffurth JN, Mainwaring P, Harland S, Flaig TW, Hutson TE, Cheng T, Patterson H, Hainsworth JD, Ryan CJ, Sternberg CN, Ellard SL, Flechon A, Saleh M, Scholz M, Efstathiou E, Zivi A, Bianchini D, Loriot Y, Chieffo N, Kheoh T, Haqq CM, Scher HI. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364(21):1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, Wongvipat J, Smith-Jones PM, Yoo D, Kwon A, Wasielewska T, Welsbie D, Chen CD, Higano CS, Beer TM, Hung DT, Scher HI, Jung ME, Sawyers CL. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324(5928):787–790. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jung ME, Ouk S, Yoo D, Sawyers CL, Chen C, Tran C, Wongvipat J. Structure-activity relationship for thiohydantoin androgen receptor antagonists for castration-resistant prostate cancer (CRPC) J Med Chem. 2010;53(7):2779–2796. doi: 10.1021/jm901488g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scher HI, Beer TM, Higano CS, Anand A, Taplin ME, Efstathiou E, Rathkopf D, Shelkey J, Yu EY, Alumkal J, Hung D, Hirmand M, Seely L, Morris MJ, Danila DC, Humm J, Larson S, Fleisher M, Sawyers CL. Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1–2 study. Lancet. 2010;375(9724):1437–1446. doi: 10.1016/S0140-6736(10)60172-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, de Wit R, Mulders P, Chi KN, Shore ND, Armstrong AJ, Flaig TW, Flechon A, Mainwaring P, Fleming M, Hainsworth JD, Hirmand M, Selby B, Seely L, de Bono JS. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367(13):1187–1197. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 15.DeVore NM, Scott EE. Structures of cytochrome P450 17A1 with prostate cancer drugs abiraterone and TOK-001. Nature. 2012;482(7383):116–119. doi: 10.1038/nature10743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O’Donnell A, Judson I, Dowsett M, Raynaud F, Dearnaley D, Mason M, Harland S, Robbins A, Halbert G, Nutley B, Jarman M. Hormonal impact of the 17α-hydroxylase/C(17,20)-lyase inhibitor abiraterone acetate (CB7630) in patients with prostate cancer. Br J Cancer. 2004;90(12):2317–2325. doi: 10.1038/sj.bjc.6601879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Richards J, Lim AC, Hay CW, Taylor AE, Wingate A, Nowakowska K, Pezaro C, Carreira S, Goodall J, Arlt W, McEwan IJ, de Bono JS, Attard G. Interactions of abiraterone, eplerenone, and prednisolone with wild-type and mutant androgen receptor: A rationale for increasing abiraterone exposure or combining with MDV3100. Cancer Res. 2012;72(9):2176–2182. doi: 10.1158/0008-5472.CAN-11-3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.http://investors.medivation.com/releasedetail.cfm?releaseid=703823.
- 19.Foster WR, Car BD, Shi H, Levesque PC, Obermeier MT, Gan J, Arezzo JC, Powlin SS, Dinchuk JE, Balog A, Salvati ME, Attar RM, Gottardis MM. Drug safety is a barrier to the discovery and development of new androgen receptor antagonists. Prostate. 2010;71(5):480–488. doi: 10.1002/pros.21263. [DOI] [PubMed] [Google Scholar]
- 20.Penning TM, Burczynski ME, Jez JM, Hung CF, Lin HK, Ma H, Moore M, Palackal N, Ratnam K. Human 3α-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the aldo-keto reductase superfamily: functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. Biochem J. 2000;351(Pt 1):67–77. doi: 10.1042/0264-6021:3510067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin HK, Jez JM, Schlegel BP, Peehl DM, Pachter JA, Penning TM. Expression and characterization of recombinant type 2 3α-hydroxysteroid dehydrogenase (HSD) from human prostate: demonstration of bifunctional 3α/17β-HSD activity and cellular distribution. Mol Endocrinol. 1997;11(13):1971–1984. doi: 10.1210/mend.11.13.0026. [DOI] [PubMed] [Google Scholar]
- 22.Chang KH, Li R, Papari-Zareei M, Watumull L, Zhao YD, Auchus RJ, Sharifi N. Dihydrotestosterone synthesis bypasses testosterone to drive castration-resistant prostate cancer. Proc Natl Acad Sci U S A. 2011;108(33):13728–13733. doi: 10.1073/pnas.1107898108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Auchus RJ. The backdoor pathway to dihydrotestosterone. Trends Endocrinol Metab. 2004;15(9):432–438. doi: 10.1016/j.tem.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 24.Chen DB, Westfall SD, Fong HW, Roberson MS, Davis JS. Prostaglandin F2α stimulates the Raf/MEK1/mitogen-activated protein kinase signaling cascade in bovine luteal cells. Endocrinology. 1998;139(9):3876–3885. doi: 10.1210/endo.139.9.6197. [DOI] [PubMed] [Google Scholar]
- 25.Matsuura K, Shiraishi H, Hara A, Sato K, Deyashiki Y, Ninomiya M, Sakai S. Identification of a principal mRNA species for human 3α-hydroxysteroid dehydrogenase isoform (AKR1C3) that exhibits high prostaglandin D2 11-ketoreductase activity. J Biochem. 1998;124(5):940–946. doi: 10.1093/oxfordjournals.jbchem.a022211. [DOI] [PubMed] [Google Scholar]
- 26.Suzuki-Yamamoto T, Nishizawa M, Fukui M, Okuda-Ashitaka E, Nakajima T, Ito S, Watanabe K. cDNA cloning, expression and characterization of human prostaglandin F synthase. FEBS Lett. 1999;462(3):335–340. doi: 10.1016/s0014-5793(99)01551-3. [DOI] [PubMed] [Google Scholar]
- 27.Reginato MJ, Krakow SL, Bailey ST, Lazar MA. Prostaglandins promote and block adipogenesis through opposing effects on peroxisome proliferator-activated receptor gamma. J Biol Chem. 1998;273(4):1855–1858. doi: 10.1074/jbc.273.4.1855. [DOI] [PubMed] [Google Scholar]
- 28.Sales KJ, Milne SA, Williams AR, Anderson RA, Jabbour HN. Expression, localization, and signaling of prostaglandin F2α receptor in human endometrial adenocarcinoma: regulation of proliferation by activation of the epidermal growth factor receptor and mitogen-activated protein kinase signaling pathways. J Clin Endocrinol Metab. 2004;89(2):986–993. doi: 10.1210/jc.2003-031434. [DOI] [PubMed] [Google Scholar]
- 29.Desmond JC, Mountford JC, Drayson MT, Walker EA, Hewison M, Ride JP, Luong QT, Hayden RE, Vanin EF, Bunce CM. The aldo-keto reductase AKR1C3 is a novel suppressor of cell differentiation that provides a plausible target for the non-cyclooxygenase-dependent antineoplastic actions of nonsteroidal anti-inflammatory drugs. Cancer Res. 2003;63(2):505–512. [PubMed] [Google Scholar]
- 30.Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM. 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPARγ. Cell. 1995;83(5):803–812. doi: 10.1016/0092-8674(95)90193-0. [DOI] [PubMed] [Google Scholar]
- 31.Kaikkonen S, Paakinaho V, Sutinen P, Levonen AL, Palvimo JJ. Prostaglandin 15d-PGJ2 inhibits androgen receptor signaling in prostate cancer cells. Mol Endocrinol. 2012;27(2):212–223. doi: 10.1210/me.2012-1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moghrabi N, Andersson S. 17β-hydroxysteroid dehydrogenases: physiological roles in health and disease. Trends Endocrinol Metab. 1998;9(7):265–270. doi: 10.1016/s1043-2760(98)00066-6. [DOI] [PubMed] [Google Scholar]
- 33.Geissler WM, Davis DL, Wu L, Bradshaw KD, Patel S, Mendonca BB, Elliston KO, Wilson JD, Russell DW, Andersson S. Male pseudohermaphroditism caused by mutations of testicular 17 β-hydroxysteroid dehydrogenase 3. Nat Genet. 1994;7(1):34–39. doi: 10.1038/ng0594-34. [DOI] [PubMed] [Google Scholar]
- 34.Bauman DR, Steckelbroeck S, Williams MV, Peehl DM, Penning TM. Identification of the major oxidative 3α-hydroxysteroid dehydrogenase in human prostate that converts 5α-androstane-3α,17β-diol to 5α-dihydrotestosterone: a potential therapeutic target for androgen-dependent disease. Mol Endocrinol. 2006;20(2):444–458. doi: 10.1210/me.2005-0287. [DOI] [PubMed] [Google Scholar]
- 35.Mohler JL, Titus MA, Bai S, Kennerley BJ, Lih FB, Tomer KB, Wilson EM. Activation of the androgen receptor by intratumoral bioconversion of androstanediol to dihydrotestosterone in prostate cancer. Cancer Res. 2011;71(4):1486–1496. doi: 10.1158/0008-5472.CAN-10-1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mohler JL, Titus MA, Wilson EM. Potential prostate cancer drug target: bioactivation of androstanediol by conversion to dihydrotestosterone. Clin Cancer Res. 2011;17(18):5844–5849. doi: 10.1158/1078-0432.CCR-11-0644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Penning TM, Burczynski ME, Jez JM, Lin HK, Ma H, Moore M, Ratnam K, Palackal N. Structure-function aspects and inhibitor design of type 5 17β-hydroxysteroid dehydrogenase (AKR1C3) Mol Cell Endocrinol. 2001;171(1–2):137–149. doi: 10.1016/s0303-7207(00)00426-3. [DOI] [PubMed] [Google Scholar]
- 38.Stanbrough M, Bubley GJ, Ross K, Golub TR, Rubin MA, Penning TM, Febbo PG, Balk SP. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006;66(5):2815–2825. doi: 10.1158/0008-5472.CAN-05-4000. [DOI] [PubMed] [Google Scholar]
- 39.Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, True LD, Nelson PS. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res. 2008;68(11):4447–4454. doi: 10.1158/0008-5472.CAN-08-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hofland J, van Weerden WM, Dits NF, Steenbergen J, van Leenders GJ, Jenster G, Schroder FH, de Jong FH. Evidence of limited contributions for intratumoral steroidogenesis in prostate cancer. Cancer Res. 2010;70(3):1256–1264. doi: 10.1158/0008-5472.CAN-09-2092. [DOI] [PubMed] [Google Scholar]
- 41.Pfeiffer MJ, Smit FP, Sedelaar JP, Schalken JA. Steroidogenic enzymes and stem cell markers are upregulated during androgen deprivation in prostate cancer. Mol Med. 2011;17(7–8):657–664. doi: 10.2119/molmed.2010.00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cai C, Chen S, Ng P, Bubley GJ, Nelson PS, Mostaghel EA, Marck B, Matsumoto AM, Simon NI, Wang H, Balk SP. Intratumoral de novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res. 2011;71(20):6503–6513. doi: 10.1158/0008-5472.CAN-11-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Downs TM, Burton DW, Araiza FL, Hastings RH, Deftos LJ. PTHrP stimulates prostate cancer cell growth and upregulates aldo-keto reductase 1C3. Cancer Lett. 2011;306(1):52–59. doi: 10.1016/j.canlet.2011.02.027. [DOI] [PubMed] [Google Scholar]
- 44.Dozmorov MG, Azzarello JT, Wren JD, Fung KM, Yang Q, Davis JS, Hurst RE, Culkin DJ, Penning TM, Lin HK. Elevated AKR1C3 expression promotes prostate cancer cell survival and prostate cell-mediated endothelial cell tube formation: implications for prostate cancer progression. BMC Cancer. 2010;10:672. doi: 10.1186/1471-2407-10-672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bydal P, Luu-The V, Labrie F, Poirier D. Steroidal lactones as inhibitors of 17β-hydroxysteroid dehydrogenase type 5: chemical synthesis, enzyme inhibitory activity, and assessment of estrogenic and androgenic activities. Eur J Med Chem. 2009;44(2):632–644. doi: 10.1016/j.ejmech.2008.03.020. [DOI] [PubMed] [Google Scholar]
- 46.Higaki Y, Usami N, Shintani S, Ishikura S, El-Kabbani O, Hara A. Selective and potent inhibitors of human 20α-hydroxysteroid dehydrogenase (AKR1C1) that metabolizes neurosteroids derived from progesterone. Chem Biol Interact. 2003;143–144:503–513. doi: 10.1016/s0009-2797(02)00206-5. [DOI] [PubMed] [Google Scholar]
- 47.Davies NJ, Hayden RE, Simpson PJ, Birtwistle J, Mayer K, Ride JP, Bunce CM. AKR1C isoforms represent a novel cellular target for jasmonates alongside their mitochondrial-mediated effects. Cancer Res. 2009;69(11):4769–4775. doi: 10.1158/0008-5472.CAN-08-4533. [DOI] [PubMed] [Google Scholar]
- 48.Brozic P, Golob B, Gomboc N, Rizner TL, Gobec S. Cinnamic acids as new inhibitors of 17β-hydroxysteroid dehydrogenase type 5 (AKR1C3) Mol Cell Endocrinol. 2006;248(1–2):233–235. doi: 10.1016/j.mce.2005.10.020. [DOI] [PubMed] [Google Scholar]
- 49.Skarydova L, Zivna L, Xiong G, Maser E, Wsol V. AKR1C3 as a potential target for the inhibitory effect of dietary flavonoids. Chem Biol Interact. 2009;178(1–3):138–144. doi: 10.1016/j.cbi.2008.10.015. [DOI] [PubMed] [Google Scholar]
- 50.Penning TM, Talalay P. Inhibition of a major NAD(P)+-linked oxidoreductase from rat liver cytosol by steroidal and nonsteroidal anti-inflammatory agents and by prostaglandins. Proc Natl Acad Sci U S A. 1983;80(14):4504–4508. doi: 10.1073/pnas.80.14.4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bauman DR, Rudnick SI, Szewczuk LM, Jin Y, Gopishetty S, Penning TM. Development of nonsteroidal anti-inflammatory drug analogs and steroid carboxylates selective for human aldo-keto reductase isoforms: potential antineoplastic agents that work independently of cyclooxygenase isozymes. Mol Pharmacol. 2005;67(1):60–68. doi: 10.1124/mol.104.006569. [DOI] [PubMed] [Google Scholar]
- 52.Steckelbroeck S, Oyesanmi B, Jin Y, Lee SH, Kloosterboer HJ, Penning TM. Tibolone metabolism in human liver is catalyzed by 3α/3β-hydroxysteroid dehydrogenase activities of the four isoforms of the aldo-keto reductase (AKR)1C subfamily. J Pharmacol Exp Ther. 2006;316(3):1300–1309. doi: 10.1124/jpet.105.091587. [DOI] [PubMed] [Google Scholar]
- 53.Byrns MC, Steckelbroeck S, Penning TM. An indomethacin analogue, N-(4-chlorobenzoyl)-melatonin, is a selective inhibitor of aldo-keto reductase 1C3 (type 2 3α-HSD, type 5 17β-HSD, and prostaglandin F synthase), a potential target for the treatment of hormone dependent and hormone independent malignancies. Biochem Pharmacol. 2008;75(2):484–493. doi: 10.1016/j.bcp.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Byrns MC, Jin Y, Penning TM. Inhibitors of type 5 17β-hydroxysteroid dehydrogenase (AKR1C3): overview and structural insights. J Steroid Biochem Mol Bio. 2011;125:95–104. doi: 10.1016/j.jsbmb.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Burczynski ME, Harvey RG, Penning TM. Expression and characterization of four recombinant human dihydrodiol dehydrogenase isoforms: oxidation of trans-7, 8-dihydroxy-7,8-dihydrobenzo[a]pyrene to the activated o-quinone metabolite benzo[a]pyrene-7,8-dione. Biochemistry. 1998;37(19):6781–6790. doi: 10.1021/bi972725u. [DOI] [PubMed] [Google Scholar]
- 56.Steckelbroeck S, Jin Y, Gopishetty S, Oyesanmi B, Penning TM. Human cytosolic 3α-hydroxysteroid dehydrogenases of the aldo-keto reductase superfamily display significant 3β-hydroxysteroid dehydrogenase activity: implications for steroid hormone metabolism and action. J Biol Chem. 2004;279(11):10784–10795. doi: 10.1074/jbc.M313308200. [DOI] [PubMed] [Google Scholar]
- 57.Guerini V, Sau D, Scaccianoce E, Rusmini P, Ciana P, Maggi A, Martini PG, Katzenellenbogen BS, Martini L, Motta M, Poletti A. The androgen derivative 5α-androstane-3β,17β-diol inhibits prostate cancer cell migration through activation of the estrogen receptor β subtype. Cancer Res. 2005;65(12):5445–5453. doi: 10.1158/0008-5472.CAN-04-1941. [DOI] [PubMed] [Google Scholar]
- 58.Adeniji AO, Twenter BM, Byrns MC, Jin Y, Winkler JD, Penning TM. Discovery of substituted 3-(phenylamino)benzoic acids as potent and selective inhibitors of type 5 17β-hydroxysteroid dehydrogenase (AKR1C3) Bioorg Med Chem Lett. 2011;21(5):1464–1468. doi: 10.1016/j.bmcl.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Adeniji AO, Twenter BM, Byrns MC, Jin Y, Chen M, Winkler JD, Penning TM. Development of potent and selective inhibitors of aldo-keto reductase 1C3 (type 5 17β-hydroxysteroid dehydrogenase) based on N-phenyl-aminobenzoates and their structure-activity relationships. J Med Chem. 2012;55(5):2311–2323. doi: 10.1021/jm201547v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sinreih M, Sosic I, Beranic N, Turk S, Adeniji AO, Penning TM, Rizner TL, Gobec S. N-Benzoyl anthranilic acid derivatives as selective inhibitors of aldo-keto reductase AKR1C3. Bioorg Med Chem Lett. 2012;22(18):5948–5951. doi: 10.1016/j.bmcl.2012.07.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jamieson SM, Brooke DG, Heinrich D, Atwell GJ, Silva S, Hamilton EJ, Turnbull AP, Rigoreau LJ, Trivier E, Soudy C, Samlal SS, Owen PJ, Schroeder E, Raynham T, Flanagan JU, Denny WA. 3-(3,4-Dihydroisoquinolin-2(1H)-ylsulfonyl)benzoic Acids: Highly potent and selective inhibitors of the type 5 17β-hydroxysteroid dehydrogenase AKR1C3. J Med Chem. 2012 doi: 10.1021/jm3007867. [DOI] [PubMed] [Google Scholar]
- 62.Yee DJ, Balsanek V, Sames D. New tools for molecular imaging of redox metabolism: development of a fluorogenic probe for 3α-hydroxysteroid dehydrogenases. J Am Chem Soc. 2004;126(8):2282–2283. doi: 10.1021/ja039799f. [DOI] [PubMed] [Google Scholar]
- 63.Guise CP, Abbattista MR, Singleton RS, Holford SD, Connolly J, Dachs GU, Fox SB, Pollock R, Harvey J, Guilford P, Donate F, Wilson WR, Patterson AV. The bioreductive prodrug PR-104A is activated under aerobic conditions by human aldo-keto reductase 1C3. Cancer Res. 2010;70(4):1573–1584. doi: 10.1158/0008-5472.CAN-09-3237. [DOI] [PubMed] [Google Scholar]
- 64.Chen M, Adeniji AO, Twenter BM, Winkler JD, Christianson DW, Penning TM. Crystal structures of AKR1C3 containing an N-(aryl)amino-benzoate inhibitor and a bifunctional AKR1C3 inhibitor and androgen receptor antagonist. Therapeutic leads for castrate resistant prostate cancer. Bioorg Med Chem Lett. 2012;22(10):3492–3497. doi: 10.1016/j.bmcl.2012.03.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brozic P, Turk S, Adeniji AO, Konc J, Janezic D, Penning TM, Lanisnik Rizner T, Gobec S. Selective inhibitors of aldo-keto reductases AKR1C1 and AKR1C3 discovered by virtual screening of a fragment library. J Med Chem. 2012;55(17):7417–7424. doi: 10.1021/jm300841n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schuster D, Kowalik D, Kirchmair J, Laggner C, Markt P, Aebischer-Gumy C, Strohle F, Moller G, Wolber G, Wilckens T, Langer T, Odermatt A, Adamski J. Identification of chemically diverse, novel inhibitors of 17β-hydroxysteroid dehydrogenase type 3 and 5 by pharmacophore-based virtual screening. J Steroid Biochem Mol Bio. 2011;125(1–2):148–161. doi: 10.1016/j.jsbmb.2011.01.016. [DOI] [PubMed] [Google Scholar]
- 67.Endo S, Matsunaga T, Kanamori A, Otsuji Y, Nagai H, Sundaram K, El-Kabbani O, Toyooka N, Ohta S, Hara A. Selective inhibition of human type-5 17β-hydroxysteroid dehydrogenase (AKR1C3) by baccharin, a component of Brazilian propolis. J Nat Prod. 2012;75(4):716–721. doi: 10.1021/np201002x. [DOI] [PubMed] [Google Scholar]
- 68.Liedtke AJ, Adeniji AO, Chen M, Byrns MC, Jin Y, Christianson DW, Marnett LJ, Penning TM. Development of potent and selective indomethacin analogues for the inhibition of AKR1C3 (type 5 17β-hydroxysteroid dehydrogenase/prostaglandin F synthase) in castrate-resistant prostate cancer. J Med Chem. 2013 doi: 10.1021/jm3017656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kandaswami C, Lee LT, Lee PP, Hwang JJ, Ke FC, Huang YT, Lee MT. The antitumor activities of flavonoids. In Vivo. 2005;19(5):895–909. [PubMed] [Google Scholar]
- 70.Krazeisen A, Breitling R, Moller G, Adamski J. Human 17β-hydroxysteroid dehydrogenase type 5 is inhibited by dietary flavonoids. Adv Exp Med Biol. 2002;505:151–161. doi: 10.1007/978-1-4757-5235-9_14. [DOI] [PubMed] [Google Scholar]
- 71.Neuhouser ML. Dietary flavonoids and cancer risk: evidence from human population studies. Nutr Cancer. 2004;50(1):1–7. doi: 10.1207/s15327914nc5001_1. [DOI] [PubMed] [Google Scholar]
- 72.Akao Y, Maruyama H, Matsumoto K, Ohguchi K, Nishizawa K, Sakamoto T, Araki Y, Mishima S, Nozawa Y. Cell growth inhibitory effect of cinnamic acid derivatives from propolis on human tumor cell lines. Biol Pharm Bull. 2003;26(7):1057–1059. doi: 10.1248/bpb.26.1057. [DOI] [PubMed] [Google Scholar]
- 73.Mishima S, Ono Y, Araki Y, Akao Y, Nozawa Y. Two related cinnamic acid derivatives from Brazilian honey bee propolis, baccharin and drupanin, induce growth inhibition in allografted sarcoma S-180 in mice. Biol Pharm Bull. 2005;28(6):1025–1030. doi: 10.1248/bpb.28.1025. [DOI] [PubMed] [Google Scholar]
- 74.Efstathiou E, Titus MA, Tsavachidou D, Hoang A, Karlou M, Wen S, Troncoso P, Ashe R, Berman CJ, Mohler J, Logothetis C. MDV3100 effects on androgen receptor (AR) signaling and bone marrow testosterone concentration modulation: A preliminary report. J Clin Oncol- ASCO Annual Meeting Proceedings (Post-Meeting Edition) 2011:abstr 4501. [Google Scholar]
- 75.Feau C, Arnold LA, Kosinski A, Zhu F, Connelly M, Guy RK. Novel flufenamic acid analogues as inhibitors of androgen receptor mediated transcription. ACS Chem Biol. 2009;4(10):834–843. doi: 10.1021/cb900143a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jackson VJ, Yosaatmadja Y, Flanagan JU, Squire CJ. Structure of AKR1C3 with 3-phenoxybenzoic acid bound. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2012;68(Pt 4):409–413. doi: 10.1107/S1744309112009049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Flanagan JU, Yosaatmadja Y, Teague RM, Chai MZ, Turnbull AP, Squire CJ. Crystal structures of three classes of non-steroidal anti-inflammatory drugs in complex with aldo-keto reductase 1C3. PLoS One. 2012;7(8):e43965. doi: 10.1371/journal.pone.0043965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lovering AL, Ride JP, Bunce CM, Desmond JC, Cummings SM, White SA. Crystal structures of prostaglandin D(2) 11-ketoreductase (AKR1C3) in complex with the nonsteroidal anti-inflammatory drugs flufenamic acid and indomethacin. Cancer Res. 2004;64(5):1802–1810. doi: 10.1158/0008-5472.can-03-2847. [DOI] [PubMed] [Google Scholar]
- 79.Chen M, Adeniji AO, Twenter BM, Liedtke AJ, Winkler JD, Marnett LJ, Christianson DW, Penning TM. Crystal structures of human 17β-hydroxysteroid dehydrogenase type 5 (AKR1C3) in complex with N-phenylanthranilic acid and indomethacin-based selective inhibitors. Endocrine Reviews. 2012;33(03_MeetingAbstracts):SAT-536. [Google Scholar]
- 80.Komoto J, Yamada T, Watanabe K, Takusagawa F. Crystal structure of human prostaglandin F synthase (AKR1C3) Biochemistry. 2004;43(8):2188–2198. doi: 10.1021/bi036046x. [DOI] [PubMed] [Google Scholar]
- 81.Byrns MC, Penning TM. Type 5 17β-hydroxysteroid dehydrogenase/prostaglandin F synthase (AKR1C3): role in breast cancer and inhibition by non-steroidal anti-inflammatory drug analogs. Chem Biol Interact. 2009;178(1–3):221–227. doi: 10.1016/j.cbi.2008.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Estébanez-Perpiñá E, Arnold LA, Nguyen P, Rodrigues ED, Mar E, Bateman R, Pallai P, Shokat KM, Baxter JD, Guy RK, Webb P, Fletterick RJ. A surface on the androgen receptor that allosterically regulates coactivator binding. Proc Natl Acad Sci U S A. 2007;104(41):16074–16079. doi: 10.1073/pnas.0708036104. [DOI] [PMC free article] [PubMed] [Google Scholar]