

Abstract
The chemokine CXCL12 and its G protein-coupled receptor (GPCR) CXCR4 are high-priority clinical targets because of their involvement in metastatic cancers (also implicated in autoimmune disease and cardiovascular disease). Because chemokines interact with two distinct sites to bind and activate their receptors, both the GPCRs and chemokines are potential targets for small molecule inhibition. A number of chemokines have been validated as targets for drug development, but virtually all drug discovery efforts focus on the GPCRs. However, all CXCR4 receptor antagonists with the exception of MSX-122 have failed in clinical trials due to unmanageable toxicities, emphasizing the need for alternative strategies to interfere with CXCL12/CXCR4-guided metastatic homing. Although targeting the relatively featureless surface of CXCL12 was presumed to be challenging, focusing efforts at the sulfotyrosine (sY) binding pockets proved successful for procuring initial hits. Using a hybrid structure-based in silico/NMR screening strategy, we recently identified a ligand that occludes the receptor recognition site. From this initial hit, we designed a small fragment library containing only nine tetrazole derivatives using a fragment-based and bioisostere approach to target the sY binding sites of CXCL12. Compound binding modes and affinities were studied by 2D NMR spectroscopy, X-ray crystallography, molecular docking and cell-based functional assays. Our results demonstrate that the sY binding sites are conducive to the development of high affinity inhibitors with better ligand efficiency (LE) than typical protein-protein interaction inhibitors (LE ≤ 0.24). Our novel tetrazole-based fragment 18 was identified to bind the sY21 site with a Kd of 24 μM (LE = 0.30). Optimization of 18 yielded compound 25 which specifically inhibits CXCL12-induced migration with an improvement in potency over the initial hit 9. The fragment from this library that exhibited the highest affinity and ligand efficiency (11: Kd = 13 μM, LE = 0.33) may serve as a starting point for development of inhibitors targeting the sY12 site.
Keywords: Chemokines, CXCL12/CXCR4 inhibitors, protein-protein interaction, metastasis, fragment-based and structure-guided drug design
Introduction
Cancer is believed to develop from the accumulation of genetic alterations. In turn, aberrant signal transduction results in several hallmarks of cancer including uncontrolled cell proliferation, evasion of apoptosis, angiogenesis and metastasis. Metastasis, the spread and growth of tumor cells to distant organ sites, is the most devastating attribute and plays a major role in patient morbidity and mortality. Chemokines are small soluble proteins (70-130 residues) that activate G-protein coupled receptors (GPCRs) and are involved in many physiological processes including cell trafficking, angiogenesis and embryogenesis [1-4]. Under normal conditions, they play pivotal roles in immune surveillance and response, inflammation, stem cell homing and other important physiological processes [5]. Their functions as chemoattractants and their effects on immune cells also underlie their involvement in many diseases such as cancer, inflammation, autoimmune and cardiovascular diseases [6, 7]. Chemokines as primary mediators of metastasis were first identified by Muller and colleagues who implicated the chemokine receptor C-X-C motif chemokine receptor 4 (CXCR4) in tumor cell trafficking [8, 9]. Different types of cancers express different chemokines and chemokine receptors [9, 10]. However, the chemokine receptor CXCR4 is the only one that is expressed by the majority of cancer types. At least 23 different cancers have been shown to express elevated levels of CXCR4 [10], sensitizing these cancers to C-X-C motif chemokine ligand 12 (CXCL12, also known as stromal cell-derived factor-1, SDF-1) gradients in distant tissues [11]. CXCL12 is constitutively expressed in the bone marrow, lungs and liver, which are common tissues of metastatic growth.
CXCL12 Inhibitors and CXCR4 Receptor Antagonists
Reagents that Modulate CXCL12/CXCR4 Interactions
In general, the three major classes of agents that can modulate the CXCL12/CXCR4 interaction are antibodies to CXCL12 or CXCR4, CXCR4 receptor antagonists, and CXCL12 inhibitors. Development of anti-CXCL12 antibodies is challenging because the chemokine sequence is nearly invariant among mammals. Therapeutic development of antibodies against CXCR4 is also very limited due to conformational heterogeneity of CXCR4 and posttranslational modifications that reduce antibody specificity and function [12].
Although GPCRs have been one of the most important drug targets for the pharmaceutical industry, representing more than 30% of all US marketed therapeutics [13], no CXCR4 antagonist has been approved for therapy of metastatic cancer. Peptide-based CXCR4 antagonists lack efficacy with poor bioavailability. In spite of the apparent success of AMD3100 (1, Plerixafor, Chart 1) for stem cell mobilization [14], all of the CXCR4 antagonists [with the exception of a bicyclam mimetic MSX-122 (2)] in clinical trials have been withdrawn because of unmanageable toxicities as of August 2008 [15]. The cardiovascular toxicities from AMD3100 are presumably related to the metal-chelating properties of bicyclam [16]. Aside from the cardiotoxicity associated with long-term administration of AMD3100, little is known of the drug-like properties of chemokine receptor antagonists or the origins of their toxicity [17].
Chart 1.

Structures of Representative Small Molecule CXCR4 Antagonists and CXCL12 Inhibitors.
Chemokines are generally viewed as “undruggable” proteins based on their size and lack of a well-defined pocket for small molecules to bind. However, recent studies have suggested otherwise, with the discovery of high-affinity inhibitors against several small proteins such as interleukin-2 and FKBP-12 [18-20]. The report of a chalcone molecule 5 binding to CXCL12 and inhibiting CXCR4 activation has further demonstrated that chemokines are viable targets for small molecule drug discovery although no structural information about chalcone binding has been revealed [21, 22]. The phosphate prodrug 6, sulfate prodrug 7 and L-seryl prodrug 8 were recently reported to overcome the poor solubility of chalcone 5 [23]. More recently, our group used a structure-guided approach to identify a series of inhibitors that bound CXCL12 and inhibit the CXCR4-mediated calcium response [24].
Known CXCR4 Antagonists are not Approved for Treatment of Metastatic Cancers
CXCR4 receptors are expressed by various solid and hematologic tumors, such as breast cancer, lung cancer, prostate cancer, and leukemia [25, 26]. In spite of advances in surgery, chemotherapy and radiotherapy over the last decades, the death rate from lung cancer has remained unchanged mainly due to cancer metastasis. Lung cancer cells express CXCR4 while stromal cells within the tumor micro-environment constitutively secrete CXCL12. Activation of CXCR4 induces lung cancer cell migration and adhesion to stromal cells, which in turn provides growth- and drug- resistance signals to the tumor cells. CXCR4 antagonists were initially developed for treatment of HIV-1 infection. At the time of their discovery in the early 1990s, the mechanism of anti-HIV activity of the CXCR4 antagonists T140 and its analogs [27, 28], AMD3100 (1) [29, 30] and ALX-4C [31], was unknown. However, discovery of the co-receptor function soon demonstrated that the activity is due to the specific binding of the antagonists to the CXCR4 receptor [31-33].
Besides peptides [28, 34, 35] and antibodies [36-38], the most advanced small-molecule CXCR4 antagonists are represented by a bicyclam AMD3100 which was originally dropped from Phase II HIV clinical trials due to cardiotoxicity, but was later approved by FDA for treatment of non-Hodgkin's lymphoma and multiple myeloma [39]. AMD3100 was reported to inhibit CXCL12-induced chemotaxis by inhibiting the site 2 interaction with CXCR4 [40]. A series of bicyclam mimetics based on AMD3100 scaffold represented by MSX-122 (2) were reported towards development of an anti-metastatic agent [15, 41]. These series of bicyclam mimetics with non-chelating pyrimidyl heterocycles were designed to overcome safety issues of AMD3100 and improve pharmacokinetic properties for oral administration [42,43]. Benzimidazole-tetrahydoquinolineamine derivatives based on AMD070 (3) were a series of CXCR4 antagonists with potent and selective in vitro inhibition of X4 viral replication by blocking fusion and viral entry into the cell [44, 45]. Another series of CXCR4 antagonists are isothiourea derivatives, which block CXCR4/CXCL12 interactions in vitro and in vivo as well as the infection of target cells by X4-tropic HIV [46]. One of the isothiourea derivatives, IT1t (4), was co-crystallized with the CXCR4 receptor at 2.5 Å resolution [47]. Other miscellaneous small molecule CXCR4 antagonists can be found in a recent review [15]. However, this avenue has proven very challenging to move into the clinic [48, 49], emphasizing the need to explore alternative strategies that target the CXCL12 chemokine directly.
Fragment-Based and Structure-Guided Study of Small-Molecule CXCL12 Inhibitors
Sulfotyrosine Recognition is Critical for the CXCL12/CXCR4 Interaction
The chemokine CXCL12 uses a “two-step, two-site” process for binding and receptor activation [50]. First, the extracellular amino-terminal domain of the receptor, a flexible ∼30-residue domain binds the conserved, disulfide stabilized core structure of the chemokine. Next, the amino terminus of the chemokine docks into a pocket within the transmembrane region of the GPCR to form a fully activated receptor complex. While amino acids at the chemokine N-terminus function as a low-affinity receptor agonist, the initial interaction provides a majority of the binding energy and much of the receptor-chemokine selectivity [51-53]. Crucial to these initial encounters are one or more sulfotyrosines (sY) on the receptor peptide [12, 54, 55]. The sulfate groups on these altered tyrosine residues, a result of post-translational modification of the receptor [56], have been shown to significantly enhance the binding affinity in chemokine recognition [57, 58]. In the case of CXCL12 the receptor affinity is improved 20-fold upon tyrosine sulfation [21]. To date, tyrosine sulfation has been biochemically characterized in a subset of the chemokine receptors, including CXCR3 [59], CXCR4 [12], CCR2B [60], CCR3 [61], CCR5 [62] and CX3CR1 [55].
Targeting the Sulfotyrosine Binding Sites for Drug Discovery
In contrast to the high-throughput type discovery method employed for the chalcone compounds, we hypothesized in silico screening could produce a higher hit rate as demonstrated for other targets [63]. The sulfotyrosine binding pockets on CXCL12 represent potential sites for small-molecule inhibitors because of their importance in receptor recognition and their unique structural features [64]. They consist of both hydrophobic and polar/charged groups that can be utilized for ligand affinity and specificity, as revealed by the NMR structure of a CXCL12 dimer bound by two CXCR4 N-terminal peptides (Fig. 1a). In the chemokine CXCL12, there are three sY binding sites: sY21, sY12 and sY7. Although members of the chemokine family share varying degrees of sequence homology (some as little as 20%), all members retain the canonical chemokine fold. Using NMR to monitor the titration of sulfotyrosine into four representative chemokines, we observed a sulfotyrosine recognition site analogous to the cleft on CXCL12 that binds sY21 of the receptor CXCR4 [65]. With an estimated 1% of all tyrosines in cell-surface and secreted human proteins modified by O-sulfation [66], inhibitor development against sY binding sites can have far reaching implications in targeting disease-related protein-protein interfaces. The discovery and optimization of a “privileged” small-molecule scaffold would be invaluable for the development of ligands against other sulfotyrosine recognition sites.
Fig. (1).
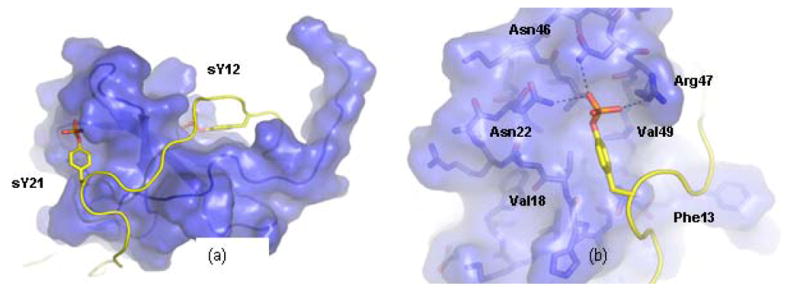
Small-Molecule CXCL12 Inhibitors Targeting the sY21 Binding Pocket
Although chalcone 5 prevents CXCL12 from binding to its CXCR4 or CXCR7 receptors [21], no structural information has ever been reported. Our recent report demonstrated that small molecule 9, identified from in silico docking studies, bound CXCL12 with a Kd = 64 μM and produced chemical shift perturbations localized to the sY21 binding site. A NMR structure of the CXCL12-inhibitor 9 complex has shown that this small molecule binds within the sY21 binding pocket and participates in both polar and nonpolar interactions that mimic CXCR4 D20 and sY21 contacts (manuscript in preparation; Fig. 2).
Fig. (2).

Design and Synthesis of a Fragment Library to Search for Better Starting Points of CXCL12 Inhibitors
A retrospective analysis of highly optimized protein inhibitors suggests maintaining a ligand efficiency (LE) ≥ 0.27-0.30 kcal/mol/non-hydrogen atom (LE: defined as the free energy of binding divided by the number of non-hydrogen atoms) [67]. However, the LE for protein-protein interaction (PPI) disruptors rarely exceeds 0.24 due to the limited number of chemical (such as buried salt bridges) and physical features (cavities) necessary for high affinity interactions [68]. The LE of compound 9 is 0.23 which is typical for PPI inhibitors. Therefore, our goal was to optimize hit 9 by advancing potency while simultaneously improving ligand efficiency. Unlike traditional hit-to-lead optimization by altering (usually adding) functionalities to improve the affinity, we decided to take a fragment-based approach to trim compound 9 in order to find a better starting molecule. The SAR analysis [24] and complex structure of compound 9-CXCL12 suggests the carboxylic acid group is essential for high-affinity binding. We envisaged its bioisostere [69, 70], a 5-substituted-1H-tetrazole group, could not only exhibit efficient binding to the sY21 pocket but would also possess improved drug-like properties as the tetrazole moiety is comparable in both size and acidity to the carboxylic acid group while being more metabolically stable [71]. In addition, the carboxylic acid was replaced with its bioisostere, 5-substituted-1H-tetrazole to maintain IP privileges. To this end, a fragment library containing nine compounds was designed (Chart 2). The design aimed to verify if the acylthiourea linker/spacer is essential. The linker/spacer was shortened from four atoms (acylthiourea) to two (amide) or three (urea) atoms. This operation keeps some molecular features such as hydrogen bond donor/acceptor unchanged while smaller fragments with higher binding potential (larger LEs) are to be discovered.
Chart 2.

The tetrazole group was used as an anchor point to bind the sY21 pocket of CXCL12 from which fragments were then synthesized with phenyl substituted groups in ortho-, meta-and para-positions to make final compounds consisting of 15 to 21 non-hydrogen atoms (MW from 203 to 280). Fragments with meta- or para-substituents are relatively linear molecules while their ortho- counterparts were expected to adopt conformations that may be too voluminous for the sY21 pocket. This design was intentional as the steric tolerance near the narrow sY21 pocket may be revealed with the set of fragments.
Fragment libraries containing only nine members were designed and synthesized as shown in Scheme 1. Ortho -(1H-tetrazol-5-yl)aniline (22), meta-(1H-tetrazol-5-yl)aniline (23) and para-(1H-tetrazol-5-yl)aniline (24) were synthesized from the corresponding commercially available aminoben-zonitriles (19-21) via a cycloaddition of the corresponding nitriles with azide in the presence of triethylamine at 110 °C using nitrobenzene as solvent [72]. The isolated yields after column chromatography (silica/DCM-MeOH, up to 15% MeOH) on a gram scale are 77% for 22, 87% for 23 and 88% for 24, respectively. N-(2-(1H-Tetrazol-5-yl)phenyl) acetamide (11), N-(3-(1H-tetrazol-5-yl)phenyl)acetamide (12) and N-(4-(1H-tetrazol-5-yl)phenyl)acetamide (14) were prepared by acetylation of the corresponding tetrazole-anilines (22-24) with 2 equivalent (0.38 mmol) of acetic anhydride in the presence of triethylamine in DMSO at room temperature for 3 hr. Products were obtained in high yields (80% for 10, 85% for 12 and 80% for 14, respectively) after column chromatography (silica/DCM-MeOH, up to 10% MeOH). N-(2-(1H-Tetrazol-5-yl)phenyl)benzamide (11), N- (3-(1H-tetrazol-5-yl)phenyl)benzamide (13) and N-(4-(1H-tetrazol-5-yl)phenyl)benzamide (15) were synthesized by treating the corresponding tetrazole-anilines (22-24) with 2 equivalent of benzoyl chloride (0.38 mmol) in the presence of triethylamine in DMF at room temperature for 3 hr. Products were obtained as solid after column chromatography (silica/DCM-MeOH, up to 10% MeOH). The isolated yields are 75% for 11, 82% for 13 and 70% for 15, respectively. The tetrazole-phenylureas (16-18) were synthesized after a mixture of 2 equivalent of phenylisocyanate (0.38 mmol) and the corresponding tetrazole-aniline (22-24) was stirred at 50 °C overnight in DMSO and after the crude product was washed with DMC three times. The yields for 1-(2-(1H-Tetrazol-5-yl)phenyl)-3-phenylurea (16), 1-(3-(1H-tetrazol-5-yl)phenyl)-3-phenylurea (17) and 1-(4-(1H-Tetrazol-5-yl)phenyl)-3-phenylurea (18) are 75%, 85% and 75%, respectively. Similarly, compound 25 was synthesized by stirring a mixture of 24 (0.2 mmol) and 1-isocyanatonaphthalene (0.4 mmol) at 50 °C overnight in DMSO followed by column chromatography (silica/DCM-MeOH, up to 10% MeOH). The purity of the final compounds ranged from 96.3% to 99.9% (Table 1). The specifications of HPLC analysis are as follows: flow rate, 1 mL/min; column, Intertsil, 2.5 μm, 4.6 × 150 mm; wavelength, 215, 254 and 280 nm; mobile phase, A: H2O with 0.1% HCO2H, B: MeOH, gradient of 30-95%B in 25 min. All compounds were fully characterized and confirmed by 1H NMR and HRMS (ESI positive).
Scheme 1.

Synthesis of fragment library.
Table 1. Structure and Activity of Fragments.
| ID | Structure | MW | HPLC (%) | Kd (μM) | LE |
|---|---|---|---|---|---|
| 9 |

|
350 | NAa | 64 ± 15 | 0.23 |
| 10 |

|
203 | 98.9 | NDb | ND |
| 11 |

|
265 | 97.7 | 13 ± 7 | 0.33 |
| 12 |

|
203 | 99.9 | ND | ND |
| 13 |

|
265 | 99.7 | 50 ± 16 | 0.29 |
| 14 |

|
203 | 98.7 | ND | ND |
| 15 |

|
265 | 98.7 | 64 ± 23 | 0.29 |
| 16 |

|
280 | 97.5 | 327 ± 92 | 0.23 |
| 17 |

|
280 | 96.3 | 126 ± 23 | 0.25 |
| 18 |

|
280 | 99.3 | 24 ± 28 | 0.30 |
Not available
Not defined.
Study CXCL12-Tetrazole Interactions Using 2D NMR Spectroscopy and Molecular Docking
2D NMR was employed to study the fragment-induced CXCL12 chemical shift perturbations and determine the binding affinity of the synthesized fragments. [U-15N]-CXCL12 expression and purification was carried out as previously described [73]. Compounds 10-18 were titrated at 0, 25, 50, 250, 800, and 1600 μM into samples containing 50 μM [U-15N]-CXCL12 in 25 mM d-MES buffer (pH 6.8), 0.02% (v/v) NaN3 and 10% (v/v) D2O; compound 18 was titrated at 0, 25, 50, 200, 800, and 1600 μM. 2D 1H-15N heteronuclear single-quantum coherence (HSQC) spectroscopy experiments were performed on a Bruker 600 MHz spectrometer equipped with a TXI triple-resonance cryoprobe at 25 °C. Data was converted and processed using NMRPipe [74]. Previously published assignments for CXCL12 [73] were transferred by visual inspection and chemical shift values were tracked using CARA [75]. Combined 1H/15NH chemical shift perturbations were calculated as ((5ΔδH)2+(ΔδNH)2)0.5, where δH and δNH are the amide proton and nitrogen chemical shifts, respectively. Equilibrium dissociation constants (Kd) were determined by non-linear fitting to a quadratic equation that accounts for non-specific binding [24].
All molecules produced a subset of chemical shift changes distinct from the DMSO control titration – indicative of a specific binding interaction. Representative 1H–15N HSQC spectra of A (11), B (18), C (12) and D (17) are shown in (Fig. 3).
Fig. (3).

CXCL12 chemical shift perturbations induced by nine fragments (10-18) are shown in (Fig. 4).
Fig. (4).

Chemical shift mapping suggests the ortho-substituents (10, 11 and 16) cluster to the β1 and β2 strands near the CXCL12 dimer interface, consistent with the hypothesis that these molecules are too voluminous to occupy the narrow sY21 pocket. We hypothesize these reflect specific interactions as the perturbations are near the binding pocket for a different CXCR4 sulfotyrosine, sY12. Although further studies will be needed to authenticate this interaction, we used molecular docking to predict potential binding poses.
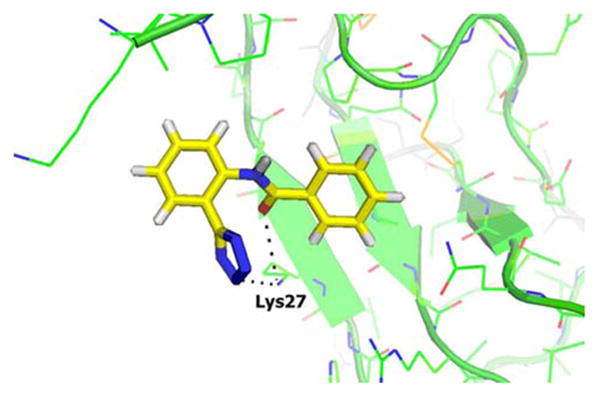
Docking was performed using DOCK3.4.54. Compounds 10, 11 and 16 were docked against 20 NMR conformations and one X-ray structure in order to account for protein flexibility and capture the best conformation. Sulfotyrosine peptide ligands were used in the NMR structures (PDB ID 2K05) to create the docking environment for both the sY12 and sY21 sites, while heparin sulfate was used to generate the docking environment for the sY12 site in the X-ray structure (PDB ID 2NWG). The predicted binding poses for each compound were visually inspected and the pose that agreed the best with the NMR perturbations was chosen. Compounds 10, 11 and 16 appear to bind to the sY12 site exploiting slightly different conformational space. As shown in (Fig. 5), docking predicts that the tetrazole in compound 11 may form hydrogen bonds with Lys27, while the amide may hydrogen bond to Lys27. Compound 10 may hydrogen bond with Arg41 and His25, while the amide may hydrogen bond with Lys27 (docking poses not shown). For compound 16, the tetrazole may form a hydrogen bond with His25, the urea nitrogen with Lys27 and the urea oxygen with Arg41 (docking poses not shown).
Fig. (5).
In contrast, the para- and meta-tetrazole fragments produce chemical shift changes near the predicted site suggesting relatively linear molecules are most compatible with the sY21 pocket. As shown in (Fig. 6), compound 18 appears to bind to the sY21 site. Docking predicts that the tetrazole in compound 18 may form hydrogen bonds with Ala19 and Asn22, while the urea nitrogen may form a hydrogen bond with Glu15 and the urea oxygen with Arg47.
Fig. (6).

It is interesting to note that several compounds induced shift perturbations in CXCL12 N-terminal residues 4-9 (Fig. 4). Unfortunately, further studies are required to gain structural insight as the N-terminus is highly flexible and its length permits contact with a ligand bound at either the dimer interface or the sY21 cleft. Non-linear fitting of chemical shift perturbations yielded affinities ranging from 13 to 327 μM (Table 1). Compounds 11 and 18 possess the highest affinities of 13 and 24 μM and exhibit corresponding LE improvements of 0.33 and 0.30, respectively. Fragments 10, 12 and 14 induced small chemical shift perturbations, resulting in large fitting errors and an inability to generate meaningful affinity values. We hypothesize the small perturbations result from the size of the acetyl group on the phenyltetrazole moiety rather than the large hydrophobic groups present in the other fragments.
As shown in (Table 1), most compounds possess improved LEs and demonstrate an enhanced binding potential compared to compound 9. The compounds with an amide linker (11, 13 and 15) uniformly demonstrated higher binding potentials than their urea counterparts (16 – 18), implying that they may be other appropriate starting points for the next round of optimization. The para-substituted molecules (14, 15 and 18) are the only set that exhibited a positive correlation between molecular weight and affinity suggesting this to be the optimal tetrazole orientation. In contrast, the affinities of ortho- and meta-substituted tetrazoles decreased 25- and 2.5-fold, respectively, from 265 to 280 Da. Overall, these results support the tetrazole moiety as an effective anchor point for targeting sulfotyrosine binding pockets and developing more efficient lead molecules.
Optimization of Tetrazole-Based CXCL12 Inhibitor 18
With compound 18 as a novel hit binding to the sY21 site of CXCL12 in hand, a subsequent library containing a dozen derivatives was designed and synthesized. In this library, both the para-substitution and urea linker were retained while different substituted aromatic functionalities replaced the benzene ring in 18. Molecular docking predicts that compound 25 binds the sY21 site similarly to that of 18 (Fig. 7). Similar to 18, compound 25 is found to not only bind in the sY21 site determined by 2D NMR (Fig. 8A – 8C) but also inhibit CXCL12-induced chemotaxis (Fig. 8D).
Fig. (7).

The docking pose of compound 25.
Fig. (8).

Tetrazole fragments bind CXCL12.
The activity of the tetrazole fragments was tested using an in vitro chemotaxis assay. THP-1 monocytes, which endogenously express the CXCR4 receptor, were incubated with CXCL12 in the absence or presence of each fragment. At 250 μM, compound 9 was unable to completely inhibit 30 nM of CXCL12-induced chemotaxis and yielded an IC50 of ∼800 μM. Although not directly comparable, compound 25 significantly diminished cell migration and inhibited chemotaxis to 10 nM CXCL12 with an IC50 = 111 ± 24 μM. Compounds 13 - 16 also significantly inhibited migration toward 10 nM CXCL12 at 250 μM. No compounds affected cell viability. To determine whether compound 25 specifically inhibits CXCL12, the chemotaxis assay was repeated with CCL2 (also known as monocyte chemoattractant protein-1, MCP-1), a distinct chemokine that activates the CCR2 receptor (data not shown). Our results indicate that compound 25 specifically inhibits CXCL12-induced migration with an improved potency compared to compound 9.
Concluding Remarks and Future Perspectives
The CXCL12/CXCR4 interaction is a high-priority clinical target because of its involvement in a broad spectrum of pathologies, including: autoimmune disease, cardiovascular disease and cancer. Current drug discovery efforts focus on identifying molecules that bind the transmembrane region of the chemokine receptor. However, antagonizing the large protein-protein interface has proven difficult for traditional high-throughput approaches and emphasizes the need for alternative strategies. We recently demonstrated the effectiveness of rationally designing small molecules that target the chemokine ligand [24]. From our published SAR analysis of initial hit 9 [24] and the unpublished complex structure with CXCL12 (manuscript in preparation), we designed a fragment library with improved affinities, efficiencies, and potencies. The best overall compound 25 retains a specific interaction with the sY21 site and exhibits a substantial improvement in potency (Fig. 8). In addition, the identification and development of compounds that bind the sY12 site provided a second target for future CXCL12-directed inhibitors and, therefore, fragment 11 serves as another starting point for ligand expansion or fragment-linkage in the development of high-affinity molecules.
Although our work focused on inhibition of CXCR4 activation, CXCL12 also interacts with C-X-C motif chemokine receptor 7 (CXCR7). CXCR7 is a non-canonical receptor that promotes arrestin recruitment and downstream signaling without activation of heterotrimeric G proteins. Following its re-classification as a chemokine receptor aberrant CXCR7 expression has been implicated in breast, lung, and B cell lymphoma [76-78]. Nonetheless characterization both in vitro and in vivo remains substantially hindered by the inability to discriminate between CXCR4- and CXCR7- mediated signaling. Indeed, two commonly used CXCR4 inhibitors (compound 1 and peptidomimetic TC14012) also activate CXCR7 [79, 80]. Several specific small molecule CXCR7 inhibitors have been identified through high throughput screening [76], but application of structure-based approaches could enhance the identification of novel antagonists [63, 81]. An obvious step in developing CXCL12 antagonists is assessing their potential targeting the CXCL12/CXCR7 interaction. Chalcone molecule 5 competes with CXCR7 binding to CXCL12 and suggests a similar inhibition may be observed using compounds targeting the sulfotyrosine binding sites [21]. Perhaps more interesting, a structural understanding of the CXCL12/CXCR7 interface may enable the development of specific inhibitors as potential therapeutics or molecular probes to dissect CXCL12 effects on respective receptors.
The success of our previous structure-based discovery of a CXCL12 antagonist [24] and its rapid optimization, reported herein, suggest the sulfotyrosine binding moiety may serve as a ‘natural small molecule’ site to guide drug discovery and development. The conservation of both sulfotyrosine binding sites and receptor tyrosine sulfation throughout the chemokine family highlights the potential for expansion of chemokine-directed discovery [65, 82]. Rational drug discovery campaigns targeting the chemokine ligand may prove more effective than traditional strategies focused at the receptor. Whereas further studies are required to gain structural information of interactions at the sY12 site of CXCL12, both NMR and mutagenesis have characterized the interface analogous to the CXCL12 sY21 recognition site for numerous chemokines [83-85]. Targeting the ligand may also enable modulation of homeostatic and pathologic processes. Chemokine signaling is generally considered to be polygamous with receptors recognizing multiple ligands. For example, although CXCL12/CXCR4 was identified as a monogamous pair, CXCR4 is now known to additionally interact with extracellular ubiquitin and MIF [86, 87]. Recent evidence suggests these systems are not redundant, but rather display signs of ligand selectively and biased agonism in which chemokine ligands signal disparate physiologic outcomes dependent upon specific structural interactions with the associate receptor [88-90]. Targeting a specific ligand may empower regulation, rather than simply elimination, of receptor activity. Futures perspective to the discovery of antagonists for CXCL12/CXCR4 interactions, may include the use of computational methodologies such as Quantitative-Structure Activity Relationships (QSAR) [91-119], especially those focused on the simultaneous classification and prediction of compounds with different biological activities [120-137], including the discovery of inhibitors for proteins associated with one or more diseases [138-143]. Alternatively, complex network theory could be used to gain a better understanding about ligand-protein and protein-protein interactions [143-152].
Acknowledgments
This research was partially supported by Moffitt Cancer Center startup funds (R.L.), Zhejiang University (G.Z. and Y.Y.), Science Technology Department of Zhejiang Province (2012C33065) and Education Department of Zhejiang Province (Y201223193) for G.Z., University of South Florida (Y.C.), NIH grants AI058072 and GM097381 (B.F.V.) and Medical College of Wisconsin Cancer Center Postdoctoral Fellowship (J.J.Z).
Footnotes
Conflict of Interest: The author(s) confirm that this article content has no conflict of interest.
References
- 1.Zou YR, Kottmann AH, Kuroda M, Taniuchi I, Littman DR. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature. 1998;393:595–599. doi: 10.1038/31269. [DOI] [PubMed] [Google Scholar]
- 2.Wang JM, Deng X, Gong W, Su S. Chemokines and their role in tumor growth and metastasis. J Immunol Methods. 1998;220:1–17. doi: 10.1016/s0022-1759(98)00128-8. [DOI] [PubMed] [Google Scholar]
- 3.Strieter RM. Chemokines: not just leukocyte chemoattractants in the promotion of cancer. Nat Immunol. 2001;2:285–286. doi: 10.1038/86286. [DOI] [PubMed] [Google Scholar]
- 4.Rossi D, Zlotnik A. The biology of chemokines and their receptors. Annu Rev Immunol. 2000;18:217–242. doi: 10.1146/annurev.immunol.18.1.217. [DOI] [PubMed] [Google Scholar]
- 5.Baggiolini M. Chemokines in pathology and medicine. J Intern Med. 2001;250:91–104. doi: 10.1046/j.1365-2796.2001.00867.x. [DOI] [PubMed] [Google Scholar]
- 6.Gerard C, Rollins BJ. Chemokines and disease. Nat Immunol. 2001;2:108–115. doi: 10.1038/84209. [DOI] [PubMed] [Google Scholar]
- 7.Proudfoot AE. Chemokine receptors: multifaceted therapeutic targets. Nat Rev Immunol. 2002;2:106–115. doi: 10.1038/nri722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, Verastegui E, Zlotnik A. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–56. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 9.Murphy PM. Chemokines and the molecular basis of cancer metastasis. N Engl J Med. 2001;345:833–835. doi: 10.1056/NEJM200109133451113. [DOI] [PubMed] [Google Scholar]
- 10.Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4:540–550. doi: 10.1038/nrc1388. [DOI] [PubMed] [Google Scholar]
- 11.Dell'Agnola C, Biragyn A. Clinical utilization of chemokines to combat cancer: the double-edged sword. Expert Rev Vaccines. 2007;6:267–283. doi: 10.1586/14760584.6.2.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Farzan M, Babcock GJ, Vasilieva N, Wright PL, Kiprilov E, Mirzabekov T, Choe H. The role of post-translational modifications of the CXCR4 amino terminus in stromal-derived factor 1 alpha association and HIV-1 entry. J Biol Chem. 2002;277:29484–29489. doi: 10.1074/jbc.M203361200. [DOI] [PubMed] [Google Scholar]
- 13.Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov. 2002;1:727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 14.Liles WC, Broxmeyer HE, Rodger E, Wood B, Hubel K, Cooper S, Hangoc G, Bridger GJ, Henson GW, Calandra G, Dale DC. Mobilization of hematopoietic progenitor cells in healthy volunteers by AMD3100, a CXCR4 antagonist. Blood. 2003;102:2728–2730. doi: 10.1182/blood-2003-02-0663. [DOI] [PubMed] [Google Scholar]
- 15.Mosley CA, Wilson LJ, Wiseman JM, Skudlarek JW, Liotta DC. Recent patents regarding the discovery of small molecule CXCR 4 antagonists. Expert Opin Ther Patents. 2009;19:23–38. doi: 10.1517/13543770802553483. [DOI] [PubMed] [Google Scholar]
- 16.De Clercq E. The bicyclam AMD3100 story. Nat Rev Drug Discov. 2003;2:581–587. doi: 10.1038/nrd1134. [DOI] [PubMed] [Google Scholar]
- 17.Horuk R. Chemokine receptor antagonists: overcoming developmental hurdles. Nat Rev Drug Discov. 2009;8:23–33. doi: 10.1038/nrd2734. [DOI] [PubMed] [Google Scholar]
- 18.Arkin MR, Randal M, DeLano WL, Hyde J, Luong TN, Oslob JD, Raphael DR, Taylor L, Wang J, McDowell RS, Wells JA, Braisted AC. Binding of small molecules to an adaptive protein-protein interface. Proc Natl Acad Sci USA. 2003;100:1603–1608. doi: 10.1073/pnas.252756299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wells J, Arkin M, Braisted A, DeLano W, McDowell B, Oslob J, Raimundo B, Randal M. Drug discovery at signaling interfaces. Ernst Schering Res Found Workshop. 2003;42:19–27. doi: 10.1007/978-3-662-05314-0_3. [DOI] [PubMed] [Google Scholar]
- 20.Shuker SB, Hajduk PJ, Meadows RP, Fesik SW. Discovering high-affinity ligands for proteins: SAR by NMR. Science. 1996;274:1531–1534. doi: 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]
- 21.Hachet-Haas M, Balabanian K, Rohmer F, Pons F, Franchet C, Lecat S, Chow KY, Dagher R, Gizzi P, Didier B, Lagane B, Kellenberger E, Bonnet D, Baleux F, Haiech J, Parmentier M, Frossard N, Arenzana-Seisdedos F, Hibert M, Galzi JL. Small neutralizing molecules to inhibit actions of the chemokine CXCL12. J Biol Chem. 2008;283:23189–23199. doi: 10.1074/jbc.M803947200. [DOI] [PubMed] [Google Scholar]
- 22.Galzi JL, Hachet-Haas M, Bonnet D, Daubeuf F, Lecat S, Hibert M, Haiech J, Frossard N. Neutralizing endogenous chemokines with small molecules Principles and potential therapeutic applications. Pharmacol Ther. 2010;126:39–55. doi: 10.1016/j.pharmthera.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gasparik V, Daubeuf F, Hachet-Haas M, Rohmer F, Gizzi P, Haiech J, Galzi JL, Hibert M, Bonnet D, Frossard N. Prodrugs of a CXC chemokine-12 (CXCL12) neutraligand prevent inflammatory reactions in an asthma model in vivo. ACS Med Chem Lett. 2012;3:10–14. doi: 10.1021/ml200017d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Veldkamp CT, Ziarek JJ, Peterson FC, Chen Y, Volkman BF. Targeting SDF-1/CXCL12 with a ligand that prevents activation of CXCR4 through structure-based drug design. J Am Chem Soc. 2010;132:7242–7243. doi: 10.1021/ja1002263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang J, Loberg R, Taichman RS. The pivotal role of CXCL12 (SDF-1)/CXCR4 axis in bone metastasis. Cancer Metastasis Rev. 2006;25:573–587. doi: 10.1007/s10555-006-9019-x. [DOI] [PubMed] [Google Scholar]
- 26.Burger JA, Stewart DJ, Wald O, Peled A. Potential of CXCR4 antagonists for the treatment of metastatic lung cancer. Expert Rev Anticancer Ther. 2011;11:621–630. doi: 10.1586/era.11.11. [DOI] [PubMed] [Google Scholar]
- 27.Nakashima H, Masuda M, Murakami T, Koyanagi Y, Matsumoto A, Fujii N, Yamamoto N. Anti-human immunodeficiency virus activity of a novel synthetic peptide, T22 ([Tyr-5,12, Lys-7]polyphemusin II): a possible inhibitor of virus- cell fusion. Antimicrob Agents Chemother. 1992;36:1249–1255. doi: 10.1128/aac.36.6.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Masuda M, Nakashima H, Ueda T, Naba H, Ikoma R, Otaka A, Terakawa Y, Tamamura H, Ibuka T, Murakami T, et al. A novel anti-HIV synthetic peptide, T-22 ([Tyr5,12,Lys7]- polyphemusin II) Biochem Biophys Res Commun. 1992;189:845–850. doi: 10.1016/0006-291x(92)92280-b. [DOI] [PubMed] [Google Scholar]
- 29.De Clercq E, Yamamoto N, Pauwels R, Baba M, Schols D, Nakashima H, Balzarini J, Debyser Z, Murrer BA, Schwartz D, et al. Potent and selective inhibition of human immunodeficiency virus (HIV)-1 and HIV-2 replication by a class of bicyclams interacting with a viral uncoating event. Proc Natl Acad Sci USA. 1992;89:5286–5290. doi: 10.1073/pnas.89.12.5286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.De Clercq E. The AMD3100 story: the path to the discovery of a stem cell mobilizer (Mozobil) Biochem Pharmacol. 2009;77:1655–1664. doi: 10.1016/j.bcp.2008.12.014. [DOI] [PubMed] [Google Scholar]
- 31.Doranz BJ, Grovit-Ferbas K, Sharron MP, Mao SH, Goetz MB, Daar ES, Doms RW, O'Brien WA. A small-molecule inhibitor directed against the chemokine receptor CXCR4 prevents its use as an HIV-1 coreceptor. J Exp Med. 1997;186:1395–1400. doi: 10.1084/jem.186.8.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schols D, Struyf S, Van Damme J, Este JA, Henson G, De Clercq E. Inhibition of T-tropic HIV strains by selective antagonization of the chemokine receptor CXCR4. J Exp Med. 1997;186:1383–1388. doi: 10.1084/jem.186.8.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murakami T, Nakajima T, Koyanagi Y, Tachibana K, Fujii N, Tamamura H, Yoshida N, Waki M, Matsumoto A, Yoshie O, Kishimoto T, Yamamoto N, Nagasawa T. A small molecule CXCR4 inhibitor that blocks T cell line-tropic HIV-1 infection. J Exp Med. 1997;186:1389–1393. doi: 10.1084/jem.186.8.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tamamura H, Xu Y, Hattori T, Zhang X, Arakaki R, Kanbara K, Omagari A, Otaka A, Ibuka T, Yamamoto N, Nakashima H, Fujii N. A low-molecular-weight inhibitor against the chemokine receptor CXCR4: a strong anti-HIV peptide T140. Biochem Biophys Res Commun. 1998;253:877–882. doi: 10.1006/bbrc.1998.9871. [DOI] [PubMed] [Google Scholar]
- 35.Tamamura H, Omagari A, Hiramatsu K, Gotoh K, Kanamoto T, Xu Y, Kodama E, Matsuoka M, Hattori T, Yamamoto N, Nakashima H, Otaka A, Fujii N. Development of specific CXCR4 inhibitors possessing high selectivity indexes as well as complete stability in serum based on an anti-HIV peptide T140. Bioorg Med Chem Lett. 2001;11:1897–1902. doi: 10.1016/s0960-894x(01)00323-7. [DOI] [PubMed] [Google Scholar]
- 36.Endres MJ, Clapham PR, Marsh M, Ahuja M, Turner JD, McKnight A, Thomas JF, Stoebenau-Haggarty B, Choe S, Vance PJ, Wells TN, Power CA, Sutterwala SS, Doms RW, Landau NR, Hoxie JA. CD4-independent infection by HIV-2 is mediated by fusin/CXCR4. Cell. 1996;87:745–756. doi: 10.1016/s0092-8674(00)81393-8. [DOI] [PubMed] [Google Scholar]
- 37.Bertolini F, Dell'Agnola C, Mancuso P, Rabascio C, Burlini A, Monestiroli S, Gobbi A, Pruneri G, Martinelli G. CXCR4 neutralization, a novel therapeutic approach for non-Hodgkin's lymphoma. Cancer Res. 2002;62:3106–3112. [PubMed] [Google Scholar]
- 38.Engl T, Relja B, Marian D, Blumenberg C, Muller I, Beecken WD, Jones J, Ringel EM, Bereiter-Hahn J, Jonas D, Blaheta RA. CXCR4 chemokine receptor mediates prostate tumor cell adhesion through alpha5 and beta3 integrins. Neoplasia. 2006;8:290–301. doi: 10.1593/neo.05694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.DiPersio JF, Uy GL, Yasothan U, Kirkpatrick P, Kirkpatrick P Plerixafor. Nat Rev Drug Discov. 2009;8:105–106. doi: 10.1038/nrd2819. [DOI] [PubMed] [Google Scholar]
- 40.Kofuku Y, Yoshiura C, Ueda T, Terasawa H, Hirai T, Tominaga S, Hirose M, Maeda Y, Takahashi H, Terashima Y, Matsushima K, Shimada I. Structural basis of the interaction between chemokine stromal cell-derived factor-1/CXCL12 and its G-protein-coupled receptor CXCR4. J Biol Chem. 2009;284:35240–35250. doi: 10.1074/jbc.M109.024851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liang Z, Zhan W, Zhu A, Yoon Y, Lin S, Sasaki M, Klapproth JM, Yang H, Grossniklaus HE, Xu J, Rojas M, Voll RJ, Goodman MM, Arrendale RF, Liu J, Yun CC, Snyder JP, Liotta DC, Shim H. Development of a unique small molecule modulator of CXCR4. PLoS One. 2012;7:e34038. doi: 10.1371/journal.pone.0034038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhan W, Liang Z, Zhu A, Kurtkaya S, Shim H, Snyder JP, Liotta DC. Discovery of small molecule CXCR4 antagonists. J Med Chem. 2007;50:5655–5664. doi: 10.1021/jm070679i. [DOI] [PubMed] [Google Scholar]
- 43.Zhu A, Zhan W, Liang Z, Yoon Y, Yang H, Grossniklaus HE, Xu J, Rojas M, Lockwood M, Snyder JP, Liotta DC, Shim H. Dipyrimidine amines: a novel class of chemokine receptor type 4 antagonists with high specificity. J Med Chem. 2010;53:8556–8568. doi: 10.1021/jm100786g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bridger GJ, Skerlj RT, Hernandez-Abad PE, Bogucki DE, Wang Z, Zhou Y, Nan S, Boehringer EM, Wilson T, Crawford J, Metz M, Hatse S, Princen K, De Clercq E, Schols D. Synthesis and structure-activity relationships of azamacrocyclic C-X-C chemokine receptor 4 antagonists: analogues containing a single azamacrocyclic ring are potent inhibitors of T-cell tropic (X4) HIV-1 replication. J Med Chem. 2010;53:1250–1260. doi: 10.1021/jm901530b. [DOI] [PubMed] [Google Scholar]
- 45.Skerlj RT, Bridger GJ, Kaller A, McEachern EJ, Crawford JB, Zhou Y, Atsma B, Langille J, Nan S, Veale D, Wilson T, Harwig C, Hatse S, Princen K, De Clercq E, Schols D. Discovery of novel small molecule orally bioavailable C-X-C chemokine receptor 4 antagonists that are potent inhibitors of T-tropic (X4) HIV-1 replication. J Med Chem. 2010;53:3376–3388. doi: 10.1021/jm100073m. [DOI] [PubMed] [Google Scholar]
- 46.Thoma G, Streiff MB, Kovarik J, Glickman F, Wagner T, Beerli C, Zerwes HG. Orally bioavailable isothioureas block function of the chemokine receptor CXCR4 in vitro and in vivo. J Med Chem. 2008;51:7915–7920. doi: 10.1021/jm801065q. [DOI] [PubMed] [Google Scholar]
- 47.Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, Hamel DJ, Kuhn P, Handel TM, Cherezov V, Stevens RC. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330:1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sun X, Cheng G, Hao M, Zheng J, Zhou X, Zhang J, Taichman RS, Pienta KJ, Wang J. CXCL12 / CXCR4 / CXCR7 chemokine axis and cancer progression. Cancer Metastasis Rev. 2010;29:709–722. doi: 10.1007/s10555-010-9256-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Horuk R. Chemokine receptor antagonists: overcoming developmental hurdles. Nat Rev Drug Discov. 2009;8:23–33. doi: 10.1038/nrd2734. [DOI] [PubMed] [Google Scholar]
- 50.Crump MP, Gong JH, Loetscher P, Rajarathnam K, Amara A, Arenzana-Seisdedos F, Virelizier JL, Baggiolini M, Sykes BD, Clark-Lewis I. Solution structure and basis for functional activity of stromal cell-derived factor-1; dissociation of CXCR4 activation from binding and inhibition of HIV-1. Embo J. 1997;16:6996–7007. doi: 10.1093/emboj/16.23.6996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gayle RB, 3rd, Sleath PR, Srinivason S, Birks CW, Weerawarna KS, Cerretti DP, Kozlosky CJ, Nelson N, Vanden Bos T, Beckmann MP. Importance of the amino terminus of the interleukin-8 receptor in ligand interactions. J Biol Chem. 1993;268:7283–7289. [PubMed] [Google Scholar]
- 52.Mizoue LS, Bazan JF, Johnson EC, Handel TM. Solution structure and dynamics of the CX3C chemokine domain of fractalkine and its interaction with an N-terminal fragment of CX3CR1. Biochemistry. 1999;38:1402–1414. doi: 10.1021/bi9820614. [DOI] [PubMed] [Google Scholar]
- 53.Doranz BJ, Orsini MJ, Turner JD, Hoffman TL, Berson JF, Hoxie JA, Peiper SC, Brass LF, Doms RW. Identification of CXCR4 domains that support coreceptor and chemokine receptor functions. J Virol. 1999;73:2752–2761. doi: 10.1128/jvi.73.4.2752-2761.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Farzan M, Mirzabekov T, Kolchinsky P, Wyatt R, Cayabyab M, Gerard NP, Gerard C, Sodroski J, Choe H. Tyrosine sulfation of the amino terminus of CCR5 facilitates HIV-1 entry. Cell. 1999;96:667–676. doi: 10.1016/s0092-8674(00)80577-2. [DOI] [PubMed] [Google Scholar]
- 55.Fong AM, Alam SM, Imai T, Haribabu B, Patel DD. CX3CR1 tyrosine sulfation enhances fractalkine-induced cell adhesion. J Biol Chem. 2002;277:19418–19423. doi: 10.1074/jbc.M201396200. [DOI] [PubMed] [Google Scholar]
- 56.Seibert C, Cadene M, Sanfiz A, Chait BT, Sakmar TP. Tyrosine sulfation of CCR5 N-terminal peptide by tyrosylprotein sulfotransferases 1 and 2 follows a discrete pattern and temporal sequence. Proc Natl Acad Sci USA. 2002;99:11031–11036. doi: 10.1073/pnas.172380899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Veldkamp CT, Seibert C, Peterson FC, Sakmar TP, Volkman BF. Recognition of a CXCR4 sulfotyrosine by the chemokine stromal cell-derived factor-1 alpha (SDF- 1alpha/CXCL12) J Mol Biol. 2006;359:1400–1409. doi: 10.1016/j.jmb.2006.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Seibert C, Veldkamp CT, Peterson FC, Chait BT, Volkman BF, Sakmar TP. Sequential tyrosine sulfation of CXCR4 by tyrosylprotein sulfotransferases. Biochemistry. 2008;47:11251–11262. doi: 10.1021/bi800965m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Colvin RA, Campanella GS, Manice LA, Luster AD. CXCR3 requires tyrosine sulfation for ligand binding and a second extracellular loop arginine residue for ligand-induced chemotaxis. Mol Cell Biol. 2006;26:5838–5849. doi: 10.1128/MCB.00556-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Preobrazhensky AA, Dragan S, Kawano T, Gavrilin MA, Gulina IV, Chakravarty L, Kolattukudy PE. Monocyte chemotactic protein-1 receptor CCR2B is a glycoprotein that has tyrosine sulfation in a conserved extracellular N-terminal region. J Immunol. 2000;165:5295–5303. doi: 10.4049/jimmunol.165.9.5295. [DOI] [PubMed] [Google Scholar]
- 61.Simpson LS, Zhu JZ, Widlanski TS, Stone MJ. Regulation of chemokine recognition by site-specific tyrosine sulfation of receptor peptides. Chem Biol. 2009;16:153–161. doi: 10.1016/j.chembiol.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Farzan M, Chung S, Li W, Vasilieva N, Wright PL, Schnitzler CE, Marchione RJ, Gerard C, Gerard NP, Sodroski J, Choe H. Tyrosine-sulfated peptides functionally reconstitute a CCR5 variant lacking a critical amino-terminal region. J Biol Chem. 2002;277:40397–40402. doi: 10.1074/jbc.M206784200. [DOI] [PubMed] [Google Scholar]
- 63.Carlsson J, Coleman RG, Setola V, Irwin JJ, Fan H, Schlessinger A, Sali A, Roth BL, Shoichet BK. Ligand discovery from a dopamine D3 receptor homology model and crystal structure. Nat Chem Biol. 2011;7:769–778. doi: 10.1038/nchembio.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Veldkamp CT, Seibert C, Peterson FC, De la Cruz NB, Haugner JC, 3rd, Basnet H, Sakmar TP, Volkman BF. Structural basis of CXCR4 sulfotyrosine recognition by the chemokine SDF-1/CXCL12. Sci Signal. 2008;1:ra4. doi: 10.1126/scisignal.1160755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ziarek JJ, Heroux MS, Veldkamp CT, Peterson FC, Volkman BF. Sulfotyrosine recognition as marker for druggable sites in the extracellular space. Inter J Mol Sci. 2011;12:3740–3756. doi: 10.3390/ijms12063740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Danan LM, Yu Z, Ludden PJ, Jia W, Moore KL, Leary JA. Catalytic Mechanism of Golgi-Resident Human Tyrosylprotein Sulfotransferase-2: A Mass Spectrometry Approach. J Am Soc Mass Spectrom. 2010;21:1633–1642. doi: 10.1016/j.jasms.2010.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hajduk PJ. Fragment-based drug design: how big is too big? J Med Chem. 2006;49:6972–6976. doi: 10.1021/jm060511h. [DOI] [PubMed] [Google Scholar]
- 68.Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 69.Herr RJ. 5-Substituted-1H-tetrazoles as carboxylic acid isosteres: medicinal chemistry and synthetic methods. Bioorg Med Chem. 2002;10:3379–3393. doi: 10.1016/s0968-0896(02)00239-0. [DOI] [PubMed] [Google Scholar]
- 70.Biot C, Bauer H, Schirmer RH, Davioud-Charvet E. 5-substituted tetrazoles as bioisosteres of carboxylic acids. Bioisosterism and mechanistic studies on glutathione reductase inhibitors as antimalarials. J Med Chem. 2004;47:5972–5983. doi: 10.1021/jm0497545. [DOI] [PubMed] [Google Scholar]
- 71.Pegklidou K, Koukoulitsa C, Nicolaou I, Demopoulos VJ. Design and synthesis of novel series of pyrrole based chemotypes and their evaluation as selective aldose reductase inhibitors. A case-of bioisosterism between a carboxylic acid moiety and that of a tetrazole. Bioorg Med Chem. 2010;18:2107–2114. doi: 10.1016/j.bmc.2010.02.010. [DOI] [PubMed] [Google Scholar]
- 72.Roh J, Artamonova TV, Vavrova K, Koldobskii GI, Hrabalek A. Practical synthesis of 5-substituted tetrazoles under microwave irradiation. Synthesis. 2009:2175–2178. [Google Scholar]
- 73.Veldkamp CT, Peterson FC, Pelzek AJ, Volkman BF. The monomer-dimer equilibrium of stromal cell-derived factor-1 (CXCL 12) is altered by pH, phosphate, sulfate, and heparin. Protein Sci. 2005;14:1071–1081. doi: 10.1110/ps.041219505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 75.Keller R. The Computer-Aided Resonance Assignment Tutorial, Cantina Verlag: Goldau. 2004 [Google Scholar]
- 76.Burns JM, Summers BC, Wang Y, Melikian A, Berahovich R, Miao Z, Penfold ME, Sunshine MJ, Littman DR, Kuo CJ, Wei K, McMaster BE, Wright K, Howard MC, Schall TJ. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J Exp Med. 2006;203:2201–2213. doi: 10.1084/jem.20052144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Miao Z, Luker KE, Summers BC, Berahovich R, Bhojani MS, Rehemtulla A, Kleer CG, Essner JJ, Nasevicius A, Luker GD, Howard MC, Schall TJ. CXCR7 (RDC1) promotes breast and lung tumor growth in vivo and is expressed on tumor-associated vasculature. Proc Natl Acad Sci USA. 2007;104:15735–15740. doi: 10.1073/pnas.0610444104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Luker KE, Lewin SA, Mihalko LA, Schmidt BT, Winkler JS, Coggins NL, Thomas DG, Luker GD. Scavenging of CXCL12 by CXCR7 promotes tumor growth and metastasis of CXCR4-positive breast cancer cells. Oncogene. 2012;31:4750–4758. doi: 10.1038/onc.2011.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kalatskaya I, Berchiche YA, Gravel S, Limberg BJ, Rosenbaum JS, Heveker N. AMD3100 is a CXCR7 ligand with allosteric agonist properties. Mol Pharmacol. 2009;75:1240–1247. doi: 10.1124/mol.108.053389. [DOI] [PubMed] [Google Scholar]
- 80.Gravel S, Malouf C, Boulais PE, Berchiche YA, Oishi S, Fujii N, Leduc R, Sinnett D, Heveker N. The peptidomimetic CXCR4 antagonist TC14012 recruits beta-arrestin to CXCR7: roles of receptor domains. J Biol Chem. 2010;285:37939–37943. doi: 10.1074/jbc.C110.147470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mysinger MM, Weiss DR, Ziarek JJ, Gravel S, Doak AK, Karpiak J, Heveker N, Shoichet BK, Volkman BF. Structure-based ligand discovery for the protein-protein interface of chemokine receptor CXCR4. Proc Natl Acad Sci USA. 2012;109:5517–5522. doi: 10.1073/pnas.1120431109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liu J, Louie S, Hsu W, Yu KM, Nicholas HB, Jr, Rosenquist GL. Tyrosine sulfation is prevalent in human chemokine receptors important in lung disease. Am J Respir Cell Mol Biol. 2008;38:738–743. doi: 10.1165/rcmb.2007-0118OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hemmerich S, Paavola C, Bloom A, Bhakta S, Freedman R, Grunberger D, Krstenansky J, Lee S, McCarley D, Mulkins M, Wong B, Pease J, Mizoue L, Mirzadegan T, Polsky I, Thompson K, Handel TM, Jarnagin K. Identification of residues in the monocyte chemotactic protein-1 that contact the MCP-1 receptor, CCR2. Biochemistry. 1999;38:13013–13025. doi: 10.1021/bi991029m. [DOI] [PubMed] [Google Scholar]
- 84.Clubb RT, Omichinski JG, Clore GM, Gronenborn AM. Mapping the binding surface of interleukin-8 complexed with an N-terminal fragment of the type 1 human interleukin-8 receptor. FEBS Lett. 1994;338:93–97. doi: 10.1016/0014-5793(94)80123-1. [DOI] [PubMed] [Google Scholar]
- 85.Mizoue LS, Sullivan SK, King DS, Kledal TN, Schwartz TW, Bacon KB, Handel TM. Molecular determinants of receptor binding and signaling by the CX3C chemokine fractalkine. J Biol Chem. 2001;276:33906–33914. doi: 10.1074/jbc.M101348200. [DOI] [PubMed] [Google Scholar]
- 86.Saini V, Marchese A, Majetschak M. CXC chemokine receptor 4 is a cell surface receptor for extracellular ubiquitin. J Biol Chem. 2010;285:15566–15576. doi: 10.1074/jbc.M110.103408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, Koenen RR, Dewor M, Georgiev I, Schober A, Leng L, Kooistra T, Fingerle-Rowson G, Ghezzi P, Kleemann R, McColl SR, Bucala R, Hickey MJ, Weber C. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med. 2007;13:587–596. doi: 10.1038/nm1567. [DOI] [PubMed] [Google Scholar]
- 88.Kohout TA, Nicholas SL, Perry SJ, Reinhart G, Junger S, Struthers RS. Differential desensitization, receptor phosphorylation, beta-arrestin recruitment, and ERK1/2 activation by the two endogenous ligands for the CC chemokine receptor 7. J Biol Chem. 2004;279:23214–23222. doi: 10.1074/jbc.M402125200. [DOI] [PubMed] [Google Scholar]
- 89.Zidar DA, Violin JD, Whalen EJ, Lefkowitz RJ. Selective engagement of G protein coupled receptor kinases (GRKs) encodes distinct functions of biased ligands. Proc Natl Acad Sci USA. 2009;106:9649–9654. doi: 10.1073/pnas.0904361106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Drury LJ, Ziarek JJ, Gravel S, Veldkamp CT, Takekoshi T, Hwang ST, Heveker N, Volkman BF, Dwinell MB. Monomeric and dimeric CXCL12 inhibit metastasis through distinct CXCR4 interactions and signaling pathways. Proc Natl Acad Sci USA. 2011;108:17655–17660. doi: 10.1073/pnas.1101133108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Roy PP, Roy K. Molecular docking and QSAR studies of aromatase inhibitor androstenedione derivatives. J Pharm Pharmacol. 2010;62:1717–1728. doi: 10.1111/j.2042-7158.2010.01154.x. [DOI] [PubMed] [Google Scholar]
- 92.Mitra I, Saha A, Roy K. Chemometric modeling of free radical scavenging activity of flavone derivatives. Eur J Med Chem. 2010;45:5071–5079. doi: 10.1016/j.ejmech.2010.08.016. [DOI] [PubMed] [Google Scholar]
- 93.Roy KK, Singh S, Saxena AK. Integration-mediated prediction enrichment of quantitative model for Hsp90 inhibitors as anti- cancer agents: 3D-QSAR study. Mol Divers. 2011;15:477–489. doi: 10.1007/s11030-010-9269-y. [DOI] [PubMed] [Google Scholar]
- 94.Pisco L, Kordian M, Peseke K, Feist H, Michalik D, Estrada E, Carvalho J, Hamilton G, Rando D, Quincoces J. Synthesis of compounds with antiproliferative activity as analogues of prenylated natural products existing in Brazilian propolis. Eur J Med Chem. 2006;41:401–407. doi: 10.1016/j.ejmech.2005.10.020. [DOI] [PubMed] [Google Scholar]
- 95.Estrada E, Uriarte E, Molina E, Simon-Manso Y, Milne GW. An integrated in silico analysis of drug-binding to human serum albumin. J Chem Inf Model. 2006;46:2709–2724. doi: 10.1021/ci600274f. [DOI] [PubMed] [Google Scholar]
- 96.Estrada E, Quincoces JA, Patlewicz G. Creating molecular diversity from antioxidants in Brazilian propolis. Combination of TOPS-MODE QSAR and virtual structure generation. Mol Divers. 2004;8:21–33. doi: 10.1023/b:modi.0000006804.97390.40. [DOI] [PubMed] [Google Scholar]
- 97.Estrada E, Vilar S, Uriarte E, Gutierrez Y. In silico studies toward the discovery of new anti-HIV nucleoside compounds with the use of TOPS-MODE and 2D/3D connectivity indices. 1. Pyrimidyl derivatives. J Chem Inf Comput Sci. 2002;42:1194–1203. doi: 10.1021/ci0255331. [DOI] [PubMed] [Google Scholar]
- 98.Estrada E, Uriarte E, Montero A, Teijeira M, Santana L, De Clercq E. A novel approach for the virtual screening and rational design of anticancer compounds. J Med Chem. 2000;43:1975–1985. doi: 10.1021/jm991172d. [DOI] [PubMed] [Google Scholar]
- 99.Speck-Planche A, Luan F, Cordeiro MN. Discovery of anti- Alzheimer agents: current ligand-based approaches toward the design of acetylcholinesterase inhibitors. Mini Rev Med Chem. 2012;12:583–591. doi: 10.2174/138955712800493744. [DOI] [PubMed] [Google Scholar]
- 100.Speck-Planche A, Kleandrova VV. QSAR and molecular docking techniques for the discovery of potent monoamine oxidase B inhibitors: Computer-aided generation of new rasagiline bioisosteres. Curr Top Med Chem. 2012;12:1734–1747. [PubMed] [Google Scholar]
- 101.Speck-Planche A, Cordeiro MN. Computer-aided drug design methodologies toward the design of anti-hepatitis C agents. Curr Top Med Chem. 2012;12:802–813. doi: 10.2174/156802612800166783. [DOI] [PubMed] [Google Scholar]
- 102.Speck-Planche A, Guilarte-Montero L, Yera-Bueno R, Rojas- Vargas JA, Garcia-Lopez A, Uriarte E, Molina-Perez E. Rational design of new agrochemical fungicides using substructural descriptors. Pest Manag Sci. 2011;67:438–445. doi: 10.1002/ps.2082. [DOI] [PubMed] [Google Scholar]
- 103.Speck-Planche A, Cordeiro MN, Guilarte-Montero L, Yera-Bueno R. Current computational approaches towards the rational design of new insecticidal agents. Curr Comput Aided Drug Des. 2011;7:304–314. doi: 10.2174/157340911798260359. [DOI] [PubMed] [Google Scholar]
- 104.Speck-Planche A, Cordeiro MN. Current drug design of anti- HIV agents through the inhibition of C-C chemokine receptor type 5. Curr Comput Aided Drug Des. 2011;7:238–248. doi: 10.2174/157340911798260287. [DOI] [PubMed] [Google Scholar]
- 105.Speck-Planche A, Scotti MT, Emerenciano VP, García-López A, Molina-Pérez E, Uriarte E. Designing novel antitrypanosomal agents from a mixed graph-theoretical substructural approach. J Comput Chem. 2010;31:882–894. doi: 10.1002/jcc.21374. [DOI] [PubMed] [Google Scholar]
- 106.Speck-Planche A, Scotti MT, de Paulo-Emerenciano V. Current pharmaceutical design of antituberculosis drugs: future perspectives. Curr Pharm Des. 2010;16:2656–2665. doi: 10.2174/138161210792389289. [DOI] [PubMed] [Google Scholar]
- 107.Speck-Planche A, Scotti MT, García-López A, Emerenciano VP, Molina-Pérez E, Uriarte E. Design of novel antituberculosis compounds using graph-theoretical and substructural approaches. Mol Divers. 2009;13:445–458. doi: 10.1007/s11030-009-9129-9. [DOI] [PubMed] [Google Scholar]
- 108.Rescigno A, Casanola-Martin GM, Sanjust E, Zucca P, Marrero-Ponce Y. Vanilloid derivatives as tyrosinase inhibitors driven by virtual screening-based QSAR models. Drug Test Anal. 2011;3:176–181. doi: 10.1002/dta.187. [DOI] [PubMed] [Google Scholar]
- 109.Marrero-Ponce Y, Siverio-Mota D, Galvez-Llompart M, Recio MC, Giner RM, Garcia-Domenech R, Torrens F, Aran VJ, Cordero-Maldonado ML, Esguera CV, de Witte PA, Crawford AD. Discovery of novel anti-inflammatory drug-like compounds by aligning in silico and in vivo screening: the nitroindazolinone chemotype. Eur J Med Chem. 2011;46:5736–5753. doi: 10.1016/j.ejmech.2011.07.053. [DOI] [PubMed] [Google Scholar]
- 110.Le-Thi-Thu H, Casanola-Martin GM, Marrero-Ponce Y, Rescigno A, Saso L, Parmar VS, Torrens F, Abad C. Novel coumarin-based tyrosinase inhibitors discovered by OECD principles-validated QSAR approach from an enlarged, balanced database. Mol Divers. 2011;15:507–520. doi: 10.1007/s11030-010-9274-1. [DOI] [PubMed] [Google Scholar]
- 111.Casañola-Martin GM, Marrero-Ponce Y, Khan MT, Khan SB, Torrens F, Perez-Jimenez F, Rescigno A, Abad C. Bond- based 2D quadratic fingerprints in QSAR studies: virtual and in vitro tyrosinase inhibitory activity elucidation. Chem Biol Drug Des. 2010;76:538–545. doi: 10.1111/j.1747-0285.2010.01032.x. [DOI] [PubMed] [Google Scholar]
- 112.Meneses-Marcel A, Rivera-Borroto OM, Marrero-Ponce Y, Montero A, Machado Tugores Y, Escario JA, Gomez Barrio A, Montero Pereira D, Nogal JJ, Kouznetsov VV, Ochoa Puentes C, Bohorquez AR, Grau R, Torrens F, Ibarra-Velarde F, Aran VJ. New antitrichomonal drug-like chemicals selected by bond (edge)-based TOMOCOMD-CARDD descriptors. J Biomol Screen. 2008;13:785–794. doi: 10.1177/1087057108323122. [DOI] [PubMed] [Google Scholar]
- 113.Marrero-Ponce Y, Meneses-Marcel A, Rivera-Borroto OM, Garcia-Domenech R, De Julian-Ortiz JV, Montero A, Escario JA, Barrio AG, Pereira DM, Nogal JJ, Grau R, Torrens F, Vogel C, Aran VJ. Bond-based linear indices in QSAR: computational discovery of novel anti-trichomonal compounds. J Comput Aided Mol Des. 2008;22:523–540. doi: 10.1007/s10822-008-9171-1. [DOI] [PubMed] [Google Scholar]
- 114.Gonzalez-Diaz H, Dea-Ayuela MA, Perez-Montoto LG, Prado-Prado FJ, Aguero-Chapin G, Bolas-Fernandez F, Vazquez-Padron RI, Ubeira FM. QSAR for RNases and theoretic-experimental study of molecular diversity on peptide mass fingerprints of a new Leishmania infantum protein. Mol Divers. 2010;14:349–369. doi: 10.1007/s11030-009-9178-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Perez-Bello A, Munteanu CR, Ubeira FM, De Magalhaes AL, Uriarte E, Gonzalez-Diaz H. Alignment-free prediction of mycobacterial DNA promoters based on pseudo-folding lattice network or star-graph topological indices. J Theor Biol. 2009;256:458–466. doi: 10.1016/j.jtbi.2008.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Santana L, Gonzalez-Diaz H, Quezada E, Uriarte E, Yanez M, Vina D, Orallo F. Quantitative structure-activity relationship and complex network approach to monoamine oxidase A and B inhibitors. J Med Chem. 2008;51:6740–6751. doi: 10.1021/jm800656v. [DOI] [PubMed] [Google Scholar]
- 117.Munteanu CR, Gonzalez-Diaz H, Magalhaes AL. Enzymes/non-enzymes classification model complexity based on composition, sequence, 3D and topological indices. J Theor Biol. 2008;254:476–482. doi: 10.1016/j.jtbi.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 118.Dea-Ayuela MA, Perez-Castillo Y, Meneses-Marcel A, Ubeira FM, Bolas-Fernandez F, Chou KC, Gonzalez-Diaz H. HP-Lattice QSAR for dynein proteins: experimental proteomics (2D-electrophoresis, mass spectrometry) and theoretic study of a Leishmania infantum sequence. Bioorg Med Chem. 2008;16:7770–7776. doi: 10.1016/j.bmc.2008.07.023. [DOI] [PubMed] [Google Scholar]
- 119.Gonzalez-Diaz H, Bonet I, Teran C, De Clercq E, Bello R, Garcia MM, Santana L, Uriarte E. ANN-QSAR model for selection of anticancer leads from structurally heterogeneous series of compounds. Eur J Med Chem. 2007;42:580–585. doi: 10.1016/j.ejmech.2006.11.016. [DOI] [PubMed] [Google Scholar]
- 120.Speck-Planche A, Luan F, Cordeiro MN. Role of ligand-based drug design methodologies toward the discovery of new anti- Alzheimer agents: futures perspectives in Fragment-Based Ligand Design. Curr Med Chem. 2012;19:1635–1645. doi: 10.2174/092986712799945058. [DOI] [PubMed] [Google Scholar]
- 121.Speck-Planche A, Kleandrova VV, Scotti MT. Fragment-based approach for the in silico discovery of multi-target insecticides. Chemometr Intell Lab Syst. 2012;111:39–45. [Google Scholar]
- 122.Speck-Planche A, Kleandrova VV, Luan F, Cordeiro MN. Chemoinformatics in multi-target drug discovery for anti-cancer therapy: in silico design of potent and versatile anti-brain tumor agents. Anticancer Agents Med Chem. 2012;12:678–685. doi: 10.2174/187152012800617722. [DOI] [PubMed] [Google Scholar]
- 123.Speck-Planche A, Kleandrova VV, Luan F, Cordeiro MN. Predicting multiple ecotoxicological profiles in agrochemical fungicides: a multi-species chemoinformatic approach. Ecotoxicol Environ Saf. 2012;80:308–313. doi: 10.1016/j.ecoenv.2012.03.018. [DOI] [PubMed] [Google Scholar]
- 124.Speck-Planche A, Kleandrova VV, Luan F, Cordeiro MN. Chemoinformatics in anti-cancer chemotherapy: multi-target QSAR model for the in silico discovery of anti-breast cancer agents. Eur J Pharm Sci. 2012;47:273–279. doi: 10.1016/j.ejps.2012.04.012. [DOI] [PubMed] [Google Scholar]
- 125.Speck-Planche A, Kleandrova VV, Luan F, Cordeiro MN. A ligand-based approach for the in silico discovery of multi-target inhibitors for proteins associated with HIV infection. Mol Biosyst. 2012;8:2188–2196. doi: 10.1039/c2mb25093d. [DOI] [PubMed] [Google Scholar]
- 126.Speck-Planche A, Kleandrova VV, Luan F, Cordeiro MN. Rational drug design for anti-cancer chemotherapy: multi-target QSAR models for the in silico discovery of anti-colorectal cancer agents. Bioorg Med Chem. 2012;20:4848–4855. doi: 10.1016/j.bmc.2012.05.071. [DOI] [PubMed] [Google Scholar]
- 127.Speck-Planche A, Kleandrova VV. In silico design of multi- target inhibitors for C-C chemokine receptors using substructural descriptors. Mol Divers. 2012;16:183–191. doi: 10.1007/s11030-011-9337-y. [DOI] [PubMed] [Google Scholar]
- 128.Speck-Planche A, Kleandrova VV, Rojas-Vargas JA. QSAR model toward the rational design of new agrochemical fungicides with a defined resistance risk using substructural descriptors. Mol Divers. 2011;15:901–909. doi: 10.1007/s11030-011-9320-7. [DOI] [PubMed] [Google Scholar]
- 129.Speck-Planche A, Kleandrova VV, Luan F, Cordeiro MN. Multi-target drug discovery in anti-cancer therapy: fragment-based approach toward the design of potent and versatile anti-prostate cancer agents. Bioorg Med Chem. 2011;19:6239–6244. doi: 10.1016/j.bmc.2011.09.015. [DOI] [PubMed] [Google Scholar]
- 130.Speck-Planche A, Kleandrova VV, Luan F, Cordeiro MN. Fragment-based QSAR model toward the selection of versatile anti-sarcoma leads. Eur J Med Chem. 2011;46:5910–5916. doi: 10.1016/j.ejmech.2011.09.055. [DOI] [PubMed] [Google Scholar]
- 131.Tenorio-Borroto E, Penuelas Rivas CG, Vasquez Chagoyan JC, Castanedo N, Prado-Prado FJ, Garcia-Mera X, Gonzalez-Diaz H. ANN multiplexing model of drugs effect on macrophages; theoretical and flow cytometry study on the cytotoxicity of the antimicrobial drug G1 in spleen. Bioorg Med Chem. 2012;20:6181–6194. doi: 10.1016/j.bmc.2012.07.020. [DOI] [PubMed] [Google Scholar]
- 132.Prado-Prado FJ, Garcia-Mera X, Gonzalez-Diaz H. Multi-target spectral moment QSAR versus ANN for antiparasitic drugs against different parasite species. Bioorg Med Chem. 2010;18:2225–2231. doi: 10.1016/j.bmc.2010.01.068. [DOI] [PubMed] [Google Scholar]
- 133.Perez-Montoto LG, Santana L, Gonzalez-Diaz H. Scoring function for DNA-drug docking of anticancer and antiparasitic compounds based on spectral moments of 2D lattice graphs for molecular dynamics trajectories. Eur J Med Chem. 2009;44:4461–4469. doi: 10.1016/j.ejmech.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Munteanu CR, Magalhaes AL, Uriarte E, Gonzalez-Diaz H. Multi-target QPDR classification model for human breast and colon cancer-related proteins using star graph topological indices. J Theor Biol. 2009;257:303–311. doi: 10.1016/j.jtbi.2008.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Concu R, Dea-Ayuela MA, Perez-Montoto LG, Prado-Prado FJ, Uriarte E, Bolas-Fernandez F, Podda G, Pazos A, Munteanu CR, Ubeira FM, Gonzalez-Diaz H. 3D entropy and moments prediction of enzyme classes and experimental-theoretic study of peptide fingerprints in Leishmania parasites. Biochim Biophys Acta. 2009;1794:1784–1794. doi: 10.1016/j.bbapap.2009.08.020. [DOI] [PubMed] [Google Scholar]
- 136.Concu R, Dea-Ayuela MA, Perez-Montoto LG, Bolas-Fernandez F, Prado-Prado FJ, Podda G, Uriarte E, Ubeira FM, Gonzalez-Diaz H. Prediction of enzyme classes from 3D structure: a general model and examples of experimental-theoretic scoring of peptide mass fingerprints of Leishmania proteins. J Proteome Res. 2009;8:4372–4382. doi: 10.1021/pr9003163. [DOI] [PubMed] [Google Scholar]
- 137.Prado-Prado FJ, Gonzalez-Diaz H, de la Vega OM, Ubeira FM, Chou KC. Unified QSAR approach to antimicrobials. Part 3: first multi-tasking QSAR model for input-coded prediction, structural back-projection, and complex networks clustering of antiprotozoal compounds. Bioorg Med Chem. 2008;16:5871–5880. doi: 10.1016/j.bmc.2008.04.068. [DOI] [PubMed] [Google Scholar]
- 138.Speck-Planche A, Kleandrova VV, Luan F, Cordeiro MN. In silico discovery and virtual screening of multi-target inhibitors for proteins in Mycobacterium tuberculosis. Comb Chem High Throughput Screen. 2012;15:666–673. doi: 10.2174/138620712802650487. [DOI] [PubMed] [Google Scholar]
- 139.Speck-Planche A, Cordeiro MNDS. Application of Bioinformatics for the search of novel anti-viral therapies: Rational design of anti-herpes agents. Curr Bioinform. 2011;6:81–93. [Google Scholar]
- 140.Prado-Prado F, Garcia-Mera X, Abeijon P, Alonso N, Caamano O, Yanez M, Garate T, Mezo M, Gonzalez-Warleta M, Muino L, Ubeira FM, Gonzalez-Diaz H. Using entropy of drug and protein graphs to predict FDA drug-target network: theoretic-experimental study of MAO inhibitors and hemoglobin peptides from Fasciola hepatica. Eur J Med Chem. 2011;46:1074–1094. doi: 10.1016/j.ejmech.2011.01.023. [DOI] [PubMed] [Google Scholar]
- 141.Gonzalez-Diaz H, Prado-Prado F, Sobarzo-Sanchez E, Haddad M, Maurel Chevalley S, Valentin A, Quetin-Leclercq J, Dea-Ayuela MA, Teresa Gomez-Munos M, Munteanu CR, Jose Torres-Labandeira J, Garcia-Mera X, Tapia RA, Ubeira FM. NL MIND-BEST: A web server for ligands and proteins discovery-Theoretic-experimental study of proteins of Giardia lamblia and new compounds active against Plasmodium falciparum. J Theor Biol. 2011;276:229–249. doi: 10.1016/j.jtbi.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 142.Gonzalez-Diaz H, Prado-Prado F, Garcia-Mera X, Alonso N, Abeijon P, Caamano O, Yanez M, Munteanu CR, Pazos A, Dea-Ayuela MA, Gomez-Munoz MT, Garijo MM, Sansano J, Ubeira FM. MIND-BEST: Web Server for Drugs and Target Discovery; Design, Synthesis, and Assay of MAO-B Inhibitors and Theoretical-Experimental Study of G3PDH Protein from Trichomonas gallinae. J Proteome Res. 2011;10:1698–1718. doi: 10.1021/pr101009e. [DOI] [PubMed] [Google Scholar]
- 143.Vina D, Uriarte E, Orallo F, Gonzalez-Diaz H. Alignment-free prediction of a drug-target complex network based on parameters of drug connectivity and protein sequence of receptors. Mol Pharm. 2009;6:825–835. doi: 10.1021/mp800102c. [DOI] [PubMed] [Google Scholar]
- 144.Estrada E. Generalized walks-based centrality measures for complex biological networks. J Theor Biol. 2010;263:556–565. doi: 10.1016/j.jtbi.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 145.Estrada E. Universality in protein residue networks. Biophys J. 2010;98:890–900. doi: 10.1016/j.bpj.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Estrada E, Hatano N. Communicability in complex networks. Phys Rev E Stat Nonlin Soft Matter Phys. 2008;77:036111. doi: 10.1103/PhysRevE.77.036111. [DOI] [PubMed] [Google Scholar]
- 147.Estrada E, Bodin O. Using network centrality measures to manage landscape connectivity. Ecol Appl. 2008;18:1810–1825. doi: 10.1890/07-1419.1. [DOI] [PubMed] [Google Scholar]
- 148.Gonzalez-Diaz H. Network topological indices, drug metabolism, and distribution. Curr Drug Metab. 2010;11:283–284. doi: 10.2174/138920010791514162. [DOI] [PubMed] [Google Scholar]
- 149.Vilar S, Gonzalez-Diaz H, Santana L, Uriarte E. A network- QSAR model for prediction of genetic-component biomarkers in human colorectal cancer. J Theor Biol. 2009;261:449–458. doi: 10.1016/j.jtbi.2009.07.031. [DOI] [PubMed] [Google Scholar]
- 150.Prado-Prado FJ, Uriarte E, Borges F, Gonzalez-Diaz H. Multi-target spectral moments for QSAR and Complex Networks study of antibacterial drugs. Eur J Med Chem. 2009;44:4516–4521. doi: 10.1016/j.ejmech.2009.06.018. [DOI] [PubMed] [Google Scholar]
- 151.Prado-Prado FJ, Ubeira FM, Borges F, Gonzalez-Diaz H. Unified QSAR & network-based computational chemistry approach to antimicrobials. II. Multiple distance and triadic census analysis of antiparasitic drugs complex networks. J Comput Chem. 2009;31:164–173. doi: 10.1002/jcc.21292. [DOI] [PubMed] [Google Scholar]
- 152.Prado-Prado FJ, Martinez de la Vega O, Uriarte E, Ubeira FM, Chou KC, Gonzalez-Diaz H. Unified QSAR approach to antimicrobials. 4. Multi-target QSAR modeling and comparative multi-distance study of the giant components of antiviral drug-drug complex networks. Bioorg Med Chem. 2009;17:569–575. doi: 10.1016/j.bmc.2008.11.075. [DOI] [PubMed] [Google Scholar]