

Abstract
Oxidative stress contributes to neurodegeneration in Huntington's disease (HD). However, the origins of oxidative stress in HD remain unclear. Studies in HD transgenic models suggest involvement of mitochondrial dysfunction, which would lead to overproduction of reactive oxygen species (ROS). Impaired mitochondria complexes occur in late stages of HD but not in presymptomatic or early-stage HD patients. Thus, other mechanisms may account for the earliest source of oxidative stress caused by endogenous mutant huntingtin. Here, we report that decreased levels of a major intracellular antioxidant glutathione coincide with accumulation of ROS in primary HD neurons prepared from embryos of HD knock-in mice (HD140Q/140Q), which have human huntingtin exon 1 with 140 CAG repeats inserted into the endogenous mouse huntingtin gene. Uptake of extracellular cysteine through the glutamate/cysteine transporter EAAC1 is required for de novo synthesis of glutathione in neurons. We found that, compared with wild-type neurons, HD neurons had lower cell surface levels of EAAC1 and were deficient in taking up cysteine. Constitutive trafficking of EAAC1 from recycling endosomes relies on Rab11 activity, which is defective in the brain of HD140Q/140Q mice. Enhancement of Rab11 activity by expression of a dominant-active Rab11 mutant in primary HD neurons ameliorated the deficit in cysteine uptake, increased levels of intracellular glutathione, normalized clearance of ROS, and improved neuronal survival. Our data support a novel mechanism for oxidative stress in HD: Rab11 dysfunction slows trafficking of EAAC1 to the cell surface and impairs cysteine uptake, thereby leading to deficient synthesis of glutathione.
Introduction
Compelling data support a critical role for oxidative stress in the pathogenesis of Huntington's disease (HD), a disorder caused by polyglutamine expansion in huntingtin (Htt). However, mechanisms for origins of oxidative stress in HD are unclear. Oxidative stress occurs with overproduction of reactive oxygen species (ROS), or reduction in the antioxidant capacity, or both. Impaired functions of mitochondrial complexes, which would cause overproduction of ROS, occur in late stages but not in presymptomatic or grade I HD patients (Guidetti et al., 2001), suggesting that sources of oxidative stress independent of mitochondria exist in early HD.
Glutathione (GSH) is a major antioxidant in the brain (Dringen, 2000) and essential for protecting cellular constituents against ROS-induced damage by reacting with ROS through its free thiol group (Schulz et al., 2000; Ballatori et al., 2009). After reaction with ROS, GSH is oxidized into GSSG (glutathione disulfide), which can be converted back to GSH for reuse by GSH reductase, a flavoprotein that uses NADPH as the electron source. A rise in ROS usually stimulates a compensatory increase in GSH synthesis to maintain the normal redox balance. Maintenance of normal GSH levels is vital for neuronal survival (Li et al., 1997; Nicole et al., 1998; Wüllner et al., 1999). In the brain, glial cells store high levels of GSH and release GSH into the extracellular space (Dringen and Hamprecht, 1998). However, neurons cannot take up extracellular GSH (Aoyama et al., 2008) and need de novo synthesis that requires uptake of the rate-limiting precursor cysteine from the extracellular space (Dringen, 2000; Aoyama et al., 2008) through the neuronal Na+-dependent glutamate transporter EAAC1 (EAAT3) (Shanker et al., 2001; Aoyama et al., 2006). Knock-out of EAAC1 causes oxidative stress in neurons and age-dependent neurodegeneration, which can be rescued by administration of membrane permeable GSH precursor N-acetylcysteine (Aoyama et al., 2006). EAAC1 locates mainly in the cytoplasm (Rothstein et al., 1994; Conti et al., 1998; He et al., 2000; Nieoullon et al., 2006) and undergoes constitutive recycling that requires normal Rab11 activity (González et al., 2007).
Decrease of GSH levels has been reported to occur in peripheral plasma (Klepac et al., 2007) and erythrocytes (Zanella et al., 1980) of HD patients. Postmortem brains of HD patients have levels of GSH similar to those of non-HD controls (Sian et al., 1994). However, study of brain tissues does not allow discrimination between neurons and glial cells because in the HD brain astrocytic storage of high levels of GSH (Dringen and Hamprecht, 1998) can mask changes in neurons (Selkoe et al., 1982; Myers et al., 1991). Therefore, it is important to know whether mutant Htt disturbs GSH levels selectively in neurons. In this study, we examined levels of GSH in primary cortical neurons prepared from embryos of a knock-in mouse model of HD (HD140Q/140Q) and wild-type (WT) mice. We found that decreased levels of GSH and increased levels of ROS simultaneously occurred in HD140Q/140Q cortical neurons. We propose that glutathione dysregulation in HD140Q/140Q neurons is linked to a deficit in Rab11-dependent trafficking of EAAC1.
Materials and Methods
Plasmid DNA.
The cDNA encoding Rab11 (BC010722) was amplified by PCR, purified from agarose gel using QIAEX II kit (QIAGEN), and digested with BamHI/XhoI. After purification from agarose gel, the digested Rab11 cDNA fragment was cloned into the BamHI/XhoI sites of the HA-pcDNA3 plasmid. Rab11 mutant Rab11Q70L was generated by PCR-based site-specific mutagenesis. All Rab11 sequences were verified by DNA sequencing. For cloning of Rab11Q70L into the CSCW2-pgk lentivirus vector, HA-Rab11Q70L-pcDNA3 was digested with BamHI and XhoI to obtain Rab11Q70L, which was subcloned into the BamHI/XhoI sites of the pENTR1A plasmid. Rab11Q70L was then released from Rab11Q70L-pENTR1A after treatment with NheI and XhoI. This NheI/XhoI Rab11Q70L fragment was cloned into the NheI/XhoI sites of the CSCW2-pgk vector that encoded enhanced green fluorescent protein (EGFP). The two genes are independently expressed via a bicistronic regulation.
Generation of lentivirus and viral titration.
CSCW2-pgk with cDNA encoding EGFP or Rab11Q70L-CSCW2-pgk-EGFP was packaged into lentivirus. 293T producer cells were maintained in DMEM high-glucose media supplemented with 10% fetal calf serum (Atlanta Biologicals), l-glutamine (Invitrogen), and sodium pyruvate (Invitrogen) without antibiotics in a 37°C cell culture incubator (Thermo Fisher Scientific). The day before transfection, 7 × 106 of 293T cells were plated in a 15 cm dish and cultured as above. Transfection was performed according to the manufacturer's instructions (Invitrogen). For each transfection, 6.75 μg of CSCW2-pgk or Rab11Q70L-CSCW2-pgk plasmid DNA, 6 μg of pCMV-dR8.2 dvpr, 0.75 μg of pCMV-VSV-G, and 243 μl of FuGene 6 (QIAGEN) were used to prepare packaging mixtures. After applying packaging mixtures, 293T cells were cultured at 37°C as above for 24 h and changed into fresh culture media. Forty-eight hours later, culture media were collected and filtrated through a 0.45 μm filter to remove cell debris. Filtrated culture media were centrifuged at 4°C, 24,000 rpm in a T50.2 rotor (Beckman Coulter) for 1.5 h. The viral pellet was resuspended in 0.2 ml of PBS containing 0.5% BSA. Titers of virus were determined using a HIV-1 p24 antigen ELISA kit (ZeptoMetrix). Viral titers were 1.83 × 108 pg ml−1 for lenti-Rab11Q70L/EGFP and 1.1 × 108 pg ml−1 for lenti-EGFP.
Preparation of primary cortical neurons and infection of lentivirus.
Cortices were dissected from the brains of embryos of embryonic day 16 (E16) mice (WT and HD140Q/140Q), dissected free of meninges and other tissue, and incubated in PBS containing antibiotics (penicillin, streptomycin, and neomycin) and 0.25% trypsin for 10 min at 37°C (all reagents were purchased from Invitrogen, except where indicated). After incubation, cells were washed twice in PBS supplemented with calcium and magnesium, and then dissociated in Neurobasal medium containing B27 supplement, N2 supplement, 25 μm mercaptoethanol and l-glutamine (NBM), seeded in plates (BD Biosciences) with or without glass coverslips (Warner Instruments), and cultured in serum-free NBM media. Plates and glass coverslips were precoated with poly-l-lysine. Twenty-four to 48 h after plating, primary cultures were treated with cytosine arabinoside (AraC) for 24 h to inhibit proliferation of glial cells. Then, fresh NBM media was added to the cultures. Quantitative analysis of the primary cultures was performed 4 d after AraC treatment to determine the presence of astrocytes (GFAP labeling), microglia (CD68 labeling), and neurons (β-III-tubulin labeling). Results showed that 99.5% of the cells were neurons.
Primary neurons were infected with 30 multiplicities of infection of lentivirus expressing EGFP or Rab11Q70L/EGFP or not for indicated times as previously reported (Li et al., 2009a). Experiments were conducted when EGFP expression in >90% of neurons was confirmed with a fluorescence microscope.
Measurement of glutathione.
Measurement of GSH was conducted according to supplier instructions (BioVision Research Products). In brief, primary neuronal cultures were lysed in assay buffer (BioVision Research Products) at 8 d in vitro (DIV8) and collected into a 1.5 ml tube. After removing nuclei by centrifugation at 4°C, 14,000 rpm for 5 min in a table centrifuge (Eppendorf), 20 μl of perchloric acid (PCA) was immediately added to 60 μl of postnuclear supernatant (S1), mixed, and incubated on ice for 5 min. After a centrifugation at 4°C, 14,000 rpm for 5 min, 40 μl of PCA-preserved supernatants were mixed with 20 μl of 3N KOH for neutralization of the pH, incubated on ice for 5 min, and centrifuged at 4°C, 14,000 rpm for 5 min. Ten microliters of the neutralized sample was diluted with assay buffer to a final volume of 90 μl, mixed with 10 μl of o-phthalaldehyde (OPA) probe, and incubated at room temperature for 40 min. Fluorescence intensities of samples and standards were read on a fluorescence plate reader equipped with excitation/emission wavelength of 340/450 nm. Data were represented as nanomoles per milligram protein.
Measurements of reactive oxygen species in living neurons.
Primary WT and HD140Q/140Q neurons from E16 embryonic days were plated at 400,000 cells/ml on 24-well plates and cultured as above. At DIV8, neurons with or without virus were used for measuring reactive oxygen species as described previously (Valencia and Morán, 2001). In brief, neurons were changed into fresh complete medium and incubated with 2 μg ml−1 5-(and-6)-carboxy-2′,7′-dichlorofluorescein diacetate (carboxy-DCFDA) (Invitrogen) at 37°C for 30 min. After incubation, neurons were washed in prewarmed PBS and imaged using a Bio-Rad Radiance 2100 confocal laser-scanning microscope with krypton–argon laser. Images were acquired through a 60× Nikon Plan Fluor Apo objective (numerical aperture, 1.4) on an inverted Nikon Eclipse TE300 fluorescence microscope using 488 nm excitation and 515 nm emission wavelengths. Each field with at least two neurons was imaged. The focus was adjusted by visualizing neurites. After adjustment of focus, images were immediately collected. At least three different fields of neurons were obtained per culture dish and used for quantification.
Images were analyzed using NIH ImageJ. The soma and neural processes were manually tracked by tracing the edge of a cell body or a selected neurite. The pixels in the equivalent area adjacent to the cell body or process were also measured. The fluorescence intensity in the area outside the soma or neurites was subtracted from the measurement of fluorescence intensity in the cell body or neurite. Three criteria were used for selecting a neurite: the neurite was in focus, had a clear point of origin from the soma, and was well separated from other processes. The average area of all examined neurons for each condition was used for normalizing the fluorescence intensity per area of the soma region for each neuron. Data are represented as the mean average intensity per neuron with SD. The average length of all examined processes for each condition was used to normalize the fluorescence intensity for each process. Data are represented as the mean total intensity per length of a process with SD.
Determination of cell viability.
Cell viability was determined by the ability of neurons to convert 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) into DMSO-soluble formazan blue crystals. Primary WT and HD140Q/140Q neurons from E16 embryonic days were plated at 800,000 cells/ml on 24-well plates and cultured as above. At DIV4, primary neurons were changed into fresh complete NBM and infected with lentivirus expressing EGFP alone or Rab11Q70L and EGFP for 7 d, respectively. At DIV11, neurons were changed into fresh complete NBM and incubated with 0.5 mg ml−1 of MTT in complete NBM for 15 min at 37°C. After MTT incubation, the medium was carefully removed. Formazan blue was dissolved in 100% DMSO. Absorbance was read at 540 nm on a multiwell spectrophotometer. The mean absorbance unit of WT neurons (no infection) or WT neurons expressing EGFP alone (viral infection) was used for calculating the percentage of MTT conversion by neurons under the corresponding condition (no infection or viral infection). The mean percentage of WT or WT-EGFP for each condition with SD was graphed.
Cysteine uptake assay.
Primary neurons in six-well plates were infected with virus or not after AraC treatment and routinely cultured. Five days after infection of virus, primary neurons were washed twice in prewarmed PBS and cultured in PBS for 1 h at 37°C. l-[14C]Cysteine uptake was performed at room temperature for 30 s in PBS containing 1% BSA. The final concentrations of l-[14C]cysteine were 0.1, 1, 5, 10, and 50 μm, respectively, and the final concentration of DTT was 25 mm. l-[14C]Cystine (20 μCi/ml; PerkinElmer) was stored at −80°C after adding DTT to a final concentration of 50 mm DTT. Without addition of DTT to l-[14C]cystine, radioactivity was hardly detectable in the cell lysates after uptake was performed.
To determine that primary neuronal cultures take up cysteine through amino acid transporters instead of the glutamate–cystine exchanger system, cysteine uptake was conducted at room temperature for 60 s in 50 mm HEPES-K, pH 7.4, 146 mm NaCl, 5 mm MgCl2, and 0.2 m sucrose in the presence or the absence of 1 mm dl-threo-β-benzyloxyaspartate (TBOA) (Tocris Bioscience), and in 50 mm HEPES-K, pH 7.4, 100 mm KCl, 5 mm MgCl2, and 0.2 m sucrose. To determine the contribution of EAAT2/GLT-1 to neuronal uptake of cysteine, cysteine uptake was performed in the absence or the presence of 100 μm dihydrokainate (DHK) (Tocris Bioscience). Before cysteine uptake, primary neurons were cultured in PBS for 1 h in a cell culture incubator and washed twice in 50 mm HEPES-K, pH 7.4, 5 mm MgCl2, 1 mm DTT, and 0.2 m sucrose. The concentration of l-[14C]cysteine was 2 μm, and the concentration of DTT was 10 mm. After cysteine uptake for indicated times, primary neurons were washed three times in cold PBS and lysed in 0.2 ml of cold 50 mm HEPES-K, pH 7.4, 100 mm KCl, 2 mm EDTA, and 1 mm DTT containing 1% Triton X-100 (TX-100) on ice for 15 min. Twenty microliters of lysates were used for scintillation count in triplicate. Data were represented as cpm per milligram of protein per second.
Biotinylation of cell surface proteins.
Biotinylation of proteins at cell surfaces was conducted according to the manufacturer's instructions. Briefly, primary WT and HD140Q/140Q cortical neurons at DIV8 with or without viral infection at DIV4 were extensively washed in cold PBS, pH 8.0. After incubation in cold PBS on ice for 15 min, cells were changed into fresh cold PBS containing 1 mm sulfo-NHS-SS-biotin (Pierce) and incubated on ice for 4 h. After extensive washes in cold PBS, cells were incubated in 0.1 mm Tris-Cl, pH 8.0, to quench nonreacted sulfo-NHS-SS-biotin on ice for 30 min and lysed in lysis buffer (50 mm HEPES, pH 7.4, 200 mm NaCl, 10 mm MgCl2, 1% TX-100, and protease inhibitors). Crude lysates were cleared by centrifugation at 4°C, 14,000 rpm for 10 min. The resulting supernatants were incubated with preequilibrated NeutrAvidin agarose (Pierce) at 4°C with rotation for at least 2 h. After incubation, samples were centrifuged at 4°C, 1000 rpm for 1 min. The resulting supernatants (intracellular proteins: unbiotinylated) were precipitated with chloroform/methanol and resuspended in sample buffer for SDS-PAGE and Western blot analysis. NeutrAvidin agarose bound with cell surface proteins (biotinylated) were washed three times in lysis buffer and boiled in sample buffer for SDS-PAGE and Western blot analysis.
The band density of biotinylated EAAC1 (biotinylated) from each set (WT vs HD) of experiments was measured using SigmaScan Pro or NIH ImageJ. At the exposure necessary to detect the signal of biotinylated EAAC1 (biotinylated), the signal for intracellular EAAC1 (unbiotinylated) was saturated. Therefore, biotinylated EAAC1 values were normalized against intracellular actin to obtain equal input for each set of experiments. Normalized values of biotinylated EAAC1 were used to calculate percentage of biotinylated EAAC1 in HD140Q/140Q neurons relative to WT neurons.
SDS-PAGE and Western blot.
SDS-PAGE and Western blot analyses were performed as described previously (Li et al., 2009a). In brief, samples were boiled in SDS-PAGE sample buffer for 5 min and loaded on a Tris-glycine gel. After electrophoresis, proteins were transferred onto nitrocellulose blots. The membranes were blocked with 5% nonfat milk in TBS containing 0.05% Tween 20 and incubated with antibodies. Concentrations of the primary antisera used for Western blot were anti-EAAC1 (1:500; Millipore Bioscience Research Reagents) and anti-actin (1:500; Sigma-Aldrich). Peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories) were diluted at 1:5000. All antisera were diluted in TBS containing 1% BSA and 0.05% Tween 20. Blots were developed using enhanced ECL (Pierce).
Immunohistochemistry.
After two washes in PBS, primary neurons on coverslips were fixed in 4% paraformaldehyde/PBS at room temperature for 15 min, quenched in 50 mm NH4Cl containing 0.1% Triton X-100, washed twice in PBS containing 0.1% Triton X-100, blocked with 1% BSA in PBS containing 0.05% Tween 20, and labeled with primary antibodies. After the primary antibody step, targeted proteins were visualized by incubation of cells on coverslips with secondary antibodies conjugated with BODIPY FL (Invitrogen) or Cy-3 (Jackson ImmunoResearch Laboratories). Primary antibodies against Rab11 (1:500; ABR–Affinity BioReagents) and EAAC1 (1:100; Millipore Bioscience Research Reagents) were used. Secondary antibodies including Hoechst 33258 (Invitrogen) were diluted at 1:1000. All antisera were diluted in PBS containing 1% BSA and 0.05% Tween 20. Microscopy was performed using 60× or 100× oil Nikon Plan Apo objective mounted on an inverted Nikon Eclipse TE300 fluorescent microscope. Individual images for each wavelength (405, 488, and 568 nm) were collected separately using a Bio-Rad 2100 LaserSharp confocal system equipped with krypton–argon and blue diode lasers. The scanned images from each fluorescence channel were merged using PhotoShop.
Fluorescence intensity of immunoreactive EAAC1 signals in soma and neurites of neurons and the corresponding background signals were captured with confocal microscopy and measured using NIH ImageJ. The average size of soma of WT (n = 30) and HD140Q/140Q neurons (n = 30) in the samples was not different. The average area of all examined neurons (WT plus HD, 60 neurons) was used for normalizing the EAAC1 fluorescence intensity in the soma and neurites of neurons. The mean intensity of EAAC1 was graphed.
Statistical analysis.
Two-tailed Student's t test was performed to determine statistical significance between two research groups. One-way ANOVA and post hoc analysis were used for determination of statistical significance for multiple comparisons.
Results
A decline in levels of GSH in HD neurons precedes neuronal death
We examined GSH levels in primary WT and HD140Q/140Q mouse cortical neurons after culture for DIV8 using a GSH assay. This assay takes advantage of the selective reaction of OPA with GSH. OPA itself has a very low fluorescence but generates strong fluorescence signal after reaction with GSH. We found that HD140Q/140Q neurons had significantly lower levels of GSH compared with WT neurons (n = 3, mean ± SD, in nanomoles per milligram protein per minute, WT vs HD140Q/140Q: 10 ± 1.1 vs 7.2 ± 0.7; p < 0.01).
Since loss of intracellular GSH can cause neurodegeneration in vitro (Li et al., 1997; Nicole et al., 1998; Wüllner et al., 1999), we looked for clues of cell death of primary HD140Q/140Q neurons. MTT conversion assay showed that at DIV8 when GSH levels have already declined, HD140Q/140Q cortical neurons did not manifest significant death relative to WT neurons (mean ± SD of percentage of WT, WT vs HD140Q/140Q: 100 ± 3.5 vs 102.8 ± 4.4%; Student's t test, no significance) (Fig. 1). However, the percentage of MTT conversion by HD140Q/140Q neurons relative to WT neurons dropped to 66.7% at DIV11 (mean ± SD of percentage of WT, WT vs HD140Q/140Q: 100 ± 4.6 vs 66.7 ± 4.3%; Student's t test, p < 0.00001) (Fig. 1), indicating significant death of HD neurons at DIV11. Thus, a decline in GSH levels precedes neuronal death and may be a pathogenic mechanism in HD140Q/140Q mice.
Figure 1.

Reduction of GSH levels precedes neuronal death in HD. Neuron-enriched primary cultures were used for MTT conversion, which positively correlates with survival, to determine cell viability at DIV8 and DIV11 as described in Materials and Methods. Four separate experiments were performed using neurons prepared from embryos of four WT and four HD pregnant mice. In each experiment, MTT conversion was performed in each of a six-well culture plate containing three wells of WT neurons and three wells of HD neurons. The average production of formazan in absorbance units in the WT neuron cultures was used for calculating the percentage of survival in HD neuron cultures (n = 4 experiments; mean ± SD; Student's t test: ns, no significance; *p < 0.00001).
HD neurons have elevated levels of reactive oxygen species
ROS are normal by-products of oxidative phosphorylation and quickly removed by antioxidants, including GSH. Having shown that HD neurons had decreased levels of GSH, we next examined whether levels of ROS are different in HD140Q/140Q neurons compared with WT neurons at DIV8. Carboxy-DCFDA is a cell-permeable nonfluorescent dye that is oxidized into highly green fluorescent DCF in the presence of oxidants, including ROS. The intensity of fluorescence of DCF generated from carboxy-DCFDA is proportional to the amount of ROS in cells (Pérez-Severiano et al., 2004; Kirkland et al., 2007; Gerstner et al., 2008; Vaughn and Deshmukh, 2008). We found that both WT and HD140Q/140Q neurons exhibited DCF signal in the somata and neurites. The DCF signal in somata was much greater than in neurites (Fig. 2). Quantification of the fluorescence intensity revealed significantly higher levels of DCF signal in the somata and neurites of HD140Q/140Q neurons compared with those of WT neurons (n = 28 WT and 24 HD neurons, mean ± SD in average fluorescence intensity in the soma per neuron, WT vs HD140Q/140Q: 147 ± 39.7 vs 192 ± 27.0; p < 0.001; n = 33 WT and 21 HD neurons, mean ± SD in average fluorescence intensity in the process per length of a process, WT vs HD140Q/140Q: 0.068 ± 0.049 vs 0.208 ± 0.123; p < 0.001). The increase in DCF fluorescence intensity in combination with lower levels of GSH in HD140Q/140Q neurons (Fig. 1) indicate that the presence of mutant Htt caused a redox imbalance—reduction in GSH and accumulation of ROS—and oxidative stress.
Figure 2.

Elevation of ROS levels in HD neurons. To detect reactive oxygen species, live cortical neurons were incubated with carboxy-DCFDA at 37°C for 30 min at DIV8. After washes in PBS, live neurons were imaged using a confocal microscope as described in Materials and Methods. Shown are examples of confocal images of live neurons containing DCF fluorescence. Scale bar, 25 μm.
HD neurons are deficient in taking up cysteine
Next, we looked for a mechanism of deficient GSH in HD cortical neurons. We examined uptake of cysteine, which is required for de novo synthesis of GSH in neurons. Uptake of radiolabeled l-cysteine was conducted at room temperature for 30 s. In the presence of low concentrations of l-cysteine (0.1, 1, or 5 μm), both WT and HD140Q/140Q neuron-enriched cultures took up cysteine with a similar rate. However, HD140Q/140Q neuron-enriched cultures took up significantly less cysteine than WT neuronal cultures did with the concentration of l-cysteine at 10 and 50 μm (mean ± SD of cpm per milligram protein per second, WT vs HD140Q/140Q: 10 μm, 775 ± 86.6 vs 296 ± 43.3; 50 μm, 945 ± 88.6 vs 511 ± 44.6; p < 0.01) (Fig. 3A). Thus, insufficient supply of cysteine can contribute to low GSH levels in HD neurons.
Figure 3.

Deficient uptake of cysteine and reduced cell surface levels of EAAC1 in HD neurons. A, At DIV8, primary cortical neurons in six-well culture plates were used for performing l-[14C]cysteine uptake at room temperature for 30 s with l-[14C]cysteine concentration of 0.1, 1, 5, 10, and 50 μm, respectively, and harvested for preparation of cell lysates. The amount of l-[14C]cysteine in cell lysates for each cysteine concentration was determined by scintillation count in triplicate and represented as mean cpm per milligram per second with SD (two-tailed Student's t test, *p < 0.01). WT neurons were prepared from three pregnant mice, and HD neurons were prepared from three pregnant mice. B, Biotinylation of cell surface proteins of neurons in a 60 mm culture dish was conducted as in Materials and Methods. Biotinylated cell surface proteins and the corresponding unbiotinylated intracellular proteins were analyzed by SDS-PAGE and Western blot with indicated antibodies. Shown is one blot analysis of three experiments. The broad migration of EAAC1 signal, which is also found in previous studies (Grewer et al., 2000), may be attributable to glycosylation. Biotinylated EAAC1 at the cell surface (arrow) is detected at a longer exposure of Western blot film (middle panel).
The inhibitor TBOA markedly inhibits l-cysteine uptake in cortical neurons (Chen and Swanson, 2003). Addition of TBOA to the uptake buffer reduced uptake of l-cysteine by >90% in WT and HD cultures (supplemental Fig. S1A, available at www.jneurosci.org as supplemental material). As previously shown for primary cortical neurons, cysteine uptake through glutamate transporters is Na-dependent (Chen and Swanson, 2003). The glial glutamate–cystine exchanger is a sodium-independent transporter that exchanges intracellular glutamate for extracellular cystine and is absent in neurons (Aoyama et al., 2008). Replacing sodium with potassium in the uptake buffer reduced l-cysteine uptake by >90% in both WT and HD neuron-enriched cultures (supplemental Fig. S1A, available at www.jneurosci.org as supplemental material). Glial cells were sparse (<0.5%) in our neuron-enriched cultures. The requirement for sodium to take up cysteine suggests that the glutamate–cystine exchanger is not contributing to the deficient uptake of cysteine in HD neurons.
Cultured neurons have been reported to express glial glutamate transporter EAAT2/GLT-1 (Chen et al., 2002, 2004; Chen and Swanson, 2003). To address the contribution of EAAT2/GLT-1 to cysteine uptake in our neuronal cultures, we treated neurons with EAAT2/GLT-1 selective inhibitor, DHK, at a concentration of 100 μm as previously described (Chen and Swanson, 2003). DHK treatment caused a similar and small reduction in uptake of cysteine in WT and HD neurons compared with untreated cultures (n = 3; mean ± SD of percentage of untreatment, WT vs HD140Q/140Q: 90 ± 16 vs 88 ± 7.9%; one-tailed Student's t test, p = 0.44), suggesting that in our cultures EAAT2/GLT1 transporter does not contribute significantly to cysteine uptake. Our results with DHK differ from those of Chen and Swanson (2003) who reported that 100 μm DHK reduced cysteine uptake to 60–65% of untreated primary neuronal cultures. However, they indicated that their neuronal cultures have ∼4% glial cells, whereas our cultures have only 0.5% cells positive for glial markers. In cultures with more glia, cysteine uptake would occur through glial localized EAAT2/GLT1 transporter and be more inhibited by the presence of DHK.
Specific reduction of EAAC1 at the cell surface in HD neurons
The neuronal transporter EAAC1 was originally identified as a glutamate transporter. It was later determined that EAAC1 is the primary route for cysteine uptake (Aoyama et al., 2006), with 10- to 20-fold more affinity to cysteine than GLT-1 or GLAST (glutamate–aspartate transporter) (Zerangue and Kavanaugh, 1996). Since HD140Q/140Q cortical neurons had impaired uptake of cysteine and cortical neurons express EAAC1 (Conti et al., 1998), we investigated whether the distribution of EAAC1 at cell surfaces is altered in HD140Q/140Q neurons by an in vitro biotinylation assay, which is widely used to study cell surface levels of proteins including overexpressed EAAC1 (González et al., 2007). To inhibit endocytosis of biotinylated EAAC1, the biotinylation assay was performed on ice so that biotinylated EAAC1 maximally represented the steady-state levels of EAAC1 at the cell surface. Western blot analysis showed that the signal of intracellularly localized actin was detected mainly in the unbiotinylated samples (Fig. 3B, bottom panel, unbiotinylated), suggesting that the biotinylation reagent did not enter the cytoplasm. The levels of actin signal were comparable in WT and HD samples, indicating that similar numbers of WT and HD neurons were used in the assay. Consistent with a predominantly intracellular localization of EAAC1 (Rothstein et al., 1994; Conti et al., 1998; He et al., 2000; Nieoullon et al., 2006), we found that the majority of EAAC1 was unbiotinylated (Fig. 3B, top and middle panels). Immunoreactive signals of unbiotinylated EAAC1 in HD140Q/140Q neurons appeared comparable with those in WT neurons (Fig. 3B, top panel), suggesting that mutant Htt does not change the protein level of EAAC1. At optimal exposure of Western blot films for detecting intracellular EAAC1 (Fig. 3B, top panel), the signal for biotinylated (cell surface) EAAC1 was not detected in either WT or HD neurons. However, when the exposure time was prolonged (Fig. 3B, middle panel), levels of biotinylated (cell surface) EAAC1 were detected in both WT and HD samples but were significantly lower in HD140Q/140Q neurons than those in WT neurons. These data suggest that HD neurons have less EAAC1 distributed at the cell surface.
Endogenous EAAC1 recycles through Rab11-positive endosomes and accumulates in HD neurons
Exogenously expressed EAAC1 recycles through Rab11-positive recycling endosomes (González et al., 2007). To determine whether endogenous EAAC1 also recycles through Rab11-positive endosomes, we double labeled WT and HD140Q/140Q neurons that were cultured on glass coverslips with anti-EAAC1 and anti-Rab11 antibodies. In agreement with previous observations in normal primary hippocampal and cortical neurons (Coco et al., 1997; González et al., 2007), we found that, in both WT and HD140Q/140Q cortical neurons, immunoreactivity of endogenous EAAC1 occurred mainly at punctate structures in the cytoplasm of soma and neurites. Immunoreactive endogenous EAAC1 and endogenous Rab11 colocalized in some structures (Fig. 4), suggesting that, like overexpressed mycEAAC1, endogenous EAAC1 traffics through Rab11-positive endosomes. Immunoreactive EAAC1 was more concentrated in somata of HD140Q/140Q cortical neurons than in somata of WT neurons (Fig. 4). Quantification of fluorescent signals revealed a significant increase in somata of HD140Q/140Q cortical neurons compared with WT neurons (n = 30 WT and 30 HD140Q/140Q neurons; mean ± SD in fluorescence intensity, WT vs HD: 52 ± 3.8 vs 62 ± 4.9; Student's t test, p < 0.0001). Together with biotinylation data in Figure 3, it is suggested that, compared with WT neurons, HD neurons have less EAAC1 on the cell surface and more in the cytoplasm.
Figure 4.

EAAC1 traffics through Rab11-positive endosomes and accumulates in the cytoplasm of HD neurons. EAAC1 and Rab11 colocalize in WT and HD neurons. Primary cortical neurons prepared from WT and HD140Q/140Q mouse embryos were fixed and processed for immunolabeling with antibodies against EAAC1 (green) and Rab11 (red). The merged signals show areas of colocalization in yellow. Shown are confocal images of one neuron from a WT culture and one neuron from a HD culture. The arrowhead points to accumulation of immunoreactive EAAC1 in the soma of an HD140Q/140Q neuron. The boxed regions contain part of a neurite and are enlarged below. The arrows in the merged photos indicate loci along the neurite where there is colabeling of EAAC1 and Rab11 (yellow).
Expression of dominant-active Rab11 increases cysteine levels and intracellular GSH in HD neurons
The cell surface abundance of mycEAAC1 in glioma cells relies on normal Rab11 activity (González et al., 2007). Rab11 activation is diminished in the brain of HD140Q/140Q knock-in mice (Li et al., 2009a) and in skin fibroblasts established from HD patients (Li et al., 2009b). Therefore, we asked whether increasing Rab11 activity by expressing dominant-active Rab11Q70L could increase cell surface targeting of EAAC1 and consequently rescue the deficit in cysteine uptake and elevate levels of GSH in HD neurons. Expression of Rab11Q70L/EGFP or EGFP alone did not alter the protein level of EAAC1 in WT and HD neurons (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). However, when compared with expression of EGFP alone, viral expression of dominant-active Rab11Q70L significantly increased levels of biotinylated EAAC1 in HD neurons (mean ± SD, percentage of WT-EGFP, HD-EGFP vs HD-Rab11Q70L: 57 ± 13 vs 97 ± 11; p = 0.016) (Fig. 5A). We also observed a slight but not significant increase of biotinylated EAAC1 in WT neurons after expression of Rab11Q70L compared with expression of EGFP alone (mean ± SD, percentage of WT-EGFP, WT-Rab11Q70L: 116 ± 10.8; p = 0.1244) (Fig. 5A). In normal cells, there may be a mechanism for controlling the cell surface abundance of proteins within a certain range. Nonetheless, the biotinylation data suggest that enhancement of Rab11 activity can improve cell surface targeting of EAAC1 in neurons.
Figure 5.

Increase of the Rab11 activity accelerates EAAC1 trafficking, improves cysteine uptake, and elevates GSH levels in primary neurons. Primary cortical neurons in six-well culture plates were infected with lentivirus expressing EGFP alone or lentivirus bicistronically expressing Rab11Q70L/EGFP for 4 d and used for analyses. A, Expression of Rab11Q70L increases cell surface levels of EAAC1. Biotinylation of cell surface proteins and quantification of biotinylated EAAC1 were conducted as in Figure 3B. The left panel shows blot analysis of biotinylated samples from one of three experiments, and the right panel illustrates quantification of biotinylated EAAC1 after expression of EGFP alone or Rab11Q70L/EGFP (n = 3 wells of neurons for each; mean ± SD, percentage of WT-EGFP; Student's t test: ns, no significance; *p < 0.05). WT neurons were prepared from three pregnant mice, and HD neurons were prepared from three pregnant mice. B, Expression of Rab11Q70L enhances cysteine uptake. l-[14C]cysteine uptake was performed, counted in triplicate for each cysteine concentration, and represented as mean cpm per milligram per second with SD as in Figure 3A. WT neurons were prepared from three pregnant mice, and HD neurons were prepared from three pregnant mice. The data in Figure 3A were plotted here as WT-no virus and HD-no virus, respectively, to facilitate the direct comparison. C, Effect of expressing Rab11Q70L on levels of GSH in primary neurons. After infection with virus, primary cortical neurons were lysed to measure levels of glutathione. The average concentration in nanomoles per milligram protein was plotted (n = 3; mean ± SD; one-way ANOVA: *p < 0.01, **p < 0.00001). To facilitate the direct comparison, data described in Results (GSH) were plotted here as no-virus. Neurons were studied in one set of cultures from embryos of WT and HD pregnant mice with each genotype in three culture dishes.
In accordance with elevation of EAAC1 at cell surfaces, expression of Rab11Q70L enhanced both WT and HD140Q/140Q cortical neurons to take up cysteine in the presence of 10 or 50 μm cysteine compared with neurons without viral infection or with viral expression of EGFP alone (mean ± SD, cpm per milligram protein per second, one-way ANOVA, WT neurons: p < 0.001 for both 10 and 50 μm cysteine; HD140Q/140Q: p < 0.00001 for both 10 and 50 μm cysteine) (Fig. 5B). Cysteine uptake was elevated to the level of WT in HD neurons after viral expression of Rab11Q70L (Fig. 5B). There were also significantly higher levels of intracellular GSH in the presence of Rab11Q70L/EGFP compared with expression of EGFP alone or no viral infection in both WT neurons (sixfold increase) and HD neurons (threefold increase) (mean ± SD, nanomoles per milligram protein, one-way ANOVA, p < 0.00001 for both WT-Rab11Q70L and HD-Rab11Q70L) (Fig. 5C). We noticed that there was no linear relationship between enhanced cysteine uptake and increased GSH levels on expression of Rab11Q70L in primary neurons. This is because constitutive expression of Rab11Q70L over several days might lead to cumulative buildup of GSH inside of neurons, which cannot be reflected by a transient measurement of cysteine uptake at certain time points. Collectively, increasing the activity of Rab11 accelerated targeting of EAAC1 to the cell surface normalized the uptake of cysteine and increased levels of GSH in HD neurons.
Expression of dominant-active Rab11 enhances clearance of reactive oxygen species
Next, we examined the effect of expressing dominant-active Rab11 (Rab11Q70L) on levels of ROS in WT and HD neurons. Primary neurons were infected at DIV4 with lentivirus expressing EGFP or Rab11Q70L/EGFP and stained with carboxy-DCFDA at DIV8. The signals for EGFP were low and comparable in WT and HD neurons (data not shown). After treatment with carboxy-DCFDA, fluorescence signals increased markedly in cell bodies of WT and HD neurons. The increase in fluorescence compared with that of EGFP alone was defined as the DCF fluorescence. In the presence of EGFP alone, HD cortical neurons had higher levels of ROS (DCF signal) than WT neurons expressing EGFP alone or Rab11Q70L/EGFP (mean ± SD, fluorescence intensity in soma per neuron; one-way ANOVA, p < 0.001) (Fig. 6A,B), indicating that lentiviral expression of EGFP alone did not change the clearance of ROS in HD neurons relative to WT neurons. In the presence of Rab11Q70L expression, levels of DCF fluorescence in the somata of HD neurons significantly declined compared with HD neurons expressing EGFP alone (mean ± SD, fluorescence intensity in soma per neuron, EGFP vs Rab11Q70L: 173 ± 35.9 vs 139 ± 34.6; p < 0.01) (Fig. 6B). The ROS levels in HD neurons expressing Rab11Q70L were not significantly different compared with those in WT neurons expressing Rab11Q70L/EGFP (Fig. 6B). These data suggest that increasing Rab11 activity helps clear ROS in HD neurons.
Figure 6.
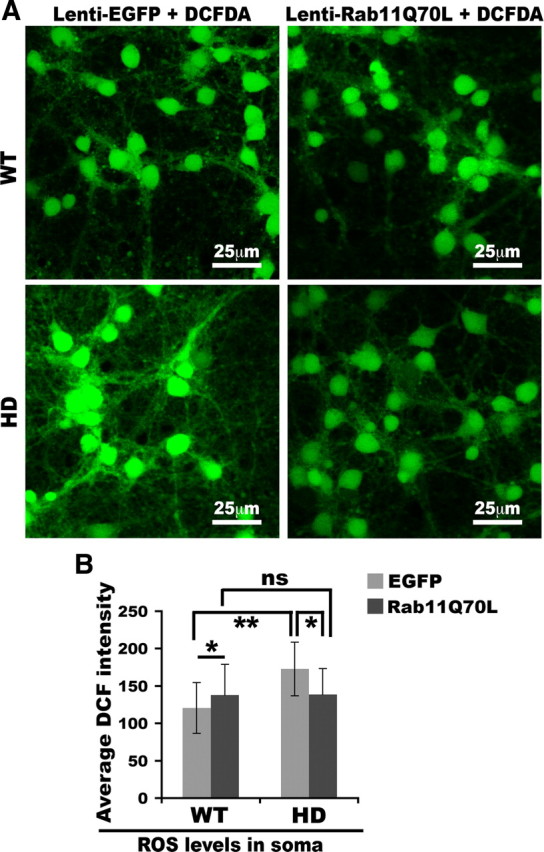
Expression of Rab11Q70L enhances clearance of ROS in HD neurons. Primary cortical neurons were infected at DIV4 with lentivirus expressing EGFP alone or lentivirus expressing Rab11Q70L/EGFP. At DIV8, virus-infected primary neurons were treat with DCFDA to assess ROS levels as described in Materials and Methods. Cells expressing EGFP alone (not shown here) had low fluorescent signals that were of comparable levels in WT and HD neurons. Thus, EGFP signals did not interfere with our quantitative analysis of DCF signals. A, Shown are examples of confocal images of DCF-labeled neurons. B, Bar graph shows quantification of intensity of fluorescence generated by carboxy-DCFDA after reaction with ROS and EGFP in the soma. Mean fluorescence intensity with SD was graphed (n = 53 WT-EGFP, 95 WT-Rab11Q70L, 46 HD-EGFP, and 84 HD-Rab11Q70L; one-way ANOVA: ns, no significance; *p < 0.01, **p < 0.001). Note that there is no significance (ns) between WT-Rab11Q70L and HD-Rab11Q70L. Neurons were from two sets of cultures from embryos of two WT and two HD pregnant mice.
WT neurons expressing Rab11Q70L had increased levels of DCF fluorescence compared with WT neurons expressing EGFP alone (mean ± SD, fluorescence intensity in soma per neuron, EGFP vs Rab11Q70L: 120 ± 34.0 vs 138 ± 40.8; p < 0.01) (Fig. 6). Increasing Rab11 activity in neurons that have normal Rab11 activity (WT neurons) may have different effects compared with effects in neurons that have diminished Rab11 activity.
Expression of dominant-active Rab11 promotes neuronal survival
We established that expression of Rab11Q70L rescues a deficit in cysteine uptake, increases GSH levels, and lowers ROS levels in HD neurons. We examined whether expression of dominant-active Rab11Q70L can promote survival of HD neurons at the steady state. The MTT assay revealed that viral expression of Rab11Q70L in neurons starting at DIV4 led to increased survival in HD neurons at DIV11 compared with neurons expressing EGFP alone (one-way ANOVA, p < 0.001) (Fig. 7). Notably, introducing Rab11Q70L into HD neurons restored the survival of HD neurons to levels of WT neurons (mean ± SD, percentage of WT-EGFP; one-way ANOVA, no significance) (Fig. 7). Thus, neuronal survival in primary HD neuronal cultures was improved by increasing Rab11 activity.
Figure 7.

Expression of Rab11Q70L promotes neuronal survival. Primary cortical neurons at DIV4 were untreated or infected with lentivirus expressing EGFP alone or lentivirus bicistronically expressing EGFP and Rab11Q70L. At DIV11, virus-treated primary neurons were used for the MTT assay to determine cell viability as in Figure 1. The average production of formazan in absorbance units from four cultures of WT neurons with infection of lenti-EGFP was used for calculating the percentage of the survival rate of WT-Rab11Q70L, HD-EGFP, and HD-Rab11Q70L neurons. The bar graph shows mean percentage with SD of surviving HD neurons relative to WT neurons infected with lenti-EGFP (n = 4; one-way ANOVA: ns, no significance; *p < 0.001).
Discussion
Indications of oxidative stress are apparent in HD, but the origins of oxidative stress are not established. GSH is a major antioxidant in the brain (Dringen, 2000). It is a tripeptide formed by glutamic acid, cysteine, and glycine. Depletion of intracellular GSH leads to death of neuronal cells in vitro (Li et al., 1997; Nicole et al., 1998; Wüllner et al., 1999), suggesting that maintenance of normal levels of GSH is essential for neuronal survival. Neurons require de novo synthesis of GSH, which involves two enzymatic reactions and the uptake of the rate-limiting precursor cysteine from the extracellular space (Dringen, 2000; Aoyama et al., 2008). Neurons take up cysteine via the neuronal Na+-dependent glutamate transporter EAAC1 (Shanker et al., 2001; Aoyama et al., 2006). In this study, we show that HD neurons had insufficient supply of the essential substrate cysteine for de novo synthesis of GSH because of aberrant trafficking of EAAC1 and accumulated intracellular ROS before the detection of cell death and/or mitochondrial dysfunction. These findings provide support for a novel mechanism of oxidative stress in HD neurons with normally expressed full-length mutant Htt protein and suggest that oxidative stress is an important pathogenic factor in HD.
Under normal circumstances, an overproduction of ROS usually elicits a compensatory increase in cysteine uptake for generation of more GSH, in which the cysteine residue provides the thiol group to inactivate ROS. In contrast, we found cysteine uptake to be reduced in HD140Q/140Q neurons compared with WT neurons, thereby impairing inactivation of ROS. Hence, our data suggest that deficient levels of GSH in HD neurons arise from an inadequate supply of cysteine for GSH synthesis, thereby causing intracellular accumulation of ROS in neurons. Of course, we cannot rule out the possibility that deficient GSH synthesis and ROS overproduction are independent events occurring contemporaneously in HD140Q/140Q neurons. Even so, deficient GSH would contribute to an increased ROS content.
In eukaryotic cells including neurons, the mitochondrion is one of the major consumers of GSH because of its role in generating ROS (Starkov, 2008). A moderate decline in GSH levels caused by a deficit in cysteine uptake could over time escalate oxidative stress in all subcellular organelles that rely on the antioxidant function of GSH, including mitochondria. We found here by MTT assays that mitochondria dysfunction occurred later than the decline in GSH levels in primary neuron-enriched cultures (Fig. 1). Mitochondria dysfunction occurs in advanced HD but not in the early stages of the disease (Guidetti et al., 2001). Rodents and primates treated with 3-nitropropionic acid (3-NP) to compromise the mitochondrial function show phenotypes reminiscent of advanced HD in humans (Beal et al., 1993; Brouillet et al., 1993, 1995). These observations suggest that impairment of mitochondrial function is important in the progression of the disease. The appearance of mitochondrial dysfunction later in the disease may be the result of a steady buildup of ROS and increase in oxidative stress from a chronic deficiency in GSH synthesis. It will be important to know in future studies what subcellular organelle(s) and molecules are affected by deficient synthesis of GSH in the early stages of HD as a way to identify therapeutic targets.
Wyttenbach et al. (2002) showed that expression of Htt exon-1 with elongated CAG repeats in cells induces an elevation in ROS levels and a decline in GSH levels. They proposed that the excess of ROS occurs because mutant Htt exon-1 causes mitochondria dysfunctions, which are major ROS generators (Benchoua et al., 2006; Solans et al., 2006; Fukui and Moraes, 2007; Oliveira et al., 2007; Starkov, 2008). They suggest that the depletion of GSH is a downstream effect of excess ROS. Based on our analysis of primary neurons that express endogenous full-length mutant Htt, we suggest that mutant Htt causes a deficiency in GSH synthesis because of insufficient supply of cysteine and thereby elevates ROS. The HD cell model used by Wyttenbach et al. (2002) may be more representative of a model of late-stage HD when mitochondria are known to be involved. In our model in primary neurons the changes observed in the presence of endogenous full-length mutant Htt might be more representative of early stages of HD.
EAAC1 is the main route for neurons to take up cysteine (Aoyama et al., 2006). Thus, any factor that causes a reduction in cell surface EAAC1 can lower cysteine uptake and subsequently affect GSH levels. It is unlikely that the decrease of cysteine uptake in HD140Q/140Q neurons results from a decrease in the overall protein level of EAAC1 because WT and HD140Q/140Q neurons had comparable levels of EAAC1 in the cytoplasm, where EAAC1 is mainly localized (Rothstein et al., 1994; Conti et al., 1998; He et al., 2000). Unlike glial glutamate transporters in glial cells, only a small proportion of EAAC1 in neurons is present at the cell surface (Rothstein et al., 1994; Conti et al., 1998; He et al., 2000). This distribution pattern of EAAC1 is similar to that of transferrin receptor, which constitutively recycles through Rab11-positive recycling endosomes. In agreement with findings using overexpressed EAAC1 (González et al., 2007), we show that the trafficking of endogenous EAAC1 is also regulated through Rab11-positive endosomes. In HD140Q/140Q cortical neurons, immunoreactive EAAC1 was accumulated in the soma compared with those of WT cortical neurons, suggesting that HD140Q/140Q neurons have a deficit in the regulation of EAAC1 trafficking.
Recent studies report that the glial transporter EAAT-2/GLT-1 is also present in neurons where it can mediate cysteine uptake (Chen et al., 2002, 2004; Chen and Swanson, 2003). We found that treatment with the EAAT2/GLT-1 selective inhibitor DHK (100 μm) had a similar and relatively modest inhibitory effect on cysteine uptake in WT and HD neurons. Thus, it is unlikely that EAAT-2/GLT-1 has a significant contribution to defective cysteine uptake in HD neurons. Furthermore, EAAT2/GLT-1 has 10- to 20-fold lower affinity for cysteine than does EAAC1 (Zerangue and Kavanaugh, 1996). Overall, our data suggest that the deficient uptake of cysteine results from improper targeting of EAAC1 to the cell surface of HD neurons.
That impaired endosomal recycling of EAAC1 compromises the capacity of HD neurons to synthesize GSH is a novel pathogenic mechanism to explain oxidative stress in HD. Interestingly, mutations in vps13A, a protein involved in endosomal recycling, causes autosomal recessive chorea acanthocytosis (Rampoldi et al., 2001; Ueno et al., 2001), a late-onset progressive neurodegenerative disorder closely resembling HD. EAAC1 trafficking requires normal Rab11 activity (González et al., 2007). We have observed defective Rab11 activation in HD (Li et al., 2009a,b). Accordingly, introducing dominant-active Rab11Q70L into HD cortical neurons normalized cysteine uptake, increased GSH synthesis, enhanced removal of ROS, and improved neuronal survival. Expression of Rab11Q70L in HD neurons could not restore cysteine uptake or GSH to the same levels in WT neurons expressing Rab11Q70L. It is possible that HD neurons may have deficits in other signaling pathways, which can regulate cell surface targeting of EAAC1 from endosomal compartments. Such signaling pathways include protein kinase A, protein kinase C, phosphatidylinositol 3-kinase and glial factors (Davis et al., 1998; Guillet et al., 2005; Akiduki and Ikemoto, 2008; Lortet et al., 2008). Future studies will be needed to know whether these signaling pathways are impaired in HD neurons.
How dominant-active Rab11Q70L provides the beneficial effect to HD neurons is not clear. One possibility is that enhancement of Rab11 activity accelerated the overall recycling rate of EAAC1 (Fig. 5A). It is known that Rab11 regulates trafficking of a variety of other critical proteins and cholesterol, so that other Rab11 effects might be involved. Brain proteins that are known Rab11-dependent cargos include transferrin receptor (iron uptake), brain-enriched Na+/H+ exchanger NHE5 (cell volume and pH maintenance) (Diering et al., 2009), glycine transporter GLYT2 (glycine uptake) (Núñez et al., 2009), glucose transporter 1 (glucose uptake) (Wieman et al., 2007), and AMPA receptor (long-term potentiation) (Park et al., 2004). The presence of Rab11Q70L in HD neurons may also improve functions related to these proteins.
We found that HD cortical neurons had a deficit in cysteine uptake. The involvement of insufficient cysteine supply in HD pathogenesis has indirect support. Treatment of R6/2 mice with cystamine significantly improves both motor performance and survival (Dedeoglu et al., 2002; Bailey and Johnson, 2006). Cystamine is an organic disulfide formed from the dimer of cysteine, cystine, through decarboxylation. Recent studies show that administration of cystamine raises cysteine levels in the brain of both R6/2 mice (Fox et al., 2004) and transgenic mice expressing full-length mutant Htt (YAC128 mice) (Pinto et al., 2005). Cystamine also protects 3-NP-induced mitochondrial dysfunction in immortal Hdh111Q striatal cells (Mao et al., 2006). Furthermore, treatment of rats with N-acetylcysteine, a membrane-permeable form of cysteine, before administration of 3-NP significantly reduces 3-NP-induced striatal injury (Fontaine et al., 2000). Regardless of the mechanisms of action, approaches that increase cysteine levels in neurons may be effective for HD.
In summary, our study supports the idea that the presence of mutant Htt causes a deficit in targeting of the neuronal glutamate/cysteine transporter EAAC1 to the cell surface and this in turn impairs uptake of extracellular cysteine and hampers generation of glutathione to handle ROS in neurons. Given that EAAC1 also mediates uptake of glutamate, we predict that HD GABAergic neurons, including medium spiny neurons, may be defective in synthesis of inhibitory neurotransmitter GABA with glutamate as the substrate. Our study suggests that impaired recycling of EAAC1 is attributable to deficient Rab11 activity. We have previously shown that mutant Htt inhibits guanine nucleotide exchange on Rab11 in brains of young HD knock-in mice (Li et al., 2009a). Manipulation of Rab11 activity may be beneficial for slowing the progression of HD. Future studies will be necessary to investigate ways to enhance Rab11 activity in HD neurons in vivo.
Footnotes
This work was supported by National Institutes of Health Grant NS038194 (N.A.) and the Huntington's Disease Society of America (M.D.). X.L. was supported by a John J. Wasmuth Postdoctoral Fellowship from the Hereditary Disease Foundation. A.V. was supported by fellowships from Fundacion Mexico en Harvard, A.C., and the Hereditary Disease Foundation. This publication was made possible by Grant 5P30 DK32520 from the National Institute of Diabetes and Digestive and Kidney Diseases. We thank Dr. Brian Healy of the Massachusetts General Hospital Biostatistics Center for his advice on the data analysis.
The authors declare no competing financial interests.
References
- Akiduki S, Ikemoto MJ. Modulation of the neural glutamate transporter EAAC1 by the addicsin-interacting protein ARL6IP1. J Biol Chem. 2008;283:31323–31332. doi: 10.1074/jbc.M801570200. [DOI] [PubMed] [Google Scholar]
- Aoyama K, Suh SW, Hamby AM, Liu J, Chan WY, Chen Y, Swanson RA. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat Neurosci. 2006;9:119–126. doi: 10.1038/nn1609. [DOI] [PubMed] [Google Scholar]
- Aoyama K, Watabe M, Nakaki T. Regulation of neuronal glutathione synthesis. J Pharmacol Sci. 2008;108:227–238. doi: 10.1254/jphs.08r01cr. [DOI] [PubMed] [Google Scholar]
- Bailey CD, Johnson GV. The protective effects of cystamine in the R6/2 Huntington's disease mouse involve mechanisms other than the inhibition of tissue transglutaminase. Neurobiol Aging. 2006;27:871–879. doi: 10.1016/j.neurobiolaging.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Ballatori N, Krance SM, Notenboom S, Shi S, Tieu K, Hammond CL. Glutathione dysregulation and the etiology and progression of human diseases. Biol Chem. 2009;390:191–214. doi: 10.1515/BC.2009.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal MF, Brouillet E, Jenkins BG, Ferrante RJ, Kowall NW, Miller JM, Storey E, Srivastava R, Rosen BR, Hyman BT. Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci. 1993;13:4181–4192. doi: 10.1523/JNEUROSCI.13-10-04181.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benchoua A, Trioulier Y, Zala D, Gaillard MC, Lefort N, Dufour N, Saudou F, Elalouf JM, Hirsch E, Hantraye P, Déglon N, Brouillet E. Involvement of mitochondrial complex II defects in neuronal death produced by N-terminus fragment of mutated huntingtin. Mol Biol Cell. 2006;17:1652–1663. doi: 10.1091/mbc.E05-07-0607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouillet E, Jenkins BG, Hyman BT, Ferrante RJ, Kowall NW, Srivastava R, Roy DS, Rosen BR, Beal MF. Age-dependent vulnerability of the striatum to the mitochondrial toxin 3-nitropropionic acid. J Neurochem. 1993;60:356–359. doi: 10.1111/j.1471-4159.1993.tb05859.x. [DOI] [PubMed] [Google Scholar]
- Brouillet E, Hantraye P, Ferrante RJ, Dolan R, Leroy-Willig A, Kowall NW, Beal MF. Chronic mitochondrial energy impairment produces selective striatal degeneration and abnormal choreiform movements in primates. Proc Natl Acad Sci U S A. 1995;92:7105–7109. doi: 10.1073/pnas.92.15.7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Aoki C, Mahadomrongkul V, Gruber CE, Wang GJ, Blitzblau R, Irwin N, Rosenberg PA. Expression of a variant form of the glutamate transporter GLT1 in neuronal cultures and in neurons and astrocytes in the rat brain. J Neurosci. 2002;22:2142–2152. doi: 10.1523/JNEUROSCI.22-06-02142.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Mahadomrongkul V, Berger UV, Bassan M, DeSilva T, Tanaka K, Irwin N, Aoki C, Rosenberg PA. The glutamate transporter GLT1a is expressed in excitatory axon terminals of mature hippocampal neurons. J Neurosci. 2004;24:1136–1148. doi: 10.1523/JNEUROSCI.1586-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Swanson RA. The glutamate transporters EAAT2 and EAAT3 mediate cysteine uptake in cortical neuron cultures. J Neurochem. 2003;84:1332–1339. doi: 10.1046/j.1471-4159.2003.01630.x. [DOI] [PubMed] [Google Scholar]
- Coco S, Verderio C, Trotti D, Rothstein JD, Volterra A, Matteoli M. Non-synaptic localization of the glutamate transporter EAAC1 in cultured hippocampal neurons. Eur J Neurosci. 1997;9:1902–1910. doi: 10.1111/j.1460-9568.1997.tb00757.x. [DOI] [PubMed] [Google Scholar]
- Conti F, DeBiasi S, Minelli A, Rothstein JD, Melone M. EAAC1, a high-affinity glutamate tranporter, is localized to astrocytes and gabaergic neurons besides pyramidal cells in the rat cerebral cortex. Cereb Cortex. 1998;8:108–116. doi: 10.1093/cercor/8.2.108. [DOI] [PubMed] [Google Scholar]
- Davis KE, Straff DJ, Weinstein EA, Bannerman PG, Correale DM, Rothstein JD, Robinson MB. Multiple signaling pathways regulate cell surface expression and activity of the excitatory amino acid carrier 1 subtype of Glu transporter in C6 glioma. J Neurosci. 1998;18:2475–2485. doi: 10.1523/JNEUROSCI.18-07-02475.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedeoglu A, Kubilus JK, Jeitner TM, Matson SA, Bogdanov M, Kowall NW, Matson WR, Cooper AJ, Ratan RR, Beal MF, Hersch SM, Ferrante RJ. Therapeutic effects of cystamine in a murine model of Huntington's disease. J Neurosci. 2002;22:8942–8950. doi: 10.1523/JNEUROSCI.22-20-08942.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diering GH, Church J, Numata M. Secretory carrier membrane protein 2 regulates cell-surface targeting of brain-enriched Na+/H+ exchanger NHE5. J Biol Chem. 2009;284:13892–13903. doi: 10.1074/jbc.M807055200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dringen R. Metabolism and functions of glutathione in brain. Prog Neurobiol. 2000;62:649–671. doi: 10.1016/s0301-0082(99)00060-x. [DOI] [PubMed] [Google Scholar]
- Dringen R, Hamprecht B. Glutathione restoration as indicator for cellular metabolism of astroglial cells. Dev Neurosci. 1998;20:401–407. doi: 10.1159/000017337. [DOI] [PubMed] [Google Scholar]
- Fontaine MA, Geddes JW, Banks A, Butterfield DA. Effect of exogenous and endogenous antioxidants on 3-nitropionic acid-induced in vivo oxidative stress and striatal lesions: insights into Huntington's disease. J Neurochem. 2000;75:1709–1715. doi: 10.1046/j.1471-4159.2000.0751709.x. [DOI] [PubMed] [Google Scholar]
- Fox JH, Barber DS, Singh B, Zucker B, Swindell MK, Norflus F, Buzescu R, Chopra R, Ferrante RJ, Kazantsev A, Hersch SM. Cystamine increases l-cysteine levels in Huntington's disease transgenic mouse brain and in a PC12 model of polyglutamine aggregation. J Neurochem. 2004;91:413–422. doi: 10.1111/j.1471-4159.2004.02726.x. [DOI] [PubMed] [Google Scholar]
- Fukui H, Moraes CT. Extended polyglutamine repeats trigger a feedback loop involving the mitochondrial complex III, the proteasome and huntingtin aggregates. Hum Mol Genet. 2007;16:783–797. doi: 10.1093/hmg/ddm023. [DOI] [PubMed] [Google Scholar]
- Gerstner B, DeSilva TM, Genz K, Armstrong A, Brehmer F, Neve RL, Felderhoff-Mueser U, Volpe JJ, Rosenberg PA. Hyperoxia causes maturation-dependent cell death in the developing white matter. J Neurosci. 2008;28:1236–1245. doi: 10.1523/JNEUROSCI.3213-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González MI, Susarla BT, Fournier KM, Sheldon AL, Robinson MB. Constitutive endocytosis and recycling of the neuronal glutamate transporter, excitatory amino acid carrier 1. J Neurochem. 2007;103:1917–1931. doi: 10.1111/j.1471-4159.2007.04881.x. [DOI] [PubMed] [Google Scholar]
- Grewer C, Watzke N, Wiessner M, Rauen T. Glutamate translocation of the neuronal glutamate transporter EAAC1 occurs within milliseconds. Proc Natl Acad Sci U S A. 2000;97:9706–9711. doi: 10.1073/pnas.160170397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidetti P, Charles V, Chen EY, Reddy PH, Kordower JH, Whetsell WO, Jr, Schwarcz R, Tagle DA. Early degenerative changes in transgenic mice expressing mutant huntingtin involve dendritic abnormalities but no impairment of mitochondrial energy production. Exp Neurol. 2001;169:340–350. doi: 10.1006/exnr.2000.7626. [DOI] [PubMed] [Google Scholar]
- Guillet BA, Velly LJ, Canolle B, Masmejean FM, Nieoullon AL, Pisano P. Differential regulation by protein kinases of activity and cell surface expression of glutamate transporters in neuron-enriched cultures. Neurochem Int. 2005;46:337–346. doi: 10.1016/j.neuint.2004.10.006. [DOI] [PubMed] [Google Scholar]
- He Y, Janssen WG, Rothstein JD, Morrison JH. Differential synaptic localization of the glutamate transporter EAAC1 and glutamate receptor subunit GluR2 in the rat hippocampus. J Comp Neurol. 2000;418:255–269. [PubMed] [Google Scholar]
- Kirkland RA, Saavedra GM, Franklin JL. Rapid activation of antioxidant defenses by nerve growth factor suppresses reactive oxygen species during neuronal apoptosis: evidence for a role in cytochrome c redistribution. J Neurosci. 2007;27:11315–11326. doi: 10.1523/JNEUROSCI.3590-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klepac N, Relja M, Klepac R, Hećimović S, Babić T, Trkulja V. Oxidative stress parameters in plasma of Huntington's disease patients, asymptomatic Huntington's disease gene carriers and healthy subjects: a cross-sectional study. J Neurol. 2007;254:1676–1683. doi: 10.1007/s00415-007-0611-y. [DOI] [PubMed] [Google Scholar]
- Li X, Sapp E, Chase K, Comer-Tierney LA, Masso N, Alexander J, Reeves P, Kegel KB, Valencia A, Esteves M, Aronin N, Difiglia M. Disruption of Rab11 activity in a knock-in mouse model of Huntington's disease. Neurobiol Dis. 2009a;36:374–383. doi: 10.1016/j.nbd.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Standley C, Sapp E, Valencia A, Qin ZH, Kegel KB, Yoder J, Comer-Tierney LA, Esteves M, Chase K, Alexander J, Masso N, Sobin L, Bellve K, Tuft R, Lifshitz L, Fogarty K, Aronin N, DiFiglia M. Mutant huntingtin impairs vesicle formation from recycling endosomes by interfering with Rab11 activity. Mol Cell Biol. 2009b;29:6106–6116. doi: 10.1128/MCB.00420-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Maher P, Schubert D. A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron. 1997;19:453–463. doi: 10.1016/s0896-6273(00)80953-8. [DOI] [PubMed] [Google Scholar]
- Lortet S, Canolle B, Masmejean F, Nieoullon A. Plasma membrane expression of the neuronal glutamate transporter EAAC1 is regulated by glial factors: evidence for different regulatory pathways associated with neuronal maturation. Neurochem Int. 2008;52:1373–1382. doi: 10.1016/j.neuint.2008.02.007. [DOI] [PubMed] [Google Scholar]
- Mao Z, Choo YS, Lesort M. Cystamine and cysteamine prevent 3-NP-induced mitochondrial depolarization of Huntington's disease knock-in striatal cells. Eur J Neurosci. 2006;23:1701–1710. doi: 10.1111/j.1460-9568.2006.04686.x. [DOI] [PubMed] [Google Scholar]
- Myers RH, Vonsattel JP, Paskevich PA, Kiely DK, Stevens TJ, Cupples LA, Richardson EP, Jr, Bird ED. Decreased neuronal and increased oligodendroglial densities in Huntington's disease caudate nucleus. J Neuropathol Exp Neurol. 1991;50:729–742. doi: 10.1097/00005072-199111000-00005. [DOI] [PubMed] [Google Scholar]
- Nicole A, Santiard-Baron D, Ceballos-Picot I. Direct evidence for glutathione as mediator of apoptosis in neuronal cells. Biomed Pharmacother. 1998;52:349–355. doi: 10.1016/S0753-3322(99)80001-8. [DOI] [PubMed] [Google Scholar]
- Nieoullon A, Canolle B, Masmejean F, Guillet B, Pisano P, Lortet S. The neuronal excitatory amino acid transporter EAAC1/EAAT3: does it represent a major actor at the brain excitatory synapse? J Neurochem. 2006;98:1007–1018. doi: 10.1111/j.1471-4159.2006.03978.x. [DOI] [PubMed] [Google Scholar]
- Núñez E, Pérez-Siles G, Rodenstein L, Alonso-Torres P, Zafra F, Jiménez E, Aragón C, López-Corcuera B. Subcellular localization of the neuronal glycine transporter GLYT2 in brainstem. Traffic. 2009;10:829–843. doi: 10.1111/j.1600-0854.2009.00911.x. [DOI] [PubMed] [Google Scholar]
- Oliveira JM, Jekabsons MB, Chen S, Lin A, Rego AC, Gonçalves J, Ellerby LM, Nicholls DG. Mitochondrial dysfunction in Huntington's disease: the bioenergetics of isolated and in situ mitochondria from transgenic mice. J Neurochem. 2007;101:241–249. doi: 10.1111/j.1471-4159.2006.04361.x. [DOI] [PubMed] [Google Scholar]
- Park M, Penick EC, Edwards JG, Kauer JA, Ehlers MD. Recycling endosomes supply AMPA receptors for LTP. Science. 2004;305:1972–1975. doi: 10.1126/science.1102026. [DOI] [PubMed] [Google Scholar]
- Pérez-Severiano F, Santamaría A, Pedraza-Chaverri J, Medina-Campos ON, Ríos C, Segovia J. Increased formation of reactive oxygen species, but no changes in glutathione peroxidase activity, in striata of mice transgenic for the Huntington's disease mutation. Neurochem Res. 2004;29:729–733. doi: 10.1023/b:nere.0000018843.83770.4b. [DOI] [PubMed] [Google Scholar]
- Pinto JT, Van Raamsdonk JM, Leavitt BR, Hayden MR, Jeitner TM, Thaler HT, Krasnikov BF, Cooper AJ. Treatment of YAC128 mice and their wild-type littermates with cystamine does not lead to its accumulation in plasma or brain: implications for the treatment of Huntington disease. J Neurochem. 2005;94:1087–1101. doi: 10.1111/j.1471-4159.2005.03255.x. [DOI] [PubMed] [Google Scholar]
- Rampoldi L, Dobson-Stone C, Rubio JP, Danek A, Chalmers RM, Wood NW, Verellen C, Ferrer X, Malandrini A, Fabrizi GM, Brown R, Vance J, Pericak-Vance M, Rudolf G, Carrè S, Alonso E, Manfredi M, Németh AH, Monaco AP. A conserved sorting-associated protein is mutant in chorea-acanthocytosis. Nat Genet. 2001;28:119–120. doi: 10.1038/88821. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, Nash N, Kuncl RW. Localization of neuronal and glial glutamate transporters. Neuron. 1994;13:713–725. doi: 10.1016/0896-6273(94)90038-8. [DOI] [PubMed] [Google Scholar]
- Schulz JB, Lindenau J, Seyfried J, Dichgans J. Glutathione, oxidative stress and neurodegeneration. Eur J Biochem. 2000;267:4904–4911. doi: 10.1046/j.1432-1327.2000.01595.x. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Salazar FJ, Abraham C, Kosik KS. Huntington's disease: changes in striatal proteins reflect astrocytic gliosis. Brain Res. 1982;245:117–125. doi: 10.1016/0006-8993(82)90344-4. [DOI] [PubMed] [Google Scholar]
- Shanker G, Allen JW, Mutkus LA, Aschner M. The uptake of cysteine in cultured primary astrocytes and neurons. Brain Res. 2001;902:156–163. doi: 10.1016/s0006-8993(01)02342-3. [DOI] [PubMed] [Google Scholar]
- Sian J, Dexter DT, Lees AJ, Daniel S, Agid Y, Javoy-Agid F, Jenner P, Marsden CD. Alterations in glutathione levels in Parkinson's disease and other neurodegenerative disorders affecting basal ganglia. Ann Neurol. 1994;36:348–355. doi: 10.1002/ana.410360305. [DOI] [PubMed] [Google Scholar]
- Solans A, Zambrano A, Rodríguez M, Barrientos A. Cytotoxicity of a mutant huntingtin fragment in yeast involves early alterations in mitochondrial OXPHOS complexes II and III. Hum Mol Genet. 2006;15:3063–3081. doi: 10.1093/hmg/ddl248. [DOI] [PubMed] [Google Scholar]
- Starkov AA. The role of mitochondria in reactive oxygen species metabolism and signaling. Ann N Y Acad Sci. 2008;1147:37–52. doi: 10.1196/annals.1427.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno S, Maruki Y, Nakamura M, Tomemori Y, Kamae K, Tanabe H, Yamashita Y, Matsuda S, Kaneko S, Sano A. The gene encoding a newly discovered protein, chorein, is mutated in chorea-acanthocytosis. Nat Genet. 2001;28:121–122. doi: 10.1038/88825. [DOI] [PubMed] [Google Scholar]
- Valencia A, Morán J. Role of oxidative stress in the apoptotic cell death of cultured cerebellar granule neurons. J Neurosci Res. 2001;64:284–297. doi: 10.1002/jnr.1077. [DOI] [PubMed] [Google Scholar]
- Vaughn AE, Deshmukh M. Glucose metabolism inhibits apoptosis in neurons and cancer cells by redox inactivation of cytochrome c. Nat Cell Biol. 2008;10:1477–1483. doi: 10.1038/ncb1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieman HL, Wofford JA, Rathmell JC. Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol Biol Cell. 2007;18:1437–1446. doi: 10.1091/mbc.E06-07-0593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wüllner U, Seyfried J, Groscurth P, Beinroth S, Winter S, Gleichmann M, Heneka M, Löschmann P, Schulz JB, Weller M, Klockgether T. Glutathione depletion and neuronal cell death: the role of reactive oxygen intermediates and mitochondrial function. Brain Res. 1999;826:53–62. doi: 10.1016/s0006-8993(99)01228-7. [DOI] [PubMed] [Google Scholar]
- Wyttenbach A, Sauvageot O, Carmichael J, Diaz-Latoud C, Arrigo AP, Rubinsztein DC. Heat shock protein 27 prevents cellular polyglutamine toxicity and suppresses the increase of reactive oxygen species caused by huntingtin. Hum Mol Genet. 2002;11:1137–1151. doi: 10.1093/hmg/11.9.1137. [DOI] [PubMed] [Google Scholar]
- Zanella A, Izzo C, Meola G, Mariani M, Colotti MT, Silani V, Pellegata G, Scarlato G. Metabolic impairment and membrane abnormality in red cells from Huntington's disease. J Neurol Sci. 1980;47:93–103. doi: 10.1016/0022-510x(80)90028-3. [DOI] [PubMed] [Google Scholar]
- Zerangue N, Kavanaugh MP. Interaction of l-cysteine with a human excitatory amino acid transporter. J Physiol. 1996;493:419–423. doi: 10.1113/jphysiol.1996.sp021393. [DOI] [PMC free article] [PubMed] [Google Scholar]