

Abstract
The dynamics of cerebrospinal fluid (CSF) tau and Aβ biomarkers over time in Alzheimer’s disease (AD) patients from prodromal pre-symptomatic to severe stages of dementia have not been clearly defined and recent studies, most of which are cross-sectional, present conflicting findings. To clarify this issue, we analyzed the longitudinal CSF tau and Aβ biomarker data from 142 of the AD Neuroimaging Initiative (ADNI) study subjects [18 AD, 74 mild cognitive impairment (MCI), and 50 cognitively normal subjects (CN)]. Yearly follow-up CSF collections and studies were conducted for up to 48 months (median = 36 months) for CSF Aβ1–42, phosphorylated tau (p-tau181), and total tau (t-tau). An unsupervised analysis of longitudinal measurements revealed that for Aβ1–42 and p-tau181 biomarkers there was a group of subjects with stable longitudinal CSF biomarkers measures and a group of subjects who showed a decrease (Aβ1–42, mean = −9.2 pg/ml/year) or increase (p-tau181, mean = 5.1 pg/ml/year) of these biomarker values. Low baseline Aβ1–42 values were associated with longitudinal increases in p-tau181. Conversely, high baseline p-tau181 values were not associated with changes in Aβ1–42 levels. When the subjects with normal baseline biomarkers and stable concentrations during follow-up were excluded, the expected time to reach abnormal CSF levels and the mean AD values was significantly shortened. Thus, our data demonstrate for the first time that there are distinct populations of ADNI subjects with abnormal longitudinal changes in CSF p-tau181 and Aβ1–42 levels, and our longitudinal results favor the hypothesis that Aβ1–42 changes precede p-tau181 changes.
Keywords: Alzheimer’s disease, Amyloid beta, Tau, Cerebrospinal fluid, Longitudinal, Dementia, Mild cognitive impairment
Introduction
Recent discoveries have led to a revised hypothetical Alzheimer’s disease (AD) progression model which is characterized by sequential and overlapping changes in chemical and imaging AD biomarkers that reflect the underlying pathological changes in the brains of subjects at different stages in the onset and progression of AD [12]. Whereas these recent publications have described the longitudinal magnetic resonance imaging (MRI) changes and some studies describing serial parenchymal Aβ amyloid imaging measurements using positron emission tomography (PET) ligands, the data regarding cerebrospinal fluid (CSF) measurements of Aβ1–42, phosphorylated tau (p-tau181), and total tau (t-tau) are limited and most studies are cross-sectional or are based on only a few measurements per longitudinally followed subjects over limited periods of time [4, 5, 14, 17, 18, 21, 29]. Therefore, most efforts to confirm the CSF biomarker model proposed by Jack et al. [12] have been made using statistical synchronization techniques in sporadic AD cases [41] and cross-sectional data from subjects with autosomal dominant AD mutations with a known age of clinical dementia onset [1, 9, 27]. Recently, the Australian Imaging Biomarker and Lifestyle study of aging (AIBL) and the Mayo Clinic Study of aging have reported the temporal dynamics of Aβ amyloid deposition in the brain using Pittsburg B-compound (PiB) combined with positron emission tomography (PiB-PET) imaging [14, 37, 38]. Based on their results, Villemagne et al. proposed that in the groups with and without pathological levels of Aβ amyloid deposition there were two subgroups of subjects, i.e. one consisting of subjects who showed no changes and a second group that showed an increased deposition of Aβ amyloid over time. These results have not been validated in other PET or CSF based studies of Aβ pathology, and measures of CSF t-tau and p-tau181 were not incorporated into the studies of Villemagne et al.
Although lumbar punctures to obtain CSF for biomarker measurements are mildly invasive, they are considered relatively safe, acceptable to patients and controls [23, 24], and sampling CSF offers the possibility to measure several biomarkers at the time of lumbar puncture while CSF aliquots can be stored in biobanks for future studies of other biomarkers. Three proteins are currently regarded as established CSF biomarkers of AD pathology: Aβ1–42, t-tau and p-tau181. In our study, we sought to analyze the CSF biomarker dynamics in a cohort that included cognitively normal (CN), mild cognitive impairment (MCI) and early stage AD subjects that had their CSF sampled repeatedly annually up to a follow-up of 48 months.
Methods
Participants
Data used in the preparation of this article, data file UPENNBIOMK4.csv, were downloaded from the AD Neuroimaging Initiative (ADNI) database 5 March 2013. The ADNI was launched in 2004 by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering (NIBIB), the Food and Drug Administration, private pharmaceutical companies and non-profit organizations, which has been extensively reviewed elsewhere [40] (see additional information in http://www.adni-info.org and supplementary material). Here, we studied 142 subjects (50 CN, 74 MCI and 18 AD) belonging to ADNI 1 (see supplementary material and previous description of the cohort [25]) who volunteered to participate in an add-on longitudinal CSF biomarker study and who provided at least three longitudinal CSF samples after baseline CSF sampling (Tables 1, 2). 56 subjects did not give five CSF samples but continued the clinical follow-ups, 54 subjects were lost to follow-up before 48 months and 32 subjects gave CSF samples up to the fourth year’s visit. MCI diagnosis was established based on the Petersen criteria [25, 26] and the AD diagnosis was based on the National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer’s Disease and Related Disorders Association criteria for probable AD [20, 25].
Table 1.
Demographic and biomarker summary by diagnostic category at baseline
| Whole sample
|
p value | Mixture model sample
|
p value | |||||
|---|---|---|---|---|---|---|---|---|
| CN (n = 50) | MCI (n = 74) | AD (n = 18) | CN (n = 38) | MCI (n = 45) | AD (n = 6) | |||
| Age at baselinea | 76.2 (4.8) | 75.1(6.3) | 73.6 (6.6) | 0.25 | 76.2 (4.9) | 73.7 (6.6) | 73.8 (6.7) | 0.09 |
| Gender (male %) | 54.0 % | 72.6 % | 33.3 % | 0.006 | 47.4 % | 72.7 % | 33.3 % | 0.017 |
| APOE ε4 (%) | 22 % | 50.7 % | 77.8 % | <0.0001 | 26.3 % | 58.1 % | 83.3 % | <0.0001 |
| Education (years)a | 15.9 (3.0) | 16.1 (2.6) | 15.2 (2.9) | 0.30 | 15.5 (3.0) | 16.4 (2.8) | 14.7 (2.9) | 0.21 |
| ADAS-Coga | 9.1 (3.8) | 18.4 (6.0) | 28.1 (7.8) | <0.0001 | 9.0 (3.9) | 18.4 (6.0) | 31.6 (8.0) | <0.0001 |
| Aβ1–42 (pg/ml)b | 229.0 (156.3–254.8) | 141.0 (122.8–177.5) | 134.0 (113.0–146.0) | <0.0001 | 229 (150.3–260.0) | 142.5 (122.0–232.5) | 134.0 (125.8–134.8) | 0.0007 |
| T-tau (pg/ml)b | 66.0 (55.0–89.0) | 86.0 (62.0–129.0) | 129.5 (93.8–176) | <0.0001 | 68.0 (55.3–90.8) | 87.5 (65.8–123) | 134.0 (81.0–136.0) | 0.011 |
| P-tau181 (pg/ml)b | 22.5 (16.3–30.8) | 32.0 (23.0–41.0) | 56.5 (34.5–57.3) | <0.0001 | 23.0 (16.3–30.8) | 28.5 (21.5–40.0) | 36.0 (35.0–37.0) | 0.011 |
AD Alzheimer’s disease, ADAS-Cog Alzheimer’s disease Assessment Scale-cognitive subscale, CN cognitively normal, MCI mild cognitive impairment
Mean (Standard deviation)
Median (1st and 3rd quartiles)
Table 2.
Diagnoses for subjects with available CSF measurements in each visit
| Baseline CN | Baseline MCI | Baseline AD | |
|---|---|---|---|
| Baseline | 50 CN | 74 MCI | 18 AD |
| 12 Months | 48 CN 1 MCI | 1 CN 56 MCI 13 AD | 17 AD |
| 24 Months | 26 CN 2 MCI | 3 CN 31 MCI 20 AD | 18 AD |
| 36 Months | 28 CN 4 MCI | 3 CN 17 MCI 16 AD 1 Not available |
6 AD |
| 48 Months | 13 CN 1 MCI | 1 CN 5 MCI 10 AD | 2 AD |
AD Alzheimer’s disease, CN cognitively normal, MCI mild cognitive impairment
Biomarker collection and analysis
Baseline CSF samples were obtained in the morning after an overnight fast and processed as previously described [30, 31] (http://www.adni-info.org/). Briefly, CSF was collected into polypropylene collection tubes or syringes provided to each site, then transferred into polypropylene transfer tubes without any centrifugation step followed by freezing on dry ice within 1 h after collection, and shipped overnight to the ADNI Biomarker Core laboratory at the University of Pennsylvania Medical Center on dry ice. Aliquots (0.5 ml) were prepared from these samples after thawing (1 h) at room temperature and gentle mixing. The aliquots were stored in bar code-labeled polypropylene vials at −80 °C. Aβ1–42, t-tau, and p-tau181 were measured using the multiplex xMAP Luminex platform (Luminex Corp, Austin, TX) with Innogenetics (INNO-BIA AlzBio3; Ghent, Belgium; for research use-only reagents) immunoassay kit-based reagents. The capture monoclonal antibodies used were 4D7A3 for Aβ1–42, AT120 for t-tau and AT270 for p-tau181. The analyte-specific detector antibodies were HT7, for tau, and 3D6, for the N-terminus of Aβ (immunoassay performance details as described in Shaw et al. [31]). Fresh, never before thawed 0.5-mL aliquots for each subject’s set of longitudinal time points were analyzed on the same 96-well plate in the same analytical run for this study to minimize run-to-run and reagent kit lot sources of variation. Within-run coefficient of variation (%CV) for duplicate samples ranged from 2.5 to 5.9 % for Aβ1–42, 2.2–6.3 % for t-tau and 2.0–6.5 % for p-tau181 and the inter-run %CV for CSF pool samples ranged from 5.1 to 14 % for Aβ1–42, 2.7–11.2 % for t-tau and 3.3–11.5 % for p-tau181.
Statistical analysis
For the univariate group comparisons, ANOVA and Chi square tests were applied for quantitative and qualitative variables, respectively, and the Kruskall–Wallis test was applied for non-normally distributed variables. CSF biomarker changes for the finite mixture model analysis were calculated subtracting the CSF baseline visit values from last available measurement of the studied subjects, sampled 36–48 months after the baseline CSF draw (n = 89). We selected this interval to maximize the number of subjects and optimize the time between visits to analyze the changes during a longer period and capture changes and patterns that might be only found for longer follow-up periods. We hypothesized that the population of subjects that have normal CSF baseline values is a mixture of subjects who will show at least two distinct longitudinal profiles, namely those who will stay stable and those who will decrease (for Aβ1–42) or increase (for t-tau and p-tau181) their measured values indicating that parenchymal deposition of tau and Aβ into tangles and plaques, respectively, is taking place. Therefore, a finite mixture model [10, 11] was fitted to the CSF biomarker change values and the Bayesian information criterion was used to select the best model. In addition, we fitted power models [2] to estimate the yearly CSF changes associated with a specific CSF value and we derived the expected CSF biomarker changes using the modified Euler method [33].
We constructed a mixed-effects model [15] to quantify patterns of CSF biomarkers longitudinally from time of entry into the study. Our model specified the intercept and the regression coefficient for the follow-up time as subject-specific random variables. Thus, follow-up time was treated as a random effect in addition to being treated as a fixed-effect (see below). The population mean coefficient for the follow-up time was estimated by averaging across the subject-specific regression coefficients for the follow-up time. Follow-up time was treated as a continuous variable. Fixed effects in the model included follow-up time, baseline clinical diagnosis, APOE ε4 presence, age at baseline and baseline biomarkers. The interaction of clinical diagnosis and APOE with follow-up time was included. A second mixed-effects model studied the association of baseline and longitudinal biomarker values with the AD Assessment Scale-cognitive subscale (ADAS-Cog) scores in a model adjusted for age at baseline, clinical diagnosis at baseline and education (fixed factors) and the intercept and the regression coefficient for the follow-up time were included as subject-specific random variables. The interaction of clinical diagnosis and APOE genotype with follow-up time was also included in this model. All statistics tests were two-sided. The local significance level was set to 0.05. p values ≤0.05 were considered significant. No adjustment for multiple testing was performed. Therefore, an overall significance level is not determined and results with p values close to the significance threshold should be considered exploratory.
Results
Demographic data
Median follow-up time between the baseline and last CSF sample was 35.8 months. Table 1 provides demographic information for the study cohort and the number of measurements and diagnoses at each visit is summarized in Table 2. Biomarker concentration values at the different visits, stratified by baseline clinical diagnosis, are represented in Fig. 1 and Supplementary Fig. 1. Education and age were similar across the three baseline diagnosis groups, but gender, Alzheimer’s disease Assessment Scale-cognitive subscale (ADAS-Cog), APOE genotype and CSF values differed between groups.
Fig. 1.

Longitudinal group changes with 95 % confidence intervals for Aβ1–42 (a), t-tau (c) and p-tau181 (e). CSF levels are displayed based on clinical diagnosis at baseline [red cognitively normal subjects (CN), green mild cognitive impairment subjects (MCI) subjects, blue Alzheimer’s disease (AD) subjects]. Individual subject longitudinal changes for Aβ1–42 (b), t-tau (d) and p-tau181 (f) CSF levels with colors identifying the baseline clinical category
Longitudinal changes in CSF Aβ1–42, t-tau and p-tau181
We first analyzed the data to determine if there were different rates of CSF biomarker changes for Aβ1–42, t-tau and p-tau181 during the onset and progression of AD. We selected subjects whose last CSF sample was obtained between 36 and 48 months after the first baseline visit CSF sample (n = 89) and subtracted the baseline visit value from the last visit value. These absolute changes in biomarker concentrations during follow-up were plotted versus the corresponding baseline visit values (Fig. 2). The local regression (LOESS) is represented for each of the different biomarkers. Aβ1–42 values showed a u-shaped distribution showing a faster decrease during follow-up in the middle range of baseline values, whereas p-tau181 values showed a faster increase in subjects with abnormal baseline values. T-tau values overlapped with the dashed line in Fig. 2 indicating that they remained stable during follow-up. In addition, subjects with high Aβ1–42 values at the baseline visit (normal range) showed a large scatter in the y-axis (change during follow-up) (Fig. 2a). T-tau and p-tau181 changes showed a modest correlation (r = −0.41, p = 0.0001), but Aβ1–42 changes did not correlate with t-tau changes (r = −0.04, p = 0.69) or p-tau181 changes (r = −0.13, p = 0.23). In this ADNI cohort, only one subject with normal baseline visit Aβ1–42 values had one or more APOE ε4 copies (Fig. 2a).
Fig. 2.

Baseline visit Aβ1–42 (a), t-tau (b) and p-tau181 (c) CSF values (x-axis) against their yearly change (y-axis) during a follow-up of 36–48 months for cognitively normal subjects (CN in red), mild cognitive impairment subjects (MCI in green) subjects and Alzheimer’s disease (AD in blue) subjects. Open versus closed circles indicate the absence or presence of an APOE ε4 allele in each diagnostic group
Finite mixture modeling of CSF biomarker changes
The large scatter of Aβ1–42 changes centered in the group with normal baseline values suggests the possibility that there are two or more populations of subjects with different rates of Aβ1–42 change. To test this hypothesis, we fitted a finite mixture model to the Aβ1–42 biomarker changes independent of baseline visit values. For this analysis, yearly Aβ1–42 changes based on the last visit were calculated by dividing the absolute biomarker change by the follow-up time measured in years for each subject. As shown in Fig. 3a, changes in CSF Aβ1–42 levels showed a bimodal distribution with a group of values centered at −0.5 pg/ml/year [Aβ1–42 stable group (Aβ-SG); 49 subjects had abnormal baseline values and 20 had normal baseline values] and a second group of values centered at −9.2 pg/ml/year [Aβ1–42 decrease group (Aβ-DG); 6 subjects had abnormal baseline values and 14 subjects had normal baseline values]. T-tau change showed a unimodal normal distribution centered at 1.7 pg/ml/year (Fig. 3b). Finally, p-tau181 change showed bimodal distribution with a group of values centered at 1.5 pg/ml/year [P-tau181 stable group (PT-SG); 22 had abnormal baseline p-tau181 values and 24 had had normal baseline p-tau181 values] and a second group centered at 5.1 pg/ml/year [P-tau181 increase group (PT-IG); 34 had abnormal baseline p-tau181 values and 6 had had normal baseline p-tau181 values] (Fig. 3c). We present the clinical and demographic characteristics of these groups in Supplementary Tables 2 and 3.
Fig. 3.
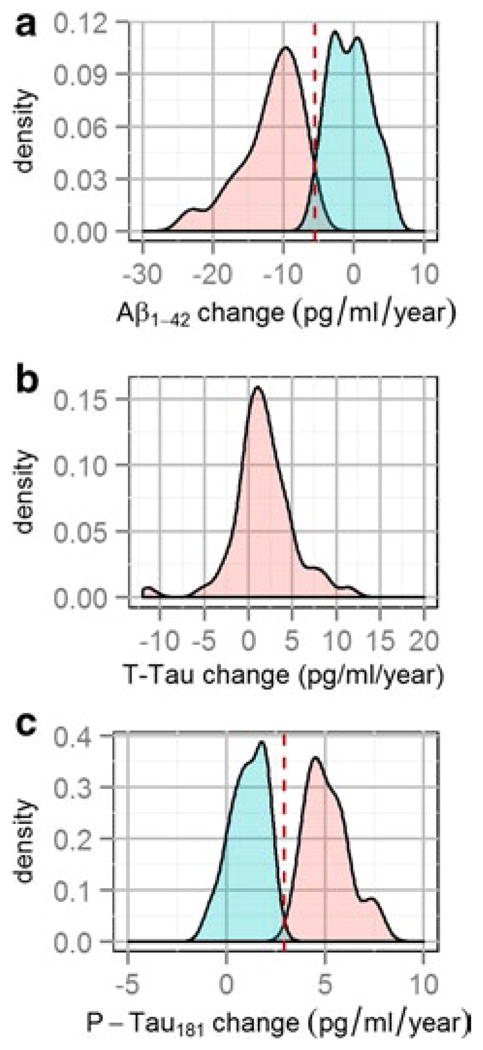
Finite mixture modeling of of Aβ1–42 (a), t-tau (b) and p-tau181 (c) yearly changes
To test the chronology of Aβ1–42 and p-tau181 changes, we asked if baseline abnormal values of Aβ1–42 predicted longitudinal changes of p-tau181 and t-tau and vice versa, in age, gender and APOE-adjusted linear regression models. Only the presence of abnormal baseline Aβ1–42 values predicted the increase of p-tau181 during follow-up (t = −2.7, p = 0.009), but neither baseline p-tau181 (t = −0.8, p = 0.43) nor t-tau (t = −1.6, p = 0.13) baseline visit values predicted changes of Aβ1–42 values (for the tau analyses predicting Aβ1–42 changes the Aβ-SG subjects with abnormal baseline values were excluded). We asked if our analysis could identify which Aβ1–42 values predicted a faster increase of p-tau181, but with the available data points we were not able to obtain a reliable estimate (data not shown).
Modeling of longitudinal CSF changes
We estimated the functions that modeled the yearly change of CSF Aβ1–42 and p-tau181 based on their baseline visit values (Fig. 4a, b). For each of the biomarkers we calculated two models; the first one was based on all the subjects (blue lines, filled and empty circles) and the second model (red lines, only filled circles) took into account the results of the finite mixture model, i.e. we excluded subjects with normal baseline visit biomarker values who did not show biomarker changes during follow-up. We developed the second model to project changes in subjects who are changing to avoid the averaging subjects who remain stable during follow-up. For Aβ1–42 values, the time to reach the abnormal cutpoints, i.e. 192 pg/ml, starting at 295 pg/ml was 18.6 years if the model with all subjects was used (blue), whereas this time shortened to 8.6 years when the model that excluded the Aβ-SG (empty circles, red line) was applied (Fig. 4c). Similarly in the p-tau181 model, the time to reach the p-tau181 pathological threshold shortened from 6.2 to 2.9 years when PT-SG (empty circles) was excluded (Fig. 4d). The time to reach the median Aβ1–42 values present in the AD group was 35.6 years for the model with all the subjects and 26.3 years for the model that excludes the Aβ-SG with normal Aβ1–42 baseline values. For p-tau181, the expected times were 10 (all subjects) and 6 years (excluding PT-SG), respectively. Supplementary Tables 4 and 5 list the expected values for each year in each model.
Fig. 4.

Aβ1–42 (a) and p-tau181 (b) yearly changes versus baseline values. The open circles represent subjects with normal baseline values who remain stable during follow-up and the filled circles represent subjects with abnormal baseline values and/or changes during follow-up. The blue line models all the subjects (i.e. filled and closed circles) while the red line only the subjects with abnormal baseline values and/or changes during follow-up (filled circles). Panels c and d illustrate the estimated trajectories of Aβ1–42 (c) and p-tau181 (d) and the estimated time to reach the AD cutpoint threshold for CSF Aβ1–42 and p-tau181 based on the model that includes all subjects (blue) or only those represented by the filled (red) circles with abnormal baseline values or changes during follow-up
Longitudinal CSF analysis using mixed-effects model analysis
We then quantified the yearly changes applying a mixed-effects model analysis that analyzed the association of different predictors with baseline values and longitudinal changes of the three studied CSF biomarkers (n = 142, Table 3). The predictors were clinical diagnosis, age at baseline and APOE. For the model that studied Aβ1–42 we included baseline p-tau181 values as a predictor, whereas in the models that studied t-tau and p-tau181 we included baseline Aβ1–42 as predictor to study the association between these biomarkers.
Table 3.
Mixed-effects model adjusting for baseline biomarkers: effects of baseline diagnostic group and follow-up time
| Diagnosis MCI
|
Diagnosis AD
|
APOE ε4
|
Baseline Aβ1–42
|
Baseline p-Tau181
|
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Baseline | Change | Baseline | Change | Baseline | Change | Baseline | Change | Baseline | Change | |
| Aβ1–42 | t = −2.8; p = 0.006 | t = 0.6.; p = 0.54 | t = −2.1; p = 0.03 | t = 0.6; p = 0.54 | t = −4.9; p < 0.0001 | t = 1.0; p = 0.31 | – | – | t = −4.5; p < 0.0001 | t = 0.6; p = 0.52 |
| T-Tau | t = 2.0; p = 0.046 | t = −0.5; p = 0.62 | t = 2.6; p = 0.009 | t = −2.2; p = 0.03 | t = 1.7; p = 0.08 | t = 0.48; p = 0.63 | t = −2.3; p = 0.02 | t = −0.78; p = 0.44 | – | – |
| P-Tau181 | t = 1.2; p = 0.25 | t = 0.4; p = 0.67 | t = 1.1; p = 0.25 | t = 0.1; p = 0.90 | t = 1.9; p = 0.06 | t = −0.3; p = 0.80 | t = −4.3; p < 0.0001 | t = −2.2; p = 0.03 | – | – |
AD Alzheimer’s disease, MCI mild cognitive impairment
MCI and AD diagnoses were associated with lower and higher levels of Aβ1–42 and t-tau at baseline, respectively. On the other hand, baseline clinical diagnosis was not associated with baseline p-tau181. However, MCI and AD clinical diagnoses were significantly associated with increased p-tau181 levels when baseline Aβ1–42 was not included in the model (data not shown). Aβ1–42 (t = −3.3, p = 0.001) and p-tau181 (t = 3.1, p = 0.002) showed a significant decrease and increase in the serial measurements, respectively; however clinical diagnostic groups did not differ in the rate of change (Table 3). T-tau values showed a non-significant increase in the serial CSF measurements (t = 1.4, p = 0.15) and the AD group showed a significantly decreased rate compared to CN subjects. Although presence of one or more APOE ε4 copies was associated with baseline Aβ1–42 values and a trend existed for the association with t-tau and p-tau181, it did not show any association with longitudinal changes of the studied biomarkers. Baseline Aβ1–42 values predicted p-tau181 increases during follow-up, but neither baseline t-tau nor p-tau181 levels predicted Aβ1–42 longitudinal changes.
Longitudinal cognitive analysis using mixed-effects model analysis
Low baseline Aβ1–42 (t = −3.1, p = 0.002) and high baseline t-tau (t = 2.2, p = 0.030) and p-tau181 (t = 2.8, p = 0.005) values were associated with cognitive decline during follow-up, as measured by the ADAS-Cog, while changes in these biomarker values (Aβ1–42: t = 0.69, p = 0.49; t-tau: t = −0.99, p = 0.33; p-tau181: t = 26, p = 0.80) were not associated with cognitive change in the model adjusted for age, education and baseline diagnosis. Last, we tested the association between the groups defined by Aβ1–42 baseline values and their longitudinal changes (Fig. 3b) and cognitive changes. In this analysis, the only group which showed a cognitive decline during follow-up was the one that comprised subjects who had abnormal baseline Aβ1–42 values that did not further decrease, because the plateau was reached (t = 2.55, p = 0.011). Supplementary Tables 6–8 summarize the presence of longitudinal changes in biomarkers stratifying by baseline values and longitudinal clinical diagnosis.
Discussion
Using an unsupervised analysis we detected two distinct populations of ADNI subjects with normal baseline visit Aβ1–42 and p-tau181 values, who showed different rates of change during serial CSF measurements. Based on these results, the timeframe to reach the abnormal Aβ1–42 and p-tau181 cutpoints [30] was shorter in the model that excluded subjects with normal baseline and longitudinally stable CSF values, and the trajectories did not follow a sigmoid trajectory. In addition, changes in p-tau181 occurred in a shorter period than changes in Aβ1–42. Low baseline Aβ1–42 values predicted a greater p-tau181 increase during the follow-up period here, but high baseline p-tau181 values were not predictive of any Aβ1–42 changes. Baseline values, but not longitudinal changes of biomarker levels, were associated with cognitive changes during follow-up.
Our unsupervised finite mixture model analysis detected the presence of two distinct trends in longitudinally measured CSF Aβ1–42, and p-tau181 biomarkers in ADNI subjects with normal baseline values. A similar approach has been applied to define AD diagnostic cutpoints based on CSF biomarker values [7] and for the analysis of PIB-PET longitudinal changes [37]. Our findings agree with the notion that a population of individuals with normal baseline biomarker values consists of two subgroups, namely those who will remain stable and those whose CSF Aβ and tau biomarker values change by becoming abnormal which, in the later situation, we interpret to reflect the progressive deposition of Aβ into amyloid plaques and the formation neurofibrillary tangles composed of pathological tau in the brains of these ADNI subjects [34]. When the Aβ–SG subjects with normal baseline values were excluded, the model did not follow a sigmoid function and the time to reach abnormal Aβ1–42 levels shortened from 18.6 to 8.6 years. Similar results were observed for p-tau181 although the time to reach the abnormal cutpoints was much shorter, namely 6.2 and 2.9 years for the model that included all subjects and the one that excluded the PT–SG, respectively. Recently, two cross-sectional studies in patients with autosomal dominant familial AD (FAD) mutations and a known expected time to onset of dementia have indicated that Aβ1–42 CSF differences between carriers and non-carriers of pathogenic FAD mutations are already present two decades before the onset of dementia [1, 27]. However, it is not known if these results can be extrapolated to sporadic AD cases, as it has been shown that the mechanisms responsible for the accumulation of Aβ deposits differ in autosomal dominant FAD and sporadic AD cases [19, 32]. Four studies have described longitudinal Aβ deposition in the brain using PIB-PET imaging in a larger number of cases [13, 14, 37, 38] with a mean follow-up of 1.5–2 years, although the last AIBL study had a mean follow-up of 3.8 years. Villain et al. [37] described “Aβ accumulators” and “Aβ non-accumulators” in their study which showed a u-shaped distribution for measures of Aβ change when plotted against baseline values and they reported a higher percentage of “Aβ accumulators” in subjects classified as PIB positive. In our ADNI cohort, the highest percentage of subjects in the Aβ-DG had normal CSF Aβ1–42 values at baseline (χ2 = 7.9, p = 0.005, 41 and 13 % Aβ-DG in those with normal and abnormal CSF Aβ1–42 values, respectively). This might be attributed to clinical differences in the subjects recruited for the two cohorts (i.e. comprised mostly of CN subjects in the study by Villain et al. and MCI in our study), differences in cutpoints for the assessment of Aβ amyloid pathology, or the fact that PIB-PET detects insoluble fibrillar Aβ, whereas the CSF Aβ1–42 assay measures soluble non-fibrillar species of Aβ1–42 although a high correlation between measures of Aβ plaque burden and CSF Aβ1–42 levels has been reported [16, 36]. Villemagne et al. [39] described an association between the APOE ε4 allele and a greater Aβ accumulation, although their analysis was neither adjusted for baseline diagnosis or other covariates, and when they stratified by clinical diagnosis the association was not significant. A nearly significant association was observed in a more recent study, but the model was not adjusted for baseline PIB-PET values which are higher in subjects with one or more copies of the APOE ε4 allele [38]. Indeed, Jack et al. [14] recently modeled PIB-PET, and when the model they used to study the rate of Aβ amyloid deposition was adjusted for baseline PIB-PET values, the effect of the APOE ε4 allele was not significant in agreement with our results. Our results favor the hypothesis that harboring at least one APOE ε4 allele advances in time the onset of Aβ accumulation in line with the effect observed on age of clinical onset of AD [6].
Notably, the model described by Villain et al. [37] describes an exponential increase in PIB-PET values without a plateau and the model by Jack et al. proposed a sigmoid-shaped increase with a plateau, which was also present in the study by Villemagne [38]. However, in our study, when we included all ADNI samples with 3 years of follow-up (including subjects with normal baseline values who showed no changes), we found a logarithmic shaped curve for changes in the levels of Aβ1–42 and or p-tau181, but when we excluded the ADNI subjects with normal values and stable course the shape changed for both biomarkers. The plateau only persisted for the Aβ1–42 model, whereas the p-tau181 model did not reach a plateau when the PT-SG subjects with normal baseline value were excluded (Fig. 4d). In our study, the AD group was under-represented, especially for the longer follow-up. Therefore, our sample might not have had enough AD subjects with a long follow-up to assess the p-tau181 changes which take place at a later stage than the Aβ1–42 changes. These observations emphasize the importance of the process of building accurate models of biomarker trajectories of the systematic collection of longitudinal biomarker data within individual cognitively normal subjects and long-term follow-up through the various stages of progression to mild impairment and dementia.
When we studied the association between baseline and longitudinal changes of these CSF biomarkers, we found an association between abnormal baseline Aβ1–42 values and p-tau181 increases during follow-up, whereas neither baseline t-tau nor p-tau181 was associated with an Aβ1–42 decrease during follow-up. T-tau and p-tau181 changes were moderately correlated, but none of these changes were associated with Aβ1–42 changes. This favors the hypothesis that pathological changes in Aβ1–42 level precede those for p-tau181 although is it well known that tau deposits in brain as neurofibrillary tangles many years before Aβ deposits as amyloid plaques, although the reasons for these discrepancies in biomarker versus pathology data are not clear [8].
Clinical diagnosis was associated with baseline of Aβ1–42, t-tau and p-tau181 (the latter only in the model that was not adjusted for baseline Aβ1–42 values), and the APOE genotype was associated with baseline Aβ1–42 (a trend was present for t-tau and p-tau181). However, the only association with longitudinal changes was a decrease in t-tau in AD subjects.
Information from longitudinal CSF studies that also include t-tau and p-tau181 measurements is more limited. Buchhave [5] reported that in subjects followed for up to 9.2 years baseline Aβ1–42 values did not differ between early and late MCI converters. On the other hand, early converters showed higher baseline t-tau and p-tau181 values than late converters. These results suggest the pathological changes in Aβ1–42 levels plateau before or around the onset of the dementia in AD patients. Several studies have compared baseline values with a second CSF measurement obtained 2–3 years later [4, 21, 29]. These studies mainly compared changes based on clinical diagnosis; however, the use of clinical diagnosis that is not neuropathologically validated might be prone to false positive results because of the assumption that all MCI and AD cases will have only AD pathology, but this assumption is incorrect in 7–17 % of AD patients followed to autopsy while the percentage of those with MCI who do or do not have AD pathology is less clear [3, 22, 28, 35]. In addition, baseline biomarker levels have been shown to be the strongest predictor of dementia in our study and in previous PET studies [14]. Some of these CSF biomarker studies reported increases in t-tau and p-tau181 [4] were larger in AD subjects [29], while most studies described a decrease of Aβ1–42 values although Bouwman et al. [4] described an increase. Seppälä reported that only the presence of an APOE ε4/ε4 genotype (and not ε3/ε4) was associated with Aβ1–42 decreases or p-tau181 increases. Mattsson [18] followed a cohort of 30 MCI subjects and collected three CSF samples that were separated 2 years from each other, and although their study mainly focused on classification of clinical progression of MCI subjects, they found no significant changes in CSF values over time, except for p-tau which showed an increase in some subjects.
In our study, we analyzed CSF biomarker dynamics based on their baseline value and also studied the association between CSF tau and Aβ biomarkers. However, when we studied the association with baseline clinical diagnosis in the mixed-effect models, clinical diagnosis was not associated overall with longitudinal changes in CSF biomarker values except a decrease in t-tau in the AD group. However, in the absence of neuropathological validation of the clinical diagnosis, it is not possible to ascertain that these subjects do not have an underlying cause for the cognitive impairment other than or in addition to AD [35], but the presence of AD-like CSF signature in CN subjects indicates that CSF biomarker changes precede clinical changes and there might be a temporal dissociation between both.
There are conflicting results regarding the association between longitudinal biomarker changes and the APOE ε4 genotype. In our sample, only one subject with one or more APOE ε4 alleles had normal baseline Aβ1–42 values, and most subjects with one or more APOE ε4 alleles were in the Aβ-SG with abnormal Aβ1–42 baseline values. These results point to the fact that the APOE ε4 allele can be associated with a decrease of Aβ1–42 at younger ages, in line with the clinical observation that individuals with an APOE ε4 allele have a younger onset of dementia. Our sample is composed of elderly subjects so, therefore, we cannot confirm if the presence of an APO ε4 is allele associated with changes starting at a younger age, a faster rate of change or both. That said, in our analysis we found that baseline biomarker values were the strongest predictors of the longitudinal cognitive changes. Therefore, differences reported in previous studies may arise from the sampling of subjects at different stages of the process and the composition of the studied cohort (age and distribution of cognitive groups in the sample). In order to assess the specific longitudinal changes associated with APOE ε4 allele and dissociate these changes from a difference in baseline biomarker values an analytical adjustment is necessary. This was the case in the recent study published by Jack et al. [14] that described how the association between an APOE ε4 genotype and longitudinal change in PIB-PET deposition became not significant when the model was adjusted for baseline values.
Lastly, a study by Le Bastard confirmed that in CN and AD subjects CSF levels of biomarkers are stable over several weeks [17] and, therefore, changes over time can be expected to reflect progression of the underlying AD neuropathology and not fluctuation of the biomarkers. Finally, in our study, CSF tau and Aβ biomarker changes were not associated with changes in the ADAS-Cog scale during follow-up, and of the three groups defined based on baseline and longitudinal Aβ1–42 changes, only the Aβ-SG with abnormal baseline Aβ1–42 levels showed worse cognitive outcomes during follow-up, which is consistent with the actual disease model [12].
In conclusion, our results on the analysis of longitudinal data on CSF tau and Aβ show that (1) CSF Aβ1–42 changes precede p-tau181 changes; (2) in the normal range of CSF biomarker values, there are distinctive subpopulations of subjects with a different longitudinal course; (3) factoring this variable modifies the expected modeling of biomarker changes. However, further studies of longitudinal changes in CSF tau and Aβ are needed and it is especially important to study longitudinal changes in CSF tau and Aβ in younger populations to establish when these CSF biomarker start to become abnormal and establish how changes are linked mechanistically to the APOE ε4 genotype as well last to the clinical manifestations of accumulating AD pathology.
Supplementary Material
Acknowledgments
This study was supported in part by the NIH/NIZ (AG10124 and AG24904). JQT is the William Maul Measey-Truman G. Schnabel, Jr., Professor of Geriatric Medicine and Gerontology and JBT is supported in part by a Grant of the Alfonso Martín Escudero Foundation. We would like to thank Xiaoyan Han, M.S. for her help with the statistical programming. J.B.T., S.X.X., J.Q.T. and LMS have no conflicts of interest. Data collection and sharing for this project were funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; BioClinica, Inc.; Biogen Idec Inc.; Bristol-Myers Squibb Company; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; GE Healthcare; Innogenetics, N.V.; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Medpace, Inc.; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Synarc Inc.; and Takeda Pharmaceutical Company. The Canadian Institutes of Health Research are providing funds to support ADNI clinical sites in Canada. Private sector contributions are Rev November 7, 2012 facilitated by the Foundation for the National Institutes of Health (http://www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of California, Los Angeles. This research was also supported by NIH Grants P30 AG010129 and K01 AG030514.
Footnotes
For the Alzheimer’s Disease Neuroimaging Initiative
Data used in preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.ucla.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at http://adni.loni.ucla.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf.
Electronic supplementary material The online version of this article (doi:10.1007/s00401-013-1151-4) contains supplementary material, which is available to authorized users.
Contributor Information
Jon B. Toledo, Department of Pathology and Laboratory Medicine, Institute on Aging, Center for Neurodegenerative Disease Research, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA 19104, USA
Sharon X. Xie, Department of Biostatistics and Epidemiology, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA 19104, USA
John Q. Trojanowski, Department of Pathology and Laboratory Medicine, Institute on Aging, Center for Neurodegenerative Disease Research, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA 19104, USA
Leslie M. Shaw, Email: Les.Shaw@uphs.upenn.edu, Department of Pathology and Laboratory Medicine, Institute on Aging, Center for Neurodegenerative Disease Research, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA 19104, USA
References
- 1.Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, Holtzman DM, Santacruz A, Buckles V, Oliver A, Moulder K, Aisen PS, Ghetti B, Klunk WE, McDade E, Martins RN, Masters CL, Mayeux R, Ringman JM, Rossor MN, Schofield PR, Sperling RA, Salloway S, Morris JC. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med. 2012;367(9):795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bates DM, Chambers JM. Nonlinear models. In: Hastie TJ, Chambers JM, editors. Statistical Models. S. Wadsworth & Brooks/Cole; Pacific Grove, California: 1992. [Google Scholar]
- 3.Beach TG, Monsell SE, Phillips LE, Kukull W. Accuracy of the clinical diagnosis of Alzheimer disease at National Institute on Aging Alzheimer Disease Centers, 2005–2010. J Neuropathol Exp Neurol. 2012;71(4):266–273. doi: 10.1097/NEN.0b013e31824b211b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bouwman FH, van der Flier WM, Schoonenboom NS, van Elk EJ, Kok A, Rijmen F, Blankenstein MA, Scheltens P. Longitudinal changes of CSF biomarkers in memory clinic patients. Neurology. 2007;69(10):1006–1011. doi: 10.1212/01.wnl.0000271375.37131.04. 10.1212/01. wnl.0000271375.37131.04. [DOI] [PubMed] [Google Scholar]
- 5.Buchhave P, Minthon L, Zetterberg H, Wallin AK, Blennow K, Hansson O. Cerebrospinal fluid levels of beta-amyloid 1–42, but not of tau, are fully changed already 5 to 10 years before the onset of Alzheimer dementia. Arch Gen Psychiatry. 2012;69(1):98–106. doi: 10.1001/archgenpsychiatry.2011.155. [DOI] [PubMed] [Google Scholar]
- 6.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261(5123):921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 7.De Meyer G, Shapiro F, Vanderstichele H, Vanmechelen E, Engelborghs S, De Deyn PP, Coart E, Hansson O, Minthon L, Zetterberg H, Blennow K, Shaw L, Trojanowski JQ. Diagnosis-independent Alzheimer disease biomarker signature in cognitively normal elderly people. Arch Neurol. 2010;67(8):949–956. doi: 10.1001/archneurol.2010.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duyckaerts C. Tau pathology in children and young adults: can you still be unconditionally baptist? Acta Neuropathol. 2011;121(2):145–147. doi: 10.1007/s00401-010-0794-7. [DOI] [PubMed] [Google Scholar]
- 9.Fleisher AS, Chen K, Quiroz YT, Jakimovich LJ, Gomez MG, Langois CM, Langbaum JB, Ayutyanont N, Roontiva A, Thiyyagura P, Lee W, Mo H, Lopez L, Moreno S, Acosta-Baena N, Giraldo M, Garcia G, Reiman RA, Huentelman MJ, Kosik KS, Tariot PN, Lopera F, Reiman EM. Florbetapir PET analysis of amyloid-beta deposition in the presenilin 1 E280A autosomal dominant Alzheimer’s disease kindred: a cross-sectional study. Lancet Neurol. 2012;11(12):1057–1065. doi: 10.1016/S1474-4422(12)70227-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gruen B, Leisch F. Fitting finite mixtures of generalized linear regressions in R. Comput Stat Data Anal. 2007;51(11):5247–5252. doi: 10.1016/j.csda.2006.08.014. [DOI] [Google Scholar]
- 11.Hastie T, Tibshirani R, Friedman J. The elements of statistical learning. Springer; New York: 2001. [Google Scholar]
- 12.Jack CR, Jr, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, Shaw LM, Vemuri P, Wiste HJ, Weigand SD, Lesnick TG, Pankratz VS, Donohue MC, Trojanowski JQ. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12(2):207–216. doi: 10.1016/S1474-4422(12)70291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jack CR, Jr, Lowe VJ, Weigand SD, Wiste HJ, Senjem ML, Knopman DS, Shiung MM, Gunter JL, Boeve BF, Kemp BJ, Weiner M, Petersen RC. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer’s disease: implications for sequence of pathological events in Alzheimer’s disease. Brain. 2009;132(Pt 5):1355–1365. doi: 10.1093/brain/awp062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jack CR, Jr, Wiste HJ, Lesnick TG, Weigand SD, Knopman DS, Vemuri P, Pankratz VS, Senjem ML, Gunter JL, Mielke MM, Lowe VJ, Boeve BF, Petersen RC. Brain beta-amyloid load approaches a plateau. Neurology. 2013;80(10):890–896. doi: 10.1212/WNL.0b013e3182840bbe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laird NM, Ware JH. Random-effects models for longitudinal data. Biometrics. 1982;38(4):963–974. [PubMed] [Google Scholar]
- 16.Landau SM, Lu M, Joshi AD, Pontecorvo M, Mintun MA, Trojanowski JQ, Shaw LM, Jagust WJ Alzheimer’s Disease Neuroimaging Initiative. Comparing PET imaging and CSF measurements of Aβ. Ann Neurol. 2013 doi: 10.1002/ana.23908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Le Bastard N, Aerts L, Sleegers K, Martin JJ, Van Broeckhoven C, De Deyn PP, Engelborghs S. Longitudinal stability of cerebrospinal fluid biomarker levels: fulfilled requirement for pharmacodynamic markers in Alzheimer’s disease. J Alzheimers Dis. 2013;33(3):807–822. doi: 10.3233/JAD-2012-110029. [DOI] [PubMed] [Google Scholar]
- 18.Mattsson N, Portelius E, Rolstad S, Gustavsson M, Andreasson U, Stridsberg M, Wallin A, Blennow K, Zetterberg H. Longitudinal cerebrospinal fluid biomarkers over four years in mild cognitive impairment. J Alzheimers Dis. 2012;30(4):767–778. doi: 10.3233/JAD-2012-120019. [DOI] [PubMed] [Google Scholar]
- 19.Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science. 2010;330(6012):1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s disease. Neurology. 1984;34(7):939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 21.Mollenhauer B, Bibl M, Trenkwalder C, Stiens G, Cepek L, Steinacker P, Ciesielczyk B, Neubert K, Wiltfang J, Kretzschmar HA, Poser S, Otto M. Follow-up investigations in cerebrospinal fluid of patients with dementia with Lewy bodies and Alzheimer’s disease. J Neural Transm. 2005;112(7):933–948. doi: 10.1007/s00702-004-0235-7. [DOI] [PubMed] [Google Scholar]
- 22.Mufson EJ, Binder L, Counts SE, DeKosky ST, de Toledo-Morrell L, Ginsberg SD, Ikonomovic MD, Perez SE, Scheff SW. Mild cognitive impairment: pathology and mechanisms. Acta Neuropathol. 2012;123(1):13–30. doi: 10.1007/s00401-011-0884-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peskind E, Nordberg A, Darreh-Shori T, Soininen H. Safety of lumbar puncture procedures in patients with Alzheimer’s disease. Curr Alzheimer Res. 2009;6(3):290–292. doi: 10.2174/156720509788486509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peskind ER, Riekse R, Quinn JF, Kaye J, Clark CM, Farlow MR, Decarli C, Chabal C, Vavrek D, Raskind MA, Galasko D. Safety and acceptability of the research lumbar puncture. Alzheimer Dis Assoc Disord. 2005;19(4):220–225. doi: 10.1097/01.wad.0000194014.43575.fd. [DOI] [PubMed] [Google Scholar]
- 25.Petersen RC, Aisen PS, Beckett LA, Donohue MC, Gamst AC, Harvey DJ, Jack CR, Jr, Jagust WJ, Shaw LM, Toga AW, Trojanowski JQ, Weiner MW. Alzheimer’s Disease Neuroimaging Initiative (ADNI): clinical characterization. Neurology. 2010;74(3):201–209. doi: 10.1212/WNL.0b013e3181cb3e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E. Mild cognitive impairment: clinical characterization and outcome. Arch Neurol. 1999;56(3):303–308. doi: 10.1001/archneur.56.3.303. [DOI] [PubMed] [Google Scholar]
- 27.Reiman EM, Quiroz YT, Fleisher AS, Chen K, Velez-Pardo C, Jimenez-Del-Rio M, Fagan AM, Shah AR, Alvarez S, Arbelaez A, Giraldo M, Acosta-Baena N, Sperling RA, Dickerson B, Stern CE, Tirado V, Munoz C, Reiman RA, Huentelman MJ, Alexander GE, Langbaum JB, Kosik KS, Tariot PN, Lopera F. Brain imaging and fluid biomarker analysis in young adults at genetic risk for autosomal dominant Alzheimer’s disease in the presenilin 1 E280A kindred: a case-control study. Lancet Neurol. 2012;11(12):1048–1056. doi: 10.1016/S1474-4422(12)70228-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schneider JA, Arvanitakis Z, Leurgans SE, Bennett DA. The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann Neurol. 2009;66(2):200–208. doi: 10.1002/ana.21706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seppala TT, Koivisto AM, Hartikainen P, Helisalmi S, Soininen H, Herukka SK. Longitudinal changes of CSF biomarkers in Alzheimer’s disease. J Alzheimers Dis. 2011;25(4):583–594. doi: 10.3233/JAD-2011-101911. [DOI] [PubMed] [Google Scholar]
- 30.Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC, Blennow K, Soares H, Simon A, Lewczuk P, Dean R, Siemers E, Potter W, Lee VM, Trojanowski JQ. Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann Neurol. 2009;65(4):403–413. doi: 10.1002/ana.21610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shaw LM, Vanderstichele H, Knapik-Czajka M, Figurski M, Coart E, Blennow K, Soares H, Simon AJ, Lewczuk P, Dean RA, Siemers E, Potter W, Lee VM, Trojanowski JQ. Qualification of the analytical and clinical performance of CSF biomarker analyses in ADNI. Acta Neuropathol. 2011;121(5):597–609. doi: 10.1007/s00401-011-0808-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sinha S, Anderson JP, Barbour R, Basi GS, Caccavello R, Davis D, Doan M, Dovey HF, Frigon N, Hong J, Jacobson-Croak K, Jewett N, Keim P, Knops J, Lieberburg I, Power M, Tan H, Tatsuno G, Tung J, Schenk D, Seubert P, Suomensaari SM, Wang S, Walker D, Zhao J, McConlogue L, John V. Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature. 1999;402(6761):537–540. doi: 10.1038/990114. [DOI] [PubMed] [Google Scholar]
- 33.Stoer J, Bulirsch R. Introduction to numerical analysis. Springer; Berlin: 2002. [Google Scholar]
- 34.Tapiola T, Alafuzoff I, Herukka SK, Parkkinen L, Hartikainen P, Soininen H, Pirttila T. Cerebrospinal fluid {beta}-amyloid 42 and tau proteins as biomarkers of Alzheimer-type pathologic changes in the brain. Arch Neurol. 2009;66(3):382–389. doi: 10.1001/archneurol.2008.596. 10.1001/ar chneurol.2008.596. [DOI] [PubMed] [Google Scholar]
- 35.Toledo JB, Brettschneider J, Grossman M, Arnold SE, Hu WT, Xie SX, Lee VM, Shaw LM, Trojanowski JQ. CSF biomarkers cutoffs: the importance of coincident neuropathological diseases. Acta Neuropathol. 2012;124(1):23–35. doi: 10.1007/s00401-012-0983-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Toledo JB, Vanderstichele H, Figurski M, Aisen PS, Petersen RC, Weiner MW, Jack CR, Jr, Jagust W, Decarli C, Toga AW, Toledo E, Xie SX, Lee VM, Trojanowski JQ, Shaw LM. Factors affecting Abeta plasma levels and their utility as biomarkers in ADNI. Acta Neuropathol. 2011;122(4):401–413. doi: 10.1007/s00401-011-0861-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Villain N, Chetelat G, Grassiot B, Bourgeat P, Jones G, Ellis KA, Ames D, Martins RN, Eustache F, Salvado O, Masters CL, Rowe CC, Villemagne VL. Regional dynamics of amyloid-beta deposition in healthy elderly, mild cognitive impairment and Alzheimer’s disease: a voxelwise PiB-PET longitudinal study. Brain. 2012;135(Pt 7):2126–2139. doi: 10.1093/brain/aws125. [DOI] [PubMed] [Google Scholar]
- 38.Villemagne VL, Burnham S, Bourgeat P, Brown B, Ellis KA, Salvado O, Szoeke C, Macaulay SL, Martins R, Maruff P, Ames D, Rowe CC, Masters CL. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol. 2013;12(4):357–367. doi: 10.1016/S1474-4422(13)70044-9. [DOI] [PubMed] [Google Scholar]
- 39.Villemagne VL, Pike KE, Chetelat G, Ellis KA, Mulligan RS, Bourgeat P, Ackermann U, Jones G, Szoeke C, Salvado O, Martins R, O’Keefe G, Mathis CA, Klunk WE, Ames D, Masters CL, Rowe CC. Longitudinal assessment of Abeta and cognition in aging and Alzheimer disease. Ann Neurol. 2011;69(1):181–192. doi: 10.1002/ana.22248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weiner MW, Veitch DP, Aisen PS, Beckett LA, Cairns NJ, Green RC, Harvey D, Jack CR, Jagust W, Liu E, Morris JC, Petersen RC, Saykin AJ, Schmidt ME, Shaw L, Siuciak JA, Soares H, Toga AW, Trojanowski JQ. The Alzheimer’s disease Neuroimaging initiative: a review of papers published since its inception. Alzheimers Dement. 2012;8(1 Suppl):S1–S68. doi: 10.1016/j.jalz.2011.09.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang E, Farnum M, Lobanov V, Schultz T, Verbeeck R, Raghavan N, Samtani MN, Novak G, Narayan V, DiBernardo A. Quantifying the pathophysiological timeline of Alzheimer’s disease. J Alzheimers Dis. 2011;26(4):745–753. doi: 10.3233/JAD-2011-110551. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.