

Abstract
Stress can either enhance or suppress immune functions depending on a variety of factors such as duration of stressful condition. Chronic stress has been demonstrated to exert a significant suppressive effect on immune function. However, the mechanisms responsible for this phenomenon remain to be elucidated. Here, male C57BL/6 mice were placed in a 50-ml conical centrifuge tube with multiple punctures to establish a chronic restraint stress model. Serum IL-10 levels, IL-10 production by the splenocytes, and activation of STAT3 in the mouse spleen were assessed. We demonstrate that IL-10/STAT3 axis was remarkably activated following chronic stress. Moreover, TLR4 and p38 MAPK play a pivotal role in the activation of IL-10/STAT3 signaling cascade. Interestingly, blocking antibody against IL-10 receptor and inhibition of STAT3 by STAT3 inhibitor S3I-201 attenuates stress-induced lymphocyte apoptosis. Inhibition of IL-10/STAT3 dramatically inhibits stress-induced reduction in IL-12 production. Furthermore, disequilibrium of Th1/Th2 cytokine balance caused by chronic stress was also rescued by blocking IL-10/STAT3 axis. These results yield insight into a new mechanism by which chronic stress regulates immune functions. IL-10/STAT3 pathway provides a novel relevant target for the manipulation of chronic stress-induced immune suppression.
Keywords: TLR4, STAT3, p38, Chronic stress, Apoptosis, Immune suppression
1. Introduction
Stress, a common life event, has been implicated as a risk factor for the onsets of immune suppression, cardiovascular diseases, functional syndromes, tumors and mood disorders (Cohen et al., 2007; Gu et al., 2012; Lamkin et al., 2012; Lucini and Pagani, 2011). The strong regulatory effects of stress on immune system have been demonstrated by accumulating evidence and suggested to play a central role in the pathogenesis of stress-related diseases (Dragos and Tanasescu, 2010; Kiecolt-Glaser et al., 2002; Reiche et al., 2004). In contrast to acute stress, chronic stress induces a suppressive effect on the bodily defense system (Dragos and Tanasescu, 2010). Our previous studies have shown that reduction of splenocyte numbers occurs in chronic restraint stress (Yin et al., 2000; Zhang et al., 2008). A recent study indicates that increased number and function of regulatory T cells also contribute to the immune suppression caused by chronic stress (Kim et al., 2012). It has been reported that the level of signal transducer and activator of transcription 3 (STAT3) is increased in the mouse liver following immobilization (Hernandez et al., 2000). However, whether STAT3 contributes to stress-induced immune suppression is not known yet.
TLR-mediated signaling pathways play pivotal roles both in the innate and adaptive immune responses. Upon ligand recognition, TLRs, including TLR4, trigger multiple downstream pathways such as NF-κB and MAPKs, which subsequently combine to regulate the production of cytokines including TNF-α, IL-12 and IL-6 (Takeuchi and Akira, 2010). However, excessive inflammatory responses result in tissue damage. To avoid this, the secretion of IL-10 is also induced by the above signals and then IL-10 mediates a negative feedback loop which keeps pro-inflammatory cytokine production under control (Bode et al., 2012; Kim et al., 2008; Liew et al., 2005; Park et al., 2005).
IL-10, a well-known anti-inflammatory cytokine, is produced by a range of cells such as T cells, B cells, macrophages and dendritic cells (DCs). Its expression is regulated by multiple signaling molecules, including p38 MAPK and ERK (Saraiva and O’Garra, 2010). The main biological function of IL-10 appears to block the production of inflammatory mediators especially in response to TLR signals (Murray, 2006). Though IL-10 is essential for limiting host inflammatory response and maintaining immune homeostasis, a growing body of evidence supports the critical role of IL-10 in the progression of cancers and infectious diseases due to its capability of inducing immune suppression (Hamidullah et al., 2012; Redford et al., 2011; Sato et al., 2011; Wilson and Brooks, 2011). Binding of IL-10 to the IL-10R results in activation of JAK1 which then initiates STAT3 phosphorylation. STAT3 has been demonstrated to be a key effector molecule of IL-10 action and its activation is necessary for the IL-10-regulated anti-inflammatory effects (Lang et al., 2002; Williams et al., 2004).
STAT3, a key member of Stat family, can be activated by multiple cytokine pathways such as IL-10 signaling (El Kasmi et al., 2006). Growing evidence suggests that STAT3 has emerged as a strong negative regulator of immune functions. Kortylewski et al. (Kortylewski et al., 2005) have reported that STAT3 ablation leads to enhanced cytolytic activity of natural killer cells and increased production of IL-12 from DCs in the tumor microenvironment. Mice devoid of STAT3 in macrophages/ neutrophils are extra sensitive to endotoxin shock due to increased production of pro-inflammatory cytokines such as TNF-α, IL-1 and IL-6 (Takeda et al., 1999). Besides, the STAT3-deficient mice also exhibit excessive Th1 response (Takeda et al., 1999). Indeed, these changes result from disruption of IL-10 responsiveness, thus further supporting the essential role of STAT3 in the IL-10-mediated anti-inflammatory response. Conversely, IL-10-increased STAT3 activity in macrophages leads to impaired antigen-specific T cell responses (Cheng et al., 2003).
The objective of this study is to evaluate whether IL-10/STAT3 signaling pathway plays a role in the chronic stress-induced immune suppression. We also identified the modulatory role of TLR4 and p38 in the activation of IL-10/STAT3 following chronic restraint stress.
2. Methods
2.1. Animals
TLR2 knockout (KO) mice, TLR4 KO mice (both in the C57BL/6 background) and wild type (WT) C57BL/6 mice were purchased from the Jackson Laboratory (Bar Harbor, ME). All mice were maintained in the Division of Laboratory Animal Resources at East Tennessee State University (ETSU), a facility accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care (AAALAC). All animal studies were approved by the ETSU Committee on Animal Care.
2.2. Experimental model of restraint stress
6–8 week-old male mice were subjected to an established chronic physical restraint stress (Yin et al., 2000; Zhang et al., 2008). Briefly, mice were placed in a 50-ml conical centrifuge tube with multiple punctures to allow ventilation. Mice were held horizontally in the tubes for 12 h. Control littermates were kept in their original cage without food and water for 12 h. After physical restraint, mice were sacrificed by CO2 asphyxiation.
2.3. Experimental protocols
To investigate the role of p38 MAPK in the regulation of IL-10/STAT3 pathway in our system, p38 inhibitor SB203580 (1 mg/ kg body weight, Calbiochem, La Jolla, CA) was given by intraperitoneal (i.p.) injection 1 h before the initiation of stress. To determine the role of IL-10/STAT3 signaling cascade in the chronic stress-induced immune suppression, anti-IL-10R antibody (0.2 mg/ kg, 0.8 mg/ kg or 2 mg/ kg body weight, BioLegend, San Diego, CA) or STAT3 inhibitor S3I-201 (10 mg/ kg body weight, Calbiochem) were given i.p. 2 h before the mice were subjected to chronic restraint stress.
2.4. Western blot analysis
Cell lysates were prepared and immunoblots were performed as described previously (Hua et al., 2007). In some experiments, nuclear extracts from mouse spleens were also obtained by using NE-PER® Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific, Rockford, IL). Samples containing equal amounts of protein were separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis and transferred onto Hybond ECL membranes (Amersham Pharmacia, NJ). The ECL membranes were incubated overnight at 4°C with the appropriate primary antibody [anti-TLR2 (Santa Cruz Biotechnology, Santa Cruz, CA), anti-TLR4 (Abcam, Cambidge, MA), anti-phospho-STAT3 (Tyr705), anti-STAT3, anti-phospho-p38 MAPK, anti-p38 MAPK, anti-phospho-JNK, anti-JNK, anti-phospho-ERK, anti-ERK, anti-cleaved-caspase-3, anti-caspase-3, anti-Lamin B1, anti-GAPDH (Cell Signaling Technology, Beverly, MA)] followed by incubation with peroxidase-conjugated secondary antibodies (Cell Signaling Technology). The blot was exposed to the SuperSignal West Dura Extended Duration substrate (Pierce Biotechnology, Rockford, IL). The signals were quantified by scanning densitometry using a Bio-Image Analysis System (Bio-Rad).
2.5. Electrophoretic mobility shift assay (EMSA)
Nuclear proteins were prepared as described in Western blot analysis. EMSA was performed using a STAT3 EMSA kit (Affymetrix, Santa Clara, CA) according to the manufacturer’s instructions. Briefly, 2 μg of nuclear protein was incubated with a biotin-labeled transcription factor Probe for 30 min at 15°C. The protein/DNA complexes were separated on a 6.0% non denaturing polyacrylamide gel and transferred to positively charged nylon membranes (Biodyne® B, Pall Corporation, Pensacola, FL). After the oligos were fixed using a UV crosslinker for 3 min, the signals on the membranes were detected using strepatvidin-HRP and chemiluminescent substrate.
2.6. Histopathology and immunohistochemistry (IHC)
Mouse spleens were fixed in 10% buffered formalin and embedded in paraffin. Sections were stained with hematoxylin & eosin (H&E) using standard procedures. IHC was performed with a Diaminobenzidine (DAB) Histochemistry kit (Molecular Probes, Invitrogen, CA) according to the manufacturer’s instructions. In brief, after antigen retrieval with 1mM EDTA (pH 8.0), deparraffinized sections were incubated in 1% Blocking Reagent solution for 1h at room temperature and then labeled with anti-phospho-STAT3 (Tyr705) (Cell Signaling Technology) diluted 1:50 in SignalStain® Antibody Diluent (Cell Signaling Technology) overnight at 4°C. As a negative control, tissues were also stained with the diluent reagent alone. After washing, sections were incubated with biotinylated anti-mouse IgG (H+L) (Vector Laboratories, Burlingame, CA) and subsequently with HRP conjugate. Finally, the signal was developed with DAB substrate and counterstained with hematoxylin.
2.7. Detection of apoptosis by TUNEL assay
Sections of mouse spleens were prepared as described in IHC assay. TUNEL staining for apoptotic nuclei was performed using an In Situ Cell Death Detection kit (Roche Diagnostic, IN). Briefly, sections were exposed for 10 min to a permeabilization solution (0.1% Triton X-100, 0.1% sodium citrate). After washing, 50 μl of TUNEL reaction mixture was placed on the sections and then incubated in a humidified atmosphere for 60 min at 37°C. 50 μl of substrate solution was added following convert-AP incubation. Finally, sections were counterstained with haematoxylin. Sections were examined with the light microscope using 40 × objective. The percentage of apoptotic cells was calculated (TUNEL-positive cells/ total cells) and averaged across at least five randomly chosen microscopic fields for each slide.
2.8. Enzyme linked immunosorbent assay (ELISA)
Isolated mouse splenocytes were seeded at a final concentration of 2.5×106 cells/ml in 96-well plates with or without immobilized anti-CD3 (50 μl of 5 μg/ ml in PBS for 24 h at 4°C, BD Biosciences Pharmingen, San Diego, CA) and soluble anti-CD28 (1 μg/ ml, BD Biosciences Pharmingen). The supernatants were collected after 24 h (IFN-γ), 48 h (IL-10, IL-12, IL-2 and TNF-α) or 72 h (IL-4) of cultivation. To determine the serum level of IL-10, blood was collected from all experimental and control mice immediately after stress. Samples were allowed to clot for 2 h at room temperature before centrifugation for 20 min at 2000×g. Then serum was removed and stored at −20°C for subsequent ELISA assay. The amount of cytokines (IFN-γ, IL-10, IL-2, TNF-α and IL-4) was examined by using a Quantikine Mouse ELISA kit (R&D Systems, MN) (Zhang et al., 2008). IL-12 was quantified using an ELISA kit from Thermo Scientific.
2.9. Isolation of RNA and real-time quantitative RT-PCR
Total RNA was isolated from mouse spleens using a RNeasy Plus Mini Kit (QIAGEN Sciences, MD). The real-time PCR was performed as described previously (Yin et al., 2006; Zhang et al., 2008). Briefly, one microgram of RNA from each sample was used for reverse transcription and synthesis of cDNA using a Reaction Ready™ first strand cDNA synthesis kit (SABiosciences, Frederick, MD). PCR was performed using RT2 real-time™ SYBR Green Fluorescien PCR Master Mix (SABiosciences). GAPDH expression was used as internal control. The primer sequences used were as follows: IL-10 forward 5′-TGC TAA CCG ACT CCT TAA TGC AGG AC-3′, IL-10 reverse 5′-CCT TGA TTT CTG GGC CAT GCT TCT C-3′, GAPDH forward 5′-TGA CCA CAG TCC ATG CCA TC-3′, GAPDH reverse 5′-GAT GGG GGT TAC ACA GGC AG-3′.
2.10. Statistical analysis
The results were presented as means ± S.D. The data were analyzed using one-way analysis of variance and Student’s t-test. A value of p < 0.05 was considered to be statistically significant.
3. Results
3.1. Chronic stress induces STAT3 phosphorylation
Upon activation by tyrosine phosphorylation, STAT3 proteins dimerize, translocate to the nucleus and drive transcription by binding to specific DNA target sites (Levy and Darnell, 2002; Mankan and Greten, 2011). In the first experiment, we prepared nuclear extracts from mouse spleens and tested whether STAT3 is phosphorylated in response to chronic stress. As shown in Fig. 1A, the nuclear level of tyrosine-phosphoylated STAT3 in the mouse spleen dramatically increased in a time dependent manner following chronic stress. Total STAT3 was expressed at comparable amounts in all groups when compared with the loading control Lamin B1. Spleens from stressed mice exhibited higher phospho-STAT3 reactivity by IHC than those from control mice (Fig. 1B). Moreover, phospho-STAT3 was detected in the nuclei from stressed mice, consistent with the elevation of nuclear phophorylated STAT3 level as shown by Western blot analysis. Besides, immunostaining also revealed that the phospho-STAT3-positive cells were predominantly located in the marginal zone of the spleen, as confirmed by corresponding H&E staining.
Fig. 1.
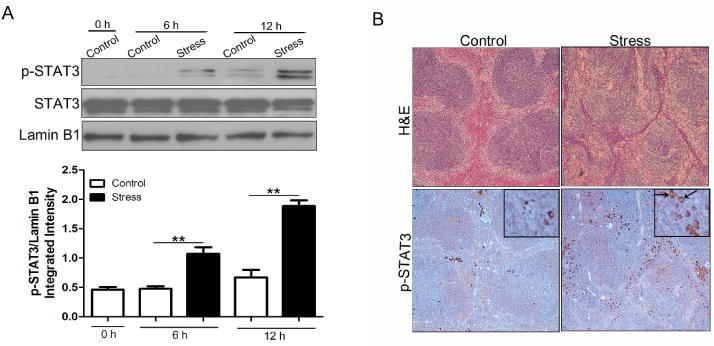
STAT3 phosphorylation is induced by chronic stress. WT C57BL/6 mice (n = 5 per group) were subjected to chronic stress. (A) After stress of the indicated time periods, spleens were harvested. Levels of total and phosphorylated STAT3 in the nucleus were determined by immunoblotting. Lamin B1 is shown as a loading control. **p <0.01 compared with indicated groups. (B) After 12 h of stress, WT mouse spleens were harvested and fixed in 10% buffered formalin. Sections (5 μm thick) were stained with hematoxylin and eosin (100×) (upper panels). IHC (lower panels) was performed on adjacent sections with anti-phospho-STAT3 (Tyr 705) Ab and hematoxylin was used as a counterstain (full panels, 100×; insets, 400×). Arrows indicate cells with positive-phospho-STAT3 staining in the nucleus. The data are representative of three independent experiments.
3.2. Chronic stress-induced STAT3 phosphorylation is through TLR4
We have reported that TLR4 deficiency inhibits chronic restraint stress-induced immune suppression (Zhang et al., 2008). In this study, Western blot analysis shows that TLR4 expression was considerably enhanced after chronic stress (Fig. 2A). In contrast, expression of TLR2, another key member of TLR family, was significantly decreased, which is in agreement with the findings from our previous study (Li et al., 2011). It has been demonstrated that TLR4 stimulation by lipopolysaccharide (LPS) induces STAT3 phosphoylation in the liver and hypothalamus (Yamawaki et al., 2009). Based on this notion and our findings of parallel changes of TLR4 and phosphorylated STAT3 expression, we speculated that TLR4 might be involved in the regulation of STAT3 activity in chronic stress. As expected, TLR4 deficiency abrogated stress-induced augmentation of STAT3 phosphorylation in the nucleus (Fig. 2B). However, there was no significant difference in the level of phospho-STAT3 between WT mice and TLR2 KO mice following chronic stress, suggesting that TLR4, but not TLR2, is required for chronic stress-induced STAT3 phosphorylation.
Fig. 2.

TLR4 mediates STAT3 phosphorylation induced by chronic stress. (A) WT mice (n = 5 per group) were sacrificed after 12 h of chronic stress. Cellular lysates were extracted from mouse spleens. Expression of TLR4 and TLR2 were determined by Western blot. GAPDH is shown as a loading control. (B) Age-matched WT and TLR4 KO as well as TLR2 KO mice (n = 5 per group) were subjected to chronic stress for 12 h. Levels of phospho-STAT3 in the nucleus were examined by Western blot. *p < 0.05; **p < 0.01; ***p < 0.001 compared with indicated groups.
3.3. p38 MAPK participates in chronic stress-induced STAT3 phosphorylation
The observation that TLR4 expression was increased in chronic stress forced us to focus on MAPKs, which are important downstream targets of TLR signals and exert strong regulatory effects on immune responses (Rincon and Davis, 2009; Zhu and Mohan, 2010). As shown in Fig. 3A, stressed mice displayed enhanced activation of p38 MAPK, whereas phospho-JNK and phospho-ERK were both diminished following chronic stress. To define whether TLR4 contributed to p38 MAPK activation, phospho-p38 was also assessed in TLR4-deficient mice following chronic stress. Fig. 3B shows that stress-induced phosphorylation of p38 MAPK was blocked by TLR4 deficiency, suggesting that TLR4 is crucial for p38 activation following chronic stress.
Fig. 3.
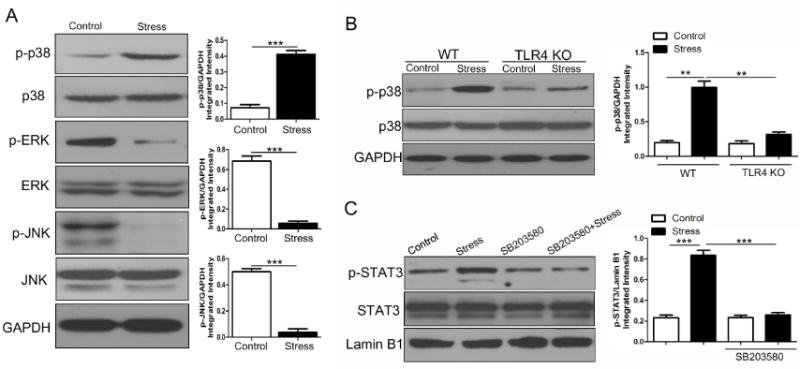
p38 MAPK activity is required for chronic stress-induced STAT3 phophorylation. (A) WT mice (n = 5 per group) were sacrificed after 12 h of chronic stress and cellular lysates were isolated. Levels of phospho-p38 MAPK, phospho-JNK and phospho-ERK were examined by immunoblotting with specific Abs. (B) Age-matched WT and TLR4 KO mice (n = 5 per group) were subjected to chronic stress for 12 h, and cellular lysates were extracted from mouse spleens. Levels of phospho-p38 MAPK were determined by immunoblotting. (C) WT mice (n = 5 per group) were administered with or without p38 MAPK inhibitor SB203580 (1 mg/ kg body weight, i.p.) 1 h before the mice were subjected to chronic stress for 12 h. Mouse spleens were harvested and nuclear extracts were isolated. Levels of phospho-STAT3 and total STAT3 in the nucleus were examined by immunobloting. **p < 0.01; ***p < 0.001 compared with indicated groups.
Inhibition of p38 prevents STAT3 phosphorylation at the site of tyrosine 705 (Riebe et al., 2011; Wolfle et al., 2011). Moreover, TLR4, the upstream regulator of p38, was shown here to be contributable to STAT3 activation. To further evaluate the role of p38 MAPK in the enhancement of STAT3 activity during chronic stress, p38 inhibitor SB203580 was introduced into experiments. Pretreatment with SB203580 significantly inhibited nuclear accumulation of tyrosine-phosphorylated STAT3 in stressed mice (Fig. 3C). These results suggest that p38 MAPK, a target of TLR4 signal, is critical for chronic stress-induced STAT3 phosphorylation.
3.4. IL-10 mediates STAT3 phosphorylation in chronic restraint stress
STAT3 can be activated by multiple cytokine signals, which is highlighted by IL-10 and IL-6 (Ahmed and Ivashkiv, 2000; Braun et al., 2013; El Kasmi et al., 2006). In this study, we evaluated IL-10 and IL-6 release from the splenocytes in vitro. Interestingly, splenocytes from stressed mice produced higher amounts of IL-10 than those from control mice (Fig. 4A), whereas IL-6 secretion was not affected (data not shown). Similar results were obtained when we analyzed the serum level of IL-10 (Fig. 4B). We also examined whether there was a change of IL-10 at the transcriptional level. As expected, the level of IL-10 mRNA in the mouse spleen was significantly increased in stressed mice as shown by quantitative RT-PCR (Fig. 4C).
Fig. 4.
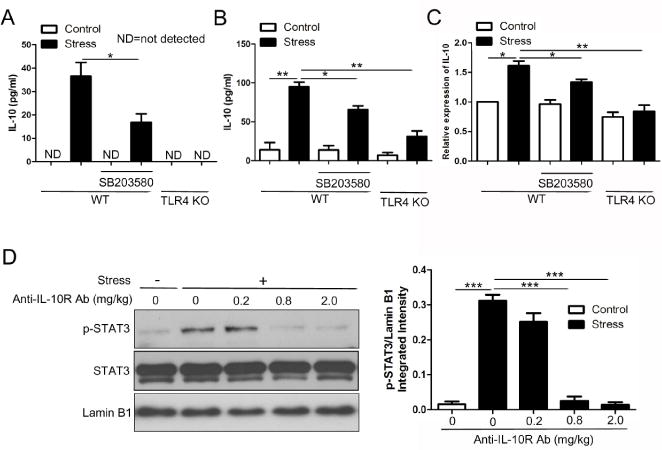
IL-10 mediates STAT3 phosphorylation induced by chronic stress. Age-matched WT and TLR4 KO mice were subjected to restraint stress for 12 h. SB203580 was injected in WT mice 1 h before the initiation of stress. After stress, serum samples were collected. Simultaneously, splenocytes were isolated and cultured in vitro for 48 h, and then cell-free supernatants were harvested. Levels of IL-10 in the supernatants (A) and sera (B) were determined by ELISA. Total RNA was isolated from mouse spleens, and IL-10 mRNA levels were determined by quantitative RT-PCR (C). Data from A to C are mean ± SD from 5 mice per group. *p < 0.05; **p < 0.01 compared with indicated groups. (D) WT mice (n = 5 per group) were administered with or without anti-IL-10R Ab at the indicated doses 2 h before the initiation of stress. At 12 h of stress, nuclear extracts were isolated from mouse spleens. Levels of phospho-STAT3 as well as total STAT3 in the nucleus were examined by immunobloting. *p < 0.05; **p < 0.01; ***p < 0.001 compared with indicated groups.
To gain insight into the mechanisms responsible for IL-10 alteration, we evaluated the influence of TLR4 deficiency on IL-10 expression. As shown in Fig. 4A–C, stress-induced increase of IL-10 was remarkably suppressed by TLR4 deficiency both at the transcriptional and protein levels. Since p38 MAPK is a mediator of TLR4 signal, it’s necessary to assess the role of p38 in the regulation of IL-10 production. Inhibition of p38 by SB203580 resulted in less amounts of IL-10 protein and mRNA compared with untreated stressed mice (Fig. 4A–C). These data indicates that chronic stress-induced IL-10 production is dependent on the TLR4/p38 signaling. However, SB203580 failed to completely blocked IL-10 induction by stress as TLR4 deficiency did, suggesting that the other pathway coordinately bridges TLR4 signal and IL-10 production in chronic stress.
Binding of IL-10 to the IL-10R initiates JAK/STAT3 signaling pathway. In this study, mice were treated with anti-IL-10R antibody at different doses 2 h before the initiation of stress to determine whether inhibition of IL-10 signal would influence STAT3 activation in response to chronic stress. Fig. 4D shows that administration of anti-IL-10R antibody dose-dependently attenuated stress-induced augmentation of nuclear STAT3 phosphorylation. It has been shown that both IL-10 and IL-6 could induce STAT3 phosphorylation at Tyr705 (Ahmed and Ivashkiv, 2000). However, in our animal model system, administration of anti-IL-6R antibody failed to reverse the alteration of STAT3 phosphorylation following chronic stress (data not shown). Taken together, these results reveal that activation of TLR4/p38 signaling up-regulates the production of IL-10, which in turn enhances STAT3 phosphorylation following chronic stress.
3.5. STAT3 function is regulated through TLR4/p38/IL-10 signaling pathway in chronic stress
To verify that STAT3 was activated by chronic stress, STAT3-DNA-binding activity was assessed by EMSA. We found that the level of tyrosine-phosphorylated STAT3 in the nucleus increased in a time dependent manner following chronic stress, and a change in the expression of phospho-STAT3 started at 3 h of stress (Fig. 5A, upper panel). However, a significant increase of STAT3-DNA-binding activity was first detected at 6 h of stress as shown by EMSA (Fig. 5A, bottom panel), demonstrating that functional alteration of STAT3 occurred in chronic stress and lagged behind elevation of phospho-STAT3 nuclear localization. There was no significant difference of STAT3-DNA-binding activity at 12 h between control mice and stressed mice, although the accumulation of phospho-STAT3 in the nucleus was dramatically induced by chronic stress at this time point. Thus, our results suggest that STAT3 was functionally activated transiently but not persistently during chronic stress.
Fig. 5.
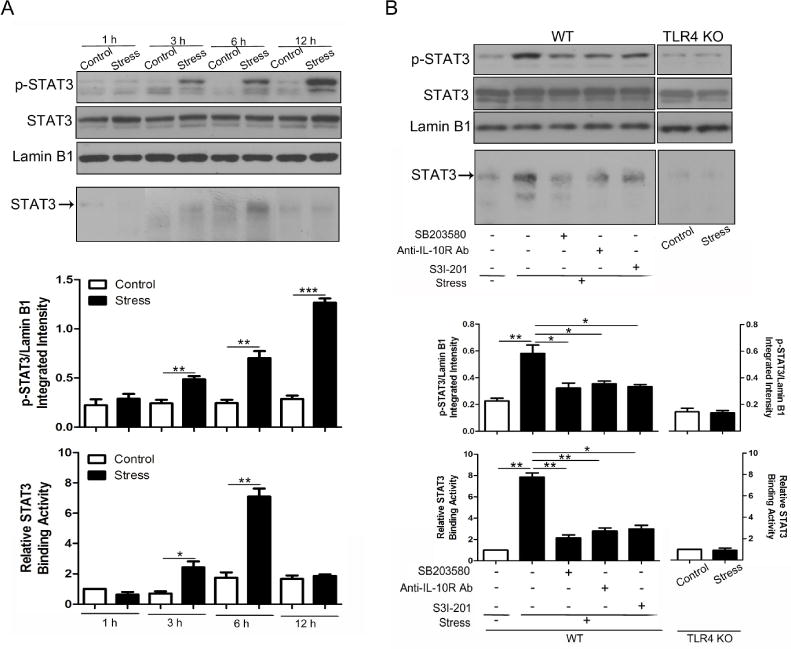
STAT3 is activated through the TLR4/p38/IL-10 pathway in chronic stress. (A) WT mice (n = 5 per group) were subjected to chronic stress for indicated time periods. Nuclear extracts were isolated from mouse spleens. Levels of phospho-STAT3 and total STAT3 in the nucleus were examined by immunobloting (upper panel). DNA-binding activity of STAT3 was determined by EMSA (bottom panel). (B) Age-matched WT and TLR4 KO mice (n = 5 per group) were subjected to restraint stress for 6 h. WT mice were pre-treated with SB203580 (1 mg/ kg body weight, i.p.), anti-IL-10R Ab (2 mg/ kg body weight, i.p.) or STAT3 inhibitor S3I-201 (10 mg/ kg body weight, i.p.) for 1 h, 2 h, and 2 h, respectively, before the initiation of stress. Levels of phospho-STAT3 and STAT3 (upper panel) and DNA-binding activity of STAT3 (bottom panel) were determined as in A. *p < 0.05; **p < 0.01; ***p < 0.001 compared with indicated groups.
Since TLR4 deficiency, SB203580 and anti-IL-10R antibody all exhibited inhibitory effects on STAT3 phosphorylation as shown before, we tested whether they had similar effects on the DNA-binding activity of STAT3. We subjected WT and TLR4 KO mice to chronic stress for 6 h. In addition, WT mice were also pre-treated with or without p38 inhibitor or anti-IL-10R antibody before the initiation of stress. We found that stress-induced enhancement of STAT3 phosphorylation (Fig. 5B, upper panel) and DNA-binding activity (Fig. 5B, bottom panel) was both considerably suppressed by inhibition of p38, blockage of IL-10 signal and TLR4 deficiency. Besides, administration of STAT3 inhibitor S3I-201, which would be utilized in the following experiments, was also shown to exert an inhibitory effect on the phosphorylation and activation of STAT3. Collectively, STAT3 is functionally regulated by chronic stress via the TLR4/p38/IL-10 pathway.
3.6. Chronic stress induces splenocyte apoptosis through IL-10/STAT3
We and others have reported that chronic stress negatively regulates immune functions (Kim et al., 2012; Sesti-Costa et al., 2012; Yin et al., 2000). The involvement of splenocyte apoptosis in chronic restraint stress has been determined in our previous studies (Yin et al., 2000; Zhang et al., 2008). In the current study, we discovered the mechanisms underlying the apoptotic cell death of splenocytes. As shown in Fig. 6A, administration of SB203580, anti-IL-10R antibody and S3I-201 all significantly attenuated stress-induced reduction of splenocytes. The level of cleaved caspase-3 and the percentage of apoptotic cells in the mouse spleen were both remarkably elevated in stressed mice (Fig. 6B and C). Importantly, these alterations were greatly prevented by pretreatment of SB203580, anti-IL-10R antibody and S3I-201 (Fig. 6B and C). Therefore, these data reveal that chronic stress induces splenocyte apoptosis dependent on p38, IL-10 and STAT3.
Fig. 6.
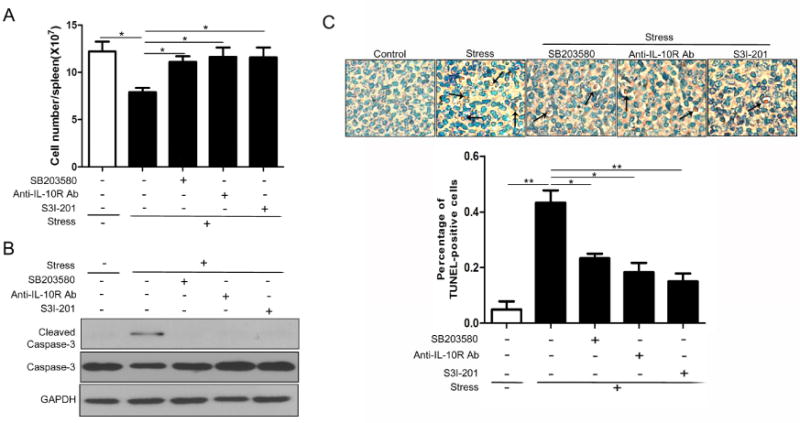
Inhibition of IL-10/STAT3 protects splenocytes against apoptosis induced by chronic stress. WT mice (n = 5 per group) were pre-treated with SB203580, anti-IL-10R Ab or STAT3 inhibitor S3I-201 as in Figure 5B, before the mice were subjected to chronic stress for 12 h. Total splenocytes were enumerated with a hemocytometer (A). Cellular lysates were isolated from mouse spleens and levels of total and cleaved caspase-3 were determined by Western blot (B). Apoptotic cells in spleens were determined by TUNEL assay (C). Cell number and TUNEL data are represented as mean ± SD. *p < 0.05; **p < 0.01 compared with indicated groups.
3.7. IL-10/STAT3 mediates chronic stress-induced immune suppression
IL-12 is an important mediator in the differentiation of Th1 cells (Trinchieri, 2003; Trinchieri et al., 2003). Conversely, IL-12 has a suppressive action on Th2 differentiation and IL-4 production. Fig. 7A shows that there was a significant decrease in the amount of IL-12 produced by splenocytes from stressed mice, consistent with the Th2 dominance in chronic stress we have previously shown (Li et al., 2011; Zhang et al., 2008). IL-10 and STAT3 both exert inhibitory effects on IL-12 production (Corinti et al., 2001; Kortylewski et al., 2005; Pestka et al., 2004). As expected, administration of anti-IL-10R antibody and S3I-201 reversed stress-reduced secretion of IL-12. Next, we examined the effect of p38 inhibition on the release of IL-12. We found that stress-induced decrease of IL-12 was not rescued by the p38 inhibitor.
Fig. 7.
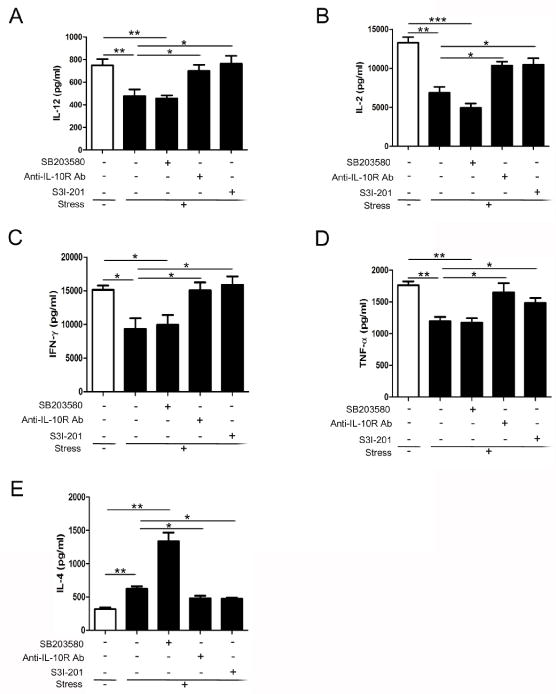
Inhibition of IL-10/STAT3 rescues chronic stress-induced immune suppression. WT mice were pre-treated with SB203580, anti-IL-10R Ab or STAT3 inhibitor S3I-201 as in Figure 5B, before the mice were subjected to chronic stress for 12 h. Splenocytes were isolated and cultured in vitro with (IL-2, TNF-α, IFN-γ, and IL-4) or without (IL-12) the stimulation of anti-CD3/anti-CD28 Abs. Levels of IL-12 (48 h) (A); IL-2 (48 h) (B); TNF-α (48 h) (C); IFN-γ (24 h) (D); and IL-4 (72 h) (E) secreted into the media were determined by ELISA. Data are expressed as mean ± SD from 5 mice per group. *p < 0.05; **p < 0.01; ***p < 0.001 compared with indicated groups.
We further determined the disequilibrium of Th1/Th2 cytokines in chronic stress. As shown in Fig. 7C–F, splenocytes from stressed mice produced higher level of Th2 cytokine IL-4 but lower levels of Th1 cytokine IL-2, IFN-γ and TNF-α compared with those from control mice. On the basis of our observation that IL-12 production was modified by anti-IL-10R antibody and S3I-201, we hypothesized that the balance of Th1/Th2 cytokines would also be regulated by IL-10/STAT3 signaling. Our results show that blockage of IL-10 signal and inhibition of STAT3 significantly repressed the alterations of Th1 and Th2 cytokine secretion induced by chronic stress (Fig. 7C–F). However, inhibition of p38, the upstream activator of IL-10/STAT3, did not interrupt Th1 cytokine secretion response to chronic stress. Intriguingly, the level of IL-4 was markedly increased by SB203580 administration in stressed mice. These results lead to the suggestion that chronic stress induces disequilibrium of Th1/Th2 cytokine balance through mechanisms dependent on IL-10/STAT3 signaling.
4. Discussion
In the present study, we discovered a new mechanism by which chronic stress induces immune suppression. The production of IL-10, a critical anti-inflammatory cytokine, was enhanced both at transcriptional and protein levels in stressed mice. The activity of STAT3, the key effector molecule of IL-10 signal, was also remarkably promoted during chronic stress. Additionally, TLR4/p38 played a crucial role in the activation of IL-10/STAT3 signaling. Of great importance was that IL-10/STAT3 inhibition blocked splenocyte apoptosis and disequilibrium of Th1/Th2 cytokine balance, the two significant consequences of chronic stress. Our findings suggest that IL-10/STAT3 axis mediates chronic stress-induced immune suppression.
Chronic stress has been confirmed to suppress immune functions. However, the mechanisms responsible for the regulatory effect still remain to be identified. Tyrosine phosphorylation is required for STAT3 dimerization and translocation to the nucleus, where it promotes the transcription of multiple genes (Levy and Darnell, 2002; Mankan and Greten, 2011). Western blot analysis in the current study displays a time dependent increase of nuclear phospho-STAT3 expression in stressed mice, suggesting that STAT3 is activated in response to chronic stress. However, expression of total STAT3 in the nucleus as well as in the whole cell lysates (data not shown) was not affected. The discrepancy with the previous report (Hernandez et al., 2000) might be due to different restraint stress models and also different tissues. Additionally, chronic stress induced a striking increase in the number of phospho-STAT3-positive cells as shown by IHC. Interestingly, corresponding H&E staining further confirmed that the cells with positive reaction to anti-phospho-STAT3 (Tyr705) antibody were mainly confined to the marginal zone of the mouse spleen. The type of positively stained cells by IHC will be investigated in our future work. Here, we would first focus on the factors contributing to STAT3 activation during chronic stress.
We have previously reported that TLR4 deficiency inhibits the reduction of splenocytes and the changes of Th1/Th2 cytokine secretion in response to chronic stress (Zhang et al., 2008). In the present study, we found that stressed mice exhibited an up-regulation of TLR4 expression, indicating that TLR4 signaling pathway is positively modulated in chronic stress. C-reactive protein (CRP), an inflammatory marker, has been reported to induce TLR4 expression and activation to initiate inflammatory response (Liu et al., 2010). Since higher levels of CRP have been found in stressed animals (Clougherty et al., 2010; Lu et al., 2013), TLR4 activation in chronic stress might be associated with elevated CRP. The effect of CRP on TLR4 activation in chronic stress will be investigated in future. MAPKs, especially p38 and JNK, play prominent roles both in the innate and adaptive immune responses (Rincon and Davis, 2009). It has been demonstrated that MAPKs are important mediators of TLR signaling pathways (Takeuchi and Akira, 2010). Interestingly, we observed that the level of phophorylated p38 MAPK was significantly increased, whereas the activities of JNK and ERK were both diminished in stressed mice. These results implicated that p38 MAPK plays a unique role that is distinct from that of JNK and ERK following chronic stress. Moreover, the enhanced p38 activity was abolished by TLR4 deficiency, confirming that TLR4 is the upstream activator of p38 in chronic stress. Several lines of evidence support that activation of TLR4/p38 signaling triggers the production of pro-inflammatory cytokines, and the release of anti-inflammatory cytokine IL-10 is coordinately induced by this signal to mediate a feedback inhibition loop that limits inflammatory responses (Bode et al., 2012; Kim et al., 2008; Zheng et al., 2012). We show here that in response to chronic stress IL-10 was also up-regulated via the TLR4/p38 signaling. IL-10 is predominantly produced by T cells, macrophages, and DCs responding to TLR stimulation (Murray, 2006). It has been reported that the T cell-specific IL-10 mutant mice kept normal innate responses to TLR4 ligand LPS (Roers et al., 2004). Therefore, macrophages and DCs might be responsible for the production of IL-10 during stress. These types of cells mainly reside in the marginal zone in the mouse spleen (Cesta, 2006). Our published work revealed that the hypothalamo-pituitary-adrenal (HPA) axis is unlikely to be involved in modulating splenocyte reduction in our established restraint stress animal model (Yin et al., 2000). Thus, we did not further evaluate the effect of the HPA axis in the same restraint stress animal model on the IL-10/STAT3 axis. However, we could not exclude the contribution by the sympathoadrenomedullary (SAM) system which is activated in stressful situation (Reiche et al., 2004), as catecholamines have been demonstrated to promote the production of IL-10 by innate immune cells (Elenkov and Chrousos. 2002).
It has been observed that TLR4 stimulation by LPS induces STAT3 phosphorylation mainly through MyD88, an adaptor protein of TLR4 (Yamawaki et al., 2009). Consistently, inhibition of p38, the downstream target of TLR4 signal, resulted in diminished expression of phospho-STAT3 at the site of tyrosine (Riebe et al., 2011; Wolfle et al., 2011). In the present study, TLR4 deficiency and p38 inhibition suppressed stress-enhanced STAT3 phosphorylation and DNA-binding activity. These data provide the link between TLR4/p38 and STAT3 in chronic stress. It’s unlikely that TLR2 participates in the regulation of STAT3 function in our system, as TLR2 expression was down-regulated following chronic stress and TLR2 deficiency failed to disturb STAT3 phosphorylation. STAT3 is activated through various cytokines including IL-10 (Ahmed and Ivashkiv, 2000; El Kasmi et al., 2006). Here, we observed that IL-10 was up-regulated after chronic stress. Importantly, blockade of IL-10 signal remarkably attenuated stress-induced enhancement of STAT3 phosphorylation, as well as DNA-binding activity. Taken together, TLR4/p38/IL-10 mediates STAT3 activation following chronic stress.
IL-10 has been recognized as a strong anti-inflammatory cytokine that has a critical role in limiting inflammatory responses and preventing tissue damage (Murray and Smale, 2012). STAT3 is the crucial molecule mediating the anti-inflammatory effect of IL-10 (Murray, 2006; Murray and Smale, 2012). Growing evidence suggests that IL-10/STAT3 axis exhibits an potent immunosuppressive effect by antagonizing the activities of macrophages and DCs (Chan et al., 2010; Melillo et al., 2010). An interesting question is whether IL-10/STAT3 plays roles in chronic stress-induced immune suppression. Our previous studies have demonstrated an induction of splenocyte apoptosis by chronic stress (Yin et al., 2000; Zhang et al., 2008). In the current study, we discovered that administration of anti-IL-10R antibody or S3I-201 reversed stress-induced reduction of splenocyte numbers, cleavage of caspase-3 and increase of apoptotic cells. These results suggest that chronic stress triggers splenocyte apoptosis through IL-10/STAT3 pathway. Besides, inhibition of p38, the activator of IL-10/STAT3, also exhibited a similar protective effect. In contrast, a previous study showed that STAT3-deficient memory CD8+ T cells have a shortened life-span and reduced expression of BCL-6 which is required for memory CD8+ T cell development (Cui et al., 2011). This discrepancy could be explained by that STAT3 signaling in different cell types plays different roles in the regulation of cell survival or apoptosis.
Disequilibrium of Th1/Th2 cytokine is a significant consequence of chronic stress. IL-12 is a crucial immunoregulatory cytokine in the maintenance of Th1/Th2 cytokine balance (Langrish et al., 2004; Wills-Karp, 2001). In this study, a decrease of IL-12 production was observed in stressed mice, which might help to explain the shift from Th1 towards a Th2 cytokine profile demonstrated in chronic stress. The expression of IL-12 is regulated by multiple factors such as IL-10 (Trinchieri, 2003). Previous studies suggested that IL-10 exerts a suppressive effect on IL-12 production (Corinti et al., 2001; Pattison et al., 2012). Incubation with LPS induces a significant increase of IL-12 synthesis in STAT3-deficient cells compared with WT DCs, suggesting that STAT3, the executor of IL-10 signal, also has a negative regulatory action on the release of IL-12 (Iwata-Kajihara et al., 2011). Consistently, our findings show that inhibition of IL-10/STAT3 significantly attenuated stress-reduced IL-12 secretion from lymphocytes. This finding suggests that activation of IL-10/STAT3 axis might be responsible for the decrease of IL-12 following chronic stress. It seems to be a paradox that TLR4 signaling pathway is activated in chronic stress, while IL-12 levels decreased. Oppositely, these results highlight the leading role of the IL-10/STAT3-mediated negative feedback mechanism in TLR4 signaling pathway at least in chronic stress situation.
Administration of anti-IL-10 antibody augments the percentage of IFN-γ-positive T cells but reduces that of IL-4-positive cells (Corinti et al., 2001). Mice devoid of STAT3 show excessive Th1 cell response suggested by increased IFN-γ production (Takeda et al., 1999). Thus, we evaluated the role of IL-10/STAT3 in the disruption of Th1/Th2 cytokine balance induced by chronic stress. As predicted, blockade of IL-10/STAT3 inhibited stress-induced changes in Th1 and Th2 cytokine production. Collectively, these data support that IL-10/STAT3 signaling pathway mediates chronic stress-induced immune suppression. Based on the regulatory action of IL-10/STAT3 on the production of IL-12, IL-10/STAT3 might regulate the Th1/Th2 responses by suppressing IL-12 secretion in chronic stress. Interestingly, p38 is located upstream of IL-10/STAT3, whereas inhibition of p38 MAPK did not rescue stress-induced disruption of Th1/Th2 cytokine profile and reverse alteration of IL-12 expression as anti-IL-10R antibody and S3I-201 did. More strikingly, inhibition of p38 by administration of SB203580 in unstressed mice remarkably increased the level of IL-4 but decreased the levels of IL-2, IFN-γ, TNF-α and IL-12 compared with untreated control mice (data not shown), suggesting that endogenous p38 and IL-10/STAT3 play opposite roles in the regulation of T cell responses and functions of antigen-presenting cells. Indeed, p38 has been demonstrated to be a positive regulator in the production of IL-12, which in turn differentially regulates Th1 and Th2 function (Kang et al., 2005; Korhonen et al., 2012). But following chronic stress, p38 MAPK signal simultaneously exerts inhibitory effect on IL-12 production indirectly by activating IL-10/STAT3 signaling. Moreover, the indirect regulating pathway played a leading role in our system, since IL-12 release was suppressed in chronic stress. Therefore, the net influence of p38 activation following chronic stress is unfavorable to Th1 response but favorable to Th2 response. Inhibition of p38 affected the two regulating pathways equally and thus failed to rescue the decreased IL-12 and disrupted Th1/Th2 cytokine balance in stressed mice.
In the current study, stressed mice exhibit an activation of TLR4/p38/IL-10/STAT3 systems in the spleens. The types of splenocyte subsets (i.e. CD4+ T cells, CD8+ T cells, B cells) involved in this mechanism will be addressed in our future research work using immunofluorescence and flow cytometry (i.e. intracellular staining). In addition, immune responses might be differentially modulated by chronic stress accordingly to different tissues analyzed and mouse strains employed. Thus, the studies would be extended to determine whether similar mechanisms responsible for immune suppression in chronic stress occur in other tissues and strains except for C57BL/6 mouse spleens.
5. Conclusions
In summary, we show that IL-10/STAT3 axis is activated by the TLR4/p38 pathway following chronic stress. IL-10/STAT3 then triggers lymphocyte apoptosis, represses the production of proinflammatory cytokine IL-12 and induces a shift of Th1/Th2 cytokine towards a Th2 dominance. Our study provides evidence that IL-10/STAT3 mediates chronic stress-induced immune suppression and identifies a possible therapeutic target for stress induced-immune suppression and infectious diseases.
Highlights.
A new mechanism involving IL-10/STAT3 pathway was disclosed for chronic stress-induced immune suppression.
Acknowledgments
This work was supported in part by National Institute of Health grants NIGM094740 and NIDA020120 and ETSU Major Research Grant 82061 grant to D. Yin. This work was also supported in part by the National Natural Science Foundation of China 81372299 to Q. Li. This work was also supported in part by the Integrated Chinese and Western Medical Research Foundation 2012Z-Y17 from Hubei Provincial Health Department to D. Hu.
Abbreviations
- TLR4
Toll-like receptor 4
- WT
wild-type
- KO
knockout
- DC
dendritic cell
- IHC
immunohistochemistry
- Th1
T helper-1
- Th2
T helper 2
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmed ST, Ivashkiv LB. Inhibition of IL-6 and IL-10 signaling and Stat activation by inflammatory and stress pathways. J Immunol. 2000;165:5227–5237. doi: 10.4049/jimmunol.165.9.5227. [DOI] [PubMed] [Google Scholar]
- Bode JG, Ehlting C, Haussinger D. The macrophage response towards LPS and its control through the p38(MAPK)-STAT3 axis. Cell Signal. 2012;24:1185–1194. doi: 10.1016/j.cellsig.2012.01.018. [DOI] [PubMed] [Google Scholar]
- Braun DA, Fribourg M, Sealfon SC. Cytokine Response Is Determined by Duration of Receptor and Signal Transducers and Activators of Transcription 3 (STAT3) Activation. J Biol Chem. 2013;288:2986–2993. doi: 10.1074/jbc.M112.386573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesta MF. Normal structure, function, and histology of the spleen. Toxicol Pathol. 2006;34:455–465. doi: 10.1080/01926230600867743. [DOI] [PubMed] [Google Scholar]
- Chan LL, Cheung BK, Li JC, Lau AS. A role for STAT3 and cathepsin S in IL-10 down-regulation of IFN-gamma-induced MHC class II molecule on primary human blood macrophages. J Leukoc Biol. 2010;88:303–311. doi: 10.1189/jlb.1009659. [DOI] [PubMed] [Google Scholar]
- Cheng F, Wang HW, Cuenca A, Huang M, Ghansah T, Brayer J, Kerr WG, Takeda K, Akira S, Schoenberger SP, Yu H, Jove R, Sotomayor EM. A critical role for Stat3 signaling in immune tolerance. Immunity. 2003;19:425–436. doi: 10.1016/s1074-7613(03)00232-2. [DOI] [PubMed] [Google Scholar]
- Clougherty JE, Rossi CA, Lawrence J, Long MS, Diaz EA, Lim RH, McEwen B, Koutrakis P, Godleski JJ. Chronic social stress and susceptibility to concentrated ambient fine particles in rats. Environ Health Perspect. 2010;118:769–775. doi: 10.1289/ehp.0901631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, Janicki-Deverts D, Miller GE. Psychological stress and disease. Jama. 2007;298:1685–1687. doi: 10.1001/jama.298.14.1685. [DOI] [PubMed] [Google Scholar]
- Corinti S, Albanesi C, la Sala A, Pastore S, Girolomoni G. Regulatory activity of autocrine IL-10 on dendritic cell functions. J Immunol. 2001;166:4312–4318. doi: 10.4049/jimmunol.166.7.4312. [DOI] [PubMed] [Google Scholar]
- Cui W, Liu Y, Weinstein JS, Craft J, Kaech SM. An interleukin-21-interleukin-10-STAT3 pathway is critical for functional maturation of memory CD8+ T cells. Immunity. 2011;35:792–805. doi: 10.1016/j.immuni.2011.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragos D, Tanasescu MD. The effect of stress on the defense systems. J Med Life. 2010;3:10–18. [PMC free article] [PubMed] [Google Scholar]
- El Kasmi KC, Holst J, Coffre M, Mielke L, de Pauw A, Lhocine N, Smith AM, Rutschman R, Kaushal D, Shen Y, Suda T, Donnelly RP, Myers MG, Jr, Alexander W, Vignali DA, Watowich SS, Ernst M, Hilton DJ, Murray PJ. General nature of the STAT3-activated anti-inflammatory response. J Immunol. 2006;177:7880–7888. doi: 10.4049/jimmunol.177.11.7880. [DOI] [PubMed] [Google Scholar]
- Elenkov IJ, Chrousos GP. Stress hormones, proinflammatory and antiinflammatory cytokines, and autoimmunity. Ann N Y Acad Sci. 2002;966:290–303. doi: 10.1111/j.1749-6632.2002.tb04229.x. [DOI] [PubMed] [Google Scholar]
- Gu HF, Tang CK, Yang YZ. Psychological stress, immune response, and atherosclerosis. Atherosclerosis. 2012;223:69–77. doi: 10.1016/j.atherosclerosis.2012.01.021. [DOI] [PubMed] [Google Scholar]
- Hamidullah, Changkija B, Konwar R. Role of interleukin-10 in breast cancer. Breast Cancer Res Treat. 2012;133:11–21. doi: 10.1007/s10549-011-1855-x. [DOI] [PubMed] [Google Scholar]
- Hernandez J, Carrasco J, Belloso E, Giralt M, Bluethmann H, Kee Lee D, Andrews GK, Hidalgo J. Metallothionein induction by restraint stress: role of glucocorticoids and IL-6. Cytokine. 2000;12:791–796. doi: 10.1006/cyto.1999.0629. [DOI] [PubMed] [Google Scholar]
- Hua F, Ha T, Ma J, Li Y, Kelley J, Gao X, Browder IW, Kao RL, Williams DL, Li C. Protection against myocardial ischemia/reperfusion injury in TLR4-deficient mice is mediated through a phosphoinositide 3-kinase-dependent mechanism. J Immunol. 2007;178:7317–7324. doi: 10.4049/jimmunol.178.11.7317. [DOI] [PubMed] [Google Scholar]
- Iwata-Kajihara T, Sumimoto H, Kawamura N, Ueda R, Takahashi T, Mizuguchi H, Miyagishi M, Takeda K, Kawakami Y. Enhanced cancer immunotherapy using STAT3-depleted dendritic cells with high Th1-inducing ability and resistance to cancer cell-derived inhibitory factors. J Immunol. 2011;187:27–36. doi: 10.4049/jimmunol.1002067. [DOI] [PubMed] [Google Scholar]
- Kang BY, Kim E, Kim TS. Regulatory mechanisms and their therapeutic implications of interleukin-12 production in immune cells. Cell Signal. 2005;17:665–673. doi: 10.1016/j.cellsig.2004.12.010. [DOI] [PubMed] [Google Scholar]
- Kiecolt-Glaser JK, McGuire L, Robles TF, Glaser R. Emotions, morbidity, and mortality: new perspectives from psychoneuroimmunology. Annu Rev Psychol. 2002;53:83–107. doi: 10.1146/annurev.psych.53.100901.135217. [DOI] [PubMed] [Google Scholar]
- Kim C, Sano Y, Todorova K, Carlson BA, Arpa L, Celada A, Lawrence T, Otsu K, Brissette JL, Arthur JS, Park JM. The kinase p38 alpha serves cell type-specific inflammatory functions in skin injury and coordinates pro- and anti-inflammatory gene expression. Nat Immunol. 2008;9:1019–1027. doi: 10.1038/ni.1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HR, Moon S, Lee HK, Kang JL, Oh S, Seoh JY. Immune dysregulation in chronic stress: a quantitative and functional assessment of regulatory T cells. Neuroimmunomodulation. 2012;19:187–194. doi: 10.1159/000331586. [DOI] [PubMed] [Google Scholar]
- Korhonen R, Huotari N, Hommo T, Leppanen T, Moilanen E. The expression of interleukin-12 is increased by MAP kinase phosphatase-1 through a mechanism related to interferon regulatory factor 1. Mol Immunol. 2012;51:219–226. doi: 10.1016/j.molimm.2012.03.019. [DOI] [PubMed] [Google Scholar]
- Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S, Niu G, Kay H, Mule J, Kerr WG, Jove R, Pardoll D, Yu H. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med. 2005;11:1314–1321. doi: 10.1038/nm1325. [DOI] [PubMed] [Google Scholar]
- Lamkin DM, Sloan EK, Patel AJ, Chiang BS, Pimentel MA, Ma JC, Arevalo JM, Morizono K, Cole SW. Chronic stress enhances progression of acute lymphoblastic leukemia via beta-adrenergic signaling. Brain Behav Immun. 2012;26:635–641. doi: 10.1016/j.bbi.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang R, Patel D, Morris JJ, Rutschman RL, Murray PJ. Shaping gene expression in activated and resting primary macrophages by IL-10. J Immunol. 2002;169:2253–2263. doi: 10.4049/jimmunol.169.5.2253. [DOI] [PubMed] [Google Scholar]
- Langrish CL, McKenzie BS, Wilson NJ, de Waal Malefyt R, Kastelein RA, Cua DJ. IL-12 and IL-23: master regulators of innate and adaptive immunity. Immunol Rev. 2004;202:96–105. doi: 10.1111/j.0105-2896.2004.00214.x. [DOI] [PubMed] [Google Scholar]
- Levy DE, Darnell JE., Jr Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- Li H, Chen L, Zhang Y, Lesage G, Zhang Y, Wu Y, Hanley G, Sun S, Yin D. Chronic stress promotes lymphocyte reduction through TLR2 mediated PI3K signaling in a beta-arrestin 2 dependent manner. J Neuroimmunol. 2011;233:73–79. doi: 10.1016/j.jneuroim.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liew FY, Xu D, Brint EK, O’Neill LA. Negative regulation of toll-like receptor-mediated immune responses. Nat Rev Immunol. 2005;5:446–458. doi: 10.1038/nri1630. [DOI] [PubMed] [Google Scholar]
- Liu N, Liu JT, Ji YY, Lu PP. C-reactive protein triggers inflammatory responses partly via TLR4/IRF3/NF-κB signaling pathway in rat vascular smooth muscle cells. Life Sci. 2010;87:367–374. doi: 10.1016/j.lfs.2010.07.012. [DOI] [PubMed] [Google Scholar]
- Lu XT, Liu YF, Zhao L, Li WJ, Yang RX, Yan FF, Zhao YX, Jiang F. Chronic psychological stress induces vascular inflammation in rabbits. Stress. 2013;16:87–98. doi: 10.3109/10253890.2012.676696. [DOI] [PubMed] [Google Scholar]
- Lucini D, Pagani M. From stress to functional syndromes: an internist’s point of view. Eur J Intern Med. 2011;23:295–301. doi: 10.1016/j.ejim.2011.11.016. [DOI] [PubMed] [Google Scholar]
- Mankan AK, Greten FR. Inhibiting signal transducer and activator of transcription 3: rationality and rationale design of inhibitors. Expert Opin Investig Drugs. 2011;20:1263–1275. doi: 10.1517/13543784.2011.601739. [DOI] [PubMed] [Google Scholar]
- Melillo JA, Song L, Bhagat G, Blazquez AB, Plumlee CR, Lee C, Berin C, Reizis B, Schindler C. Dendritic cell (DC)-specific targeting reveals Stat3 as a negative regulator of DC function. J Immunol. 2010;184:2638–2645. doi: 10.4049/jimmunol.0902960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray PJ. Understanding and exploiting the endogenous interleukin-10/STAT3-mediated anti-inflammatory response. Curr Opin Pharmacol. 2006;6:379–386. doi: 10.1016/j.coph.2006.01.010. [DOI] [PubMed] [Google Scholar]
- Murray PJ, Smale ST. Restraint of inflammatory signaling by interdependent strata of negative regulatory pathways. Nat Immunol. 2012;13:916–924. doi: 10.1038/ni.2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JM, Greten FR, Wong A, Westrick RJ, Arthur JS, Otsu K, Hoffmann A, Montminy M, Karin M. Signaling pathways and genes that inhibit pathogen-induced macrophage apoptosis–CREB and NF-kappaB as key regulators. Immunity. 2005;23:319–329. doi: 10.1016/j.immuni.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Pattison MJ, Mackenzie KF, Arthur JS. Inhibition of JAKs in macrophages increases lipopolysaccharide-induced cytokine production by blocking IL-10-mediated feedback. J Immunol. 2012;189:2784–2792. doi: 10.4049/jimmunol.1200310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestka S, Krause CD, Sarkar D, Walter MR, Shi Y, Fisher PB. Interleukin-10 and related cytokines and receptors. Annu Rev Immunol. 2004;22:929–979. doi: 10.1146/annurev.immunol.22.012703.104622. [DOI] [PubMed] [Google Scholar]
- Redford PS, Murray PJ, O’Garra A. The role of IL-10 in immune regulation during M. tuberculosis infection. Mucosal Immunol. 2011;4:261–270. doi: 10.1038/mi.2011.7. [DOI] [PubMed] [Google Scholar]
- Reiche EM, Nunes SO, Morimoto HK. Stress, depression, the immune system, and cancer. Lancet Oncol. 2004;5:617–625. doi: 10.1016/S1470-2045(04)01597-9. [DOI] [PubMed] [Google Scholar]
- Riebe C, Pries R, Schroeder KN, Wollenberg B. Phosphorylation of STAT3 in head and neck cancer requires p38 MAPKinase, whereas phosphorylation of STAT1 occurs via a different signaling pathway. Anticancer Res. 2011;31:3819–3825. [PubMed] [Google Scholar]
- Rincon M, Davis RJ. Regulation of the immune response by stress-activated protein kinases. Immunol Rev. 2009;228:212–224. doi: 10.1111/j.1600-065X.2008.00744.x. [DOI] [PubMed] [Google Scholar]
- Roers A, Siewe L, Strittmatter E, Deckert M, Schlüter D, Stenzel W, Gruber AD, Krieg T, Rajewsky K, Müller W. T cell-specific inactivation of the interleukin 10 gene in mice results in enhanced T cell responses but normal innate responses to lipopolysaccharide or skin irritation. J Exp Med. 2004;200:1289–1297. doi: 10.1084/jem.20041789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraiva M, O’Garra A. The regulation of IL-10 production by immune cells. Nat Rev Immunol. 2010;10:170–181. doi: 10.1038/nri2711. [DOI] [PubMed] [Google Scholar]
- Sato T, Terai M, Tamura Y, Alexeev V, Mastrangelo MJ, Selvan SR. Interleukin 10 in the tumor microenvironment: a target for anticancer immunotherapy. Immunol Res. 2011;51:170–182. doi: 10.1007/s12026-011-8262-6. [DOI] [PubMed] [Google Scholar]
- Sesti-Costa R, Ignacchiti MD, Chedraoui-Silva S, Marchi LF, Mantovani B. Chronic cold stress in mice induces a regulatory phenotype in macrophages: correlation with increased 11beta-hydroxysteroid dehydrogenase expression. Brain Behav Immun. 2012;26:50–60. doi: 10.1016/j.bbi.2011.07.234. [DOI] [PubMed] [Google Scholar]
- Takeda K, Clausen BE, Kaisho T, Tsujimura T, Terada N, Forster I, Akira S. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. 1999;10:39–49. doi: 10.1016/s1074-7613(00)80005-9. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–146. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- Trinchieri G, Pflanz S, Kastelein RA. The IL-12 family of heterodimeric cytokines: new players in the regulation of T cell responses. Immunity. 2003;19:641–644. doi: 10.1016/s1074-7613(03)00296-6. [DOI] [PubMed] [Google Scholar]
- Williams L, Bradley L, Smith A, Foxwell B. Signal transducer and activator of transcription 3 is the dominant mediator of the anti-inflammatory effects of IL-10 in human macrophages. J Immunol. 2004;172:567–576. doi: 10.4049/jimmunol.172.1.567. [DOI] [PubMed] [Google Scholar]
- Wills-Karp M. IL-12/IL-13 axis in allergic asthma. J Allergy Clin Immunol. 2001;107:9–18. doi: 10.1067/mai.2001.112265. [DOI] [PubMed] [Google Scholar]
- Wilson EB, Brooks DG. The role of IL-10 in regulating immunity to persistent viral infections. Curr Top Microbiol Immunol. 2011;350:39–65. doi: 10.1007/82_2010_96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfle SJ, Strebovsky J, Bartz H, Sahr A, Arnold C, Kaiser C, Dalpke AH, Heeg K. PD-L1 expression on tolerogenic APCs is controlled by STAT-3. Eur J Immunol. 2011;41:413–424. doi: 10.1002/eji.201040979. [DOI] [PubMed] [Google Scholar]
- Yamawaki Y, Kimura H, Hosoi T, Ozawa K. MyD88 plays a key role in LPS-induced Stat3 activation in the hypothalamus. Am J Physiol Regul Integr Comp Physiol. 2009;298:R403–410. doi: 10.1152/ajpregu.00395.2009. [DOI] [PubMed] [Google Scholar]
- Yin D, Tuthill D, Mufson RA, Shi Y. Chronic restraint stress promotes lymphocyte apoptosis by modulating CD95 expression. J Exp Med. 2000;191:1423–1428. doi: 10.1084/jem.191.8.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin D, Zhang Y, Stuart C, Miao J, Zhang Y, Li C, Zeng X, Hanley G, Moorman J, Yao Z, Woodruff M. Chronic restraint stress modulates expression of genes in murine spleen. J Neuroimmunol. 2006;177:11–17. doi: 10.1016/j.jneuroim.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Woodruff M, Zhang Y, Miao J, Hanley G, Stuart C, Zeng X, Prabhakar S, Moorman J, Zhao B, Yin D. Toll-like receptor 4 mediates chronic restraint stress-induced immune suppression. J Neuroimmunol. 2008;194:115–122. doi: 10.1016/j.jneuroim.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Guo Z, He W, Yang Y, Li Y, Zheng A, Li P, Zhang Y, Ma J, Wen M, Yang M, An H, Ji G, Yu Y. Ephedrine hydrochloride protects mice from LPS challenge by promoting IL-10 secretion and inhibiting proinflammatory cytokines. Int Immunopharmacol. 2012;13:46–53. doi: 10.1016/j.intimp.2012.03.005. [DOI] [PubMed] [Google Scholar]
- Zhu J, Mohan C. Toll-like receptor signaling pathways–therapeutic opportunities. Mediators Inflamm. 2010;2010:781235. doi: 10.1155/2010/781235. [DOI] [PMC free article] [PubMed] [Google Scholar]