

Abstract
Increasing evidence suggests that the non-canonical IKKs play critical roles in tumor genesis and development, leading to the notion that non-canonical IKKs may be good targets for cancer therapy. Here, we demonstrate that although TBK1 is not over-expressed or constitutively activated in some tumor cells, targeting IKKi induces the activation of TBK1. Therefore, simultaneously targeting both kinases is necessary to efficiently suppress tumor cell proliferation. We show that three TBK1/IKKi dual inhibitors, which are based on a structurally rigid 2-amino-4-(3′-cyano-4′-pyrrolidine)phenyl-pyrimidine scaffold, potently inhibit cell viability in human breast, prostate, and oral cancer cell lines. Treatment with these TBK1/IKKi dual inhibitors significantly impairs tumor development in xenograft and allograft mouse models. The anti-cancer function of these inhibitors may be due partially to their suppression of TBK1/IKKi-mediated AKT phosphorylation and VEGF expression. Most importantly, these TBK1/IKKi dual inhibitors have drug-like properties including low molecular weight, low Cytochrome P450 inhibition, and high metabolic stability. Therefore, our studies provide proof of concept for further drug discovery efforts that may lead to novel strategies and new therapeutics for the treatment of human cancer.
Keywords: TBK1, IKKi, TBK1/IKKi inhibitor, cancer, therapy
Introduction
The IKK family includes four kinase members, the canonical IKKα and IKKβ, as well as two non-canonical family members, TBK1 and IKKi. The canonical IKK complex consists of IKKα, IKKβ and a nonenzymatic regulatory component, IKKγ/NEMO 1. NF-κB is generally activated through canonical IKK-dependent phosphorylation and subsequent degradation of IκB inhibitory proteins 2. Liberated NF-κB dimers then enter the nucleus where they induce the transcription of various genes 3, 4. By contrast, TBK1 and IKKi are especially critical for antiviral responses via phosphorylation and activation of the transcription factors IRF3, IRF7, and STAT1 5, 6. Although TBK1 and IKKi are not a part of the canonical IKK complex, they were originally characterized as activators of NF-κB and are believed to target multiple NF-κB members and effectors 5. However, NF-κB activation in either TBK1 or IKKi single and double knockout models is predominantly normal, apart from minimal defects in the induction of select NF-κB target genes 7, 8, indicating that the non-canonical IKKs are sufficient, but not essential for NF-κB activation.
In addition to their role in antiviral immune responses, the non-canonical IKKs play important roles in tumor genesis and development 9–11. It was reported that TBK1 kinase activity was up-regulated and required for the survival of some transformed cells 9, 12. Silencing TBK1 expression by RNAi induced tumor cell apoptosis, while non-tumorigenic epithelial cells did not depend on TBK1 for survival 9. An integrative genomic approach showed IKKi as an oncogene in human breast cancer, and suppression of IKKi expression in breast cancer cell lines with IKKi DNA amplifications induced cell death 13. Moreover, a recent report showed that TBK1 and IKKi activated AKT by direct phosphorylation of AKT 14,15.
Based on their critical roles in tumor cell survival, the non-canonical IKKs are becoming attractive targets for cancer therapy 16–19. Here, we demonstrate that both TBK1 and IKKi are essential for tumor cell survival. Simultaneously targeting both kinases is an efficient way to suppress tumor cell proliferation. We identify selective TBK1/IKKi dual inhibitors with high potency of anti-proliferation in human cancer cell lines. Treatment of these TBK1/IKKi dual inhibitors significantly impairs tumor development in xenograft and allograft mouse models.
Materials and methods
Cell culture and materials
Human oral cancer cell lines (SCC-9, SCC-15, SCC-25), human prostate cancer cell lines (PC3, DU145, LNCaP), human breast cancer cell lines (MCF-7, MBA-MB-231, T-47D) and RAW264.7 macrophages, purchased from American Type Culture Collection (ATCC), were maintained in respective medium as recommended in protocols. Myc-CaP mouse prostate cancer cell line, which was isolated from a c-Myc transgenic mouse with prostate cancer, and can grow as tumors in immune competent FVB mice in an androgen dependent manner, was kindly provided by Charles Sawyers, MSKCC. Myc-CaP cells were cultured in RPMI-1640 medium supplemented with 10% heat-inactivated FBS, penicillin-streptomycin 100 units/ml. Lipopolysaccharides (LPS, from Escherichia coli 055:B5) were purchased from Sigma-Aldrich Co. (Saint Louis, Missouri).
TBK1/IKKi dual inhibitors
In counterscreening studies of our in-house 4-phenyl-pyrimidine based JNK inhibitors, we discovered that some JNK inhibitor candidates showed strong TBK1/IKKi inhibition (Patent: Scripps, WO2009032861 and Supplementary Figure 1). After structure modifications and SAR (structure-activity relationship) studies, we successfully generated some potent TBK1/IKKi dual inhibitors (Supplemtary Table 1 and 2). It should be noted that we realized recently that our compounds are structurally similar to those reported in the Dominex patent applications (Domainex, WO2012010826, CAS#1356225-62-1, and CAS#1354224-35-8). For cell culture experiments TBK1/IKKi dual inhibitors were dissolved in DMSO, for animal studies TBK1/IKKi dual inhibitors were dissolved in a mixture of 5%DMSO:10%Tween-80:85%H2O.
RNA interference
Cells at 50% confluency were transfected with 50 nM scrambled small interfering RNA (siRNA), TBK1 specific siRNA, or IKKi specific siRNA (Santa Cruz Biotechnology, Santa Cruz, CA) using TransIT-TKO (Mirus Bio Corp., Madison, WI) according to the manufacturer’s instructions.
MTT assay
Cell viability was examined by using the 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazoliumbromide (MTT) colorimetric assay (Molecular Probe, Life Technologies, INC, Grand Island, NY). Briefly, cells were plated in 96-well plates at a density of 3–4 × 103 cells per well, cells were treated with indicated inhibitors or vehicles for 72 hr, followed by addition of MTT for at least 4 hr. The absorbance was measured at 570/630 nm using a spectrophotometer (SpectraMax M5; Molecular Devices Crop). Cell viability (%) was calculated as follows: .
Apoptosis analysis
Apoptosis was examined by using the TUNEL assay - based ApopTag in situ Apoptosis detection kit (Chemicon, Temecula, CA) according to manufacturer’s instructions. Apoptotic cells were counted and compared between different groups.
Western blot analysis
Cell lysates were separated by SDS-PAGE and transferred to PVDF membranes. Immunostaining was performed using antibodies specific for TBK1/NAK (D1B4) Rabbit mAb, IKKi (D61F9) XP Rabbit mAb, Phospho-TBK1/NAK (Ser172) (D52C2) XP Rabbit mAb, Phospho-IKKi (Ser172) (D1B7) Rabbit mAb, IRF-3 (D83B9) Rabbit mAb, Phospho-IRF-3 (Ser396) (4D4G) Rabbit mAb, Phospho-CYLD (Ser418) Antibody, CYLD (D1A10) Rabbit mAb, Cleaved Caspase-9 (Asp330) (D2D4) Rabbit mAb, Cleaved Caspase-3 (Asp175) (5A1E) Rabbit mAb, Cleaved Caspase-7 (Asp198) (D6H1) Rabbit mAb, Cleaved PARP (Asp214) (D64E10) XP Rabbit mAb, AKT (pan) (40D4) Mouse mAb, Phospho-AKT (Thr308) (244F9) Rabbit mAb, Phospho-AKT (Ser473) (D9E) XP Rabbit mAb purchased from Cell Signaling Technology (Danvers, MA) and Cyclin D1 Antibody (M-20), β-actin purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Immunoreactive proteins were detected by chemoluminescence using the Pierce ECL Western Blotting Substrate (Thermo Scientific, Rockford, IL).
Reverse Transcription (RT)-PCR assay
Tumor tissue was homogenized with lysis buffer (RNeasy mini kit, QIAGEN Science, Germantown, MD) on ice. Total RNA was purified using RNeasy mini kit. Reverse transcription was carried out using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems by Life Technologies, Foster City, CA). The sequences of the primers were as follows: for VEGF, 5′-CTACCTCCACCATGCCAAGT-3′ (sense) and 5′-TCTCTCCTATGTGCTGGCCT-3′ (antisense); for β-actin, 5′-CATCGTCACCAACTGGGAC-3′ (sense) and 5′-CGATTTCCCGCTCGGCCGTGG-3′ (antisense).
Mouse tumor models
For SCC-9 xenograft model, eight-weeks old nu/nu mice (Charles River Laboratories) weight at 22–24 g were used. For Myc-CaP allograft model, eight-weeks old FVB male mice (Charles River Laboratories) weight at 22–24 g were used. The mice were housed with a regular 12 hr light/12 hr dark cycle and were kept in a pathogen-free environment. SCC-9 cells (1×107 cells) mixed in Matrigel, were injected into nude mice subcutaneously, Myc-CaP cells (3×106 cells) mixed in Matrigel were inoculated into FVB mice subcutaneously. One day after cell implantation, the mice were randomly divided into two groups (For SCC-9 xenograft model, N = 5 for control group and N=12 for treatment group; for Myc-CaP allograft model, N = 6 for control group and treatment group). Mice were administered ip with TBK1/IKKi kinase dual inhibitor 200A (100 mg/kg) or vehicles once a day. Tumor growth in these mice was monitored and measured every 3 days. The tumor volumes were calculated by the equation V (mm3) = a × b2/2, where “a” is the largest diameter and “b” is the perpendicular diameter.
Immunohistochemistry
Immunohistochemistry was done on SCC-9 xenograft tumor tissue frozen sections using the VECTASTAIN Elite ABC Kit (Universal) (Vector Laboratories, Burlingame, CA). The primary antibodies were mouse monoclonal VEGF (C-1) antibody (SC-7269, Santa Cruz Biotechnology) at 1:200 dilution, rabbit polyclonal phosphor-AKT (Ser 473) antibody (Cell Signaling Technology, Danvers, MA) at 1:200 dilution.
Statistical analysis
All cell MTT data were from at least three independent experiments performed in triplicate and expressed as the mean ± SD. A P-value of <0.05 between experimental and control groups were considered statistically significant. ANOVA with general linear model repeated measures were used to determine tumor volume difference among different groups over treatment time, followed by post-hoc Tukey test. The Student’s t test was also used for univariate analysis. A value of P < 0.05 were considered significant. Immunostaining was expressed as the arithmetic mean ± SD and each evaluated with an unpaired t test. Apoptotic index data were expressed as the mean number ± SD in each tumor area, and nonparametric comparisons (χ2) were made for each treatment group compared with their respective control. A value of P < 0.05 were considered statistically significant
Results
Both TBK1 and IKKi are essential for tumor cell survival
TBK1 and IKKi have been well established as regulators of the innate immune response via their ability to phosphorylate IFN regulatory transcription factors 3 and 7 (IRF3/IRF7). Recent evidence indicates that TBK1 and IKKi are also involved in promoting cell survival and tumorigenesis. To establish whether TBK1 and IKKi are constitutively activated in cancer cells, we checked the phosphorylation levels of TBK1 and IKKi in a number of cancer cell lines. We found that IKKi was expressed and phosphorylated in all cancer cell lines examined while TBK1 was selectively phosphorylated in certain cancer cell lines (Fig. 1A). The expression of p-TBK1 was very low or undetectable by western blot in human oral cancer cell line SCC-25. However, knockdown of IKKi in SCC-25 cells induced both TBK1 and p-TBK1 expression (Fig. 1B), suggesting that inhibition of IKKi leads to a compensatory expression and phosphorylation of TBK1. Consistently, although IKKi is constitutively phosphorylated in SCC-25 cells, knockdown of either IKKi or TBK1 respectively had only minor effects on cell survival while knockdown of both TBK1 and IKKi significantly inhibited cell proliferation (Fig. 1C). These results suggest that both TBK1 and IKKi are essential for cancer cell survival, inhibiting either one is not enough to inhibit cancer cell proliferation. Thus, simultaneously targeting both TBK1 and IKKi is necessary for efficient suppression of cancer cell growth.
Figure 1. Both TBK1 and IKKi are essential for cancer cell survival.

(A) Western blot analysis of the expression of p-IKKi, IKKi, p-TBK1, TBK1 in indicated cell lines.
(B) Western blot analysis of the expression of IKKi, p-TBK1, TBK1 in oral cancer cell line SCC-25 transfected with scrambled or IKKi-siRNA for 72 hr.
(C) Cell proliferation analysis of SCC-25 cells transfected with indicated siRNAs. The results are present as the means ± SD of one representative experiment (from three independent experiments), performed in triplicate. Statistically significant differences are indicated. (*) P < 0.05; (**) P < 0.01. The knockdown efficiency was verified by western blot.
Identification of selective TBK1 and IKKi dual inhibitors
To demonstrate that the dual inhibition of TBK1 and IKKi is an effective and safe way to suppress tumor growth, we generated highly potent TBK1/IKKi dual inhibition compounds which are based on a structurally rigid 2-amino-4-(3′-cyano-4′-pyrrolidine)phenyl-pyrimidine scaffold. In counterscreening studies of our in-house 4-phenyl-pyrimidine based JNK inhibitors, we discovered that some of the JNK inhibitor candidates showed strong TBK1/IKKi inhibition (Supplementary Figure 1). After structure modifications and structure-activity relationship (SAR) studies, we successfully developed compounds with significant reduction in anti-JNK activity while keeping a strong TBK1/IKKi inhibition (Supplementary Table 1 and 2). Compounds SR8185, 200A, and 200B exhibited low nanomolar activities against TBK1 and IKKi in enzyme assays and had good polar surface area (PSA) values as well (Fig. 2A and Supplementary Table 1). Counterscreening studies demonstrated that these compounds are selective against ALK, CDK4, CDK5, CK1d, JNK, IKKβ, IKKα, ROCK, PKA, p38 (Supplementary Table 2). Remarkably, SAR studies demonstrated that there was no need for a heavy halo-substitution (iodo- or bromo-) on the molecule in order to obtain high potency and selectivity, and this heavy halo-substitution is required in several published structures for TBK1/IKKi inhibition 17, 18. The inhibition profile of a compound against cytochrome P450 enzymes (CYPs) is also an important property to evaluate in drug development. As shown in Supplementary Table 1, inhibitor 200A showed an activity against four selected P450 isoforms with IC50 values estimated to be > 1000-fold higher than its activity against IKKi/TBK1, which indicates that 200A is a promising lead for further drug discovery efforts. The stability of these inhibitors in human and rat microsomes was also assessed. Among these three inhibitors 200A had the best stability.
Figure 2. The structure and the kinase inhibition of TBK1/IKKi dual inhibitors.
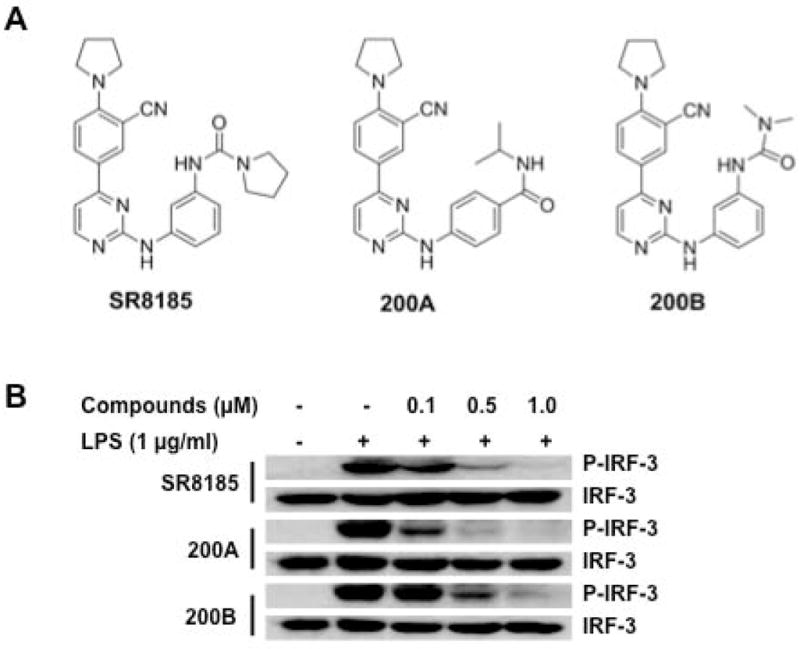
(A) Structures of selected TBK1/IKKi dual inhibitors - SR8185, 200A and 200B.
(B) Western blot analysis of IRF3 and p-IRF-3 (Ser396) in RAW264.7 cells treated with different concentrations of indicated TBK1/IKKi dual inhibitors for 1 hr, followed by incubation with LPS (1 μg/ml) for 1 hr.
To investigate the potency of TBK1/IKKi kinase inhibition of these new compounds in living cells, we tested whether LPS-induced activation of IRF3 in macrophages can be blocked by the treatment of these compounds20, 21. We verified that LPS treatment induced phosphorylation of IRF3 in RAW264.7 cells as expected (Supplementary Figure 2). RAW264.7 cells were then treated with SR8185, 200A, or 200B at different concentrations for 1hr, followed by LPS treatment for 1 hr, cells then were collected for western blot analysis. We found that treatment with these compounds reduced or completely blocked LPS-induced phosphorylation of IRF-3 (Fig. 2B). It was noted that compound 200A displayed the best inhibition. These results suggest that these compounds might exert their cellular activity via inhibiting TBK1/IKKi kinase.
TBK1/IKKi dual inhibitors suppress cancer cell proliferation in vitro
To test the anti-cancer potency of these TBK1/IKKi kinase dual inhibitors, we treated a group of cancer cell lines including oral cancer cells (SCC-9, SCC-15, SCC-25), prostate cancel cells (PC3, DU145, LNCaP, Myc-CaP), and breast cancer cells (MCF-7, MDA-MB-231, T-47D) with different concentrations of these compounds for 3 days, and the cell viability was determined by MTT assay. We found that these TBK1/IKKi kinase dual inhibitors have a potent anti-proliferative effect on all cancer cell lines examined although the sensitivity of cancer cells to these dual inhibitors are various among different cell lines (Fig. 3). These compounds displayed potent cytotoxic activities on cancer cell lines, with IC50 values in low micromolar range (Supplementary Table 3). Among these compounds, 200A presented high anti-proliferative activity in all tested cancer cell lines, with the lowest IC50.
Figure 3. TBK1/IKKi dual inhibitors suppress cancer cell proliferation.

(A–C) Oral cancer cell lines, (D–G) prostate cancer cell lines, and (H–J) breast cancer cell lines were treated with different concentrations of indicated TBK1/IKKi dual inhibitors for 72 hr, cell proliferation was analyzed by MTT assay. Cell viability (%) was calculated as (OD570−OD630 of the samples/OD570−OD630 of the control) ×100 (%).
The results are the means ± SD of one representative experiment (from three independent experiments), performed in triplicate.
Cyclin D1, whose activity is required for cell cycle G1/S transition, is a well-known marker for cell cycle progression and proliferation. Consistent with the anti-proliferation function, TBK1/IKKi dual inhibitors reduced Cyclin D1 expression in SSC-9 cells in a time-dependent manner (Supplementary Figure 3).
TBK1/IKKi dual inhibitors induce cancer cell apoptosis in vitro
MTT assays showed strong anti-proliferation potency of these TBK1/IKKi kinase dual inhibitors in different cancer cell lines. To further investigate the cytotoxicity of these dual inhibitors, SCC-9 cells were treated with 1.0 μM of TBK1/IKKi kinase dual inhibitor 200A for 72 hr, and then the effect on apoptosis was measured. We found that the treatment of TBK1/IKKi kinase dual inhibitor 200A induced significant apoptosis in SCC-9 cells (Fig. 4A). Consistently, the expression of the pro-apoptotic proteins was significantly increased in a time and dose -dependent manner when SCC-9 cells were exposed to TBK1/IKKi kinase dual inhibitor 200A (cleaved caspase-3 and cleaved PARP: P<0.01) (Fig. 4B and 4C).
Figure 4. TBK1/IKKi dual inhibitors induce apoptosis in cancer cells.

(A) Apoptotic cells in SCC-9 cells, treated with 1.0 μM 200A for 72 hr, were identified by TUNEL staining (red). Cells were counterstained with DAPI (blue).
(B) Western blot analysis of apoptotic protein in SCC-9 cells treated with 1.0 μM 200A for different time.
(C) Western blot analysis of apoptotic protein in SCC-9 cells treated with 200A at different concentrations for 24 hr.
The results in (B) and (C) are the means ± SD of one representative experiment from three independent experiments.
TBK1/IKKi dual inhibitors suppress tumor growth in mouse models
Based on the anti-proliferative effect of the TBK1/IKKi dual inhibitors on cancer cells in vitro and other biochemical characteristics, we chose compound 200A to test the anti-tumor growth efficacy of TBK1/IKKi dual inhibitors in mouse models. Nu/nu mice were inoculated subcutaneously (s.c.) in both flanks with 2 × 107 SSC-9 cells. One day after cell inoculation, mice were randomly divided into two groups. One group was treated with TBK1/IKKi dual inhibitor 200A (ip, 100mg/Kg, once per day), and another group was treated with vehicles. Tumor growth was monitored and measured every three days. Tumor volumes were calculated by the equation V (mm3) = a × b2/2, where “a” is the largest diameter and “b” is the perpendicular diameter (Fig. 5A). Thirty-four days later, mice were sacrificed and tumors were collected, weighed (Fig. 5B), and photographed (Fig. 5C). Notably, the treatment of 200A significantly suppressed the growth of SSC-9 xenograft tumor in nude mice.
Figure 5. TBK1/IKKi dual inhibitors suppress tumor growth in mouse models.

(A–C) 1×107 SCC-9 cells were inoculated into nu/nu mice subcutaneously. One day after cell implantation, mice were randomly divided into two groups. One group was administered ip with TBK1/IKKi kinase dual inhibitor 200A (100 mg/kg, once a day), another group treated with vehicles. Tumor growth was monitored and measured every three days (A). The tumor volumes were calculated by the equation V (mm3) = a × b2/2, where “a” is the largest diameter and “b” is the perpendicular diameter. Thirty-four days later, mice were sacrificed and tumors were removed, weighed (B), and photographed (C). Note: Xenograft tumors No.11 and 12 regressed after 200A treatment.
(D–F) 3×106 Myc-CaP cells were inoculated into FVB mice subcutaneously. One day after cell implantation, mice were randomly divided into two groups. One group was administered ip with TBK1/IKKi kinase dual inhibitor 200A (100 mg/kg, once a day), another group treated with vehicles. Tumor growth was monitored and measured every three days (D). Eighteen days later, mice were sacrificed and tumors were removed, weighed (E), and photographed (F).
The results in (A), (B), (D), and (E) are the mean ± SD (* P < 0.05; ** P < 0.01).
In another mouse model, we employed the Myc-CaP mouse prostate cancer cell line. This cell line was isolated from a c-Myc transgenic mouse with prostate cancer, and can grow as tumors in immune competent FVB mice in an androgen dependent manner. 3×106 Myc-CaP cells were inoculated s.c. into FVB mice. One day after cell implantation, mice were randomly divided into two groups. One group was administered ip with 200A (100 mg/kg, once a day), and another group treated with vehicles. Tumor growth was monitored and measured every three days (Fig. 5D). Eighteen days later, mice were sacrificed and tumors were collected, weighed (Fig. 5E), and photographed (Fig. 5F). Similar to that seen with the SCC-9 xenograft model described above, the treatment of TBK1/IKKi kinase dual inhibitor 200A significantly slowed down the growth of Myc-CaP allograft tumors in FVB mice.
It should be noted that there were no apparent signs of toxicity in both Nu/Nu and FVB mice treated with 200A, as evidenced by body weight and other life symptom monitoring. Therefore, TBK1/IKKi dual inhibitor 200A showed a safe and potent anti-tumor effect in tumor mouse models. Thus, the data indicate that simultaneous inhibition of both TBK1 and IKKi might be an efficient way to suppress tumor development.
TBK1/IKKi dual inhibitors suppress the expressions of p-CYLD and p-AKT, and angiogenesis
It has been reported that phosphorylation of the tumor suppressor CYLD by IKKi promotes cell transformation 22. To examine whether TBK1/IKKi dual inhibitors suppress CYLD phosphorylation, SCC-9 cells were treated with different TBK1/IKKi dual inhibitors for 1 hr, and cells were collected for western blot. We found that SCC-9 cells treated with TBK1/IKKi dual inhibitors had less p-CYLD expression than that in controls (Fig. 6A).
Figure 6. Reduced expression of p-CYLD, Cyclin D1, p-AKT, and VEGF in TBK1/IKKi dual inhibitor-treated tumor cells or tissues.

(A) Western blot analysis of CYLD and p-CYLD (Ser418) in SCC-9 cells cells treated with 1.0 μM of indicated TBK1/IKKi dual inhibitors for 1 hr.
(B) Western blot analysis of total and phosphorylated AKT (S473, T308) in SCC-9 cells treated with different concentration of 200A for 30 min.
(C) Western blot analysis of AKT and p-AKT in tumor tissue from mice treated with or without 200A.
(D) Immunohistochemical staining of p-AKT in tumor tissue from mice treated with or without 200A. Tissues were counterstained with hematoxylin (blue) (Magnification 400 X). The relative p-AKT expression shown in right panel is present as the mean ± SD (* p<0.05).
(E) RT-PCR analysis of VEGF in tumor tissue from mice treated with or without 200A.
(F) Immunohistochemical staining of VEGF in SCC-9 xenograft tumors from mice treated with or without 200A. Tissues were counterstained with hematoxylin (blue) (Magnification 400 X). The relative VEGF expression shown in right panel is present as the mean ± SD (* P < 0.05).
AKT plays a pivotal role in fundamental cellular functions such as cell proliferation and survival. Recently it was reported that both TBK1 and IKKi phosphorylate AKT 14, 15, 23, 24. To investigate whether TBK1/IKKi dual inhibitors affect AKT phosphorylation, SCC-25 cells were treated with different doses of 200A for 30 min and cells were then collected for western blot. We found that treatment with 200A decreased p-AKT expression in a dose-dependent manner (Fig. 6B). Similarly, western blot and immunohistochemistry analysis showed that treatment with TBK1/IKKi dual inhibitor 200A led to significant reduction of p-AKT expression in xenograft tumor tissues (Fig. 6C and 6D). These results suggest that the anti-tumor effect of TBK1/IKKi dual inhibitors may be via their suppression of AKT activation.
It has been reported that AKT1 regulates pathological angiogenesis, vascular maturation and permeability in vivo 25. Since our results showed TBK1/IKKi dual inhibitors reduced AKT activation in both cancer cell lines and tumor tissues, we asked whether TBK1/IKKi dual inhibitors affect VEGF expression and angiogenesis in tumors. We therefore used RT-PCR and immunohistochemistry to examine VEGF expression in tumors from 200A- treated and control mice. We found that tumor tissues from 200A-treated mice had much less VEGF expression compared with untreated groups (Fig. 6E and 6F). Altogether, these results suggest that the anti-cancer function of TBK1/IKKi dual inhibitors may be mediated by their suppression of AKT activation, leading to impaired angiogenesis and therefore the inhibition of tumor growth.
Discussion
Accumulating evidence support that the non-canonical IKKs, both TBK1 and IKKi, play critical roles in tumor genesis and development 9–11, leading to the notion that non-canonical IKKs may be good targets for cancer therapy. In the present study, we have generated potent TBK1/IKKi selective dual inhibitors which are based on a structurally rigid 2-amino-4-(3′-cyano-4′-pyrrolidine)phenyl-pyrimidine scaffold. Many of these TBK1/IKKi dual inhibitors exhibited high potency of anti-cell proliferation in human breast, prostate, and oral cancer cell lines. Treatment with these TBK1/IKKi dual inhibitors significantly impairs tumor development in xenograft and allograft models. Most importantly, these TBK1/IKKi dual inhibitors have drug-like properties including low molecular weight, low cytochrome P450 (CYPs) inhibition, and high metabolic stability.
There are several reasons why we prefer TBK1 and IKKi dual inhibitors rather than single isoform-selective inhibitors for the treatment of cancer: A) Necessity of double TBK1-IKKi inhibition. Consistent with another report 6, our results demonstrate that even though TBK1 may not be over-expressed or constitutively activated in some tumor cells, targeting IKKi will induce the activation of TBK1. Therefore, simultaneously targeting both TBK1 and IKKi is necessary to suppress tumor cell proliferation. Therefore, TBK1 and IKKi dual inhibitors represent new opportunities for drug development. B) Safe and effective targets. IKKi knockout mice are viable, fertile and largely normal 26. Silencing TBK1 expression by RNAi was found to induce tumor cell apoptosis, while non-tumorigenic epithelial cells do not depend on TBK1 for survival 9. Therefore, targeting TBK1 and IKKi might be a safe and effective means of suppressing cancer development that could yield few side effects related to conventional cancer therapy and avoid notorious side effects associated with complete IKKβ/NF-κB inhibition. It was reported that although the non-canonical IKKs are not essential for IκB degradation, TBK1/IKKi phosphorylate key targets downstream of the canonical IKK complex (including NF-κB itself), and induce a group of well-characterized NF-κB target genes 27, 28. However, NF-κB activation in either TBK1 or IKKi single and double knockout models is predominantly normal, apart from minimal defects in the induction of select NF-κB target genes 7, 8, indicating that the non-canonical IKKs are sufficient, but not essential for NF-κB activation. Our compounds selectively inhibit TBK1 and IKKi without affecting IKKβ activity. Thus, application of these compounds will not significantly block NF-κB activation (data not shown), and therefore will avoid the notorious side effects associated with complete IKKβ/NF-κB inhibition. It should be noted that the insufficient blockage of NF-κB should not affect the anti-cancer efficacy of TBK1 and IKKi dual inhibition as the contribution of both TBK1 and IKKi to cancer development may not be mainly through transiently activated NF-κB. We recently reported that the cause and maintenance of constitutive NF-κB activation are different from those of its transient activation 29, 30. It is the constitutively activated (rather than the transiently activated) NF-κB that plays a key role in the maintenance of the malignancy of cancer cells 29, 30, thus, it will be interesting to examine if TBK1 and IKKi dual inhibition blocks constitutively activated NF-κB in cancer cells while sparing NF-κB transient activation in immune cells.
The non-canonical IKKs are well established as regulators of the innate immune response via their abilities to phosphorylate IRF-3/7 transcription factors. However, the role and the mechanism of non-canonical IKKs in mediating inflammatory interferon responses are not related to their function in cancer 10. It was reported that IKKi was up-regulated in breast cancer cells by CK-2 31, and that IKKi regulates NF-κB target genes through phosphorylation of p65 at Ser536 32. It was also reported that TBK1 suppressed apoptosis in K-Ras-transformed cells through c-Rel and by up-regulation of Bcl-xL 11. Recently, TBK1 and IKKi have been shown to directly phosphorylate AKT 14, 15, 23, 24. Consistently, we found that our TBK1/IKKi dual inhibitor significantly suppressed the expression of p-AKT in cancer cells and in tumor tissues, suggesting that the anti-tumor effect of TBK1/IKKi dual inhibitors may be, at least partially, via their suppression of AKT activity.
Our results demonstrate that TBK1/IKKi dual inhibitors down-regulate the VEGF expression in tumor tissues, suggesting that activation of non-canonical IKKs may promote oncogenic angiogenesis. It has been proposed that angiogenesis usually get full robustness in tumorigenesis when complex positive feedback loop signaling systems become integrative 33. A cancer hypoxic microenvironment generates positive loops inducing formation of the vascular functional shunts 33. AKT1, whose function is related to pathological angiogenesis, vascular maturation and permeability 25, is located at the upstream angiogenic locus of the integrative robustness. Our results show that TBK1/IKKi dual inhibitors inhibit AKT activation in both cancer cell lines and tumor tissues. Thus, the effect of TBK1/IKKi dual inhibitors on VEGF expression and angiogenesis in tumors may be mediated by their suppression of AKT activity.
Finally, we would like to mention that although the present study is focused on characterizing the anti-cancer function of TBK1/IKKi dual inhibitors, these dual inhibitors may also be suitable to the development of novel therapeutics for other non-cancer diseases, such as glaucoma and rheumatoid arthritis.
Supplementary Material
Novelty and impact.
Our studies demonstrate anti-tumor effects of TBK1/IKKi dual inhibitors, and provide proof of concept for further drug discovery efforts that may lead to novel strategies and efficient therapeutics for the treatment of human cancer.
Acknowledgments
We thank Dr. Antonio L Amelio (Scripps) for critical review of the manuscript. This work was supported by grants from the United States Department of Defense (W81XWH-09-1-0533), National Institute of Health (1R01CA140956-01, 1R21NS073098-01), and monies from the ThinkPinkKids Foundation to J.L.L., by a postdoctoral trainee fellowship from the Frenchman’s Creek Women For Cancer Research to J.H.J., Research Fund for International Young Scientist from China Natural Science Foundation to J.H.J. and S.J.P., and fund from Hunan Provincial Innovation Foundation For Postgraduate (CX2011B066) and a scholarship from China Scholarship Council to J. L.
Footnotes
There are no conflicts
References
- 1.Rothwarf DM, Karin M. The NF-kappa B activation pathway: a paradigm in information transfer from membrane to nucleus. Sci STKE. 1999;1999:RE1. doi: 10.1126/stke.1999.5.re1. [DOI] [PubMed] [Google Scholar]
- 2.Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol. 2000;18:621–63. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- 3.Greten FR, Karin M. NF-kB: Linking Inflammation and Immunity to Cancer Development and Progression. Nature Reviews Immunology. 2005;5:749–59. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 4.Luo JL, Kamata H, Karin M. IKK/NF-kappaB signaling: balancing life and death--a new approach to cancer therapy. J Clin Invest. 2005;115:2625–32. doi: 10.1172/JCI26322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shen RR, Hahn WC. Emerging roles for the non-canonical IKKs in cancer. Oncogene. 2011;30:631–41. doi: 10.1038/onc.2010.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clement JF, Meloche S, Servant MJ. The IKK-related kinases: from innate immunity to oncogenesis. Cell Res. 2008;18:889–99. doi: 10.1038/cr.2008.273. [DOI] [PubMed] [Google Scholar]
- 7.Bonnard M, Mirtsos C, Suzuki S, Graham K, Huang J, Ng M, Itie A, Wakeham A, Shahinian A, Henzel WJ, Elia AJ, Shillinglaw W, et al. Deficiency of T2K leads to apoptotic liver degeneration and impaired NF-kappaB-dependent gene transcription. EMBO J. 2000;19:4976–85. doi: 10.1093/emboj/19.18.4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hemmi H, Takeuchi O, Sato S, Yamamoto M, Kaisho T, Sanjo H, Kawai T, Hoshino K, Takeda K, Akira S. The roles of two IkappaB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J Exp Med. 2004;199:1641–50. doi: 10.1084/jem.20040520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chien Y, Kim S, Bumeister R, Loo YM, Kwon SW, Johnson CL, Balakireva MG, Romeo Y, Kopelovich L, Gale M, Jr, Yeaman C, Camonis JH, et al. RalB GTPase-mediated activation of the IkappaB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell. 2006;127:157–70. doi: 10.1016/j.cell.2006.08.034. [DOI] [PubMed] [Google Scholar]
- 10.Boehm JS, Zhao JJ, Yao J, Kim SY, Firestein R, Dunn IF, Sjostrom SK, Garraway LA, Weremowicz S, Richardson AL, Greulich H, Stewart CJ, et al. Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell. 2007;129:1065–79. doi: 10.1016/j.cell.2007.03.052. [DOI] [PubMed] [Google Scholar]
- 11.Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, Frohling S, Chan EM, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–12. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bodemann BO, White MA. Ral GTPases and cancer: linchpin support of the tumorigenic platform. Nat Rev Cancer. 2008;8:133–40. doi: 10.1038/nrc2296. [DOI] [PubMed] [Google Scholar]
- 13.Boehm JS, Zhao JJ, Yao J, Kim SY, Firestein R, Dunn IF, Sjostrom SK, Garraway LA, Weremowicz S, Richardson AL, Greulich H, Stewart CJ, et al. Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell. 2007;129:1065–79. doi: 10.1016/j.cell.2007.03.052. [DOI] [PubMed] [Google Scholar]
- 14.Xie X, Zhang D, Zhao B, Lu MK, You M, Condorelli G, Wang CY, Guan KL. IkappaB kinase epsilon and TANK-binding kinase 1 activate AKT by direct phosphorylation. Proc Natl Acad Sci U S A. 2011;108:6474–9. doi: 10.1073/pnas.1016132108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ou YH, Torres M, Ram R, Formstecher E, Roland C, Cheng T, Brekken R, Wurz R, Tasker A, Polverino T, Tan SL, White MA. TBK1 directly engages Akt/PKB survival signaling to support oncogenic transformation. Mol Cell. 2011;41:458–70. doi: 10.1016/j.molcel.2011.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bamborough P, Christopher JA, Cutler GJ, Dickson MC, Mellor GW, Morey JV, Patel CB, Shewchuk LM. 5-(1H-Benzimidazol-1-yl)-3-alkoxy-2-thiophenecarbonitriles as potent, selective, inhibitors of IKK-epsilon kinase. Bioorg Med Chem Lett. 2006;16:6236–40. doi: 10.1016/j.bmcl.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 17.Wang T, Block MA, Cowen S, Davies AM, Devereaux E, Gingipalli L, Johannes J, Larsen NA, Su Q, Tucker JA, Whitston D, Wu J, et al. Discovery of azabenzimidazole derivatives as potent, selective inhibitors of TBK1/IKKepsilon kinases. Bioorg Med Chem Lett. 2012;22:2063–9. doi: 10.1016/j.bmcl.2012.01.018. [DOI] [PubMed] [Google Scholar]
- 18.Clark K, Plater L, Peggie M, Cohen P. Use of the pharmacological inhibitor BX795 to study the regulation and physiological roles of TBK1 and IkappaB kinase epsilon: a distinct upstream kinase mediates Ser-172 phosphorylation and activation. J Biol Chem. 2009;284:14136–46. doi: 10.1074/jbc.M109.000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McIver EG, Bryans J, Birchall K, Chugh J, Drake T, Lewis SJ, Osborne J, Smiljanic-Hurley E, Tsang W, Kamal A, Levy A, Newman M, et al. Synthesis and structure-activity relationships of a novel series of pyrimidines as potent inhibitors of TBK1/IKKepsilon kinases. Bioorg Med Chem Lett. 2012;22:7169–73. doi: 10.1016/j.bmcl.2012.09.063. [DOI] [PubMed] [Google Scholar]
- 20.Solis M, Romieu-Mourez R, Goubau D, Grandvaux N, Mesplede T, Julkunen I, Nardin A, Salcedo M, Hiscott J. Involvement of TBK1 and IKKepsilon in lipopolysaccharide-induced activation of the interferon response in primary human macrophages. Eur J Immunol. 2007;37:528–39. doi: 10.1002/eji.200636090. [DOI] [PubMed] [Google Scholar]
- 21.Gatot JS, Gioia R, Chau TL, Patrascu F, Warnier M, Close P, Chapelle JP, Muraille E, Brown K, Siebenlist U, Piette J, Dejardin E, et al. Lipopolysaccharide-mediated interferon regulatory factor activation involves TBK1-IKKepsilon-dependent Lys(63)-linked polyubiquitination and phosphorylation of TANK/I-TRAF. J Biol Chem. 2007;282:31131–46. doi: 10.1074/jbc.M701690200. [DOI] [PubMed] [Google Scholar]
- 22.Hutti JE, Shen RR, Abbott DW, Zhou AY, Sprott KM, Asara JM, Hahn WC, Cantley LC. Phosphorylation of the Tumor Suppressor CYLD by the Breast Cancer Oncogene IKK epsilon Promotes Cell Transformation. Molecular Cell. 2009;34:461–72. doi: 10.1016/j.molcel.2009.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gulen MF, Bulek K, Xiao H, Yu M, Gao J, Sun L, Beurel E, Kaidanovich-Beilin O, Fox PL, Dicorleto PE, Wang JA, Qin J, et al. Inactivation of the Enzyme GSK3alpha by the Kinase IKKi Promotes AKT-mTOR Signaling Pathway that Mediates Interleukin-1-Induced Th17 Cell Maintenance. Immunity. 2012;37:800–12. doi: 10.1016/j.immuni.2012.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo JP, Coppola D, Cheng JQ. IKBKE protein activates Akt independent of phosphatidylinositol 3-kinase/PDK1/mTORC2 and the pleckstrin homology domain to sustain malignant transformation. J Biol Chem. 2011;286:37389–98. doi: 10.1074/jbc.M111.287433. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.Chen J, Somanath PR, Razorenova O, Chen WS, Hay N, Bornstein P, Byzova TV. Akt1 regulates pathological angiogenesis, vascular maturation and permeability in vivo. Nat Med. 2005;11:1188–96. doi: 10.1038/nm1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tenoever BR, Ng SL, Chua MA, McWhirter SM, Garcia-Sastre A, Maniatis T. Multiple functions of the IKK-related kinase IKKepsilon in interferon-mediated antiviral immunity. Science. 2007;315:1274–8. doi: 10.1126/science.1136567. [DOI] [PubMed] [Google Scholar]
- 27.Fujita F, Taniguchi Y, Kato T, Narita Y, Furuya A, Ogawa T, Sakurai H, Joh T, Itoh M, Delhase M, Karin M, Nakanishi M. Identification of NAP1, a regulatory subunit of IkappaB kinase-related kinases that potentiates NF-kappaB signaling. Mol Cell Biol. 2003;23:7780–93. doi: 10.1128/MCB.23.21.7780-7793.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buss H, Dorrie A, Schmitz ML, Hoffmann E, Resch K, Kracht M. Constitutive and interleukin-1-inducible phosphorylation of p65 NF-{kappa}B at serine 536 is mediated by multiple protein kinases including I{kappa}B kinase (IKK)-{alpha}, IKK{beta}, IKK{epsilon}, TRAF family member-associated (TANK)-binding kinase 1 (TBK1), and an unknown kinase and couples p65 to TATA-binding protein-associated factor II31-mediated interleukin-8 transcription. J Biol Chem. 2004;279:55633–43. doi: 10.1074/jbc.M409825200. [DOI] [PubMed] [Google Scholar]
- 29.Rokavec M, Wu W, Luo JL. IL6-Mediated Suppression of miR-200c Directs Constitutive Activation of Inflammatory Signaling Circuit Driving Transformation and Tumorigenesis. Mol Cell. 2012 doi: 10.1016/j.molcel.2012.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rokavec M, Luo JL. The transient and constitutive inflammatory signaling in tumorigenesis. Cell Cycle. 2012;11:2587–8. doi: 10.4161/cc.21139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eddy SF, Guo S, Demicco EG, Romieu-Mourez R, Landesman-Bollag E, Seldin DC, Sonenshein GE. Inducible IkappaB kinase/IkappaB kinase epsilon expression is induced by CK2 and promotes aberrant nuclear factor-kappaB activation in breast cancer cells. Cancer Res. 2005;65:11375–83. doi: 10.1158/0008-5472.CAN-05-1602. [DOI] [PubMed] [Google Scholar]
- 32.Adli M, Baldwin AS. IKK-i/IKKepsilon controls constitutive, cancer cell-associated NF-kappaB activity via regulation of Ser-536 p65/RelA phosphorylation. J Biol Chem. 2006;281:26976–84. doi: 10.1074/jbc.M603133200. [DOI] [PubMed] [Google Scholar]
- 33.Radisavljevic Z. AKT as locus of cancer angiogenic robustness and fragility. J Cell Physiol. 2013;228:21–4. doi: 10.1002/jcp.24115. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.