

Abstract
The recent formalization of clinical criteria for PD with dementia (PD-D) codifies many studies on this topic, including those assessing biological correlates. These studies show that the emergence of PD-D occurs on the background of severe dopamine deficits with the main pathological drivers of cognitive decline being a synergistic effect between α -synuclein and Alzheimer's disease pathology. The presence of these pathologies correlates with a marked loss of limbic and cortically projecting dopamine, noradrenaline, serotonin and acetylcholine neurons, although the exact timing of these relationships remains to be determined. Genetic factors, such as triplications in the α-synuclein gene, lead to a clear increased risk of PD-D, while others, such as parkin mutations, are associated with a reduced risk of PD-D. The very recent formalization of clinical criteria for PD with mild cognitive impairment (PD-MCI) allows only speculation on its biological and genetic bases. Critical assessment of animal models shows that chronic low dose MPTP treatment in primates recapitulates PD-MCI over time, enhancing the current biological concept of PD-MCI as having enhanced dopamine deficiency in frontostriatal pathways as well as involvement of other neurotransmitter systems. Data from other animal models support multiple transmitter involvement in cognitive impairment in PD. While dopamine dysfunction has been highlighted because of its obvious role in PD, the role of the other neurotransmitter systems, neurodegenerative pathologies and genetic factors in PD-MCI remain to be fully elucidated.
Keywords: Parkinson's disease dementia, genetic risk, neuropathology, neurotransmitters, preclinical models
Introduction
The neurobiological basis for Parkinson's disease (PD) is degeneration of nigrostriatal dopamine neurons and the pathological deposition of the protein α-synuclein in intraneuronal Lewy inclusions within vulnerable populations of neurons in the brain (Figure 1A-D).1 The pathologic changes in PD occur gradually and progressively over many years with a significant period of clinically silent cellular dysfunction and, for some populations of neurons, cell death.2 It has also become apparent that in the elderly such pathologies often occur on a background of age-related pathologies, which will be discussed in more detail below. This has resulted in the revised neuropathological criteria for differentiating PD with and without additional Alzheimer type pathology by the incorporation of a probabilistic statement about the likelihood that each different pathology contributes to a cognitive disorder (Table 1).1 The initiating neurobiological bases for such dysfunction are uncertain in many instances and will not be discussed in detail, but are known to include a variety of factors based on the relatively large numbers of genes now known to be causative for PD.3
Figure 1. Tissue histopathology of PD and PD-D.
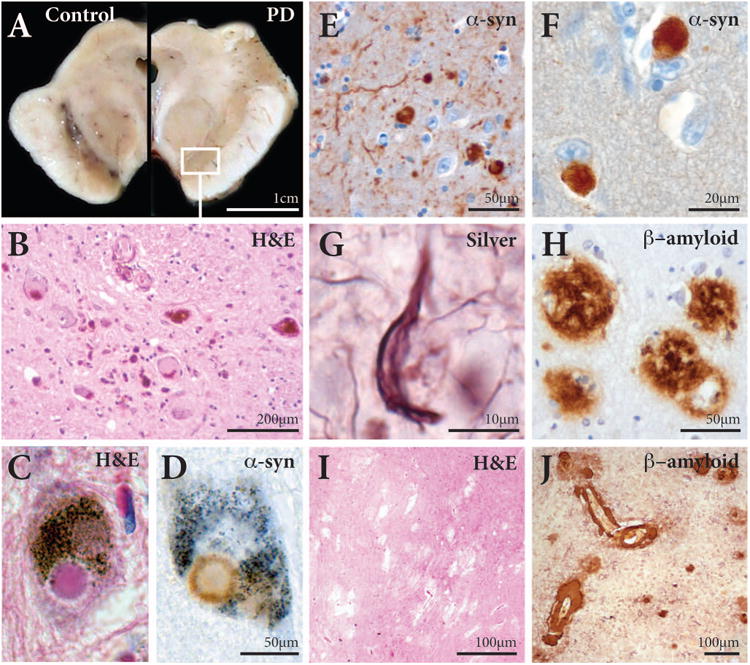
A Transverse section through the midbrain of a control (at left) showing the darkly pigmented substantia nigra in the ventral aspect of the midbrain, while the pigmented neurons in this structure are lost in patients with PD (at right).
B Higher magnification (box in A) of a haematoxylin and eosin (H&E) stained section through the substantia nigra showing only a few pigmented neurons remaining with many smaller phagocytic microglia.
C,D Higher magnification of a H&E stained (C) and an α-synuclein-immunoreactive (D) pigmented neuron in the substantia nigra of a PD patient containing a Lewy body.
E,F α-Synuclein immunoreactive Lewy bodies and Lewy neurites in the amygdala (E) and anterior cingulate cortex (F) of a patient with PD-D.
G Silver-stained neurofibrillary tangle in the cortex of a patient with PD-D.
H β-amyloid-immunoreactive plaques in the cortex of a patient with PD-D.
I Vascular ischaemic tissue damage identified in a H&E stained section of the globus pallidus in a patient with PD-D.
J β-amyloid-immunoreactive congophilic angiopathy in the cortex of a patient with PD-D.
Table 1. Probability of different pathologies contributing to a cognitive disorder1.
| NIA-Reagan Alzheimer type pathology | ||||
|---|---|---|---|---|
| NFT stage I-II/ CERAD possible | NFT stage III-IV/ CERAD probable | NFT stage V-VI/ CERAD definite | ||
| Lewy body type | Brainstem | Low | Low | Low |
| Transitional | High | Intermediate | Low | |
| Diffuse | High | High | Intermediate | |
NIA=National Institute of Aging; NFT=neurofibrillary tangle; CERAD=Center for Education and Research on Alzheimer's Disease
To discuss the neurobiological basis of PD dementia (PD-D) necessitates a brief discussion of the type of cases required to be analyzed. The MDS published a review and clinical criteria for PD-D in 2007 with the core features being a clinical diagnosis of established PD and a dementia syndrome with insidious onset and slow progression largely meeting DSM IV criteria for dementia (Table 2).4 The pathological validity of and neurobiological basis for this criterion will be evaluated further below.
Table 2. Criteria for PD-D4.
| Core Features | Associated Features | Exclusions | |
|---|---|---|---|
|
| |||
| Probable PD-D |
|
|
|
|
| |||
| Possible PD-D |
|
|
|
In 2012 the MDS used a similar process to determine that mild cognitive impairment was common in PD (∼25% in those without PD-D), was clinically heterogeneous, and increased the risk of progression to dementia.5. In establishing a diagnostic criteria for mild cognitive impairment in PD (PD-MCI)(Table 3)5 that progresses to PD-D, the number of cases in any pathological series of end-stage cases are likely to be smaller than the number with PD-D. These newer criteria for PD-MCI are still evolving, eg. establishing cutoffs for neuropsychological scores and the true prevalence of single domain subtype.6 Thus, it may be early to discuss fully the neurobiological bases and underlying pathology for PD-MCI, but some attempt will be made in the following sections.
Table 3. Criteria for PD-MCI5.
| Inclusion | Exclusion | |
|---|---|---|
|
| ||
| PD-MCI single domain |
|
|
|
| ||
| PD-MCI multiple domain |
|
|
Neuropathology
The pathologic substrate for PD-D and PD-MCI appears to be heterogeneous and includes Lewy bodies, Alzheimer's disease (AD) pathology, cerebrovascular disease, and other findings (Figure 1).7-12 Data and thoughts regarding the role of Lewy bodies appear to have changed with the introduction of α-synuclein immunostaining and data presented here only considers studies using these methods.
Role of Lewy-type α-synucleinopathy in PD-D
The most compelling evidence to date suggests that Lewy-related pathology (LRP) (data using α-synuclein staining, Figure 1D-F) is the most important factor in the development of cognitive impairment in PD. Most studies compared LRP with AD pathology with less written on other pathologic findings. One study of 45 PD cases assessing cognitive impairment from retrospective chart reviews showed that the severity of cognitive impairment correlated with cortical Lewy bodies, especially in the frontal and cingulate gyrus, even when AD was not present.13 In a study of 22 PD-D cases two did not have cortical Lewy bodies, 13 (59%) had marked cortical Lewy bodies, and 7 (32%) had both cortical Lewy bodies and AD.8 The marker that most correlated with dementia was the cortical Lewy bodies.8 In a study of 12 PD-D cases there was a 10× increase in neocortical and limbic LRP compared to non-demented PD cases and only one case met NIA-Reagan criteria for AD.14 Neocortical Lewy bodies correlated with both senile plaques and neurofibrillary tangles and two cases had multiple infarcts.14 While a study of 18 PD-D cases found that none met neuropathological criteria for AD.15 A larger study found that 28/51 PD-D cases had concurrent AD, but that clinically the cases with and without AD were not distinguishable, suggesting the LRP was the more critical factor.10 Neocortical LRP was also found to correlate with dementia while AD did not in a study of 129 PD cases (not all of whom had dementia), although those cases with dementia had higher neurofibrillary tangle pathology.16 Finally, a recent study of 92 PD-D and 48 non-demented PD cases found the severity of cortical LRP was the factor that most correlated with dementia.12 In a community based study of 872 autopsies, 103 had LRP with a neocortical distribution associated with increased odds of dementia and lower and more rapid decline in all cognitive domains, while a limbic distribution specifically associated with lower and more rapid decline in visuospatial skills.17 These relationships were not modified by AD pathology.17 It should be noted that not all patients with cortical LRP will have dementia,12, 16, 18 although in a study that excluded AD and assessed 41 PD cases, Lewy body densities in the temporal lobe were significantly higher in cases with PD-D compared to PD without dementia.19 This differentiation was not observed in frontal or limbic cortical regions.19
Role of AD pathology in PD-D
There are widely conflicting reports regarding the co-occurrence of AD pathology (Figure 1G,H) in PD-D. Neuritic plaque pathology was greater in PD-D cases compared with non-demented cases in one study.9 Some studies found only 1/1214 and 3/1720 PD-D cases met NIA-Reagan criteria for AD while other studies found a much higher occurrence 18/41,21 7/22,8 18/48,12 and 28/51.10 Meeting criteria for AD may not be the key however, as there appears to be a positive correlation between neocortical LRP and both senile plaques and neurofibrillary tangles.14 There is also a correlation between neurofibrillary tangles and dementia in PD.8, 12 A recent study quantitatively assessing these cortical pathologies identified that the combination of LRP and AD pathologies most robustly correlate with PD-D.22 The issue of a synergistic role needs further research, although the deposition of α-synuclein promotes the intracellular aggregation of tau23 and β-amyloid24 in cell models.
Role of other pathologies
The role of other pathologic findings in causing cognitive impairment in PD remains unclear. As the majority of PD autopsy series have a mean age of cases being >75 years old, the presence of other pathologic findings is extremely common and thus controlling for pathologic changes of aging are critical. While a recent study showed that there is a reduced prevalence of small vessel disease in PD compared to matched controls,25 the role of vascular changes (Figure 1I,J), including infarcts, is one of the most difficult issues when studying PD-D. One study found that 8/18 PD cases had vascular changes.15 Lower rates were found in multiple other studies including 2/17 PD-D cases,20 2/12 may have had multiple infarcts contributing to the dementia,14 and 5/40 PD-D cases had significant cerebrovascular disease.26 In a study of 25 non-demented PD and 25 PD-D cases that excluded concurrent AD or hippocampal sclerosis, PD-D correlated with a the cumulative burden of cerebral white matter disease.11 So the role of vascular changes remains unclear.
Cerebral amyloid angiopathy (CAA, Figure 1J), not uncommon in aged controls, was associated with PD-D in one study.12 CAA had a positive correlation to LRP while vascular pathology was negatively correlated with LRP in a second study.27 Finally, CAA was more common in PD than controls, and was more common in PD cases with concurrent AD than those without AD.28
Hippocampal sclerosis (HS), the loss of neurons and presence of gliosis in the CA1 region of the hippocampus and subiculum,29 may be found in ∼10% of AD cases.30 In PD-D one study found HS in only 1/17 cases,20 a second study found no correlation between HS and PD-D,12 and a third study found 4.4% of 561 AD cases had HS while 5.3% of 131 PD-D cases had HS (no controls were studied).31 The role of other pathologies, including argyrophilic grains and TDP-43 are even less well studied.
PD-MCI pathology
There is scant neuropathological data on PD-MCI, but the data available suggest a similar underlying heterogeneity of pathology to that described above. Aarsland et al.15 reported three of four PD cases with cognitive impairment, but not dementia, had neocortical Lewy bodies while one did not. Adler et al.7 studied eight cases with PD-MCI (4 amnestic MCI-memory only, 4 non-amnestic MCI), mean age 82.8 yrs (range 74-89), and mean PD duration 11.4 yrs (range 2-25 yrs). Using Beach et al. Unified LB staging (ULBSS) criteria,32 three cases were brainstem-predominant (stage 2a), three were brainstem-limbic predominant (stage 3), and two were neocortical Lewy body stage (stage 4).7 Half had moderate neuritic plaques and two met NIA-Reagan criteria for AD (both having amnestic MCI).7 Jellinger also reported 8 PD-MCI cases, four amnestic and four non-amnestic MCI.33 Two were ULBSS stage 2a, five were stage 3, and one was stage 4, while AD pathology was also heterogeneous.33 Both studies found variable amounts of cerebral amyloid angiopathy (CAA).7, 33 Given this limited amount of data the roles of each type of pathology in PD-MCI remains unclear.
Severity and timing of pathologies
As autopsy studies are by definition cross-sectional, piecing together the severity and timing of pathologic changes causing cognitive impairment is not easy. However, at present there are few neuroimaging studies that have confirmation of the underlying pathologies discussed above that associate with PD-D,34 and as structural and functional changes are biomarkers of AD pathology,35 it remains to be determined whether current neuroimaging studies assessing such changes are capturing PD-specific changes or those associated with additional pathologies common in patients with PD-D (see above).
The assessment of pathological studies shows that when dividing PD-D cases based on disease duration, those with a shorter duration of disease prior to death had an older age of onset and were more likely to have concurrent AD, but even in this group it was only 10% (1/10) of the cases.26 All PD-D groups had either limbic or limbic and neocortical stage Lewy bodies.26 There is some data that suggests chronological aging may be an important driving factor in the onset of dementia in patients with PD,36 a factor also related to the prevalence of AD pathology in patients with dementia.37-39
A number of studies suggest there is a more rapid disease progression in PD-D cases with coexistent AD.22 Mean survival from onset of PD went from a 10.1 years in cases without AD to 4.5 years in those with AD.40, 41 Studies have shown that PD-D cases with AD may have older age of PD onset,10, 12, 22, 40, 41 older age of death, and shorter disease duration,10, 12, 40, 41 but this is not always the case.8
Summary of Neuropathological Findings in PD-D
While this has been a discussion of the role of individual pathologies in causing dementia in PD, it is very likely that there is a synergistic effect between α-synuclein pathology, age and other pathologies, especially AD pathology,42, 43 that is the main driver of cognitive decline in PD.
Neurotransmitter Systems
Multiple neurotransmitter deficits have long been emphasized as of major importance in PD and particularly in underlying the cognitive deficits.44, 45 At present changes to dopamine medication use and cholinesterase inhibitors are considered the most practical management options for PD-D,46, 47 highlighting the involvement of these systems in PD-D.
Dopamine systems and PD-D
The majority of dopamine neurons in the brain are found in three main midbrain dopamine regions which project to the basal ganglia (nigrostriatal system), limbic regions (mesolimbic system) and cortical regions (mesocortical system)(Figure 2).48, 49 The largest group of dopamine neurons (A9 group) are found in the substantia nigra, an integral part of the basal ganglia that provides a feedback loop to the striatum important for the control of actions and thoughts. The ventrolateral A9 neurons that project to the putamen degenerate early prior to Lewy body formation in PD,50 with increasing degeneration of other A9 nigrostriatal neurons relating to increasing motor deficits as the disease progresses,51, 52 deficits which are initially ameliorated by dopamine replacement therapies53 and enhanced by the use of dopamine blocking drugs.54 A number of histopathological52, 55 and imaging56-58 studies show that the severity of A9 dopamine loss is independent of dementia in PD.
Figure 2. Dopamine pathways affected in PD and PD-D.

Red outline=substantia nigra (SN) which contains both dopamine neurons in the pars compacta that give rise to the nigrostriatal projections, and GABA neurons in the pars reticulata which innervate the thalamus. Dotted red line=ventrolateral substantia nigra (VLa SN) which is selectively damaged in patients with PD. Yellow outline=ventral tegmental area (VTA) which contains both dopamine and non-dopamine neurons that project to limbic and cortical regions. Dotted orange outline=medial SN and VTA which give rise to mesolimbic projections affected in patients with PD-D. cp=cerebral peduncle, N. acc=nucleus accumbens, R=red nucleus.
Mesolimbic dopamine neurons are found in the medial substantia nigra (A9 neurons) and nearby ventral tegmental area (A10 neurons).59 There is greater degeneration of medial A9 dopamine neurons in PD-D,60-62 and potentially related to this cell loss are findings in the caudate nucleus and ventral striatum of a loss of presynaptic dopamine in PD-D63 as well as a correlation between the density of postsynaptic D1 dopamine receptors and cognitive impairment.64 These medial mesolimbic A9 dopamine neurons play an important role in behavioural selection and impulsivity,59 and enhanced function in mesolimbic/ventral striatal dopaminergic circuits following dopamine agonist use may contribute to impulse control disorders in PD.53, 65
The ventral tegmental area A10 dopamine neurons are considered integral in both reward behaviors (mesolimbic) and cognitive functions (mesocortical),49 but are less studied in patients with PD. In humans the A10 dopamine neurons are spread over a large rostrocaudal region of the midbrain and are difficult to identify with certainty in single sections.48 Only a single serial section study has assessed the mesocortical A10 dopamine neurons proper and revealed limited degeneration in the A10 dopamine cell groups in 7 PD patients, 5 with PD-D.66 Despite this, in vivo imaging suggests a reduction in cortical dopamine in patients with PD-D,63 indicating a functional rather than structural depletion of dopamine in the mesocortical system. The effects of dopamine replacement therapy on mesocortical dopamine function is still poorly understood.67, 68
Other monoamine systems and PD-D
There is less data examining the extent to which the loss of brain noradrenaline contributes to PD-D, although severe lesions of the A6 noradrenaline neurons in the locus coeruleus that innervate the entire forebrain are frequent in PD-D (Figure 3).55, 62, 69 This marked degeneration of A6 noradrenaline locus coeruleus neurons in PD-D correlates with a marked loss of forebrain Ch4 cholinergic neurons in the nucleus basalis (see below).55, 62 In older adults without a clinical diagnosis of PD, the density of A6 noradrenaline neurons in the locus coeruleus correlates with the density of A9 dopamine neurons and the severity of mild parkinsonian features.70 In early PD there is an upregulation of noradrenaline locus coeruleus transmission, possibly affecting the thalamus,71 to compensate for the loss of striatal dopamine,58, 72 with significant reductions in forebrain noradrenaline by end-stage PD.58, 71, 73 Importantly, improved cognitive function has been shown following administration of a noradrenaline reuptake inhibitor.74
Figure 3. Noradrenaline pathways affected in PD and PD-D.

The A6 noradrenaline neurons innervate most of the brain including the substantia nigra (SN) and thalamus, pathways affected in patients with PD, as well as the cholinergic nucleus basalis (NBM), limbic and cortical regions, pathways affected in PD-D. N. acc=nucleus accumbens.
There are two major serotonin cell groups that project to the forebrain, the B6/7 neurons in the dorsal raphe nucleus which project largely to the striatum and cortex, and the B5/8 neurons in the median raphe nucleus which project largely to the cortex and hippocampus (Figure 4).75 Although data are limited, there appears to be no loss of B6/7 serotonin neurons but a variable loss of B5/8 serotonin neurons by end-stage PD76, and no reduction in striatal serotonin transporter early in PD77, 78 but more marked reductions in the caudate nucleus compared to the putamen by the endstage.79, 80 There do not appear to be any major changes to serotonin receptors in PD.81-83 There is a correlation between the reduction in striatal dopamine transporter and increased levels of striatal serotonin transporter in early PD, indicating a potential initial compensatory mechanism.78 However, as the disease progresses, some serotonin structures degenerate,80 most markedly in the B5/8 system. Whether this degeneration relates to the onset or expression of cognitive impairment has not been evaluated. However, in PD-D there is an increase in cortical serotonin turnover84 and serotonin 2A receptors,85 suggestive of reduced serotonin innervation and consistent with the late loss of B5/8 serotonin neurons.76
Figure 4. Serotonin pathways affected in PD and PD-D.
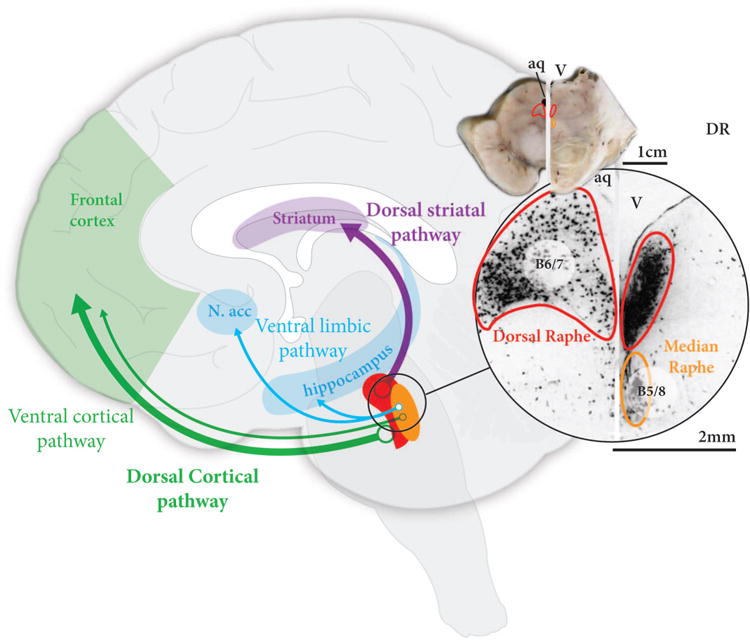
The two major serotonin nuclei with projections to the forebrain are the dorsal raphe containing B6/7 neurons, located in the periaqueductal grey matter of the midbrain, and the median raphe nucleus containing B5/8 neurons, located in the midline of the upper pons. The dorsal raphe projects to the striatum and cortex, while the median raphe projects to limbic regions and cortex. Only the median raphe is affected in PD, but whether this is associated with dementia or not is not clear. aq=aqueduct, N. acc=nucleus accumbens, V=fourth ventricle.
Acetylcholine systems and PD-D
There are three main sources of acetylcholine in the brain - striatal interneurons, cortically-projecting Ch4 neurons in the nucleus basalis, and thalamic-projecting Ch5 neurons in the pedunculopontine nucleus (Figure 5).86, 87 Striatal interneurons are not affected in PD, but there is ample evidence that the other neuronal groups are affected in PD.86, 87 On average there is a 40% loss of Ch5 pedunclopontine neurons which correlates with the degree of degeneration in the A9 dopamine cell group,88 and also with the severity of motor impairment.89
Figure 5. Acetylcholine pathways affected in PD and PD-D.
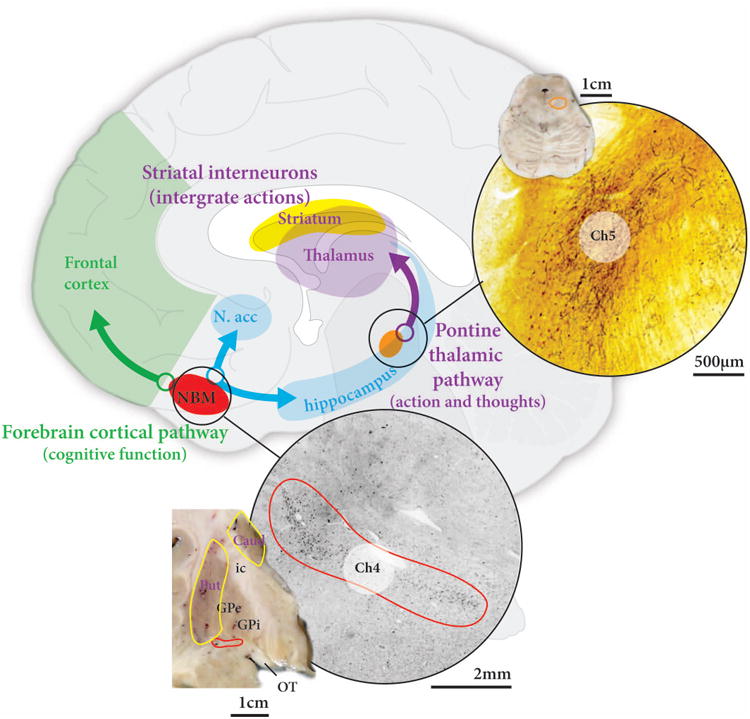
The two major acetylcholine nuclei with projections to the forebrain are the nucleus basalis containing Ch4 neurons, located in the basal forebrain, and the pedunculopontine nucleus containing Ch5 neurons, located at the junction of the midbrain and pons. The pedunculopontine nucleus projects to the thalamus and is affected in PD, while the nucleus basalis projects to limbic regions and cortex and is affected in PD-D. Caud=caudate nucleus, GPe=external globus pallidus, GPi=internal globus pallidus, ic=internal capsule, OT=optic tract, Put=putamen.
Degeneration of Ch4 nucleus basalis neurons is observed in most end-stage cases of PD, but is most severe in PD-D.55, 62 Ch4 cell loss correlates with reductions in cortical choline acetyltransferase,55, 62 and the reduction in cortical choline acetyltransferase correlates with the extent of cognitive impairment.64, 82 Imaging studies confirm the widespread loss of cortical acetylcholine in PD-D.86, 87 Importantly, anticholinergic medications accelerate cognitive impairment in PD,90 while cholinesterase inhibitors have beneficial effects in PD-D.47
Neurotransmitters in PD-MCI
A number of studies suggest that neurotransmitter dysregulation rather than frank neurodegeneration is key to PD-MCI.44, 45, 91, 92 In particular, dopamine deficiency in frontostriatal pathways are considered to play a key role, as discussed above, with new evidence linking dopamine function and hippocampal long-term potentiation to cognitive impairment in PD.93, 94 Reduced neocortical acetylcholinesterase independently relates to lower cognitive performance in non-demented PD patients.95, 96 The role of the other neurotransmitter systems discussed above in PD-MCI remain less certain, although if PD-MCI develops into PD-D, then disruption to these system must at least begin to appear in patients with PD-MCI.
Severity and timing of neurotransmitter disruption
The data above suggest that the loss of dopamine occurs prior to the loss of the other neurotransmitter systems in PD, and that increased compensatory neurotransmitter activity initially occurs in regions affected by the deficit in dopamine. Whether such increased activity or a reduced compensatory capacity drives subsequent degeneration remains to be determined, but it is of interest that degeneration of the dopamine system occurs more rapidly in older compared with younger PD patients.97
Summary of Neurotransmitter Changes in PD-D
The emergence of PD-D occurs on the background of severe dopamine deficits, and correlates with a marked loss of limbic and cortically projecting dopamine, noradrenaline, serotonin and acetylcholine neurons (Figure 6), although the relationship between such forms of degeneration remains to be determined.
Figure 6. Summary of neurotransmitter pathways affected in PD and PD-D.

Brain regions important for both movement and cognition are the neocortex, striatum, thalamus and limbic brain regions. These brain regions receive modulating neurotransmitter input from dopamine, noradrenaline, serotonin and acetylcholine nuclei located in the brainstem and basal forebrain. Patients with PD have significant degeneration of the dopamine neurons in the lateral substantia nigra (SN), acetylcholine neurons in the pedunculopontine nucleus (PPN) and noradrenaline neurons in the locus coeruleus (LC). In PD-D there is additional degeneration of acetylcholine neurons in the nucleus basalis (NBM), dopamine neurons in the medial SN, serotonin neurons in the median raphe (MR) and noradrenaline neurons in the LC. In addition, dysfunction without degeneration may occur in other neurotransmitter pathways (not shown).
Genetic Risk Factors
While large genome-wide association studies have identified a variety of genes contributing to sporadic PD,98 similar large studies have not been performed for PD-D and particularly for PD-MCI as their definitions have only recently been operationalized (Tables 2&3). This means that the genetics underlying PD-D and PD-MCI are incompletely understood, although there is a substantial literature on smaller cohorts that clearly demonstrates the importance of genetic factors for cognitive impairment (CI) in PD (Table 4). Evidence comes both from studies examining cognition in familial forms of PD and from studies of PD and non-PD associated genetic risk factors.
Table 4. Genes associated with cognitive impairment (CI) in PD.
| Familial PD | Sporadic PD | |
|---|---|---|
| SNCA | increased | - |
| LRRK2 | decreased | - |
| parkin | decreased | - |
| GBA | - | increased |
| MAPT H1 haplotype | - | increased |
| COMT Met/Met | - | mixed findings |
| APOE 4 | - | increased |
SNCA=α-synuclein gene, LRRK2=leucine-rich repeat kinase 2 gene, GBA=glucocerebrosidase gene, MAPT=microtubule associated protein tau gene, COMT=catechol-o-methyl transferase gene, APOE=apolipoprotein E gene
Cognition in familial PD
Reports of CI in both autosomal recessive and dominant forms of familial PD suggest that the frequency and characteristics of CI in these various familial forms of PD differ. In autosomal dominant forms of PD, the most extensively studied are those linked to mutations, duplications and triplications in the α-synuclein gene (SNCA) and to mutations in the leucine-rich repeat kinase 2 gene (LRRK2).99 All mutations in the SNCA gene have thus far been associated with CI in at least some cases, although the frequency may be lower than that observed in sporadic PD.100 However, one SNCA mutation (E46K) has been associated with PD-D in all mutation carriers during the clinical course and CI at onset of PD in at least one affected mutation carrier.101, 102 Duplications and triplications of the SNCA gene are associated with markedly different clinical characteristics, including CI.103-105 While both duplication and triplication are associated with an autosomal dominantly inherited form of PD, SCNA gene duplications are generally associated with a phenotype more consistent with late-onset sporadic PD, while triplications are associated with a substantially earlier onset of both motor symptoms and CI. Triplication cases have prominent early CI, including some with a clinical picture more similar to dementia with Lewy bodies than PD-D.106, 107 In summary, the characteristics of CI in SNCA-associated familial PD are variable and range from a lower than expected frequency of CI to prominent early PD-D (Table 4). Certain SNCA changes, specifically gene triplication and the E46K mutation appear to have an increased risk for early PD-D. There are multiple pathogenic LRRK2 mutations and they are generally associated with a clinical picture very similar to that observed in sporadic PD (Table 4). Studies of cognition in LRRK2 mutation carriers have suggested a frequency of CI that is similar to or less frequent than that observed in sporadic PD.108-111 Interestingly, a recent report suggested that asymptomatic G2019S LRRK2 mutation carriers without overt PD were more impaired on a test of executive functioning.112
Recessive forms of familial PD include those with genetic changes in the parkin, PINK1, and DJ-1 genes. These recessive forms of PD are generally characterized by early onset PD, but with a relatively slow progression of symptoms.99 Systematically collected data on cognition in PINK1 and DJ-1 is limited. A very atypical clinical picture with PD-D and motor neuron disease was reported in a family with a double mutation in DJ-1 and PD-D was reported in two cases with PINK1 deletion mutations.113, 114 On the other hand, CI in the more common parkin mutation carriers has been examined in some detail (Table 4). A comparison of parkin, LRRK2 and glucocerebrosidase (GBA) mutation carriers reported no differences between the three genetic groups on performance of the mini-mental state examination, although GBA mutation carriers subjectively reported more CI.115 In the relatively large study, early onset PD patients that were either parkin mutation heterozygote carriers, compound heterozygote/homozygyote, or non-carriers were compared on a detailed neuropsychological test battery.116 There were no significant differences with regard to neuropsychological test performance amongst the three groups (Table 4). Thus, in autosomal recessive forms of PD there is again a range of reported frequency of CI.
Cognition and PD risk genes
Two risk genes for PD, microtubule-associated protein tau (MAPT) and GBA, have been associated with CI in PD (Table 4). Despite the fact that tau pathology is not a neuropathologic feature of PD (see above), the MAPT gene has consistently been identified as a risk gene for PD in several genome-wide association studies and the H1 MAPT haplotype has been associated with increased risk for PD.98, 117, 118 Two moderately sized studies with prospective cognitive assessments have suggested that the H1 MAPT haplotype is associated with CI in PD patients.119, 120 Goris and colleagues found the H1/H1 MAPT haplotype was overrepresented in PD-D and was associated with a more rapid decline in cognitive performance, while Morley et al. found that H1/H1 carriers had lower memory scores, but they did not observe a more rapid decline in cognition than other PD patients. This latter group also reported an association between the catechol-o-methyl transferase (COMT) Met/Met haplotype and higher attention test performance, in contrast to previous reports121 (Table 4). Interestingly, a recent pathologic study, found the H1 MAPT haplotype was associated with lower tau-associated tangle pathology in PD, suggesting that the MAPT association with PD-D is not likely due to tau-associated neurofibrillary tangle pathology. Mutations in the GBA gene have also been found to increase the risk for PD.122 Although using fairly small numbers of cases, multiple studies have now suggested that GBA mutation carriers have an increased risk for PD-D.123-125
Cognition and non-PD genes
Due to the strong association between dementia risk (e.g., Alzheimer's disease) and the APOE 4 genotype, a number of investigators have examined the impact of this genotype on CI in PD. Of note, there is no evidence of an association between APOE 4 and risk for PD.126 For those PD patients carrying an APOE 4 genotype, the risk for CI appears to be increased (Table 4), although some studies have suggested that this effect is at most only modest.120, 127 Two recent neuropathologic studies have found that the APOE 4 genotype is associated with CI, even when controlling for Alzheimer's disease pathology severity (Table 4).12, 128 Irwin and colleagues compared autopsied PD and PD-D cases and found APOE 4 to be an independent predictor of PD-D (Table 4).12 Tsuang and colleagues found that APOE 4 genotype was associated with earlier onset of PD-D in autopsy cases with α-synuclein-positive Lewy body pathology (Table 4), again even when excluding cases with co-existent Alzheimer's disease.128
Summary of Genetic Risk Factor and Cognitive Impairment in PD
In summary, it is clear that genetic factors play a role in CI in PD (Table 4). Some genetic changes, even in early onset cases such as SNCA triplication, are clearly linked to substantial increased risk for CI, while others such as parkin mutation-associated autosomal recessive PD appears to be associated with reduced risk compared with sporadic PD (Table 4). Even for mutations within the same gene, there appears to be variability in CI frequency between different mutations. Given the importance of CI in PD and the apparent influence of genetic factors, further prospective study of the genetics of CI in PD is needed.
Preclinical Models
As described earlier, defining the qualitative aspects of MCI in PD has only recently received considerable attention,5 with most studies suggesting primarily a frontal or fronto-striatal nature to the cognitive deficits, particularly during the earlier stages of the disease.129-132 The cognitive domains most often affected include attention, working and short-term memory, language, visuospatial abilities, and various aspects of executive functioning.5, 133 Development and refinement of animal models of these different PD-MCI deficits would create an opportunity for studying the pathophysiology of this potentially pre-dementia state in PD and allow for pre-clinical testing of new therapies targeting PD cognitive decline. Yet, considering the potential clinical importance of PD-MCI, there has been relatively little preclinical research into this problem.
Modeling the cognitive dysfunction in PD
Can we effectively model PD cognitive dysfunction in animals? First, we need to briefly review the types of PD models currently available. These are genetic mouse models, primarily based on rare familial forms of PD (i.e., LRRK2 mutations, α-synuclein mutation/overexpression, parkin knockout); genetic rat models (i.e., LRRK2, α-synuclein mutations), adeno-associated virus (AAV)-mediated overexpression of α-synuclein,; and toxin models (primarily 6-hydroxydopamine (6-OHDA) lesions in rats; 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) lesions in mice and non-human primates).134 While all of these models have their respective strengths and weaknesses, one lingering issue with the genetic models is that they have not been able to reliably capture the neurochemical and anatomical pathology of PD (including the degeneration of A9 dopamine neurons), and these animals do not display typical parkinsonian motor signs. Although some pathology135 and neurotransmitter changes have been described in genetic models, particularly at advanced ages,136, 137 for the most part, these models have been described as modeling “pre-manifest PD”.137 Toxin-induced models, and in particular, the MPTP model, reproduces the main neurochemical and anatomical pathology of PD. Animals with MPTP-induced parkinsonism show multi-neurotransmitter defects138 (i.e., dopaminergic deficits as well as noradrenergic and serotonergic deficits) and significant degeneration of dopamine neurons, although Lewy bodies, the pathological hallmark of PD, are typically not observed. While 6-OHDA-lesioned rats can display parkinsonian motor signs, and some locomotor deficits can be detected in MPTP-treated mice, the MPTP-treated non-human primate presents with features that most closely resemble human PD.138
MPTP-treated mice have not been particularly assessed for cognitive dysfunction. Studies that have assessed aspects of cognition in rodent genetic PD models (primarily mouse models) have reported a variety of cognitive deficits including decreased spontaneous alternation in a T maze139 or Y maze,137 impaired learning and retention in the Morris water maze,135, 136, 140 and deficits in object-place recognition, operant reversal learning, and novel object recognition.137 More recently, the MitoPark mouse, a transgenic model based on mitochondrial dysfunction, has been developed and exhibits the cardinal features of PD, including adult onset of neurodegeneration, progressive decline in motor function, and presence of intraneuronal inclusions (possibly a Lewy body equivalent).141 A recent study described mild spatial learning and memory deficits in the Barnes maze and object recognition deficits that preceded the appearance of motor deficits in these animals.141 Although some of the tests mentioned above detected deficits in cognitive domains of potential relevance to PD, others may have more limited relevance to PD. For example, the Y maze, object-place recognition, novel object recognition, and the Morris water maze/Barnes maze primarily assess spatial learning and reference memory, cognitive functions not typically discussed as being affected in PD. However, deficits in spontaneous alternation testing (working/short-term memory) and reversal learning (cognitive flexibility) may be more relevant to the cognitive dysfunction often described in PD. However, caution is needed when interpreting some of these data from transgenic mouse models as many also exhibit anxiety-like behaviors that could impact performance on some of the cognitive tests used.
More recently, Hall and colleagues142 used targeted overexpression of α-synuclein in the ventral tegmental area A10 cell group (mesocorticolimbic dopamine system) and the medial septum/vertical limb of the diagonal band of Broca Ch1/2 cell groups (septohippocampal cholinergic system) in rats, achieved by direct injection of AAV vectors carrying the coding sequence for human wild-type α–synuclein, to examine the role of α–synuclein overexpression in these regions and cognitive functioning. These animals, which had mild loss of A10 dopamine neurons (approx. 24% decrease) and a 47% loss of Ch1/2 cholinergic neurons, had no spontaneous motor impairments but showed deficits in spatial learning and working memory but not in reference memory. While these results are interesting, the translational value of this model for understanding cognitive dysfunction in PD is not clear, as the A10 dopamine neurons are affected to a limited extent in PD (including in PD-D58) and although cholinergic deficits are described in advanced PD- D48, 55), they are not typically found in the septohippocampal system but are more often described in the basal forebrain Ch4 cholinergic system (see above).
Although there is no best animal model of PD, as all are only approximations of the human disease, the MPTP primate models of PD appear to reproduce most, although not all, of the clinical and pathological hallmarks of PD.138 To assess cognitive dysfunction in PD, specific non-human primate models have been created, taking advantage of the rich behavioral repertoire of non-human primates and the ability to assess their behavior on non-human primate analogs of human neuropsychological tests. The chronic low dose (CLD) MPTP model of parkinsonism was initially developed to produce cognitive impairments in non-human primates prior to the development of parkinsonian motor symptoms.143-146 These animals developed cognitive deficits analogous to those described in early PD patients, including deficits in attention (sustained and selective attention), attention set shifting, working/short-term memory, cognitive flexibility, planning and problem solving but not in reference or recognition memory.143-147 Early work was performed using a modified Wisconsin general test apparatus but more recent studies have used automated, computerized testing stations that reduce intra- and inter-animal variability, allow collection of precise timing data, and can present standardized testing based on automated human neuropsychological test paradigms. CLD MPTP animals not only showed attention, memory, and executive function deficits but also showed broad basal ganglia and extra-basal ganglia pathology, including dopamine and noradrenaline deficits at striatal and cortical levels and loss of SN dopamine neurons (with more loss in medial and ventral portions of A9) and modest loss of A10 VTA dopamine neurons, as more typically occurs in PD.148, 149 This model is also responsive to a variety of neuro-pharmacological manipulations, particularly of the nicotinic system.150
More recently, the model has been further refined to produce animals that express early-appearing frontal-like cognitive deficits upon which typical parkinsonian motor signs are superimposed.151 This model, which recapitulates both motor and cognitive deficits of PD, was recently shown to demonstrate levodopa-responsive motor deficits along with attention/working memory deficits which either did not respond positively to levodopa or were further impaired with administration of doses of levodopa that produced maximal improvement of motor symptoms.151 These data are in agreement with a number of clinical studies in which the benefits of dopamine-replacement therapy on frontal cortical-based cognitive deficits in PD have been inconsistent152, 153 and in some studies has resulted in further impairments of cognitive functioning.152, 154-156
Translational value of the models
The non-human primate model described may have the greatest translational potential in that it most reliably reproduces motor and cognitive dysfunction similar to that in PD, has neurochemical and anatomical pathology most similar to PD, and appears to respond to drug treatments in a manner similar to that described in PD (Table 5). Of the available models, the non-human primate model appears to best able to capture dysfunction in the cognitive domains most affected in PD and more accurately mimics various aspects of human PD compared to rodent models. Although the rodent models are more readily available and generally do not require the specialized expertise, housing and testing situations needed for non-human primate work, at present these models lack neurochemical and anatomical features that closely resemble the human PD condition and thus far, the cognitive deficits demonstrated in most of these models are limited and in some instances, not in cognitive domains typically affected in PD (Table 5). However, all of the models have relative strengths and weaknesses and the selection of a model for study needs to be based on the nature of the research question being asked. For example, studying cognitive deficits in a 6-OHDA-lesioned rat in which a very specific dopaminergic lesion is produced could provide useful information regarding the role of dopamine per se in certain behaviors but may have more limited relevance for developing new drug treatments for PD cognitive dysfunction, which appears to involve dopaminergic and non-dopaminergic mechanisms.
Table 5. Comparison of characteristics of PD versus primate and rodent models.
| Parkinson's Disease | CLD MPTP Monkey | Rodent Genetic PD Models | |
|---|---|---|---|
| Multi-neurotransmitter deficits | ✔ | ✔ | ✔ |
| DA neuron pathology | ✔ | ✔ | X |
| Working memory deficits | ✔ | ✔ | X/✔ |
| Attention deficits | ✔ | ✔ | X |
| Executive function deficits | ✔ | ✔ | X/✔ |
| Visuospatial deficits | ✔ | ✔ | X |
| Reference Memory deficits | X | X | ✔ |
| Recognition Memory deficits | X | X | ✔ |
| Parkinsonian Motor deficits | ✔ | ✔ | X |
| Varied effects from L-dopa | ✔ | ✔ | X |
✔ = present; X = absent
Conclusions
As criteria for PD-MCI are very recent and are based on limited underpinning biological and genetic data, further research is required to establish if such patients have more significant dopamine deficits as their PD neurodegeneration progresses. Current limited data and animal modeling supports this concept,157-159 but much more work needs to be done. At present the biological basis of PD-D is more certain, with data pointing to dominant limbic and cortical deficits. PD-D is definitely age related and associated not only with increasing α-synuclein pathology, but also with age-associated AD pathologies. In addition, there is marked loss of limbic and cortically projecting dopamine, noradrenaline, serotonin and acetylcholine neurons, although the relationship between these degenerative changes remains to be determined. Genetic factors associated with CI require further evaluation, but those that enhance α-synuclein pathology (like SNCA triplication), enhance AD pathology (like APOE 4 genotype) or enhance lysosomal deficits (like GBA mutations) are associated with enhanced CI, while those that enhance mitochondrial and/or proteosome dysfunction (like parkin mutations) are associated with reduced CI. Translation of these findings into clinical treatments will require further evaluation of the relationships and timing between these biological and genetic associations.
Acknowledgments
We wish to thank Heidi Cartwright for the preparation of the figures.
Funding sources: GMH is a Senior Principal Research Fellow of the National Health and Medical Research Council of Australia (#630434).
Full Financial Disclosures of all Authors for the Past Year: GMH receives research funds from the National Health and Medical Research Council of Australia (project grants 1008307, 1022325, 1029538; program grant 1037746; Senior Principal Research Fellowship 630434), the University of New South Wales (Goldstar award; infrastructure), Michael J Fox Foundation, Shake-It-Up Foundation, Parkinson's NSW, and the Rebecca Cooper Foundation. JBL receives research funds from Veterans Affairs, the NIH (P50NS062684, P50AG05136, KO1-AT004404), Michael J Fox Foundation, and the American Parkinson's Disease Association. JSS receives research funds from NIH (NS0555916, NS44481, ES15295, ES10975, ES021534), Michael J Fox Foundation. CHA receives research funds from the Michael J. Fox Foundation, National Institute of Neurological Disorders and Stroke, Avid Radiopharmaceuticals.
|
Stock Ownership in medically-related fields GMH – Cochlear (2004 on)&NIB Holdings (2007 on) JBL – none JSS – none CHA - none |
Intellectual Property Rights GMH – none JBL – none JSS – none CHA - none |
|
| |
|
Consultancies GMH – for committee work for the NHMRC JBL – Boehringer Ingelheim, Navidea Biopharmaceuticals, Piramal Healthcare JSS – Motac Cognition, Inc. CHA - Lilly, Merz, Novartis, Xenoport |
Expert Testimony GMH – none JBL – none JSS – none CHA - none |
|
| |
|
Advisory Boards GMH – Kolling Institute, University of Sydney&Centre for Brain and Mind Research, University of Newcastle JBL – University of Indiana Alzheimer's Disease Center, W. Garfield Weston Foundation JSS – none CHA – Bachmann Strauss Foundation |
Employment GMH – Salary from NHMRC Grant 630434&University of New South Wales JBL – Salary from Veterans Affairs, University of Washington, Cleveland Clinic Foundation JSS – Salary from Thomas Jefferson University CHA - Salary from Mayo Clinic College of Medicine |
|
| |
|
Partnerships GMH – none JBL – none JSS – none CHA - none |
Contracts GMH – none JBL – none JSS – none CHA - none |
|
| |
|
Honoraria GMH – MDS, MDPD, WFN World Congress on PD, Dopamine2013, AAIC, International frontotemporal dementia meeting, International alpha-synuclein meeting, Universidad de Navarra, Lund University, University of Queensland, Van Andel Institute JBL – University of Indiana JSS – MDS, Western University CHA – MDS, Bachmann Strauss Foundation, University of Arizona |
Royalties GMH – The Human Nervous System (first and third editions), Atlas of the Developing Mouse Brain at E17.5, P0 and P6&Non-dopamine Lesions in Parkinson's Disease JBL – none JSS – none CHA - Springer |
|
| |
|
Grants GMH – NHMRC (1008307, 1022325, 1029538, 1037746&630434), Michael J Fox Foundation, Shake-it-up Australia, Parkinson's NSW, Rebecca Cooper Foundation, University of New South Wales (Goldstar award and infrastructure). JBL – Veterans Affairs, the NIH (P50NS062684, P50AG05136, KO1-AT004404), Michael J Fox Foundation, American Parkinson's Disease Association, Jane and Lee Seidman Fund JSS – NIH (NS055916, NS044481, ES015295, ES010975, ES021534), Michael J Fox Foundation CHA - NINDS (U24 NS072026), Arizona Biomedical Research Commission (contracts 4001, 0011, 05-901 and 1001 to the Arizona Parkinson's Disease Consortium), Michael J. Fox Foundation. |
Other GMH – none JBL – none JSS – none CHA - none |
Footnotes
Author Roles: GMH – 1) A. Conception, B. Organization, C. Execution; 3) A. Writing of the first draft, B. Review and Critique.
JBL – 1) A. Conception, B. Organization, C. Execution; 3) A. Writing of the first draft, B. Review and Critique.
JSS – 1) A. Conception, B. Organization, C. Execution; 3) A. Writing of the first draft, B. Review and Critique.
CHA - 1) A. Conception, B. Organization, C. Execution; 3) A. Writing of the first draft, B. Review and Critique.
Financial Disclosure/Conflict of Interest concerning the research related to the manuscript: The authors report no conflicts of interest.
References
- 1.Dickson DW, Braak H, Duda JE, et al. Neuropathological assessment of Parkinson's disease: refining the diagnostic criteria. Lancet Neurol. 2009;8(12):1150–1157. doi: 10.1016/S1474-4422(09)70238-8. [DOI] [PubMed] [Google Scholar]
- 2.Halliday GM, McCann H. The progression of pathology in Parkinson's disease. Ann N Y Acad Sci. 2010;1184:188–195. doi: 10.1111/j.1749-6632.2009.05118.x. [DOI] [PubMed] [Google Scholar]
- 3.Singleton AB, Farrer MJ, Bonifati V. The genetics of Parkinson's disease: progress and therapeutic implications. Mov Disord. 2013;28(1):14–23. doi: 10.1002/mds.25249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Emre M, Aarsland D, Brown R, et al. Clinical diagnostic criteria for dementia associated with Parkinson's disease. Mov Disord. 2007;22(12):1689–1707. doi: 10.1002/mds.21507. [DOI] [PubMed] [Google Scholar]
- 5.Litvan I, Goldman JG, Troster AI, et al. Diagnostic criteria for mild cognitive impairment in Parkinson's disease: Movement Disorder Society Task Force guidelines. Mov Disord. 2012;27(3):349–356. doi: 10.1002/mds.24893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldman JG, Holden S, Bernard B, Ouyang B, Goetz CG, Stebbins GT. Defining optimal cutoff scores for cognitive impairment using Movement Disorder Society Task Force criteria for mild cognitive impairment in Parkinson's disease. Mov Disord. 2013 doi: 10.1002/mds.25655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Adler CH, Caviness JN, Sabbagh MN, et al. Heterogeneous neuropathological findings in Parkinson's disease with mild cognitive impairment. Acta Neuropathol. 2010;120:829–830. doi: 10.1007/s00401-010-0744-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hurtig HI, Trojanowski JQ, Galvin J, et al. Alpha-synuclein cortical Lewy bodies correlate with dementia in Parkinson's disease. Neurology. 2000;54(10):1916–1921. doi: 10.1212/wnl.54.10.1916. [DOI] [PubMed] [Google Scholar]
- 9.Jellinger KA. Pathological substrate of dementia in Parkinson's disease--its relation to DLB and DLBD. Parkinsonism Relat Disord. 2006;12(2):119–120. doi: 10.1016/j.parkreldis.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 10.Sabbagh MN, Adler CH, Lahti TJ, et al. Parkinson disease with dementia: comparing patients with and without Alzheimer pathology. Alzheimer Dis Assoc Disord. 2009;23(3):295–297. doi: 10.1097/WAD.0b013e31819c5ef4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi SA, Evidente VG, Caviness JN, et al. Are there differences in cerebral white matter lesion burdens between Parkinson's disease patients with or without dementia? Acta Neuropathol. 2009;119:147–149. doi: 10.1007/s00401-009-0620-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Irwin DJ, White MT, Toledo JB, et al. Neuropathologic substrates of Parkinson disease dementia. Ann Neurol. 2012;72(4):587–598. doi: 10.1002/ana.23659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mattila PM, Rinne JO, Helenius H, Dickson DW, Roytta M. Alpha-synuclein-immunoreactive cortical Lewy bodies are associated with cognitive impairment in Parkinson's disease. Acta Neuropathol (Berl) 2000;100(3):285–290. doi: 10.1007/s004019900168. [DOI] [PubMed] [Google Scholar]
- 14.Apaydin H, Ahlskog JE, Parisi JE, Boeve BF, Dickson DW. Parkinson disease neuropathology: later-developing dementia and loss of the levodopa response. Arch Neurol. 2002;59(1):102–112. doi: 10.1001/archneur.59.1.102. [DOI] [PubMed] [Google Scholar]
- 15.Aarsland D, Perry R, Brown A, Larsen JP, Ballard C. Neuropathology of dementia in Parkinson's disease: a prospective, community-based study. Ann Neurol. 2005;58(5):773–776. doi: 10.1002/ana.20635. [DOI] [PubMed] [Google Scholar]
- 16.Kempster PA, O'Sullivan SS, Holton JL, Revesz T, Lees AJ. Relationships between age and late progression of Parkinson's disease: a clinico-pathological study. Brain : a journal of neurology. 2010;133(Pt 6):1755–1762. doi: 10.1093/brain/awq059. [DOI] [PubMed] [Google Scholar]
- 17.Schneider JA, Arvanitakis Z, Yu L, Boyle PA, Leurgans SE, Bennett DA. Cognitive impairment, decline and fluctuations in older community-dwelling subjects with Lewy bodies. Brain. 2012;135(Pt 10):3005–3014. doi: 10.1093/brain/aws234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Colosimo C, Hughes AJ, Kilford L, Lees AJ. Lewy body cortical involvement may not always predict dementia in Parkinson's disease. J Neurol Neurosurg Psychiatry. 2003;74(7):852–856. doi: 10.1136/jnnp.74.7.852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harding AJ, Halliday GM. Cortical Lewy body pathology in the diagnosis of dementia. Acta Neuropathol. 2001;102(4):355–363. doi: 10.1007/s004010100390. [DOI] [PubMed] [Google Scholar]
- 20.Hely MA, Reid WG, Adena MA, Halliday GM, Morris JG. The Sydney multicenter study of Parkinson's disease: the inevitability of dementia at 20 years. Mov Disord. 2008;23(6):837–844. doi: 10.1002/mds.21956. [DOI] [PubMed] [Google Scholar]
- 21.Mattila PM, Roytta M, Torikka H, Dickson DW, Rinne JO. Cortical Lewy bodies and Alzheimer-type changes in patients with Parkinson's disease. Acta Neuropathol (Berl) 1998;95(6):576–582. doi: 10.1007/s004010050843. [DOI] [PubMed] [Google Scholar]
- 22.Compta Y, Parkkinen L, O'Sullivan SS, et al. Lewy- and Alzheimer-type pathologies in Parkinson's disease dementia: which is more important? Brain. 2011;134(Pt 5):1493–1505. doi: 10.1093/brain/awr031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Waxman EA, Giasson BI. Induction of intracellular tau aggregation is promoted by α-synuclein seeds and provides novel insights into the hyperphosphorylation of tau. J Neurosci. 2011;31(21):7604–7618. doi: 10.1523/JNEUROSCI.0297-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kazmierczak A, Strosznajder JB, Adamczyk A. alpha-Synuclein enhances secretion and toxicity of amyloid beta peptides in PC12 cells. Neurochem Int. 2008;53(6-8):263–269. doi: 10.1016/j.neuint.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 25.Schwartz RS, Halliday GM, Cordato DJ, Kril JJ. Small-vessel disease in patients with Parkinson's disease: a clinicopathological study. Mov Disord. 2012;27(12):1506–1512. doi: 10.1002/mds.25112. [DOI] [PubMed] [Google Scholar]
- 26.Halliday G, Hely M, Reid W, Morris J. The progression of pathology in longitudinally followed patients with Parkinson's disease. Acta Neuropathol. 2008;115(4):409–415. doi: 10.1007/s00401-008-0344-8. [DOI] [PubMed] [Google Scholar]
- 27.Ghebremedhin E, Rosenberger A, Rub U, et al. Inverse relationship between cerebrovascular lesions and severity of lewy body pathology in patients with lewy body diseases. J Neuropathol Exp Neurol. 2010;69(5):442–448. doi: 10.1097/NEN.0b013e3181d88e63. [DOI] [PubMed] [Google Scholar]
- 28.Bertrand E, Lewandowska E, Stepien T, Szpak GM, Pasennik E, Modzelewska J. Amyloid angiopathy in idiopathic Parkinson's disease. Immunohistochemical and ultrastructural study. Folia Neuropathol. 2008;46(4):255–270. [PubMed] [Google Scholar]
- 29.Amador-Ortiz C, Ahmed Z, Zehr C, Dickson DW. Hippocampal sclerosis dementia differs from hippocampal sclerosis in frontal lobe degeneration. Acta Neuropathol. 2007;113(3):245–252. doi: 10.1007/s00401-006-0183-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beach TG, Monsell SE, Phillips LE, Kukull W. Accuracy of the clinical diagnosis of Alzheimer disease at National Institute on Aging Alzheimer Disease Centers, 2005-2010. J Neuropathol Exp Neurol. 2012;71(4):266–273. doi: 10.1097/NEN.0b013e31824b211b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malek-Ahmadi M, Kahlon V, Adler CH, et al. Prevalence of hippocampal sclerossis in a clinicopathologically characterized cohort. Clin Exp Med Sci. 2013;1:317–327. doi: 10.12988/cems.2013.13026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beach TG, Adler CH, Lue L, et al. Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol. 2009;117(6):613–634. doi: 10.1007/s00401-009-0538-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jellinger KA. Neuropathology in Parkinson's disease with mild cognitive impairment. Acta Neuropathol. 2010;120(6):829–830. doi: 10.1007/s00401-010-0755-1. [DOI] [PubMed] [Google Scholar]
- 34.Duncan GW, Firbank MJ, O'Brien JT, Burn DJ. Magnetic resonance imaging: a biomarker for cognitive impairment in Parkinson's disease? Mov Disord. 2013;28(4):425–438. doi: 10.1002/mds.25352. [DOI] [PubMed] [Google Scholar]
- 35.Weiner MW, Veitch DP, Aisen PS, et al. The Alzheimer's Disease Neuroimaging Initiative: a review of papers published since its inception. Alzheimers Dement. 2013;9(5):e111–194. doi: 10.1016/j.jalz.2013.05.1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reid WG, Hely MA, Morris JG, Loy C, Halliday GM. Dementia in Parkinson's disease: a 20-year neuropsychological study (Sydney Multicentre Study) J Neurol Neurosurg Psychiatry. 2011;82(9):1033–1037. doi: 10.1136/jnnp.2010.232678. [DOI] [PubMed] [Google Scholar]
- 37.Erten-Lyons D, Dodge HH, Woltjer R, et al. Neuropathologic basis of age-associated brain atrophy. JAMA Neurol. 2013;70(5):616–622. doi: 10.1001/jamaneurol.2013.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Middleton LE, Grinberg LT, Miller B, Kawas C, Yaffe K. Neuropathologic features associated with Alzheimer disease diagnosis: age matters. Neurology. 2011;77(19):1737–1744. doi: 10.1212/WNL.0b013e318236f0cf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Monsell SE, Mock C, Roe CM, et al. Comparison of symptomatic and asymptomatic persons with Alzheimer disease neuropathology. Neurology. 2013;80(23):2121–2129. doi: 10.1212/WNL.0b013e318295d7a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jellinger KA, Seppi K, Wenning GK, Poewe W. Impact of coexistent Alzheimer pathology on the natural history of Parkinson's disease. J Neural Transm. 2002;109(3):329–339. doi: 10.1007/s007020200027. [DOI] [PubMed] [Google Scholar]
- 41.Jellinger KA. Age-associated prevalence and risk factors of Lewy body pathology in a general population. Acta Neuropathol. 2003;106(4):383–384. doi: 10.1007/s00401-003-0751-9. [DOI] [PubMed] [Google Scholar]
- 42.Pletnikova O, West N, Lee MK, et al. Abeta deposition is associated with enhanced cortical alpha-synuclein lesions in Lewy body diseases. Neurobiol Aging. 2005;26(8):1183–1192. doi: 10.1016/j.neurobiolaging.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 43.Dickson DW, Fujishiro H, Orr C, et al. Neuropathology of non-motor features of Parkinson disease. Parkinsonism Relat Disord. 2009;15(3):S1–5. doi: 10.1016/S1353-8020(09)70769-2. [DOI] [PubMed] [Google Scholar]
- 44.Kehagia AA, Barker RA, Robbins TW. Neuropsychological and clinical heterogeneity of cognitive impairment and dementia in patients with Parkinson's disease. Lancet Neurol. 2010;9(12):1200–1213. doi: 10.1016/S1474-4422(10)70212-X. [DOI] [PubMed] [Google Scholar]
- 45.Ray NJ, Strafella AP. The neurobiology and neural circuitry of cognitive changes in Parkinson's disease revealed by functional neuroimaging. Mov Disord. 2012;27(12):1484–1492. doi: 10.1002/mds.25173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hindle JV. The practical management of cognitive impairment and psychosis in the older Parkinson's disease patient. J Neural Transm. 2013;120(4):649–653. doi: 10.1007/s00702-013-0994-0. [DOI] [PubMed] [Google Scholar]
- 47.Rolinski M, Fox C, Maidment I, McShane R. Cholinesterase inhibitors for dementia with Lewy bodies, Parkinson's disease dementia and cognitive impairment in Parkinson's disease. Cochrane Database Syst Rev. 2012;3:CD006504. doi: 10.1002/14651858.CD006504.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Halliday G, Reyes S, Double K. Substantia nigra, ventral tegmental area, and retrorubral fields. In: Mai JK, Paxinos G, editors. The Human Nervous System. 3rd. London: Elsevier; 2012. pp. 441–457. [Google Scholar]
- 49.Roeper J. Dissecting the diversity of midbrain dopamine neurons. Trends Neurosci. 2013;36(6):336–342. doi: 10.1016/j.tins.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 50.Milber JM, Noorigian JV, Morley JF, et al. Lewy pathology is not the first sign of degeneration in vulnerable neurons in Parkinson disease. Neurology. 2012;79(24):2307–2314. doi: 10.1212/WNL.0b013e318278fe32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Greffard S, Verny M, Bonnet AM, et al. Motor score of the Unified Parkinson Disease Rating Scale as a good predictor of Lewy body-associated neuronal loss in the substantia nigra. Arch Neurol. 2006;63(4):584–588. doi: 10.1001/archneur.63.4.584. [DOI] [PubMed] [Google Scholar]
- 52.Rinne JO, Rummukainen J, Paljarvi L, Sako E, Molsa P, Rinne UK. Neuronal loss in the substantia nigra in patients with Alzheimer's disease and Parkinson's disease in relation to extrapyramidal symptoms and dementia. Prog Clin Biol Res. 1989;317:325–332. [PubMed] [Google Scholar]
- 53.Alobaidi H, Pall H. The role of dopamine replacement on the behavioural phenotype of Parkinson's disease. Behav Neurol. 2013;26(4):225–235. doi: 10.3233/BEN-2012-120265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Goldman JG, Holden S. Treatment of psychosis and dementia in Parkinson's disease. Curr Treat Options Neurol. 2014;16(3):281. doi: 10.1007/s11940-013-0281-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gaspar P, Gray F. Dementia in idiopathic Parkinson's disease. A neuropathological study of 32 cases. Acta Neuropathol. 1984;64(1):43–52. doi: 10.1007/BF00695605. [DOI] [PubMed] [Google Scholar]
- 56.Hilker R, Thomas AV, Klein JC, et al. Dementia in Parkinson disease: functional imaging of cholinergic and dopaminergic pathways. Neurology. 2005;65(11):1716–1722. doi: 10.1212/01.wnl.0000191154.78131.f6. [DOI] [PubMed] [Google Scholar]
- 57.Klein JC, Eggers C, Kalbe E, et al. Neurotransmitter changes in dementia with Lewy bodies and Parkinson disease dementia in vivo. Neurology. 2010;74(11):885–892. doi: 10.1212/WNL.0b013e3181d55f61. [DOI] [PubMed] [Google Scholar]
- 58.Pavese N, Rivero-Bosch M, Lewis SJ, Whone AL, Brooks DJ. Progression of monoaminergic dysfunction in Parkinson's disease: a longitudinal 18F-dopa PET study. Neuroimage. 2011;56(3):1463–1468. doi: 10.1016/j.neuroimage.2011.03.012. [DOI] [PubMed] [Google Scholar]
- 59.Reyes S, Cottam V, Kirik D, Double KL, Halliday GM. Variability in neuronal expression of dopamine receptors and transporters in the substantia nigra. Mov Disord. 2013;28(10):1351–1359. doi: 10.1002/mds.25493. [DOI] [PubMed] [Google Scholar]
- 60.Rinne JO, Rummukainen J, Paljarvi L, Rinne UK. Dementia in Parkinson's disease is related to neuronal loss in the medial substantia nigra. Ann Neurol. 1989;26(1):47–50. doi: 10.1002/ana.410260107. [DOI] [PubMed] [Google Scholar]
- 61.Paulus W, Jellinger K. The neuropathologic basis of different clinical subgroups of Parkinson's disease. J Neuropathol Exp Neurol. 1991;50(6):743–755. doi: 10.1097/00005072-199111000-00006. [DOI] [PubMed] [Google Scholar]
- 62.Zweig RM, Cardillo JE, Cohen M, Giere S, Hedreen JC. The locus ceruleus and dementia in Parkinson's disease. Neurology. 1993;43(5):986–991. doi: 10.1212/wnl.43.5.986. [DOI] [PubMed] [Google Scholar]
- 63.Ito K, Nagano-Saito A, Kato T, et al. Striatal and extrastriatal dysfunction in Parkinson's disease with dementia: a 6-[18F]fluoro-L-dopa PET study. Brain. 2002;125(Pt 6):1358–1365. doi: 10.1093/brain/awf134. [DOI] [PubMed] [Google Scholar]
- 64.Mattila PM, Roytta M, Lonnberg P, Marjamaki P, Helenius H, Rinne JO. Choline acetytransferase activity and striatal dopamine receptors in Parkinson's disease in relation to cognitive impairment. Acta Neuropathol. 2001;102(2):160–166. doi: 10.1007/s004010100372. [DOI] [PubMed] [Google Scholar]
- 65.Voon V, Mehta AR, Hallett M. Impulse control disorders in Parkinson's disease: recent advances. Curr Opin Neurol. 2011;24(4):324–330. doi: 10.1097/WCO.0b013e3283489687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McRitchie DA, Cartwright HR, Halliday GM. Specific A10 dopaminergic nuclei in the midbrain degenerate in Parkinson's disease. Exp Neurol. 1997;144(1):202–213. doi: 10.1006/exnr.1997.6418. [DOI] [PubMed] [Google Scholar]
- 67.Vaillancourt DE, Schonfeld D, Kwak Y, Bohnen NI, Seidler R. Dopamine overdose hypothesis: evidence and clinical implications. Mov Disord. 2013;28(14):1920–1929. doi: 10.1002/mds.25687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Poletti M, Bonuccelli U. Acute and chronic cognitive effects of levodopa and dopamine agonists on patients with Parkinson's disease: a review. Ther Adv Psychopharmacol. 2013;3(2):101–113. doi: 10.1177/2045125312470130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Del Tredici K, Braak H. Dysfunction of the locus coeruleus-norepinephrine system and related circuitry in Parkinson's disease-related dementia. J Neurol Neurosurg Psychiatry. 2013;84(7):774–783. doi: 10.1136/jnnp-2011-301817. [DOI] [PubMed] [Google Scholar]
- 70.Buchman AS, Nag S, Shulman JM, et al. Locus coeruleus neuron density and parkinsonism in older adults without Parkinson's disease. Mov Disord. 2012;27(13):1625–1631. doi: 10.1002/mds.25142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pifl C, Kish SJ, Hornykiewicz O. Thalamic noradrenaline in Parkinson's disease: deficits suggest role in motor and non-motor symptoms. Mov Disord. 2012;27(13):1618–1624. doi: 10.1002/mds.25109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Isaias IU, Marotta G, Pezzoli G, et al. Enhanced catecholamine transporter binding in the locus coeruleus of patients with early Parkinson disease. BMC Neurol. 2011;11:88. doi: 10.1186/1471-2377-11-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Scatton B, Javoy-Agid F, Rouquier L, Dubois B, Agid Y. Reduction of cortical dopamine, noradrenaline, serotonin and their metabolites in Parkinson's disease. Brain Res. 1983;275(2):321–328. doi: 10.1016/0006-8993(83)90993-9. [DOI] [PubMed] [Google Scholar]
- 74.Marsh L, Biglan K, Gerstenhaber M, Williams JR. Atomoxetine for the treatment of executive dysfunction in Parkinson's disease: a pilot open-label study. Mov Disord. 2009;24(2):277–282. doi: 10.1002/mds.22307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tork I. Anatomy of the serotonergic system. Ann N Y Acad Sci. 1990;600:9–34. doi: 10.1111/j.1749-6632.1990.tb16870.x. [DOI] [PubMed] [Google Scholar]
- 76.Halliday GM, Li YW, Blumbergs PC, et al. Neuropathology of immunohistochemically identified brainstem neurons in Parkinson's disease. Ann Neurol. 1990;27(4):373–385. doi: 10.1002/ana.410270405. [DOI] [PubMed] [Google Scholar]
- 77.Bedard C, Wallman MJ, Pourcher E, Gould PV, Parent A, Parent M. Serotonin and dopamine striatal innervation in Parkinson's disease and Huntington's chorea. Parkinsonism Relat Disord. 2011;17(8):593–598. doi: 10.1016/j.parkreldis.2011.05.012. [DOI] [PubMed] [Google Scholar]
- 78.Strecker K, Wegner F, Hesse S, et al. Preserved serotonin transporter binding in de novo Parkinson's disease: negative correlation with the dopamine transporter. J Neurol. 2011;258(1):19–26. doi: 10.1007/s00415-010-5666-5. [DOI] [PubMed] [Google Scholar]
- 79.Kish SJ, Tong J, Hornykiewicz O, et al. Preferential loss of serotonin markers in caudate versus putamen in Parkinson's disease. Brain. 2008;131(Pt 1):120–131. doi: 10.1093/brain/awm239. [DOI] [PubMed] [Google Scholar]
- 80.Kerenyi L, Ricaurte GA, Schretlen DJ, et al. Positron emission tomography of striatal serotonin transporters in Parkinson disease. Arch Neurol. 2003;60(9):1223–1229. doi: 10.1001/archneur.60.9.1223. [DOI] [PubMed] [Google Scholar]
- 81.Waeber C, Palacios JM. Serotonin-1 receptor binding sites in the human basal ganglia are decreased in Huntington's chorea but not in Parkinson's disease: a quantitative in vitro autoradiography study. Neuroscience. 1989;32(2):337–347. doi: 10.1016/0306-4522(89)90082-1. [DOI] [PubMed] [Google Scholar]
- 82.Perry EK, McKeith I, Thompson P, et al. Topography, extent, and clinical relevance of neurochemical deficits in dementia of Lewy body type, Parkinson's disease, and Alzheimer's disease. Ann N Y Acad Sci. 1991;640:197–202. doi: 10.1111/j.1749-6632.1991.tb00217.x. [DOI] [PubMed] [Google Scholar]
- 83.Cheng AV, Ferrier IN, Morris CM, et al. Cortical serotonin-S2 receptor binding in Lewy body dementia, Alzheimer's and Parkinson's diseases. J Neurol Sci. 1991;106(1):50–55. doi: 10.1016/0022-510x(91)90193-b. [DOI] [PubMed] [Google Scholar]
- 84.Perry EK, Marshall E, Thompson P, et al. Monoaminergic activities in Lewy body dementia: relation to hallucinosis and extrapyramidal features. J Neural Transm Park Dis Dement Sect. 1993;6(3):167–177. doi: 10.1007/BF02260919. [DOI] [PubMed] [Google Scholar]
- 85.Huot P, Johnston TH, Darr T, et al. Increased 5-HT2A receptors in the temporal cortex of parkinsonian patients with visual hallucinations. Mov Disord. 2010;25(10):1399–1408. doi: 10.1002/mds.23083. [DOI] [PubMed] [Google Scholar]
- 86.Yarnall A, Rochester L, Burn DJ. The interplay of cholinergic function, attention, and falls in Parkinson's disease. Mov Disord. 2011;26(14):2496–2503. doi: 10.1002/mds.23932. [DOI] [PubMed] [Google Scholar]
- 87.Bohnen NI, Albin RL. The cholinergic system and Parkinson disease. Behav Brain Res. 2011;221(2):564–573. doi: 10.1016/j.bbr.2009.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zweig RM, Jankel WR, Hedreen JC, Mayeux R, Price DL. The pedunculopontine nucleus in Parkinson's disease. Ann Neurol. 1989;26(1):41–46. doi: 10.1002/ana.410260106. [DOI] [PubMed] [Google Scholar]
- 89.Rinne JO, Ma SY, Lee MS, Collan Y, Roytta M. Loss of cholinergic neurons in the pedunculopontine nucleus in Parkinson's disease is related to disability of the patients. Parkinsonism Relat Disord. 2008;14(7):553–557. doi: 10.1016/j.parkreldis.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 90.Ehrt U, Broich K, Larsen JP, Ballard C, Aarsland D. Use of drugs with anticholinergic effect and impact on cognition in Parkinson's disease: a cohort study. J Neurol Neurosurg Psychiatry. 2010;81(2):160–165. doi: 10.1136/jnnp.2009.186239. [DOI] [PubMed] [Google Scholar]
- 91.Pagonabarraga J, Kulisevsky J. Cognitive impairment and dementia in Parkinson's disease. Neurobiol Dis. 2012;46(3):590–596. doi: 10.1016/j.nbd.2012.03.029. [DOI] [PubMed] [Google Scholar]
- 92.Narayanan NS, Rodnitzky RL, Uc EY. Prefrontal dopamine signaling and cognitive symptoms of Parkinson's disease. Rev Neurosci. 2013;24(3):267–278. doi: 10.1515/revneuro-2013-0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Calabresi P, Castrioto A, Di Filippo M, Picconi B. New experimental and clinical links between the hippocampus and the dopaminergic system in Parkinson's disease. Lancet Neurol. 2013;12(8):811–821. doi: 10.1016/S1474-4422(13)70118-2. [DOI] [PubMed] [Google Scholar]
- 94.Lisman JE, Grace AA. The hippocampal-VTA loop: controlling the entry of information into long-term memory. Neuron. 2005;46(5):703–713. doi: 10.1016/j.neuron.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 95.Bohnen NI, Muller ML, Kotagal V, et al. Heterogeneity of cholinergic denervation in Parkinson's disease without dementia. J Cereb Blood Flow Metab. 2012;32(8):1609–1617. doi: 10.1038/jcbfm.2012.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bohnen NI, Kaufer DI, Hendrickson R, et al. Cognitive correlates of cortical cholinergic denervation in Parkinson's disease and parkinsonian dementia. J Neurol. 2006;253(2):242–247. doi: 10.1007/s00415-005-0971-0. [DOI] [PubMed] [Google Scholar]
- 97.de la Fuente-Fernandez R, Schulzer M, Kuramoto L, et al. Age-specific progression of nigrostriatal dysfunction in Parkinson's disease. Ann Neurol. 2011;69(5):803–810. doi: 10.1002/ana.22284. [DOI] [PubMed] [Google Scholar]
- 98.Simon-Sanchez J, Schulte C, Bras JM, et al. Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat Genet. 2009;41(12):1308–1312. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Trinh J, Farrer M. Advances in the genetics of Parkinson disease. Nat Rev Neurol. 2013;9(8):445–454. doi: 10.1038/nrneurol.2013.132. [DOI] [PubMed] [Google Scholar]
- 100.Kasten M, Klein C. The many faces of alpha-synuclein mutations. Mov Disord. 2013;28(6):697–701. doi: 10.1002/mds.25499. [DOI] [PubMed] [Google Scholar]
- 101.Zarranz JJ, Alegre J, Gomez-Esteban JC, et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55(2):164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- 102.Somme JH, Gomez-Esteban JC, Molano A, Tijero B, Lezcano E, Zarranz JJ. Initial neuropsychological impairments in patients with the E46K mutation of the alpha-synuclein gene (PARK 1) Journal of the Neurological Sciences. 2011;310(1-2):86–89. doi: 10.1016/j.jns.2011.07.047. [DOI] [PubMed] [Google Scholar]
- 103.Singleton AB, Farrer M, Johnson J, et al. alpha-Synuclein locus triplication causes Parkinson's disease. Science. 2003;302(5646):841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 104.Chartier-Harlin MC, Kachergus J, Roumier C, et al. Alpha-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet. 2004;364(9440):1167–1169. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- 105.Ibanez P, Bonnet AM, Debarges B, et al. Causal relation between alpha-synuclein gene duplication and familial Parkinson's disease. Lancet. 2004;364(9440):1169–1171. doi: 10.1016/S0140-6736(04)17104-3. [DOI] [PubMed] [Google Scholar]
- 106.Fuchs J, Nilsson C, Kachergus J, et al. Phenotypic variation in a large Swedish pedigree due to SNCA duplication and triplication. Neurology. 2007;68(12):916–922. doi: 10.1212/01.wnl.0000254458.17630.c5. [DOI] [PubMed] [Google Scholar]
- 107.Muenter MD, Forno LS, Hornykiewicz O, et al. Hereditary form of parkinsonism--dementia. Ann Neurol. 1998;43(6):768–781. doi: 10.1002/ana.410430612. [DOI] [PubMed] [Google Scholar]
- 108.Belarbi S, Hecham N, Lesage S, et al. LRRK2 G2019S mutation in Parkinson's disease: a neuropsychological and neuropsychiatric study in a large Algerian cohort. Parkinsonism Relat Disord. 2010;16(10):676–679. doi: 10.1016/j.parkreldis.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 109.Ben Sassi S, Nabli F, Hentati E, et al. Cognitive dysfunction in Tunisian LRRK2 associated Parkinson's disease. Parkinsonism Relat Disord. 2012;18(3):243–246. doi: 10.1016/j.parkreldis.2011.10.009. [DOI] [PubMed] [Google Scholar]
- 110.Shanker V, Groves M, Heiman G, et al. Mood and cognition in leucine-rich repeat kinase 2 G2019S Parkinson's disease. Mov Disord. 2011;26(10):1875–1880. doi: 10.1002/mds.23746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Goldwurm S, Zini M, Di Fonzo A, et al. LRRK2 G2019S mutation and Parkinson's disease: a clinical, neuropsychological and neuropsychiatric study in a large Italian sample. Parkinsonism Relat Disord. 2006;12(7):410–419. doi: 10.1016/j.parkreldis.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 112.Thaler A, Mirelman A, Gurevich T, et al. Lower cognitive performance in healthy G2019S LRRK2 mutation carriers. Neurology. 2012;79(10):1027–1032. doi: 10.1212/WNL.0b013e3182684646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Annesi G, Savettieri G, Pugliese P, et al. DJ-1 mutations and parkinsonism-dementia-amyotrophic lateral sclerosis complex. Ann Neurol. 2005;58(5):803–807. doi: 10.1002/ana.20666. [DOI] [PubMed] [Google Scholar]
- 114.Li Y, Tomiyama H, Sato K, et al. Clinicogenetic study of PINK1 mutations in autosomal recessive early-onset parkinsonism. Neurology. 2005;64(11):1955–1957. doi: 10.1212/01.WNL.0000164009.36740.4E. [DOI] [PubMed] [Google Scholar]
- 115.Alcalay RN, Mejia-Santana H, Tang MX, et al. Self-report of cognitive impairment and mini-mental state examination performance in PRKN, LRRK2, and GBA carriers with early onset Parkinson's disease. J Clin Exp Neuropsychol. 2010;32(7):775–779. doi: 10.1080/13803390903521018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Caccappolo E, Alcalay RN, Mejia-Santana H, et al. Neuropsychological Profile of Parkin Mutation Carriers with and without Parkinson Disease: The CORE-PD Study. J Int Neuropsychol Soc. 2011;17(1):91–100. doi: 10.1017/S1355617710001190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Satake W, Nakabayashi Y, Mizuta I, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson's disease. Nat Genet. 2009;41(12):1303–1307. doi: 10.1038/ng.485. [DOI] [PubMed] [Google Scholar]
- 118.Zabetian CP, Hutter CM, Factor SA, et al. Association analysis of MAPT H1 haplotype and subhaplotypes in Parkinson's disease. Ann Neurol. 2007;62(2):137–144. doi: 10.1002/ana.21157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Goris A, Williams-Gray CH, Clark GR, et al. Tau and alpha-synuclein in susceptibility to, and dementia in, Parkinson's disease. Ann Neurol. 2007;62(2):145–153. doi: 10.1002/ana.21192. [DOI] [PubMed] [Google Scholar]
- 120.Morley JF, Xie SX, Hurtig HI, et al. Genetic influences on cognitive decline in Parkinson's disease. Mov Disord. 2012;27(4):512–518. doi: 10.1002/mds.24946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Williams-Gray CH, Hampshire A, Barker RA, Owen AM. Attentional control in Parkinson's disease is dependent on COMT val 158 met genotype. Brain. 2008;131(Pt 2):397–408. doi: 10.1093/brain/awm313. [DOI] [PubMed] [Google Scholar]
- 122.Sidransky E, Nalls MA, Aasly JO, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med. 2009;361(17):1651–1661. doi: 10.1056/NEJMoa0901281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chahine LM, Qiang J, Ashbridge E, et al. Clinical and biochemical differences in patients having Parkinson disease with vs without GBA mutations. JAMA Neurol. 2013;70(7):852–858. doi: 10.1001/jamaneurol.2013.1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Brockmann K, Srulijes K, Hauser AK, et al. GBA-associated PD presents with nonmotor characteristics. Neurology. 2011;77(3):276–280. doi: 10.1212/WNL.0b013e318225ab77. [DOI] [PubMed] [Google Scholar]
- 125.Alcalay RN, Caccappolo E, Mejia-Santana H, et al. Cognitive performance of GBA mutation carriers with early-onset PD: the CORE-PD study. Neurology. 2012;78(18):1434–1440. doi: 10.1212/WNL.0b013e318253d54b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Federoff M, Jimenez-Rolando B, Nalls MA, Singleton AB. A large study reveals no association between APOE and Parkinson's disease. Neurobiol Dis. 2012;46(2):389–392. doi: 10.1016/j.nbd.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Williams-Gray CH, Goris A, Saiki M, et al. Apolipoprotein E genotype as a risk factor for susceptibility to and dementia in Parkinson's disease. J Neurol. 2009;256(3):493–498. doi: 10.1007/s00415-009-0119-8. [DOI] [PubMed] [Google Scholar]
- 128.Tsuang D, Leverenz JB, Lopez OL, et al. APOE epsilon4 increases risk for dementia in pure synucleinopathies. JAMA Neurol. 2013;70(2):223–228. doi: 10.1001/jamaneurol.2013.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hietanen M, Teravainen H. Cognitive performance in early Parkinson's disease. Acta Neurol Scand. 1986;73(2):151–159. doi: 10.1111/j.1600-0404.1986.tb03257.x. [DOI] [PubMed] [Google Scholar]
- 130.Levin BE, Llabre MM, Weiner WJ. Cognitive impairments associated with early Parkinson's disease. Neurology. 1989;39(4):557–561. doi: 10.1212/wnl.39.4.557. [DOI] [PubMed] [Google Scholar]
- 131.Owen AM, James M, Leigh PN, et al. Fronto-striatal cognitive deficits at different stages of Parkinson's disease. Brain. 1992;115(Pt 6):1727–1751. doi: 10.1093/brain/115.6.1727. [DOI] [PubMed] [Google Scholar]
- 132.Verbaan D, Marinus J, Visser M, et al. Cognitive impairment in Parkinson's disease. J Neurol Neurosurg Psychiatry. 2007;78(11):1182–1187. doi: 10.1136/jnnp.2006.112367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Owen AM. Cognitive dysfunction in Parkinson's disease: the role of frontostriatal circuitry. Neuroscientist. 2004;10(6):525–537. doi: 10.1177/1073858404266776. [DOI] [PubMed] [Google Scholar]
- 134.Welchko RM, Leveque XT, Dunbar GL. Genetic rat models of Parkinson's disease. Parkinsons Dis. 2012;2012:128356. doi: 10.1155/2012/128356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Nuber S, Petrasch-Parwez E, Winner B, et al. Neurodegeneration and motor dysfunction in a conditional model of Parkinson's disease. J Neurosci. 2008;28(10):2471–2484. doi: 10.1523/JNEUROSCI.3040-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Freichel C, Neumann M, Ballard T, et al. Age-dependent cognitive decline and amygdala pathology in alpha-synuclein transgenic mice. Neurobiol Aging. 2007;28(9):1421–1435. doi: 10.1016/j.neurobiolaging.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 137.Magen I, Fleming SM, Zhu C, et al. Cognitive deficits in a mouse model of pre-manifest Parkinson's disease. Eur J Neurosci. 2012;35(6):870–882. doi: 10.1111/j.1460-9568.2012.08012.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Porras G, Li Q, Bezard E. Modeling Parkinson's disease in primates: The MPTP model. Cold Spring Harb Perspect Med. 2012;2(3):a009308. doi: 10.1101/cshperspect.a009308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Itier JM, Ibanez P, Mena MA, et al. Parkin gene inactivation alters behaviour and dopamine neurotransmission in the mouse. Hum Mol Genet. 2003;12(18):2277–2291. doi: 10.1093/hmg/ddg239. [DOI] [PubMed] [Google Scholar]
- 140.Zhu XR, Maskri L, Herold C, et al. Non-motor behavioural impairments in parkin-deficient mice. Eur J Neurosci. 2007;26(7):1902–1911. doi: 10.1111/j.1460-9568.2007.05812.x. [DOI] [PubMed] [Google Scholar]
- 141.Li X, Redus L, Chen C, et al. Cognitive dysfunction precedes the onset of motor symptoms in the MitoPark mouse model of Parkinson's disease. PLoS One. 2013;8(8):e71341. doi: 10.1371/journal.pone.0071341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hall H, Jewett M, Landeck N, et al. Characterization of cognitive deficits in rats overexpressing human alpha-synuclein in the ventral tegmental area and medial septum using recombinant adeno-associated viral vectors. PLoS One. 2013;8(5):e64844. doi: 10.1371/journal.pone.0064844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Schneider JS, Kovelowski CJ., 2nd Chronic exposure to low doses of MPTP. I. Cognitive deficits in motor asymptomatic monkeys. Brain Res. 1990;519(1-2):122–128. doi: 10.1016/0006-8993(90)90069-n. [DOI] [PubMed] [Google Scholar]
- 144.Schneider JS, Roeltgen DP. Delayed matching-to-sample, object retrieval, and discrimination reversal deficits in chronic low dose MPTP-treated monkeys. Brain Res. 1993;615(2):351–354. doi: 10.1016/0006-8993(93)90049-s. [DOI] [PubMed] [Google Scholar]
- 145.Roeltgen DP, Schneider JS. Task persistence and learning ability in normal and chronic low dose MPTP-treated monkeys. Behav Brain Res. 1994;60(2):115–124. doi: 10.1016/0166-4328(94)90138-4. [DOI] [PubMed] [Google Scholar]
- 146.Schneider JS, Pope-Coleman A. Cognitive deficits precede motor deficits in a slowly progressing model of parkinsonism in the monkey. Neurodegeneration. 1995;4(3):245–255. doi: 10.1016/1055-8330(95)90014-4. [DOI] [PubMed] [Google Scholar]
- 147.Decamp E, Schneider JS. Attention and executive function deficits in chronic low-dose MPTP-treated non-human primates. Eur J Neurosci. 2004;20(5):1371–1378. doi: 10.1111/j.1460-9568.2004.03586.x. [DOI] [PubMed] [Google Scholar]
- 148.Schneider JS, Yuwiler A, Markham CH. Selective loss of subpopulations of ventral mesencephalic dopaminergic neurons in the monkey following exposure to MPTP. Brain Res. 1987;411(1):144–150. doi: 10.1016/0006-8993(87)90691-3. [DOI] [PubMed] [Google Scholar]
- 149.Schneider JS. Chronic exposure to low doses of MPTP. II. Neurochemical and pathological consequences in cognitively-impaired, motor asymptomatic monkeys. Brain Res. 1990;534(1-2):25–36. doi: 10.1016/0006-8993(90)90108-n. [DOI] [PubMed] [Google Scholar]
- 150.Schneider JS. Induction and reversal of cognitive deficits in a primate model of Parkinson's disease. In: Bedard MA, Agid Y, Chouinard S, Fahn S, Korczyn A, Lesperance P, editors. Mental and Behavioral Dysfunction in Movement Disorders. New York: Humana Press; 2003. [Google Scholar]
- 151.Schneider JS, Pioli EY, Jianzhong Y, Li Q, Bezard E. Levodopa improves motor deficits but can further disrupt cognition in a macaque Parkinson model. Mov Disord. 2013;28(5):663–667. doi: 10.1002/mds.25258. [DOI] [PubMed] [Google Scholar]
- 152.Lewis SJ, Slabosz A, Robbins TW, Barker RA, Owen AM. Dopaminergic basis for deficits in working memory but not attentional set-shifting in Parkinson's disease. Neuropsychologia. 2005;43(6):823–832. doi: 10.1016/j.neuropsychologia.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 153.Kulisevsky J. Role of dopamine in learning and memory: implications for the treatment of cognitive dysfunction in patients with Parkinson's disease. Drugs Aging. 2000;16(5):365–379. doi: 10.2165/00002512-200016050-00006. [DOI] [PubMed] [Google Scholar]
- 154.Nieoullon A. Dopamine and the regulation of cognition and attention. Prog Neurobiol. 2002;67(1):53–83. doi: 10.1016/s0301-0082(02)00011-4. [DOI] [PubMed] [Google Scholar]
- 155.Williams SM, Goldman-Rakic PS. Widespread origin of the primate mesofrontal dopamine system. Cereb Cortex. 1998;8(4):321–345. doi: 10.1093/cercor/8.4.321. [DOI] [PubMed] [Google Scholar]
- 156.Nieoullon A, Coquerel A. Dopamine: a key regulator to adapt action, emotion, motivation and cognition. Curr Opin Neurol. 2003;16(2):S3–9. [PubMed] [Google Scholar]
- 157.Pedersen KF, Larsen JP, Tysnes OB, Alves G. Prognosis of mild cognitive impairment in early Parkinson disease: the Norwegian ParkWest study. JAMA Neurol. 2013;70(5):580–586. doi: 10.1001/jamaneurol.2013.2110. [DOI] [PubMed] [Google Scholar]
- 158.Williams-Gray CH, Foltynie T, Brayne CE, Robbins TW, Barker RA. Evolution of cognitive dysfunction in an incident Parkinson's disease cohort. Brain. 2007;130(Pt 7):1787–1798. doi: 10.1093/brain/awm111. [DOI] [PubMed] [Google Scholar]
- 159.Broeders M, de Bie RM, Velseboer DC, Speelman JD, Muslimovic D, Schmand B. Evolution of mild cognitive impairment in Parkinson disease. Neurology. 2013;81(4):346–352. doi: 10.1212/WNL.0b013e31829c5c86. [DOI] [PubMed] [Google Scholar]