

Abstract
Recent findings in a range of scientific disciplines are challenging the conventional wisdom regarding the etiology, classification and treatment of psychiatric disorders. This review focuses on the current state of the psychiatric diagnostic nosology and recent progress in three areas: genomics, neuroimaging, and therapeutics development. The accelerating pace of novel and unexpected findings is transforming the understanding of mental illness and represents a hopeful sign that the approaches and models that have sustained the field for the past 40 years are yielding to a flood of new data and presaging the emergence of a new and more powerful scientific paradigm.
Recent findings in genetics and genomics, neurobiology, cognitive neuroscience, neuroimaging and pharmacology are presenting an accelerating array of challenges to the conventional wisdom regarding the etiology, classification, and treatment of psychiatric conditions, and forcing a reappraisal of research methods and approaches. Psychiatry now finds itself in the midst of considerable intellectual turmoil and facing some striking contradictions. Recent successful efforts at gene discovery are validating the utility of long-suspect categorical diagnoses while simultaneously undermining foundational elements of these same diagnostic schemes. Much of the pharmaceutical industry is withdrawing from psychiatric research while antidepressants and antipsychotics continue to rank among the top selling therapeutic agents in the United States. Increased public awareness has focused national attention on the importance of the treatment of psychiatric disorders and yet the legitimacy of psychiatry as a medical discipline continues to be debated, as it has been for decades (Szasz, 1960), and a shamefully large proportion of seriously and chronically mentally ill individuals are cared for in the justice system, instead of in the healthcare system
And despite obvious and rapid scientific advances, there is widespread frustration with the overall pace of progress in understanding and treating serious psychiatric illness. The beginning of the transition from an era dominated by psychoanalytic thinking to a “medical/biological” paradigm, in the 1960s and 1970s, was accompanied by high expectations. Yet, the wave of discovery in psychopharmacology that helped drive this transition was followed by what can only be described as a fallow period. There have been strikingly few novel treatment targets for serious mental illness identified and brought to market since the serendipitous identification of lithium, anti-psychotics and anti-depressants nearly 40 years ago (Hyman, 2013). For many of the most serious and debilitating disorders, this failure has not been the result of a general lack of interest or investment, but rather a consequence of their etiological complexity and the attendant difficulties in characterizing molecular, cellular and circuit level mechanisms that translate into viable treatment targets.
In light of these realities, it might seem folly to argue the case for optimism. But, despite the halting progress, the ongoing challenges and the controversies, the near-term future of psychiatry has, in fact, never been brighter: On the one hand, a growing appreciation of the extraordinary burden, world wide, of psychiatric illness (Vos et al., 2012), coupled with changes in the landscape of health care financing in the US are opening doors to a fuller integration of mental health care with other medical disciplines. Just as importantly, recent dramatic scientific advances, including the ability to systematically and reliably identify genetic risks, efficiently edit the genome, elaborate the anatomical and molecular landscape of the developing human brain, pursue circuit level analyses in both humans and model organisms, and follow-up on therapeutic observations with an unprecedented degree of molecular resolution, are leading the field toward a tipping point. The current generation of psychiatric trainees will practice in an era of profound transformation in the understanding of and ability to treat serious mental illness.
The Ongoing Challenge of Psychiatric Diagnosis
The current intellectual challenges confronting psychiatry are clearly evident in ongoing debates over diagnostic schema. The difficulty in arriving at a widely accepted, biologically-relevant nosology reflects the still rudimentary understanding of the neural mechanisms of cognition, behavior and emotion, the even more limited understanding of the intersection of pathophysiological mechanisms with these processes, and the unique character of psychiatry as a medical discipline. The field still lacks objective measures of psychopathology and biomarkers that reliably delineate normal from disease states, and one disease state from another. Moreover, more so than in any other area of medicine, conceptions about mental health and disease remain profoundly influenced by social and cultural norms and stigma.
The field has, from its inception, confronted the difficult task of categorizing, studying and treating manifestly debilitating disorders without being able to consistently and definitively establish the properties of psychiatric illness as opposed to variations in normal human behavior. This challenge, while perhaps particularly nuanced and contentious as a consequence of addressing complex human behavior, is certainly not unique to psychiatry. Every field in medicine has relied to one degree or another on descriptive approaches and confronted the ambiguities at the boundaries of disease.
Not surprisingly, then, as the field began, in the middle of the last century to wrestle with the limitations of diagnoses based on psychoanalytic theory, as well as with the inability to identify relevant neuropathology, it turned to other areas of medicine for inspiration. Beginning in the late 1950s, Eli Robins, Samuel Guze and George Winokur led an influential school of thought focused on developing a nosological system based on the idea diagnostic validity. The authors identified key domains for defining discrete diagnoses, including: clinical description, laboratory studies, exclusion of other disorders, follow-up studies and family history(Robins and Guze, 1970). These concepts served as the basis for the development of the first large-scale efforts at creating valid categorical diagnoses which, in turn, evolved into an widely-used set of Research Diagnostic Criteria (Spitzer et al., 1978) and subsequently, became the conceptual foundation for the Diagnostic and Statistical Manual (DSM) from the 1980s onward, including the most recent version, DSM-V.
This move toward categorical definitions served multiple valuable purposes. It provided, at a critical time, a shared language for clinicians, scientists, courts, and private and public funders of healthcare, set the stage for investigations of increasingly large cohorts, established an important foundation for epidemiological studies, and served as the basis for insurance reimbursement. Nonetheless, over time, it became clear that the limitations of the approach were particularly pronounced, even when compared with syndromic diagnoses in other areas of in medicine: The persistent inability to identify biomarkers continues to dictate a wholesale reliance on the subjective experience of patients and families and their equally subjective interpretation by physicians; clinicians are ultimately forced to make categorical distinctions within domains of cognitive, behavioral and emotional functioning that are continuously distributed in the population or across subgroups of patients; and the creation of new diagnoses or the modification of existing criteria, of necessity, still relies in part on conventional wisdom and consensus as well as data. The end result is a deep and seemingly expanding chasm between clinical diagnostic classification and the advancing understanding of brain and molecular science. Moreover, in practice, the field confronts the vexing situation in which some diagnoses are so obvious that detailed, elaborate criteria are essentially irrelevant to clinical practice. While, at the same time, for many conditions, the boundaries between typical and pathological states can be sufficiently subtle and difficult to codify that even apparently modest changes in diagnostic criteria can prompt intense debate and consternation.
In an effort to address the longstanding critiques and clear limitations of categorical approaches to diagnosis, Dr. Thomas Insel, Director of the National Institute of Mental Health (NIMH), has taken the lead in developing a dimensional approach to clinical observation, codified in the new Research Domain Criteria (Insel et al., 2010). This is based on a matrix of major neural systems (specifically: negative valence systems; positive valence systems, cognitive systems, social processing systems, arousal and regulatory systems), which are assessed across multiple units of analysis (including: genes, molecules, cells, circuits, physiology, behavior and patient report). The ultimate aim of this effort is not to serve as a substitute for the DSM at present, but as a first concerted foray into a dimensional approach that has the capacity to provide a stronger foundation for research into pathophysiology, which ultimately may inform future clinical classification schemes and, eventually, help identify new targets for treatment development.
This approach promises to move the field toward a more useful integration of clinical observation and biological understanding. It is also likely that, at least for the foreseeable future, psychiatry will increasingly rely on a hybrid of categorical, dimensional and biological descriptors. In other areas of medicine, one sees a synergistic application of traditional clinical or organ-based diagnoses, molecular characterization of pathology, and dimensional diagnostic and prognostic markers. One can envision for psychiatry a similar integration of categorical diagnoses, rapidly emerging genomic and other forms of biological data, and an anatomically-informed dimensional schema, serving as a viable path forward.
Advances in Psychiatric Genetics and Genomics
Perhaps the most surprising recent observation with regard to psychiatric nosology is the extent to which current approaches have proven adequate to drive the science. In fact, despite a widespread reliance on categorical diagnoses, one of the most productive areas of psychiatric research in the last half decade has been in human genetics. Here findings are reproducibly identifying risk genes and supporting neurobiological studies illuminating molecular, cellular and circuit level pathology. In addition, these advances are prompting a reevaluation of current thinking regarding underlying mechanisms of disease and the research paradigms that will be necessary to move the field forward.
The single most important development in psychiatric genetics has been the very recent emergence of systematic and reliable approaches to gene discovery. This stands in stark contrast to the prior era in which it seemed the search for replicable genetic risks for common psychiatric disorders might prove futile. Interestingly, despite the obvious importance of now being able to establish, definitively, that specific genes carry demonstrable risk, the significance of these developments appears to not yet be uniformly appreciated in the field. The prior era yielded hundreds of reports of putative associations based on hypothesis-driven studies of select common polymorphisms (defined as alleles with a population frequency of 1 percent or greater) mapping in or near a small number of biologically-plausible candidate genes. And despite the now-widely-recognized flaws of this approach (Hirschhorn and Altshuler, 2002) underpowered, poorly-controlled, and non-replicating common variant candidate-gene studies continue to be published in the psychiatric literature, and a not insignificant share of neurobiological investigations continue to rely on highly questionable genetic data for developing hypotheses and pursuing pathological mechanisms. This despite the manifest success of genome-wide discovery methods, including genome wide association studies (GWAS) of common alleles, genome-wide studies of copy number variation (CNV), whole-exome and whole-genome sequencing, for several paradigmatic disorders.
The emergence of a new state-of-the-art in psychiatric genomics has been reviewed elsewhere (Altshuler et al., 2008; Malhotra and Sebat, 2012; Veltman and Brunner, 2012). Briefly, the development of high resolution, high-throughput, and low cost genomic technologies are increasingly allowing for the (largely) unbiased assessment of variation, both common and rare, across the human genome. Simultaneously, it has become clear that the effect sizes of genetic contributions to common psychiatric disorders, in general, are considerably smaller than expected, leading to the realization that cohort sizes had to be dramatically larger than previously anticipated. In addition, the advent of dense micro-arrays and next generation sequencing allowed for the assessment of the contribution of several classes of germ-line genetic variation that had previously either been unappreciated, for example widespread copy number variation in typical individuals, or over-looked, as is the case with regard to the potential contribution of rare de novo mutation to common psychiatric disorders.
In fact, the convergence of new genomic technologies, improved statistical methods, an elaboration of key confounds in case-control genomic studies, dramatically larger cohorts and open-access biomaterials have led to a series of truly remarkable findings of late, particularly with regard to schizophrenia, bipolar disorder, autism spectrum disorders (ASD) and other neurodevelopmental syndromes. Findings in the first two of these areas have largely been in regards to common polymorphisms, while advances in gene discovery in ASD, epilepsy, and intellectual disability have predominantly involved rare and de novo variation. This dichotomy, as well as a dearth of results so far from GWAS studies of, for example, major depressive disorder (MDD), ASD, attention deficit hyperactivity disorder (ADHD), obsessive-compulsive disorder (OCD), PTSD (Post Traumatic Stress Disorder) and Tourette disorder, has been the subject of considerable discussion. The most compelling recent evidence suggests both that the proportional contribution of common and rare risk alleles differ for different disorders (as categorically defined) and that difficulty or delay in identifying genome-wide evidence for specific common alleles for many common psychiatric conditions is likely the result of still underpowered study cohorts.
Several other notable observations have emerged from recent genomic studies that point to the challenges ahead. First, in general, the estimated scale of locus heterogeneity (i.e. the number of independent genes that have the potential to increase risk for a disorder) has been found to be exceptionally high. For example, recent exome-sequencing studies of ASD have estimated between several hundred to 1000 genes anticipate carrying risk as a consequence of just de novo point mutations (Iossifov et al., 2012; Sanders et al., 2012).
The illumination of extensive genetic heterogeneity has been accompanied by the discovery of remarkable phenotypic variability. For example, a large proportion of the copy number variation has been found to carry risks for a wide range of independent outcomes including schizophrenia, intellectual disability, ASD, specific language impairment and reduced cognitive abilities that fall within the typical range (Malhotra and Sebat, 2012; Stefansson et al., 2013). Similarly, recent findings from whole-exome sequencing in individuals with schizophrenia (Fromer et al., 2014) and epilepsy(Epi et al., 2013) have pointed to a subset of genes that carry risk that cross diagnostic boundaries including ASD and cognitive functioning, in addition to these primary diagnoses.
This overlap is not exclusive to high-effect rare variants. A recent analysis of single nucleotide polymorphism (SNP)-based heritability across five disorders (Cross-Disorder Group of the Psychiatric Genomics et al., 2013) found shared risk for schizophrenia and bipolar disorder, with less, but still significant, overlap for bipolar disorder and MDD, schizophrenia and MDD, MDD and ADHD, and ASD and schizophrenia. These findings included genome-wide significant evidence for a small number of specific SNPS carrying diagnostically divergent risks, as well as for a “polygenic signal” shared across diagnoses.
Overall, the notion that a given SNP, de novo mutation, deletion or duplication in the genome can lead to diagnoses that cut across well-accepted boundaries is a thought provoking observation. Some degree of overlap was surely anticipated for closely related conditions such as schizophrenia and schizoaffective disorder. However, the degree to which risks have been found to cross syndromes with distinctive symptomatology, natural history and treatment response, has been a surprise. Moreover, the wide range of findings across multiple studies using research diagnostic criteria, and the consistency of results across disorders and mutation classes, makes the notion that this phenomenon may be explained entirely by cryptic co-morbidity or diagnostic ambiguity, increasingly unlikely. And while there are sure to be a series of specific explanations for the “one to many” relationships now being observed between genotype and phenotype, these findings are increasingly mounting a challenge to the notion of etiological specificity.
As noted above, a foundational element in psychiatric diagnosis, dating back to Robins, Guze and Winokur (Robins and Guze, 1970) has been that a valid category must be able to identify one disorder and exclude another. This has led in some quarters to the expectation that as etiological mechanisms were elaborated, they would be found to be specific for a given phenotype and/or help to better parse current diagnostic categories. With regard to genetics, this translated into some expectation that certain genes would be closely and directly tied to subsets of complex behaviors or phenotypes. For example, that multiple genes underlying autism might parse into subgroups separately involved in coding for cognition, social communication and language functioning.
The observation that the apparently identical mutation in one gene or genomic interval can lead to psychosis or language impairment or social deficits or epilepsy, or some combination of these phenotypes certainly challenges this view. Instead, it suggests that some significant portion of genetic risk impairs fundamental (“non-diagnosis-specific”) processes in brain development and functioning, and that the emergence of diverse clinical (or subclinical) phenotypes in these cases, results from inputs other than the identified “etiological” germ-line genetic variant, even when this is found to carry relatively large risks (State and Levitt, 2011; State and Sestan, 2012). These additional factors plausibly include epigenetic mechanisms, stochastic events, environmental variables, polygenic modifiers, somatic mutation, the microbiome and immune function, among others. Clarifying the nature and relative contribution of these inputs remains a considerable challenge. However, there is little question that success in gene discovery combined with advancing “omics” technologies, an increasingly nuanced understanding of the characteristics of many of these variables, and the development of large-scale and epidemiologically-based study cohorts coupled with the ability to ascertain groups of individuals based on mutation status, irrespective of clinical diagnosis, will help resolve these critically important questions.
Moreover, with the caveat that reliable gene discovery in psychiatry is still in its infancy, the recent finding are not only challenging psychiatric nosology but also the some aspects of the conventional wisdom regarding translational neuroscience. The standard “bottom-up” paradigm in psychiatry for moving from gene discovery to the identification of treatment target has relied largely on an approach that has historically been successful in leveraging the identification of Mendelian mutations, namely knocking-out a gene of interest and exploring the resulting, and often the most overtly dramatic, phenotype(s) (Figure 1A).
Figure 1.
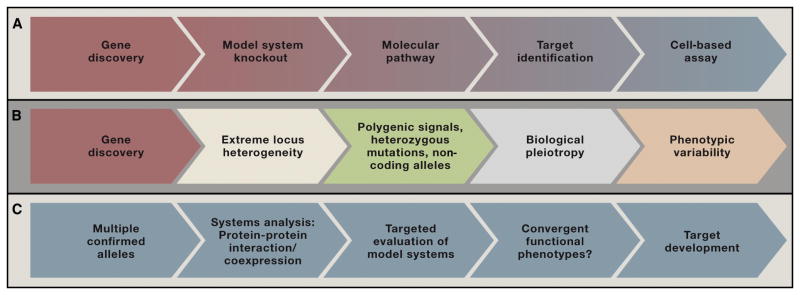
A) A standard model for the translation of genetic findings into therapeutics has been successful in Mendelian disorders and rare examples of common disorders showing Mendelian inheritance: A disease-related mutation is identified and modeled, typically via a constitutive or conditional knock-out; the molecular function and relevant pathways are identified and manipulated; a potential therapeutic target is identified. B) For many common psychiatric conditions, recent findings suggest additional complexities that confound this application of this approach: gene discovery has led to the identification of extreme locus heterogeneity; genes are often affected by heterozygous mutations and in non-coding regions; high-effect coding mutations may demonstrate biological pleiotropy with multiple roles that vary across cell types, brain region and developmental periods. C. An alternative emerging model for “bottom-up” translational work in psychiatric disorders. Multiple confirmed genes/mutations can be evaluated simultaneously, currently, for example, using protein-protein interaction and/or gene expression databases; the identification of points of overlap or intersection among genes and the identification of putative networks may be used to constrain key variables in the study of model systems, and convergent molecular, cellular and/or circuit level phenotype are sought as a prelude to target and assay development.
This approach will require some re-evaluation given: 1) The sheer number of risk alleles already identified; 2) the very small effect sizes conferred particularly by common variation; 3) the polygenic nature of risk reflected in studies of both common and rare variants (International Schizophrenia et al., 2009; Purcell et al., 2014); 4) the observation that many common risk alleles are both heterozygous and fall outside of coding regions; 5) the widely varied psychiatric outcomes emerging from the identical or similar mutations; and, 6) particularly for neurodevelopmental disorders, the common observation of biological pleiotropy, in which a single gene may encode the same or multiple proteins isoforms serving multiple functions across development (Figure 1B).
A recent spate of studies focusing on systems biological approaches has emerged in part as a response to these types challenges. The underlying rationale for such studies is that identifying points of convergence among multiple risk genes may be a necessary prelude to dissecting specific disease-related mechanisms. An initial wave of these studies have relied on protein-protein interaction databases, gene ontologies, and expression analyses in normal versus pathological tissue to organize various risk or candidate genes and identify areas of shared biology (O’Roak et al., 2012; Pinto et al., 2010; Voineagu et al., 2011) (Figure 2). More recently, studies focusing on autism and schizophrenia have attempted to incorporate developmental variables by leveraging human brain expression data from the Brainspan project (Gulsuner et al., 2013; Parikshak et al., 2013; Willsey et al., 2013).
Figure 2.

Recent examples of systems-biological approaches to interpreting genetic findings in common psychiatric disorders. A) (Adapted from Fromer et al. 2014) A recent exome sequencing study of schizophrenia used curated protein-protein databases to identify molecular pathways implicated by genes carrying non-synonymous rare de novo mutations. The proteins shown are present in the synaptic compartment of excitatory neurons and correspond in to NMDA (N-methyl-D-aspartate) and AMPA (α-Amino-3- hydroxy-5-methyl-4-isoxazolepropionic acid) receptor complexes and signaling pathways. B. (Adapted from Willsey et al 2013). An alternative recent approach restricted an initial analysis of exome data in ASD to only to those genes showing multiple de novo loss-of-function mutations in the same gene, consequently having the highest probability of being true ASD risk alleles. Expression data from the Brainspan project, capturing both anatomical and temporal dimensions of human brain development, were used to evaluate these nine seed genes. Co-expression networks representing discrete spatio-temporal windows were constructed for each of the seed genes and then evaluated for additional ASD mutations in order to identify pathology-associated networks. These in turn were found to correspond to human mid fetal prefrontal cortex and to implicate cortical excitatory neurons in deep layers (Layers 5–6). The inset illustrates that the proteins constructing these networks include genes in multiple cellular compartments and with varying functions. In this analysis, the temporal-spatial convergence was prioritized over identifying overlap in a specific cellular compartment or mapping to specific signaling pathways.
All of these approaches have revealed a marked degree of convergence, particularly given the expected scale of locus heterogeneity. This is certainly good new for those concerned that a thousand risk alleles might suggest the need for a thousand different treatments. Moreover, recent studies have broadened the view of the range of possible points of intersection for diverse sets of risk mutations to include both individual pathways and cellular compartments (e.g the synapse) as well as developmental timing and circuit-level anatomy (State and Sestan, 2012)(Figure 2).
A key issue for the future of these types of systems biological investigations is the current state of foundational resources. For example, current protein-protein interaction databases typically have only a tenuous connection with human brain or development, pathological specimens for all neuropsychiatric disorders are terribly limited, and efforts such as Brainspan, which are generating tremendously useful resources with regard to normative brain development, are still in their early stages. In this regard, the recent interim report on the Brain Research Through Advancing Innovative Technologies (BRAIN) initiative highlights priorities that will be critical for understanding the genetic contribution to complex psychiatric disorders, including the elaboration of cell specific data in developing and developed brain (http://www.nih.gov/science/brain/11252013-Interim-Report-ExecSumm.pdf.)
Clearly, recent advances in neurobiology and related sciences are already having a profoundly empowering effect on the prospects for illuminating genetic findings and clarifying the etiology of psychiatric disorders. These developments are too protean to review extensively here. Suffice it to say that advances in neuroscience are transforming the capacity to understand genetic variation and its consequence on brain structure and function: for example, the development of vastly improved genome editing technologies are allowing for the creation and study of model systems at an unprecedented scale and pace, and with the possibility of investigating multiple mutations simultaneously; new imaging techniques are offering an ability to visualize living and post-mortem tissue in profoundly more informative ways; the combination of stem cell technology and genomic editing is moving the field closer to being able to test hypothesis in a highly relevant biological context; and the emerging maps of molecular, cellular and circuit level landscape of brain in multiple species are providing a critical foundation for future studies.
Thus, despite the increasing complexity suggested by recent genetic findings, the challenges these pose to the conventional wisdom regarding diagnostic classification and research paradigms, and uneven progress across currently-defined disorders, the prospects for a transformation of our understanding of genes, brain and behavior and their roles in psychopathology have never been greater or closer at hand. This “best of times, worst of times” quality is likewise reflected in the areas of neuroimaging and therapeutics development, as discussed below.
Advances in Psychiatric Imaging
During the remarkable epoch when Kraepelin, Alzheimer, and their colleagues described features of dementia, schizophrenia, and bipolar disorder that persist in current diagnostic schema, both the opportunities and challenges associated with understanding their etiology were already apparent. The identification of profound brain atrophy combined with neural plaques and tangles in post-mortem tissue associated with the cognitive, emotional, and behavioral changes of dementia hinted that many neuropsychiatric disorders might be linked to identifiable neuropathophysiologic substrates. However, the failure of the same group of scientists using the same methods to uncover a compelling neurobiology for dementia praecox (schizophrenia) or manic-depressive disorder quickly highlighted the relatively greater complexity of the underlying neurobiology of these disorders.
Now, despite the more than 15,000 papers listed in PubMed for “magnetic resonance imaging and psychiatry” and the nearly 3000 papers listed for “positron emission tomography and psychiatry” there has been no neuroimaging biomarker for psychiatric disorders established sufficiently to guide psychiatric diagnosis or treatment. There are many factors that are likely to have contributed to this lack of progress ranging from the absence, until quite recently, of sufficiently large-scale studies designed expressly for this purpose, to the lack of a sufficiently clear understanding of the underlying biology of these disorders to guide the development of informative biomarkers.
Nonetheless, neuroimaging has emerged over the past 30 years as a critically important research strategy, leveraging an expanding array of available tools, including radiographic (i.e., x-ray based), magnetic resonance-based (i.e., MRI, DTI, fMRI, ASL, MRA, MRS), emission tomographic (i.e., PET, SPECT), near infrared spectroscopic, and electrophysiologic (electroencephalography, magnetoencephelography) approaches to evaluating human brain structure, function, and chemistry. These current technologies have proven to be useful for validating long-standing pathophysiologic hypotheses, empowering advances in cognitive neuroscience, implicating novel mechanisms in pathophysiology, and beginning to characterize macro-circuit level contributions to behavior and psychiatric disorders.
The contribution of imaging techniques to testing pathophysiological hypotheses is perhaps best exemplified in work on the role of dopamine in pathology of schizophrenia. By the mid-1970’s it was hypothesized that dopamine played a central role based on three cardinal observations: 1) amphetamine appeared to produce psychotic symptoms in humans not previously diagnosed with a psychotic disorder and to worsen symptoms in patients diagnosed with schizophrenia; 2) animal models suggested that amphetamine’s effects were mediated, at least in part, by dopamine; and 3) psychotic symptoms could be treated by depleting brain dopamine or by blocking dopamine receptors (Snyder, 1972).
While it took nearly 25 years, the advent of SPECT and PET imaging, the development of low-affinity radiotracers for the D2 dopamine receptor, and the validation of amphetamine-induced displacement of these D2 radiotracers from their receptors as an indirect measure of dopamine release, this research ultimately demonstrated: 1) increased striatal dopamine release in schizophrenia, including in medication-naïve patients; 2) that increased dopamine release was a feature of the dorsal associative striatum rather than the ventral limbic striatum or cortex; 3) that dopamine increases were associated with the level of psychotic symptoms, and were less markedly elevated in non-psychotic patients with schizophrenia; and, 4) the extent of the increased occupancy of D2 receptors by dopamine in patients correlated with the predicted optimal occupancy of these receptors by D2 receptor antagonist medications, i.e., that treatment worked by reducing dorsal striatal D2 signaling to a level similar to or even below that of healthy subjects. Further progress placed disturbances in dopamine signaling in a broader neurobiological context, particularly emphasizing primary abnormalities in glutamate signaling that might give rise to hyperactivity of dopamine inputs into the striatum(Laruelle et al., 2005).
Although functional MRI (fMRI) does not yet guide psychiatric diagnosis or treatment, it has played a key role in translational cognitive neuroscience. For example, fMRI studies have implicated amygdala-related circuits (i.e.: amgydala, ventral striatum, orbital frontal cortex, anterior cingulate, insula, anterior hippocampus) in the acquisition (Buchel and Dolan, 2000), reconsolidation (Schiller and Phelps, 2011), and extinction (LaBar et al., 1998) of human fear. This work also supported learning theory-related models involving these same circuits in anxiety disorders and provided evidence that cognitive-behavioral (Barsaglini et al., 2013) and pharmacologic (Paulus et al., 2005) treatments normalized circuit activity. Closely related circuitry were linked to addictions, to rewarding behaviors (e.g, gambling, sex) and to abused substances (e,g, alcohol, cocaine, food, opiates). Further, fMRI provided human circuit-based insights into the addiction process, i.e. intoxication, the acquisition and extinction of Pavlovian conditioned responses, stress- and cue-induced reinstatement of craving, reward-related instrumental learning, and habit formation (Fryer et al., 2013; Goldstein et al., 2009; O’Doherty et al., 2004; Potenza et al., 2012; Prevost et al., 2012; Sjoerds et al., 2013). Elements of these same circuits are involved broadly in psychopathology, such as depression-related anhedonia, the negative symptoms of schizophrenia, psychotic delusions and obsessions. In short, these studies implicated common macrocircuits across a wide range of psychiatric diagnoses without shedding light on pathology at the microcircuit, synaptic, or molecular level. As a result, the involvement of similar circuits across clinical conditions does not imply that they share a common etiology or respond to similar treatments. However, these molecular insights may emerge from studies that combine fMRI with pharmacology, genetics, and neurochemical brain imaging (MRS, PET, SPECT)
Neuroimaging has also implicated disturbances in cortical connectivity in psychiatric disability. In schizophrenia, findings include progressive reductions in cortical gray and white matter volume on MRI (Olabi et al., 2011), decreases in the integrity of white matter tracks as measured by DTI (Fitzsimmons et al., 2013), abnormalities in covariant brain activity (resting functional connectivity) (Yu et al., 2012), and deficits in signal over background activity of high frequency cortical oscillations on EEG (Uhlhaas, 2013). While some studies highlight regional changes, particularly involving prefrontal or temporal cortices, other studies highlight the existence of global changes or large-scale shifts in the pattern (Anticevic et al., 2013) or organizational structure of cortical functional connectivity. Studies of functional connectivity are paralleled by fMRI studies describing reduced activation of neural circuits sub-serving a wide range of cognitive functions and reductions of the functional connectivity in these circuits as they are activated during task performance.
Neuroimaging techniques also provide insights into links between neuro-inflammation, glial dysfunction, and glutamate synaptic dysfunction in depression (Krystal et al., 2013; Miller et al., 2009). Glia are particularly vulnerable to injury by these inflammatory processes, consistent with post-mortem data of reductions in glial populations associated with depression (Sanacora and Banasr, 2013). These glial deficits would be predicted to reduce their capacity to take up and activate glutamate, raising extra-synaptic glutamate levels. Presynaptically, this is hypothesized to suppress synaptic glutamate release via metabotropic glutamate receptors (mGluR2) and postsynaptically to cause reductions in synaptic connectivity by over-stimulating extra-synaptic NMDA receptors, especially receptors bearing the NR2B subunit (Krystal et al., 2013).
Recently, magnetic resonance spectroscopy (MRS) has emerged as a powerful tool for characterizing disturbances in the functional integrity of neuronal and glial mechanisms in energy metabolism and amino acid neurotransmission related to psychiatric disorders. 13C-MRS using 13C-labled glucose and acetate as metabolic tracers is a powerful tool for characterizing the impact of glial dysfunction and loss on brain energy metabolism and glutamate neurotransmission in animal models (Banasr et al., 2010) and for characterizing glutamatergic transmission and cellular energetics in animals (Chowdhury et al., 2012). 1H-MRS spectroscopy studies have characterized reductions in glutamatergic and GABAergic neurotransmission in depressed patients (Sanacora et al., 2000; Yuksel and Ongur, 2010). However, the interpretation of the large number of 1H-MRS studies measuring voxel glutamate and glutamine levels are less clear as these measures are not simply or directly related to the rate of glutamate neurotransmission. Thus it is not clear why ketamine, a drug known to increase glutamate release in some circuits increased glutamate but not the total of glutamate and glutamine levels in one study (Stone et al., 2012), but raised glutamine but not glutamate in another (Rowland et al., 2005). As a result, 13C-MRS combined with the use of isotopic tracers remains the only definitive spectroscopic approach to characterize neuronal and glial energy metabolism.
Finally, neuroimaging has made unique contributions to the current understanding of brain macrocircuits (Buckner et al., 2013; Craddock et al., 2013). This progress is both identifying novel features of human cortical functional organization and shedding light on links between circuitry, normal behavior, and psychiatric disorders. New insights into macrocircuits involvement in cognition, behavior, emotion, and psychiatric symptoms has played an important role in inspiring the RDoC initiative as an alternative to categorical diagnostic approaches to psychiatry.
However, neuroimaging has yet to emerge as a psychiatric diagnostic tool. Support vector machine learning has been applied to structural MRI, DTI and fMRI data with the aim of identifying patterns of results that could substitute for symptoms as a basis for psychiatric diagnoses. This is an a-theoretical approach through which a computer identifies patterns in a dependent measure that maximizes subgroup differences within any data set, i.e., categorizes subjects (Orru et al., 2012). Thus if diagnoses were based simply on discrete patterns of changes in a set of measures, for example the data points in an MRI scan, this approach could prove to be a powerful means to define diagnoses. The approach has been applied to a range of psychiatric diagnoses, including schizophrenia, bipolar disorder, and major depression (Lord et al., 2012; Mwangi et al., 2012; Schnack et al., 2014). To date, studies appear to be able to separate healthy subjects from patients diagnosed with schizophrenia with 70–90% accuracy and to separate patients with bipolar disorder from both groups with lower levels of accuracy. It does not appear, however, that these imaging techniques have the reliability or discriminative validity to improve the diagnostic process beyond clinical interviewing. However, these approaches have been applied to relatively small samples (<100/group) and much larger cohorts might yield greater sensitivity and specificity if current diagnostic categories are roughly representative of discrete patterns of changes in brain structure and function.
Of course, it is not a foregone conclusion that available neuroimaging approaches have the capacity to inform current diagnostic schemes. As described earlier, overlap in the genes contributing to a wide range of psychiatric disorders suggests at least one model in which a more general risk imparted by variations in the human genome are conferred greater symptomatic/diagnostic specificity by other factors or at other levels of analysis. What these are remain uncertain, but one possibility is that in addition to genetic overlap, there is also substantial macro-circuit level overlap underlying these conditions as well. And, it would follow that the greater the overlap in neuroimaging findings across diagnoses, the more challenging it will be to use these types of approaches for diagnostic purposes.
Current imaging techniques are also limited because they describe brain structure, function, and chemistry with far lower spatial and temporal resolution or reduced sensitivity compared to the more invasive and informative techniques that can be applied to the study of animals. The result is that human studies employing functional neuroimaging, for example, are not simply a foggy version of single unit recordings made in the animal brain. Rather these clinical research studies query the brain in ways that differ fundamentally from studies conducted in animals.
This observation points to continuing critical technical gaps in the ability of neuroimaging to characterize the structure, function, and chemistry of microcircuits, which constitutes a fundamental obstacle to the translation of knowledge across the levels of genes, proteins, cells, local networks, distributed networks, and behavior in the human brain. Failure to capture the diversity of molecular, synaptic, cellular, and microcircuit mechanisms that are currently beyond the resolution of neuroimaging and which might give rise to common disturbances in macro-circuit function is a potentially fundamental challenge to the application of neuroimaging to diagnosis and medication development.
As noted above, there may be ways to create heuristic and computational models that enable neuroscience research to traverse the knowledge gap between basic and clinical neuroscience research, perhaps through uniting neuroimaging with data from genetics, pharmacology, data from animal models, human post-mortem tissue, induced human pluripotent stem cells, cultured nasal neuroepithelial cells, and clinical data (Brennand et al., 2013; Schadt and Bjorkegren, 2012). However, these integrative strategies currently are largely exploratory in nature. Moreover, important initiatives such as the Human Connectome Project and, more recently, the BRAIN Project collectively are likely to lead to the development of new tools that will further increase study resolution and yield a more informed understanding of the relationship between neuroimaging and electrophysiological findings, and neural processing (Leopold and Maier, 2012; Logothetis, 2012). Recent, very elegant studies serve as a harbinger of these possibilities, using electrophysiological approaches directly on the surface of human brain in patients undergoing epilepsy surgery, to dissect complex human behaviors, including speech and language(Bouchard et al., 2013).
Still, it remains to be seen whether current approaches, as powerful as they are, will be adequate to inform a diagnostic system that builds from pathology at the level of the synapse or local circuit. Similarly, the exploratory nature of neuroimaging findings combined with their limitations with regards to characterization of microcircuit mechanism seem to constitute fundamental limitations in the ability to minimize or “de-risk” the exploratory nature of psychiatry drug development. Thus, it may not be surprising that neuroimaging approaches have yet to identify a new treatment mechanism for psychiatric disorders.
Psychiatric Therapeutics
The field of psychiatry and individuals with psychiatric disorders are in desperate need of treatment breakthroughs. Most classes of medications employed in psychiatry are at least 40 years old and show little or no diagnostic specificity. Moreover, patients are treated increasingly with combinations of medications from several treatment classes. It has been seven years since a drug with an arguably novel mechanism of action, the nicotine receptor partial agonist verenicline, has been approved for a psychiatric indication, namely for tobacco addiction. And, apart from the identification of clozapine pharmacotherapy for treatment-resistant symptoms schizophrenia in mid-1980s, it is difficult to recall another example of a truly novel mechanism driving the marketing of a psychiatric medication since the very early days of psychopharmacology.
Moreover, there is very limited depth of US FDA-approved treatment options for most disorders. A large number of novel treatment mechanisms have been explored by the pharmaceutical industry for the treatment of some conditions, such as schizophrenia, depression, and generalized anxiety disorder, while most other syndromes receive little industry investment. For example, cocaine or cannabis addiction have no FDA approved pharmacotherapy. There is a particular dearth of FDA approved agents for childhood disorders, in part as a consequence of knowledge gaps and in part a practical consequence of the challenges associated with studying drugs in the pediatric population.
The limited armamentarium is up against a truly enormous world-wide burden of psychiatric morbidity: for example: seven of the top 20 most disabling medical conditions in the world are illnesses treated by psychiatrists, with depression number two on this list (Vos et al., 2012). Patients diagnosed with one of these chronic disabling disorders die, on average, 25 years earlier than the general population (Lutterman et al., 2003). The top three causes of death in young adults are frequently complications of psychiatric disorders including suicide, homicide, and accidents (http://www.cdc.gov/injury/overview/data.html). In fact, it is estimated that over 20% of hospitalized patients diagnosed with bipolar disorder may eventually die by suicide. Consequently, there is tremendous urgency in identifying novel treatment targets and mechanisms in order to respond to the need for more and more effective treatments.
In the current era, where scientific discovery and innovation is occurring across a range of relevant disciplines, new treatments seem to be emerging from many levels of investigation, including clinical observations, neuroimaging findings, and animal models (Figure 3). Interestingly, despite the recent wave of discovery in genetics and genomics, one strategy that has yet to definitively succeed in psychiatry is the direct translation from genetic variation to biology to treatment, despite efforts to apply this strategy to Alzheimer disease (antibodies to Abeta protein, gamma secretase inhibitors, beta secretase inhibitors) autism (metabotropic glutamate receptor-5 antagonists). To illustrate paths that are leading to recent exciting therapeutic developments, this review will focus on the development of new rapid-acting antidepressants, which emerged from clinical observation; the development of drugs to enhance synaptic neuroplasticity to enhance the efficacy of pharmacotherapy, based on animal models; and the development of neuro-stimulation treatments that developed as a consequence of neuroimaging studies of depression.
Figure 3.

Examples of multiple paths through which new treatments may emerge in psychiatry. The archetypal rapid-acting antidepressant, the NMDA glutamate receptor (NMDA-R) antagonist ketamine, was first identified as a treatment for depression as a consequence of a clinical observation. The ability of D-cycloserine (DCS) to augment the efficacy of behavioral therapy for anxiety disorders developed from an animal model where this drug promoted fear extinction. Deep brain stimulation treatment for depression was inspired by neuroimaging studies describing dysfunction of the subgenual prefrontal cortex in depressed patients.
Rapid-acting antidepressant medications may become the first fundamentally new class of pharmacotherapies for psychiatry in several decades (Krystal et al., 2013). The prototype for this class of medications, ketamine, was identified in academia through basic science-informed human experimentation (Berman et al., 2000). Ketamine is an uncompetitive NMDA glutamate receptor antagonist that had been first studied in humans as a probe for the neurobiology of schizophrenia (Krystal et al., 2003). In the attempt to extend this work to the study of depression, and informed by preclinical descriptions of antidepressant effects of these drugs (Skolnick et al., 2009; Trullas and Skolnick, 1990), a pilot study was conducted in patients with treatment-resistant symptoms, published in 2000, that reported: clinical improvement in patients within 4 hours of a single dose, 50% of patients meeting criteria for clinical response within 24 hours, and response lasting for up to two weeks (Berman et al., 2000). This pattern stands in marked contrast to the characteristics of standard treatments for depression, which have been comprehensively described in the NIMH STAR*D study: including an seven week interval, on average, to remission in treatment-responsive patients, and a 10–15% response rate for treatment-resistant patients following medication changes in those who initially failed a trail of a serotonin uptake inhibitor (Gaynes et al., 2009).
Within a decade, there were more than a dozen replications of the therapeutic effects of ketamine without a single negative study. These documented prominent reductions in suicidal ideation, response rates in the 50%–80% range (including in electroconvulsive therapy non-responders), a third of patients sustaining response from a single dose for two weeks, and preliminary evidence that the antidepressant responses of a single dose could be sustained by repeated ketamine dosing (Aan Het Rot et al., 2012). Further, a growing list of studies of NMDA receptor antagonists or NMDA receptor negative allosteric modulators, including S-ketamine, CP101,606 (NR2B-selective), lanicemine (AZD6765, NR2B-selective), and GlyX13 (NR2B partial agonist) provided further evidence that ketamine’s effects were mediated primarily by its actions at NMDA receptors.
Preclinical insights into the antidepressant effects of ketamine have helped to identify a broader class of antidepressants that may converge on common signaling mechanisms (Krystal et al., 2013). The antidepressant effects of ketamine appear to be mediated, at least in part, by the ability to increase glutamate release, perhaps overcoming suppression of mGluR2 receptors by elevations in extrasynaptic glutamate noted earlier, and to stimulate AMPA receptors, raising BDNF levels, increasing insertion of AMPA receptor subunits into cell members, enhance signaling via Akt/mTOR, and enhancing the rapid growth of functional dendritic spines (Li et al., 2010). This process appears to be enhanced by the blockade of extrasynaptic NMDA receptors, which reduces the phosphorylation of ELF2, and disinhibits elevations in BDNF (Autry et al., 2011; Hardingham and Bading, 2010).
The articulation of these mechanisms through which ketamine may function has helped to show that other putative rapid-acting antidepressant may similarly work through a common set of mechanisms. Thus the muscarinic receptor antagonist, scopolamine, shows rapid antidepressants in human pilot studies (Drevets et al., 2013). It also enhances cortical signaling via Akt/mTOR in animals (Voleti et al., 2013). Similarly, the putative antidepressant effects of mGluR2 antagonists and AMPAkines (Lindholm et al., 2012) are dependent on enhancing AMPA receptor and perhaps also Akt/mTOR signaling (Koike et al., 2013; Yoshimizu et al., 2006). If this new class of medications reduces depression symptoms within hours instead of months and makes its possible to effectively treatment many patients who could not be reached with current treatments, it could fundamentally change the functional impact of depression in society.
Another new direction is to develop medications that enhance neuroplasticity in the service of promoting a learning-based cognitive or rehabilitative therapy rather than by directly normalizing circuit activity (Krystal et al., 2009). One might argue that drugs are poor choices to optimize circuit activity as a particular receptor might be widely distributed in the brain and represented on multiple cellular elements within a local circuit and thus produce opposing effects within local or distributed circuits. Further, drug would not be expected to approximate the kinetic binding properties of natural ligands for those receptors. Thus, drugs might be expected to introduce abnormalities in the temporal features of normal circuit function. From this perspective, it might be preferable to treat psychiatric disorders by inducing a state of increased plasticity in a circuit that one wished to modify and then behaviorally engaging that circuit in a manner to induce a desired change in its function, i.e., to extinguish a fear or enhance a cognitive function. Once the desired change in circuit function has occurred, the medication might become superfluous. For example, a growing number of studies have used low doses of a partial agonist of the glycine site of NMDA receptors, D-cycloserine, to enhance the impact of fear extinction or cognitive therapy for anxiety disorders and addictions (Norberg et al., 2008; Prisciandaro et al., 2013; Ressler et al., 2004). While D-cycloserine at these doses does not appear to be anxiolytic by itself, there is evidence that enhancing NMDA receptor-related neuroplasticity in this way both hastens clinical response in phobia, potentially increasing the efficiency and cost effectiveness of behavioral therapy, and protects against the reinstatement of fear.
An alternative approach to achieving circuit-specific functional alterations is to manipulate these circuits directly. The first circuit-based treatments in psychiatry were ablative treatments, orbital frontal lobotomy and leucotomy for refractory mood and psychotic disorders (Moniz, 1937) and, more recently, surgical and radiotherapeutic (“Gamma knife”) ablations of the anterior cingulate cortex or anterior interior capsule for medication-resistant obsessions and compulsions (Baer et al., 1995; D’Astous et al., 2013). These treatments have their roots in a rather superficial understanding of how the circuits that are disrupted by these treatments contribute to symptoms. Thus it is not surprising that lobotomy was infrequently a definitive treatment for treatment-resistant symptoms of schizophrenia or bipolar disorder and may have further compromised the capacity for self-care (Harvey et al., 1993). In contrast, ablative treatments for medication-resistant symptoms of obsessive-compulsive disorder continue to be performed in highly selected extremely disabled patients in parallel with detailed ongoing scrutiny of these procedures.
However, neurostimulation treatments, which have the advantage over neuroablative therapies of reversibility, seem the most likely to yield important new circuit-based treatments for psychiatry disorders. Current neurostimulation treatments evolved from the current “gold standard” for the treatment of depression, electroconvulsive therapy (ECT) (Cerletti, 1940; Lisanby, 2007). The neurostimulation field has developed treatment strategies that produce seizures in the target circuits and spare others that seem more closely related to side effects.
Deep brain stimulation and repeated transcranial magnetic stimulation (rTMS) may be strategies to produce this type of targeted therapeutic change in brain activity. Neuroimaging studies conducted by Mayberg and her colleagues identified a potential target for neurostimulation: the subgenual prefrontal cortex (infralimbic cortex). This area is hyperactive in depressed patients and in euthymic individuals with transiently induced sadness (Mayberg et al., 1999) and the group was aware that chronic high frequency stimulation (130 Hz) reduced network hyperactivity in Parkinson’s disease (Benabid, 2003), although the neurobiology underlying this intervention was not known.
Fortunately, high frequency stimulation to subgenual PFC showed compelling promise in clinical trials (Holtzheimer et al., 2012; Mayberg et al., 2005), offering new hope for patients suffering medication- and ECT-resistant symptoms of depression. Subsequently, the delivery of this type of stimulation to other sites in reward network, including the internal capsule, the ventral striatum, the habenula, and the median forebrain bundle, have also produced encouraging results in preliminary trials (Malone, 2010). Deep brain stimulation of the internal capsule and ventral striatum also show promise for the treatment of medication-refractory obsessive compulsive disorder (Goodman et al., 2010).
Ongoing research into deep brain stimulation constitutes an important response to the needs of patients who are refractory to currently available treatments. It nonetheless represents a considerable leap of faith in the face of the currently limited understanding of the underlying neuroscience. While neural dysfunction in depression and OCD have been characterized at very gross macrocircuit level, the precise alteration in activity in particular cell types that give rise to the symptoms of depression and to its treatment remain obscure. It is striking, for example, that small anatomical differences in electrode placement in subgenual PFC, result in substantial differences in the circuits effected by DBS (Lujan et al., 2013). Even with precise anatomical localization, neurostimulation does not yield uniform activation of brain circuits. Rather, activation of some cortical inputs would be expected to activate a given target region, but also to result in suppression of neighboring areas (Logothetis et al., 2010). Further, relatively little is known about the synaptic neurobiology of the high frequency stimulation (>100 Hz) used in deep brain stimulation studies, compared to low frequency stimulation (~1Hz) or “theta” (~10 Hz) stimulation parameters that have been used to implicate NMDA receptors and other mechanisms in long-term depression and potentiation, respectively (Malenka and Bear, 2004). In contrast, high frequency stimulation (>100 Hz) appears to induce an NMDA receptor-independent form of LTP mediated by L-type voltage-gated calcium channels (Cavus and Teyler, 1996). Given these open questions, this “high-risk, high reward” research is accompanied by scrupulous efforts to protect patients and research subjects and extremely close follow-up of clinical outcomes.
Transcranial magnetic stimulation (TMS), a non-invasive localized strategy for cortical stimulation, also shows promise as a treatment for depression and other conditions. The relative safety and tolerability of this procedure has enabled a broad range of studies of repeated TMS (rTMS) for the treatment of medication-resistant psychiatric symptoms; including the application to auditory hallucinations. Prior research indicated that auditory hallucinations associated with schizophrenia were accompanied by hyperactivity of circuits involved in auditory perception and language processing cortical regions (Silbersweig et al., 1995). Hoffman and Cavus hypothesized that activity within these circuits could be reduced by administering low frequency (1 Hz) stimulation to these regions with rTMS, modeled after long-term depression in animals (Hoffman and Cavus, 2002). Applying this strategy to treatment patients, Hoffman and colleagues produced positive findings (Hoffman et al., 2005), particularly when rTMS was delivered stereotaxically over regions that had shown hallucination-related activation on functional magnetic resonance imaging (Hoffman et al., 2007).
There are significant knowledge and technology gaps representing unaddressed grand challenges for psychiatric therapeutics research. For example, the identification of gene variants that have large effects on the risk for psychiatric disorders hold enormous promise to guide target development, but, as described above, this is not likely to be realized until the complex biology through which these variants act to alter microcircuits is elucidated. Similarly, the development of cell-based strategies for regulating brain circuit activity, such as optogenetics (Williams and Deisseroth, 2013), seems to hold significant promise for neurostimulation treatments. However, if and when the technologic hurdles are surmounted to enable the application of this technique to humans, our limited understanding of microcircuit dysregulation in psychiatric disorders may still constrain the impact of these treatments.
Conclusion
This review has of necessity sacrificed comprehensiveness for a highly selective consideration of historical trends and recent advances in psychiatry. It gave short shrift to a host of truly astounding findings in basic neuroscience, and to a range of profoundly interesting advances in areas such as epigenomics, metabalomics, proteomics, and gene-environment interactions all of which promise to further enrich and perhaps transform our understanding of psychopathology. It avoided the potentially tectonic shifts in mental health care delivery attending changes in the health care environment in the US and has neglected the critically important topics of dissemination science and the implementation of proven therapies.
Instead, we focused on current controversies over psychiatric diagnosis and on three areas of recent scientific advance that are having an immediate and profound impact on the field. Overall, while it is impossible to discount the scale of the short-term challenges present in these three areas, the longer-term prospects for the science of psychiatry are thrilling. These advances are providing a rapidly deepening understanding of complex psychiatric disorders and the accelerating pace of surprising, sometimes confusing and often contradictory findings is a hopeful sign that current scientific paradigms that have sustained the field over the past 40 to 50 years are no longer adequate to incorporate and integrate a flood of new data. As Thomas Kuhn described so elegantly(Kuhn, 1962), these anomalous findings are the harbinger of true scientific revolution. In this case of psychiatry, this cannot come too soon for patients, their families and society.
Acknowledgments
We gratefully acknowledge the Department of Veterans Affairs, the VA National Center for PTSD and its joint funding (with the Department of Defense) of the Coalition to Alleviate PTSD (CAP to JK), the National Institute on Alcohol Abuse and Alcoholism (3P50AA012870 to JK), the National Institute of Mental Health (FAST-PS to JK and R01 MH092289-01A1, R01 MH081754 and U01 MH100239 to MWS), the National Center for Advancing Translational Science (UH2TR000960-01; Clinical and Translational Science Award Grant No. UL1 RR024139 to JK), and support from the Simons Foundation Autism Research Initiative (to MWS) the State of Connecticut Department of Mental Health and Addiction Services for its support of the Abraham Ribicoff Research Facilities of the Connecticut Mental Health Center.
Footnotes
Disclosures:
Dr. Krystal has served as a scientific consultant to the following companies with compensation in the past year of more than $5,000 USD: Novartis Pharma AG, Janssen Research and Development LLC, AbbVie, Inc., Eli Lilly Corporation, and Naurex, Inc. He holds stock in Biohaven Medical Sciences. He also has the following patents and inventions: 1) dopamine and noradrenergic reuptake inhibitors in treatment of schizophrenia. Patent no: 5447948, 5 September 1995; 2) a pending patent for glutamatergic treatments for neuropsychiatric disorders (PCTWO06108055A1); 3) a pending patent for some applications of ketamine to the treatment of depression.
References
- Aan Het Rot M, Zarate CA, Jr, Charney DS, Mathew SJ. Ketamine for Depression: Where Do We Go from Here? Biol Psychiatry. 2012 doi: 10.1016/j.biopsych.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altshuler D, Daly MJ, Lander ES. Genetic mapping in human disease. Science. 2008;322:881–888. doi: 10.1126/science.1156409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anticevic A, Cole MW, Repovs G, Murray JD, Brumbaugh MS, Winkler AM, Savic A, Krystal JH, Pearlson GD, Glahn DC. Characterizing Thalamo-Cortical Disturbances in Schizophrenia and Bipolar Illness. Cereb Cortex. 2013 doi: 10.1093/cercor/bht165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng PF, Kavalali ET, Monteggia LM. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature. 2011;475:91–95. doi: 10.1038/nature10130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baer L, Rauch SL, Ballantine HT, Jr, Martuza R, Cosgrove R, Cassem E, Giriunas I, Manzo PA, Dimino C, Jenike MA. Cingulotomy for intractable obsessive-compulsive disorder. Prospective long-term follow-up of 18 patients. Arch Gen Psychiatry. 1995;52:384–392. doi: 10.1001/archpsyc.1995.03950170058008. [DOI] [PubMed] [Google Scholar]
- Banasr M, Chowdhury GM, Terwilliger R, Newton SS, Duman RS, Behar KL, Sanacora G. Glial pathology in an animal model of depression: reversal of stress-induced cellular, metabolic and behavioral deficits by the glutamate-modulating drug riluzole. Mol Psychiatry. 2010;15:501–511. doi: 10.1038/mp.2008.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barsaglini A, Sartori G, Benetti S, Pettersson-Yeo W, Mechelli A. The effects of psychotherapy on brain function: A systematic and critical review. Prog Neurobiol. 2013 doi: 10.1016/j.pneurobio.2013.10.006. [DOI] [PubMed] [Google Scholar]
- Benabid AL. Deep brain stimulation for Parkinson’s disease. Curr Opin Neurobiol. 2003;13:696–706. doi: 10.1016/j.conb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, Krystal JH. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47:351–354. doi: 10.1016/s0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- Bouchard KE, Mesgarani N, Johnson K, Chang EF. Functional organization of human sensorimotor cortex for speech articulation. Nature. 2013;495:327–332. doi: 10.1038/nature11911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennand KJ, Landek-Salgado MA, Sawa A. Modeling Heterogeneous Patients with a Clinical Diagnosis of Schizophrenia with Induced Pluripotent Stem Cells. Biol Psychiatry. 2013 doi: 10.1016/j.biopsych.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchel C, Dolan RJ. Classical fear conditioning in functional neuroimaging. Curr Opin Neurobiol. 2000;10:219–223. doi: 10.1016/s0959-4388(00)00078-7. [DOI] [PubMed] [Google Scholar]
- Buckner RL, Krienen FM, Yeo BT. Opportunities and limitations of intrinsic functional connectivity MRI. Nat Neurosci. 2013;16:832–837. doi: 10.1038/nn.3423. [DOI] [PubMed] [Google Scholar]
- Cavus I, Teyler T. Two forms of long-term potentiation in area CA1 activate different signal transduction cascades. Journal of neurophysiology. 1996;76:3038–3047. doi: 10.1152/jn.1996.76.5.3038. [DOI] [PubMed] [Google Scholar]
- Cerletti U. L’Elettroshock. Rivista Sperimentale di Frenatria. 1940;1:209–310. [Google Scholar]
- Chowdhury GM, Behar KL, Cho W, Thomas MA, Rothman DL, Sanacora G. (1)H-[(1)(3)C]-nuclear magnetic resonance spectroscopy measures of ketamine’s effect on amino acid neurotransmitter metabolism. Biol Psychiatry. 2012;71:1022–1025. doi: 10.1016/j.biopsych.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craddock RC, Jbabdi S, Yan CG, Vogelstein JT, Castellanos FX, Di Martino A, Kelly C, Heberlein K, Colcombe S, Milham MP. Imaging human connectomes at the macroscale. Nature methods. 2013;10:524–539. doi: 10.1038/nmeth.2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smoller JW, Craddock N, Kendler K, Lee PH, Neale BM, Nurnberger JI, Ripke S, Santangelo S, Sullivan PF Cross-Disorder Group of the Psychiatric Genomics C. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013;381:1371–1379. doi: 10.1016/S0140-6736(12)62129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Astous M, Cottin S, Roy M, Picard C, Cantin L. Bilateral stereotactic anterior capsulotomy for obsessive-compulsive disorder: long-term follow-up. Journal of neurology, neurosurgery, and psychiatry. 2013;84:1208–1213. doi: 10.1136/jnnp-2012-303826. [DOI] [PubMed] [Google Scholar]
- Drevets WC, Zarate CA, Jr, Furey ML. Antidepressant effects of the muscarinic cholinergic receptor antagonist scopolamine: a review. Biol Psychiatry. 2013;73:1156–1163. doi: 10.1016/j.biopsych.2012.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epi KC, Allen AS, Berkovic SF, Cossette P, Delanty N, Dlugos D, Eichler EE, Epstein MP, Glauser T, et al. Epilepsy Phenome/Genome P. De novo mutations in epileptic encephalopathies. Nature. 2013;501:217–221. doi: 10.1038/nature12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzsimmons J, Kubicki M, Shenton ME. Review of functional and anatomical brain connectivity findings in schizophrenia. Curr Opin Psychiatry. 2013;26:172–187. doi: 10.1097/YCO.0b013e32835d9e6a. [DOI] [PubMed] [Google Scholar]
- Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, Georgieva L, Rees E, Palta P, Ruderfer DM, et al. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014 doi: 10.1038/nature12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer SL, Jorgensen KW, Yetter EJ, Daurignac EC, Watson TD, Shanbhag H, Krystal JH, Mathalon DH. Differential brain response to alcohol cue distractors across stages of alcohol dependence. Biological psychology. 2013;92:282–291. doi: 10.1016/j.biopsycho.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaynes BN, Warden D, Trivedi MH, Wisniewski SR, Fava M, Rush AJ. What did STAR*D teach us? Results from a large-scale, practical, clinical trial for patients with depression. Psychiatr Serv. 2009;60:1439–1445. doi: 10.1176/ps.2009.60.11.1439. [DOI] [PubMed] [Google Scholar]
- Goldstein RZ, Craig AD, Bechara A, Garavan H, Childress AR, Paulus MP, Volkow ND. The neurocircuitry of impaired insight in drug addiction. Trends in cognitive sciences. 2009;13:372–380. doi: 10.1016/j.tics.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman WK, Foote KD, Greenberg BD, Ricciuti N, Bauer R, Ward H, Shapira NA, Wu SS, Hill CL, Rasmussen SA, et al. Deep brain stimulation for intractable obsessive compulsive disorder: pilot study using a blinded, staggered-onset design. Biol Psychiatry. 2010;67:535–542. doi: 10.1016/j.biopsych.2009.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulsuner S, Walsh T, Watts AC, Lee MK, Thornton AM, Casadei S, Rippey C, Shahin H, et al. Consortium on the Genetics of S Group PS. Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell. 2013;154:518–529. doi: 10.1016/j.cell.2013.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. 2010;11:682–696. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey PD, Mohs RC, Davidson M. Leukotomy and aging in chronic schizophrenia: a followup study 40 years after psychosurgery. Schizophr Bull. 1993;19:723–732. doi: 10.1093/schbul/19.4.723. [DOI] [PubMed] [Google Scholar]
- Hirschhorn JN, Altshuler D. Once and again-issues surrounding replication in genetic association studies. The Journal of clinical endocrinology and metabolism. 2002;87:4438–4441. doi: 10.1210/jc.2002-021329. [DOI] [PubMed] [Google Scholar]
- Hoffman RE, Cavus I. Slow transcranial magnetic stimulation, long-term depotentiation, and brain hyperexcitability disorders. American Journal of Psychiatry. 2002;159:1093–1102. doi: 10.1176/appi.ajp.159.7.1093. [DOI] [PubMed] [Google Scholar]
- Hoffman RE, Gueorguieva R, Hawkins KA, Varanko M, Boutros NN, Wu YT, Carroll K, Krystal JH. Temporoparietal transcranial magnetic stimulation for auditory hallucinations: safety, efficacy and moderators in a fifty patient sample. Biol Psychiatry. 2005;58:97–104. doi: 10.1016/j.biopsych.2005.03.041. [DOI] [PubMed] [Google Scholar]
- Hoffman RE, Hampson M, Wu K, Anderson AW, Gore JC, Buchanan RJ, Constable RT, Hawkins KA, Sahay N, Krystal JH. Probing the Pathophysiology of Auditory/Verbal Hallucinations by Combining Functional Magnetic Resonance Imaging and Transcranial Magnetic Stimulation. Cereb Cortex. 2007;17:2733–2743. doi: 10.1093/cercor/bhl183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzheimer PE, Kelley ME, Gross RE, Filkowski MM, Garlow SJ, Barrocas A, Wint D, Craighead MC, Kozarsky J, Chismar R, et al. Subcallosal cingulate deep brain stimulation for treatment-resistant unipolar and bipolar depression. Arch Gen Psychiatry. 2012;69:150–158. doi: 10.1001/archgenpsychiatry.2011.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman SE. Psychiatric drug development: diagnosing a crisis. Cerebrum : the Dana forum on brain science. 2013;2013:5. [PMC free article] [PubMed] [Google Scholar]
- Purcell SM, Wray NR, Stone JL, Visscher PM, O’Donovan MC, Sullivan PF, Sklar P International Schizophrenia C. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee YH, Narzisi G, Leotta A, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike H, Fukumoto K, Iijima M, Chaki S. Role of BDNF/TrkB signaling in antidepressant-like effects of a group II metabotropic glutamate receptor antagonist in animal models of depression. Behav Brain Res. 2013;238:48–52. doi: 10.1016/j.bbr.2012.10.023. [DOI] [PubMed] [Google Scholar]
- Krystal JH, D’Souza DC, Mathalon D, Perry E, Belger A, Hoffman R. NMDA receptor antagonist effects, cortical glutamatergic function, and schizophrenia: toward a paradigm shift in medication development. Psychopharmacology (Berl) 2003;169:215–233. doi: 10.1007/s00213-003-1582-z. [DOI] [PubMed] [Google Scholar]
- Krystal JH, Sanacora G, Duman RS. Rapid-acting glutamatergic antidepressants: the path to ketamine and beyond. Biol Psychiatry. 2013;73:1133–1141. doi: 10.1016/j.biopsych.2013.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystal JH, Tolin DF, Sanacora G, Castner SA, Williams GV, Aikins DE, Hoffman RE, D’Souza DC. Neuroplasticity as a target for the pharmacotherapy of anxiety disorders, mood disorders, and schizophrenia. Drug Discov Today. 2009;14:690–697. doi: 10.1016/j.drudis.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn TS. The structure of scientific revolutions. Chicago: University of Chicago Press; 1962. [Google Scholar]
- LaBar KS, Gatenby JC, Gore JC, LeDoux JE, Phelps EA. Human amygdala activation during conditioned fear acquisition and extinction: a mixed-trial fMRI study. Neuron. 1998;20:937–945. doi: 10.1016/s0896-6273(00)80475-4. [DOI] [PubMed] [Google Scholar]
- Laruelle M, Frankle WG, Narendran R, Kegeles LS, Abi-Dargham A. Mechanism of action of antipsychotic drugs: from dopamine D(2) receptor antagonism to glutamate NMDA facilitation. Clinical therapeutics. 2005;27(Suppl A):S16–24. doi: 10.1016/j.clinthera.2005.07.017. [DOI] [PubMed] [Google Scholar]
- Leopold DA, Maier A. Ongoing physiological processes in the cerebral cortex. Neuroimage. 2012;62:2190–2200. doi: 10.1016/j.neuroimage.2011.10.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, Li XY, Aghajanian G, Duman RS. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329:959–964. doi: 10.1126/science.1190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindholm JS, Autio H, Vesa L, Antila H, Lindemann L, Hoener MC, Skolnick P, Rantamaki T, Castren E. The antidepressant-like effects of glutamatergic drugs ketamine and AMPA receptor potentiator LY 451646 are preserved in bdnf(+)/(−) heterozygous null mice. Neuropharmacology. 2012;62:391–397. doi: 10.1016/j.neuropharm.2011.08.015. [DOI] [PubMed] [Google Scholar]
- Lisanby SH. Electroconvulsive therapy for depression. N Engl J Med. 2007;357:1939–1945. doi: 10.1056/NEJMct075234. [DOI] [PubMed] [Google Scholar]
- Logothetis NK. Intracortical recordings and fMRI: an attempt to study operational modules and networks simultaneously. Neuroimage. 2012;62:962–969. doi: 10.1016/j.neuroimage.2012.01.033. [DOI] [PubMed] [Google Scholar]
- Logothetis NK, Augath M, Murayama Y, Rauch A, Sultan F, Goense J, Oeltermann A, Merkle H. The effects of electrical microstimulation on cortical signal propagation. Nat Neurosci. 2010;13:1283–1291. doi: 10.1038/nn.2631. [DOI] [PubMed] [Google Scholar]
- Lord A, Horn D, Breakspear M, Walter M. Changes in community structure of resting state functional connectivity in unipolar depression. PLoS One. 2012;7:e41282. doi: 10.1371/journal.pone.0041282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan JL, Chaturvedi A, Choi KS, Holtzheimer PE, Gross RE, Mayberg HS, McIntyre CC. Tractography-activation models applied to subcallosal cingulate deep brain stimulation. Brain stimulation. 2013;6:737–739. doi: 10.1016/j.brs.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutterman T, Ganju V, Schacht L, Shaw R, Monihan K, Bottger R, Brunk M, Koch JR, Callahan N, Colton C, et al. DoHaH Services, editor. Sixteen-State Study on Mental Health Performance Measures. Rockville, MD: Substance Abuse and Mental Health Services Administration, Center for Mental Health Services; 2003. [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Malhotra D, Sebat J. CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell. 2012;148:1223–1241. doi: 10.1016/j.cell.2012.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone DA., Jr Use of deep brain stimulation in treatment-resistant depression. Cleveland Clinic journal of medicine. 2010;77(Suppl 3):S77–80. doi: 10.3949/ccjm.77.s3.14. [DOI] [PubMed] [Google Scholar]
- Mayberg HS, Liotti M, Brannan SK, McGinnis S, Mahurin RK, Jerabek PA, Silva JA, Tekell JL, Martin CC, Lancaster JL, et al. Reciprocal limbic-cortical function and negative mood: converging PET findings in depression and normal sadness. American Journal of Psychiatry. 1999;156:675–682. doi: 10.1176/ajp.156.5.675. [DOI] [PubMed] [Google Scholar]
- Mayberg HS, Lozano AM, Voon V, McNeely HE, Seminowicz D, Hamani C, Schwaib JM, Kennedy SH. Deep brain stimulation for treatment-resistant depression. Neuron. 2005;45:651–660. doi: 10.1016/j.neuron.2005.02.014. [DOI] [PubMed] [Google Scholar]
- Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry. 2009;65:732–741. doi: 10.1016/j.biopsych.2008.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moniz E. Am J Psychiatry. 1937;93:1379–1385. [Google Scholar]
- Mwangi B, Ebmeier KP, Matthews K, Steele JD. Multi-centre diagnostic classification of individual structural neuroimaging scans from patients with major depressive disorder. Brain. 2012;135:1508–1521. doi: 10.1093/brain/aws084. [DOI] [PubMed] [Google Scholar]
- Norberg MM, Krystal JH, Tolin DF. A Meta-Analysis of D-Cycloserine and the Facilitation of Fear Extinction and Exposure Therapy. Biol Psychiatry. 2008 doi: 10.1016/j.biopsych.2008.01.012. [DOI] [PubMed] [Google Scholar]
- O’Doherty J, Dayan P, Schultz J, Deichmann R, Friston K, Dolan RJ. Dissociable roles of ventral and dorsal striatum in instrumental conditioning. Science. 2004;304:452–454. doi: 10.1126/science.1094285. [DOI] [PubMed] [Google Scholar]
- O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olabi B, Ellison-Wright I, McIntosh AM, Wood SJ, Bullmore E, Lawrie SM. Are there progressive brain changes in schizophrenia? A meta-analysis of structural magnetic resonance imaging studies. Biol Psychiatry. 2011;70:88–96. doi: 10.1016/j.biopsych.2011.01.032. [DOI] [PubMed] [Google Scholar]
- Orru G, Pettersson-Yeo W, Marquand AF, Sartori G, Mechelli A. Using Support Vector Machine to identify imaging biomarkers of neurological and psychiatric disease: a critical review. Neuroscience and biobehavioral reviews. 2012;36:1140–1152. doi: 10.1016/j.neubiorev.2012.01.004. [DOI] [PubMed] [Google Scholar]
- Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V, Horvath S, Geschwind DH. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell. 2013;155:1008–1021. doi: 10.1016/j.cell.2013.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulus MP, Feinstein JS, Castillo G, Simmons AN, Stein MB. Dose-dependent decrease of activation in bilateral amygdala and insula by lorazepam during emotion processing. Arch Gen Psychiatry. 2005;62:282–288. doi: 10.1001/archpsyc.62.3.282. [DOI] [PubMed] [Google Scholar]
- Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potenza MN, Hong KI, Lacadie CM, Fulbright RK, Tuit KL, Sinha R. Neural correlates of stress-induced and cue-induced drug craving: influences of sex and cocaine dependence. Am J Psychiatry. 2012;169:406–414. doi: 10.1176/appi.ajp.2011.11020289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prevost C, Liljeholm M, Tyszka JM, O’Doherty JP. Neural correlates of specific and general Pavlovian-to-Instrumental Transfer within human amygdalar subregions: a high-resolution fMRI study. J Neurosci. 2012;32:8383–8390. doi: 10.1523/JNEUROSCI.6237-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prisciandaro JJ, Myrick H, Henderson S, McRae-Clark AL, Santa Ana EJ, Saladin ME, Brady KT. Impact of DCS-facilitated cue exposure therapy on brain activation to cocaine cues in cocaine dependence. Drug Alcohol Depend. 2013;132:195–201. doi: 10.1016/j.drugalcdep.2013.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, O’Dushlaine C, Chambert K, Bergen SE, Kahler A, et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature. 2014 doi: 10.1038/nature12975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ressler KJ, Rothbaum BO, Tannenbaum L, Anderson P, Graap K, Zimand E, Hodges L, Davis M. Cognitive enhancers as adjuncts to psychotherapy: use of D-cycloserine in phobic individuals to facilitate extinction of fear. Arch Gen Psychiatry. 2004;61:1136–1144. doi: 10.1001/archpsyc.61.11.1136. [DOI] [PubMed] [Google Scholar]
- Robins E, Guze SB. Establishment of diagnostic validity in psychiatric illness: its application to schizophrenia. The American journal of psychiatry. 1970;126:983–987. doi: 10.1176/ajp.126.7.983. [DOI] [PubMed] [Google Scholar]
- Rowland LM, Bustillo JR, Mullins PG, Jung RE, Lenroot R, Landgraf E, Barrow R, Yeo R, Lauriello J, Brooks WM. Effects of ketamine on anterior cingulate glutamate metabolism in healthy humans: a 4-T proton MRS study. Am J Psychiatry. 2005;162:394–396. doi: 10.1176/appi.ajp.162.2.394. [DOI] [PubMed] [Google Scholar]
- Sanacora G, Banasr M. From pathophysiology to novel antidepressant drugs: glial contributions to the pathology and treatment of mood disorders. Biol Psychiatry. 2013;73:1172–1179. doi: 10.1016/j.biopsych.2013.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanacora G, Mason GF, Krystal JH. Impairment of GABAergic transmission in depression: new insights from neuroimaging studies. Crit Rev Neurobiol. 2000;14:23–45. doi: 10.1615/critrevneurobiol.v14.i1.20. [DOI] [PubMed] [Google Scholar]
- Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadt EE, Bjorkegren JL. NEW: network-enabled wisdom in biology, medicine, and health care. Sci Transl Med. 2012;4:115rv111. doi: 10.1126/scitranslmed.3002132. [DOI] [PubMed] [Google Scholar]
- Schiller D, Phelps EA. Does reconsolidation occur in humans? Front Behav Neurosci. 2011;5:24. doi: 10.3389/fnbeh.2011.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnack HG, Nieuwenhuis M, van Haren NE, Abramovic L, Scheewe TW, Brouwer RM, Hulshoff Pol HE, Kahn RS. Can structural MRI aid in clinical classification? A machine learning study in two independent samples of patients with schizophrenia, bipolar disorder and healthy subjects. Neuroimage. 2014;84:299–306. doi: 10.1016/j.neuroimage.2013.08.053. [DOI] [PubMed] [Google Scholar]
- Silbersweig DA, Stern E, Frith C, Cahill C, Holmes A, Grootoonk S, Seaward J, McKenna P, Chua SE, Schnorr L, et al. A functional neuroanatomy of hallucinations in schizophrenia. Nature. 1995;378:176–179. doi: 10.1038/378176a0. [DOI] [PubMed] [Google Scholar]
- Sjoerds Z, de Wit S, van den Brink W, Robbins TW, Beekman AT, Penninx BW, Veltman DJ. Behavioral and neuroimaging evidence for overreliance on habit learning in alcohol-dependent patients. Translational psychiatry. 2013;3:e337. doi: 10.1038/tp.2013.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skolnick P, Popik P, Trullas R. Glutamate-based antidepressants: 20 years on. Trends Pharmacol Sci. 2009;30:563–569. doi: 10.1016/j.tips.2009.09.002. [DOI] [PubMed] [Google Scholar]
- Snyder SH. Catecholamines in the brain as mediators of amphetamine psychosis. Arch Gen Psychiatry. 1972;27:169–179. doi: 10.1001/archpsyc.1972.01750260021004. [DOI] [PubMed] [Google Scholar]
- Spitzer RL, Endicott J, Robins E. Research diagnostic criteria: rationale and reliability. Archives of general psychiatry. 1978;35:773–782. doi: 10.1001/archpsyc.1978.01770300115013. [DOI] [PubMed] [Google Scholar]
- State MW, Levitt P. The conundrums of understanding genetic risks for autism spectrum disorders. Nature neuroscience. 2011;14:1499–1506. doi: 10.1038/nn.2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- State MW, Sestan N. Neuroscience. The emerging biology of autism spectrum disorders. Science. 2012;337:1301–1303. doi: 10.1126/science.1224989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson H, Meyer-Lindenberg A, Steinberg S, Magnusdottir B, Morgen K, Arnarsdottir S, Bjornsdottir G, Walters GB, Jonsdottir G, Doyle OM, et al. CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature. 2013 doi: 10.1038/nature12818. [DOI] [PubMed] [Google Scholar]
- Stone JM, Dietrich C, Edden R, Mehta MA, De Simoni S, Reed LJ, Krystal JH, Nutt D, Barker GJ. Ketamine effects on brain GABA and glutamate levels with 1H-MRS: relationship to ketamine-induced psychopathology. Mol Psychiatry. 2012;17:664–665. doi: 10.1038/mp.2011.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szasz T. The Myth of Mental Illness. American Psychologist. 1960;15:113–118. [Google Scholar]
- Trullas R, Skolnick P. Functional antagonists at the NMDA receptor complex exhibit antidepressant actions. European Journal of Pharmacology. 1990;185:1–10. doi: 10.1016/0014-2999(90)90204-j. [DOI] [PubMed] [Google Scholar]
- Uhlhaas PJ. Dysconnectivity, large-scale networks and neuronal dynamics in schizophrenia. Curr Opin Neurobiol. 2013;23:283–290. doi: 10.1016/j.conb.2012.11.004. [DOI] [PubMed] [Google Scholar]
- Veltman JA, Brunner HG. De novo mutations in human genetic disease. Nature reviews Genetics. 2012;13:565–575. doi: 10.1038/nrg3241. [DOI] [PubMed] [Google Scholar]
- Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ, Geschwind DH. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474:380–384. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voleti B, Navarria A, Liu RJ, Banasr M, Li N, Terwilliger R, Sanacora G, Eid T, Aghajanian G, Duman RS. Scopolamine rapidly increases mammalian target of rapamycin complex 1 signaling, synaptogenesis, and antidepressant behavioral responses. Biol Psychiatry. 2013;74:742–749. doi: 10.1016/j.biopsych.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos T, Flaxman AD, Naghavi M, Lozano R, Michaud C, Ezzati M, Shibuya K, Salomon JA, Abdalla S, Aboyans V, et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2163–2196. doi: 10.1016/S0140-6736(12)61729-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SC, Deisseroth K. Optogenetics. Proc Natl Acad Sci U S A. 2013;110:16287. doi: 10.1073/pnas.1317033110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willsey AJ, Sanders SJ, Li M, Dong S, Tebbenkamp AT, Muhle RA, Reilly SK, Lin L, Fertuzinhos S, Miller JA, et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell. 2013;155:997–1007. doi: 10.1016/j.cell.2013.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimizu T, Shimazaki T, Ito A, Chaki S. An mGluR2/3 antagonist, MGS0039, exerts antidepressant and anxiolytic effects in behavioral models in rats. Psychopharmacology (Berl) 2006;186:587–593. doi: 10.1007/s00213-006-0390-7. [DOI] [PubMed] [Google Scholar]
- Yu Q, Allen EA, Sui J, Arbabshirani MR, Pearlson G, Calhoun VD. Brain connectivity networks in schizophrenia underlying resting state functional magnetic resonance imaging. Curr Top Med Chem. 2012;12:2415–2425. doi: 10.2174/156802612805289890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuksel C, Ongur D. Magnetic resonance spectroscopy studies of glutamate-related abnormalities in mood disorders. Biol Psychiatry. 2010;68:785–794. doi: 10.1016/j.biopsych.2010.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]