

Summary
Purpose
The mitogen-activated extracellular signal-related kinase kinase (MEK) is a member of the RAS/RAF/MEK/ERK signalling cascade, which is commonly activated in melanoma. Direct inhibition of MEK inhibits ERK signalling.
Methods
We conducted a multicentre, first-in-human, three-part study (dose escalation, cohort expansion, and pharmacodynamic evaluation) to evaluate the oral small-molecule MEK inhibitor trametininb (GSK1120212) in advanced cancer. Intermittent and continuous dosing regimens were evaluated. Safety and efficacy data in patients with melanoma are presented here, with exploratory analyses of available tumour tissues performed on an Illumina genotyping platform. This completed study is registered with ClinicalTrials.gov, number NCT00687622.
Findings
Ninety-seven melanoma patients, including 81 with cutaneous or unknown primary melanoma (36 BRAF-mutant, 39 BRAF wild-type, six BRAF status unknown) and 16 uveal melanoma patients were enrolled. The most common treatment-related adverse events were rash/dermatitis acneiform (80 out of 97; 82%) and diarrhoea (n=44; 45%), most of which were grade 2 or lower. No cutaneous squamous cell carcinomas were observed. Among the 36 BRAF-mutant patients, 30 were BRAF-inhibitor naïve. Among these 30 patients, 2 complete responses (CRs) and 10 partial responses (PRs) were observed (unconfirmed response rate=40%) including 2 confirmed CRs and 8 confirmed PRs (confirmed response rate=33%); the median progression-free survival was 5·7 months (95% CI, 4·0–7·4). Among the 6 BRAF-mutant patients who received prior BRAF inhibitor therapy, 1 unconfirmed PR was observed. Among 39 patients with BRAF wild-type melanoma, 4 PRs (all confirmed) were observed (confirmed response rate=10%).
Conclusions
To our knowledge, this is the first demonstration of substantial clinical activity by a MEK inhibitor in melanoma. These data suggest that MEK is a valid therapeutic target.
Introduction
Metastatic melanoma is an aggressive disease, with a median survival of less than 1 year1. Few effective systemic therapies are available. Most approved treatments, such as dacarbazine, high-dose interleukin-2, and ipilimumab have response rates (RR) of 6–20%1,2 and are associated with severe toxicities including capillary leak syndrome1 and immune-mediated issues.2
The mitogen-activated extracellular signal-related kinase kinase (MEK) is a member of the RAS/RAF/MEK/ERK (MAPK) signalling cascade, an important pathway in cell proliferation. Constitutive activation of MEK through genetic mutations results in oncogenic transformation of normal cells.3 Activating mutations within the MAPK pathway are common in melanoma. Mutations in neuroblastoma RAS viral oncogene homolog (NRAS) are observed in 10–20% of cutaneous melanomas.4,5 Serine/threonine-protein kinase B-Raf (BRAF) mutations are more common, occurring in 40–60% of cutaneous melanomas.5,6 Over 80% of BRAF mutations have substitution of valine with glutamic acid at amino acid residue 600 (V600E), while substitution with lysine (V600K) occurs in 3–20% of cases.5,6 In uveal melanoma, BRAF mutations are rare, but MAPK activating mutations in guanine nucleotide-1 binding protein q polypeptide (GNAQ) or guanine nucleotide-binding protein alpha 11 (GNA11) are common, detected in approximately 80% of cases.7,8
Recently, potent and selective BRAF inhibitors have been developed, including dabrafenib (GSK2118436)9 and vemurafenib (PLX4032, RG7204),10 with the latter receiving approval by the United States Food and Drug Administration in 2011.10 However, even among patients with BRAF-mutant melanoma, the majority will progress, and some patients have primary resistance to single-agent BRAF inhibitor therapy.
Trametinib is a reversible, selective, allosteric inhibitor of MEK1/MEK2 activation and kinase activity, with a half-maximal inhibitory concentration (IC50) of 0·7–14·9 nM for MEK1/MEK2.11 Trametinib inhibited proliferation of BRAFV600E melanoma cell lines at concentrations of 1·0–2·5 nM.11 In xenografted tumour models, trametinib demonstrated sustained suppression of pERK and tumour growth inhibition.11
We report the results of melanoma patients treated in the Phase I, first-in-human study of trametinib for patients with advanced malignancies. The main objectives included evaluation of maximum tolerated dose, safety, and antitumour activity; translational objectives included exploration of the association of tumour genetic profiles with clinical endpoints. The companion manuscript by Infante et al. reports the study design, pharmacokinetics, and pharmacodynamic results, as well as efficacy data in non-melanoma tumours of the parent study.
Methods
Study Design and Dosing
This study (NCT00687622) was sponsored by GlaxoSmithKline, and patients enrolled at ten centres in the United States. The protocol was approved by institutional review boards, and all enrolled patients provided written informed consent. This analysis of melanoma patients was part of a larger, three-part study that enrolled 206 patients with solid tumours,12 97 of whom had melanoma (see Supplementary Figure 1 and accompanying paper from Infante et al.). Part 1 identified the maximum tolerated dose of trametinib using safety, pharmacokinetic, and pharmacodynamic (PD) assessments. In Part 2, safety and efficacy of the recommended Phase II dose (RP2D) were assessed in patients with selected tumor types. Part 3 characterized the biologically active dose range of trametinib. Patients with melanoma were enrolled in all three parts of the study. Trametinib doses ranged from 0·125 mg to 4·0 mg, administered orally once daily (QD). In some instances, loading doses (Day 1 or Days 1 and 2) and run-in doses (Days 1–14) were used (Supplementary Table 1). Of the 97 melanoma patients, 93 were treated at or above the RP2D of 2·0 mg QD.12
The protocol was approved by institutional review boards, and all participants provided written informed consent.
Patients
Eligibility criteria included age ≥18 years, histologically or cytologically confirmed diagnosis of solid tumour or lymphoma, Eastern Cooperative Oncology Group (ECOG) performance status ≤1, and adequate haematological, hepatic, renal, and cardiac function. Patients with a history of retinal vein occlusion (RVO), central serous retinopathy (CSR), risk factors for RVO or CSR, or glaucoma diagnosed within 1 month prior to study entry were excluded. Patients with brain metastases were required to have received prior local treatment; patients treated with gamma knife or whole-brain radiation required a 2- or 4-week washout period, respectively. Patients previously treated with MEK inhibitors were excluded.
Study Assessments
Adverse events (AEs) were evaluated according to the National Cancer Institute's Common Terminology Criteria for Adverse Events (CTCAE) version 3.0.13 Patients receiving at least one dose of study drug were included in both safety and efficacy analyses. Disease assessments, performed at screening and every 8 weeks, were evaluated by investigators per Response Evaluation Criteria in Solid Tumors (RECIST) version 1.0.14
Tumour Genotyping
Tumour samples for genotyping were requested for all patients and were mandatory for a subset of patients, unless BRAF mutational status was provided from local assays. BRAF status was not an eligibility criterion. Submitted tumour samples were analysed at Response Genetics, Inc. (RGI; Los Angeles, CA, USA) using allele-specific PCR to identify BRAFV600E and BRAFV600K mutations. BRAF mutation-negative results are referred to as BRAF wild-type. Patients with at least one positive BRAF mutation test were considered to be BRAF-mutant. For patients whose tumour samples were not submitted to RGI, mutation status of BRAF, NRAS, GNAQ and GNA11 was reported based on local assays, if available. DNA extracted from pre-treatment tumour tissue at RGI was whole-genome amplified and then analysed on the Illumina genotyping platform (Expression Analysis, Durham, NC, USA),15 a high-throughput single nucleotide polymorphism genotyping assay designed to survey common somatic mutations and copy number alterations. BRAF and NRAS loci were further evaluated using traditional Sanger sequencing methods (Applied Biosystems 3730 DNA Analyzer and Big-Dye Chemistry; Applied Biosystems, Foster City, CA). Genetic results from local laboratories and RGI are referred to as clinical, and results from the Illumina platform as exploratory.
Statistical Analysis
As a part of a Phase I, dose-escalation/dose-expansion trial, no formal hypotheses were tested; analyses were descriptive and exploratory. Sample-size selection was based on feasibility. Exact 95% confidence intervals (CIs) for overall RRs in relevant subgroups were calculated. Progression-free survival (PFS) was defined as time from date of first dose to date of disease progression according to clinical or radiological assessment, or death due to any cause, whichever occurred earlier. PFS based on investigator-assessed data was derived and summarised using Kaplan–Meier estimates. SAS® (version 9.1) was used for all statistical analysis. This study is registered with ClinicalTrials.gov, number NCT00687622.
Role of the funding source
This study was funded, initiated, administered, and sponsored by GlaxoSmithKline, which also provided data analysis. The study was designed by the sponsor in collaboration with JRI, HAB, KF and WAM. All authors had access to all study data, contributed to data interpretation, and were responsible for the decision to publish. The manuscript was written by GSF with contributions by all authors, including employees of the sponsor. Editorial support was provided by MediTechMedia and was funded by the sponsor.
Results
Patients
Between 31 July 2008 and 05 October 2010, 206 patients were enrolled. Detailed results of the pharmacokinetic analysis, pharmacodynamic analysis, and adverse events of the parent study are described in the concurrently published manuscript by Infante et al. Ninety-seven patients with advanced melanoma were enrolled across all parts of the study (Table 1). Poor prognosis features were common, with most patients having M1c disease. Among 36 BRAF-mutant patients, the majority had a BRAFV600E mutation and prior brain metastases, and six had prior treatment with a BRAF inhibitor. Mutation status of BRAF, NRAS, GNAQ, and GNA11 was not available for all patients due to insufficient or unavailable tissue (Table 1).
Table 1. Baseline Characteristics of Melanoma Patients (n=97).
| BRAF mutant | BRAF wild-type | BRAF mutation status unknown1 | Uveal primary | |
|---|---|---|---|---|
| Number of patients | 36 | 39 | 6 | 16 |
| Age, years | ||||
| Median | 55·0 | 62 0 | 70 0 | 53 0 |
| Range | 19–75 | 27–92 | 54–77 | 36–80 |
| Sex, n (%) | ||||
| Male | 19 (53) | 27 (69) | 4 (67) | 9 (56) |
| Female | 17 (47) | 12 (31) | 2(33) | 7 (44) |
| ECOG PS, n (%)2 | ||||
| 0 | 19 (53) | 17 (44) | 3 (50) | 10 (63) |
| 1 | 17 (47) | 21 (54) | 3 (50) | 6 (38) |
| Prior lines of therapy, n (%) | ||||
| 0 | 3 (8) | 4 (10) | 1 (17) | 1 (6) |
| 1–2 | 20 (56) | 14 (36) | 4 (67) | 7 (44) |
| ≥3 | 13 (36) | 21 (54) | 1 (17) | 8 (50) |
| M status, n (%) | ||||
| M1a | 3 (8) | 5 (13) | 0 | 0 |
| M1b | 4 (11) | 7 (18) | 1 (17) | 0 |
| M1c | 26 (72) | 21 (54) | 4 (67) | 16 (100) |
| Unknown | 3 (8) | 6 (15) | 1 (17) | 0 |
| LDH > ULN, n (%) | 12 (33) | 16 (41) | 3 (50) | 10 (63) |
| Prior brain metastases, n (%) | 19 (53) | 8 (21) | 0 | 1 (6) |
| Prior BRAF therapy, n (%) | 6 (17) | NA | NA | NA |
| Mutational status, n (%) | ||||
| BRAFV600E | 19 (53) | |||
| BRAFV600K | 6 (17) | |||
| BRAF mutant, other3 | 2 (2) | |||
| BRAF mutant, not otherwise specified | 11 (31) | |||
| NRAS mutant3 | 7 | |||
| GNAQ or GNA11 mutant | 2 GNAQ 2 GNA11 | |||
ECOG PS = Eastern Cooperative Oncology Group Performance Status; LDH = lactate dehydrogenase; M = metastasis; NA = not applicable; ULN = upper limit of normal.
Mutation status unknown due to unevaluable or unavailable tissue.
One BRAF wild-type patient had an ECOG of 1 at screening and 2 on Day 1.
NRAS status was available from clinical data for 11 patients.
Treatment-related Adverse Events
The most common treatment-related AEs (≥10%) are shown (Table 2) and are concordant with those observed in the parent study. Twenty-one percent (20 out of 97) of patients had treatment-related toxicity greater than grade 2; 19% (18/97) was grade 3. Two grade 4 events, rash (1/97; 1%) and thrombocytopenia (1/97; 1%), were observed. Among patients administered the RP2D of 2 mg QD, grade 3 events of rash (2/97; 2%) and fatigue (1/97; 1%) were reported. There were no grade 4 treatment-related AEs among these patients.
Table 2. Most Common (≥10%) Drug-related Adverse Events Among All Melanoma Patients1.
| 2 mg QD (n=26) | All doses (n=97) | ||||
|---|---|---|---|---|---|
| G1 | G2 | G3 | Total | Total | |
| Skin-related toxicities2 Rash/dermatitis acneiform | 10 (38) 10 (38) | 12 (46) 10 (38) | 2 (8) 2 (8) | 24 (92) 22 (85) | 85 (88) 80 (82) |
| Diarrhoea | 9 (35) | 2 (8) | 0 | 11(42) | 44 (45) |
| Peripheral oedema | 8 (31) | 1 (4) | 0 | 9 (35) | 34 (35) |
| Fatigue | 5 (19) | 3 (12) | 1 (4) | 9 (35) | 30 (31) |
| Nausea | 3 (12) | 0 | 0 | 3 (12) | 20 (21) |
| Dry skin/chapped skin/skin fissures | 6 (23) | 2 (8) | 0 | 8 (31) | 22 (23) |
| Pruritus | 4 (15) | 0 | 0 | 4 (15) | 13 (13) |
| Vomiting | 2 (8) | 0 | 0 | 2 (8) | 12 (12) |
| Mucosal inflammation | 1 (4) | 0 | 0 | 1 (4) | 10 (10) |
All but four patients received ≥2 mg QD. These four patients are included only in the total patient population analysis. See Methods for further details.
In most patients, the observed rash was acneiform and located on the face, scalp, chest, and back. The multiple terms include: acne, dermatitis acneiform, dermatitis psoriasiform, erythema, genital rash, palmar-plantar erythrodysesthesia, rash, rash erythematous, rash follicular, rash generalised, rash macular, rash pruritic, and rash pustular.
The most common AE was rash/dermatitis acneiform (80 out of 97 patients; 82%), which in most patients was located on the face, scalp, chest, and back. No hyperkeratotic lesions or cutaneous squamous cell carcinomas were observed. Treatment-related ocular AEs were observed in ten patients (10%); all events were grade 1. The most common ocular events were visual impairment (4/97; 4%), dry eye (2/97; 2%), and blurred vision (2/97; 2%). All ocular AEs, except for dry eye, occurred at doses >2 mg QD. No RVO or CSR was reported. Treatment-related left ventricular dysfunction/ejection fraction decrease was observed in seven patients (7%); all were grade 1 (3/97; 3%) or grade 2 (4/97; 4%). Of these, one grade 1 event and two grade 2 events occurred at 2 mg QD.
Treatment-related serious AEs were observed in two patients (2%): grade 3 fatigue (1/97; 1%) and grade 3 pulmonary hypertension (1/97; 1%). Both events occurred at 2 mg QD.
AEs requiring dose reductions occurred in 22 (23%) of the 97 patients, and in three (12%) of 26 patients administered 2 mg QD. The most common cause of dose reduction was rash, which was observed in 11 patients (11%). Only one patient had a treatment-related AE (pulmonary hypertension) requiring drug withdrawal. No deaths resulted from treatment-related AEs.
Efficacy
Two complete responses (CRs) and ten partial responses (PRs) were observed among 30 BRAF-mutant, BRAF inhibitor-naïve melanoma patients (RR=40%) (confirmed RR=33%, two confirmed CRs and eight confirmed PRs; Figure 1A, Table 3). The median PFS among these patients was 5·7 months (95% CI, 4·0–7·4), and median duration of response was 5·6 months (95% CI, 5·5–11·1). Four patients (13%) received trametinib for >1 year, and two patients continued treatment for >8 weeks after disease progression per RECIST because of continued overall benefit, as permitted by the protocol (Figure 1B). Seven of the 12 responses were observed at first disease assessment, and further tumour reduction continued beyond first disease assessment. Tumour reduction was observed in 19 (63%) of the 30 patients with BRAF-mutant, BRAF inhibitor-naive melanoma. Among 16 patients dosed at 2 mg QD (including loading and run-in doses), two CRs and five PRs were observed (Figure 1A), of which both of the CRs and three PRs were confirmed. Median PFS was 7·4 months (95% CI, 1·9–9·2) among BRAF-mutant, BRAF inhibitor-naïve patients without brain metastases. Among six patients with BRAFV600K mutations, four responses (67%) were observed.
Figure 1. Efficacy data for BRAF-mutant, BRAF wild-type, and uveal melanoma patients.




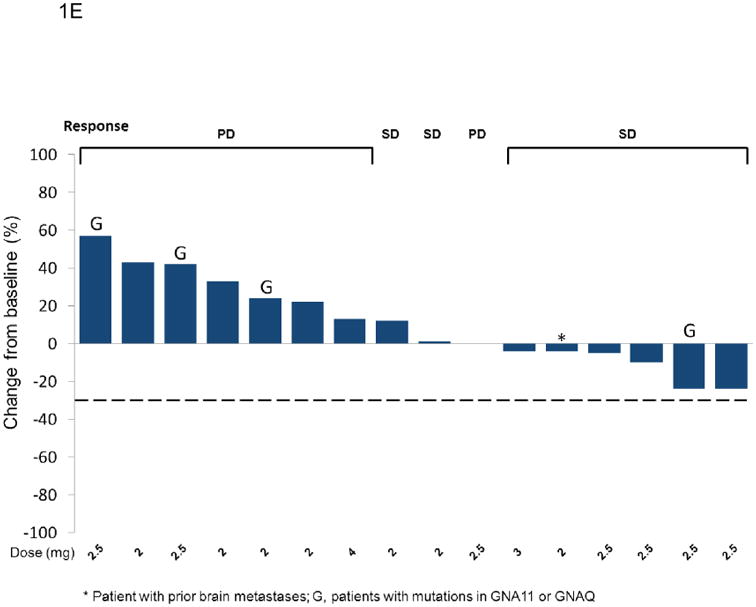

Best unconfirmed response for patients with BRAF-mutant, BRAF inhibitor-naïve melanoma (Panel A), BRAF wild-type melanoma (Panel C) and uveal melanoma (Panel E). Positive values indicate tumour growth, negative values indicate tumour reduction, and the dashed line represents the threshold for partial response by RECIST (version 1.0). Loading doses of 6 mg on Day 1, and 6 or 10 mg on Days 1 and 2 followed by 2 mg or 3 mg QD are designated 6/2, 6/6/2, and 10/10/3, respectively. Run-in doses of 0·5 and 1 mg followed by 2 mg or 2·5 mg QD are designated 0·5/2, 1/2, and 1/2·5. All other doses are 2, 2·5 or 3 mg QD. In some instances, patients had less than 20% growth in target lesions but had progressive disease for other reasons (e.g. appearance of new lesions or progression of non-target lesions). Three patients are not included in Panel A due to clinical PD (n=1), SD with no target lesions (n=1), and withdrawal prior to first disease assessment (n=1). Four patients are not included in Panel C due to clinical progression (n=3) and non-radiological assessment (n=1). In Panel E, G indicates either a GNAQ or GNA11 mutation. Panels B (BRAF-mutant melanoma), D (BRAF wild-type melanoma), and F (uveal melanoma) show duration of treatment as measured by time from first dose to time of last dose. Best unconfirmed response and clinical mutational status are indicated. The arrowheads indicate the patient was still receiving treatment at the time of data cut-off. Circles indicate time of disease progression or death, whichever came first. Black triangles indicate patients who continued on treatment for longer than 8 weeks after progression. In panel B, E indicates patients with V600E mutation and K indicates patients with a V600K mutation. Absence of an E or K indicates that there was a BRAF mutation, but the mutation was not specified.
Table 3. Tumor Response and Progression-free Survival in Melanoma Subpopulations (n=97)1.
| Melanoma patients | N | Unconfirmed response | Overall unconfirmed response rate (PR+CR) (95% CI) | Median PFS (95% CI) |
|---|---|---|---|---|
| BRAF mutant (BRAFi naïve) | 30 | 2CR (7%), 10PR (33%), 11SD (37%) | 40 (22·7, 59·4) | 5·7 (4·0, 7·4) |
| Without prior brain metastases | 14 | 1CR (7%), 4PR (29%), 4SD (29%) | 36 (12·8, 64·9) | 7.4 (1·9, 9·2) |
| With prior brain metastases | 16 | 1CR (6%), 6PR (38%), 7SD (44%) | 44 (19·8, 70·1) | 5·5 (4·0, 7·4) |
| BRAF mutant previously treated with BRAFi2 | 6 | 1PR (17%), 4SD (67%) | NA | NA |
| BRAF wild-type melanoma | 39 | 4PR (10%), 15SD (38%) | 10 (2·9, 24·2) | 2.0 (1·7, 3·7) |
| Without prior brain metastases | 31 | 4PR (13%), 13SD (42%) | 13 (2·6, 29·8) | 3.3 (1·8, 5·8) |
| With prior brain metastases | 8 | 2SD (25%) | 0 | NA |
| NRAS mutant | 7 | 2SD (29%) | 0 | NA |
| Unknown mutational status | 6 | 3SD (50%) | 0 | NA |
| Uveal | 16 | 8SD (50%) | 0 | 1·8 (1·8, 3·7) |
BRAFi = BRAF inhibitor; CR = complete response; PR = partial response; SD = stable disease. NA = not applicable, summary statistics were not provided if N <12.
Unconfirmed response based on investigator analysis. N = all treated patients.
Three patients had prior brain metastases, three did not.
Among six BRAF-mutant patients previously treated with a selective BRAF inhibitor (three with dabrafenib and three with vemurafenib), an unconfirmed PR was observed in one patient (17%) who received study treatment for 24 weeks (Table 3). Stable disease (SD) was observed in four patients (67%), including one patient who received study treatment for 37 weeks.
Among the 39 BRAF wild-type melanoma patients, four confirmed PRs (10%) were observed (Figure 1C, Table 3). In addition, ten patients (26%) remained on study for >24 weeks, and six patients (15%) were on study for >1 year (Figure 1D). The best response observed in seven patients with an NRAS mutation was SD (n=2), one of whom received treatment for 48 weeks. Of the six patients with unknown BRAF mutational status, three achieved SD, one of whom received study treatment for 22 weeks.
Among the 16 patients with uveal melanoma, two patients (13%) achieved a 24% tumour reduction, one of whom was GNAQ mutation-positive and received study treatment for >16 weeks (Figures 1E and 1F; Table 3). Stable disease for ≥16 weeks was observed in four patients (25%), including two who received treatment for >40 weeks).
Exploratory Genetic Analysis Using Illumina Genotyping
The Illumina platform evaluated mutational status and copy number status of 78 different genes commonly implicated in tumourogenesis.16 DNA from 19 patients with BRAF-mutant melanoma and 23 patients with BRAF wild-type melanoma, as determined by a local laboratory and/or RGI, were analysed using the Illumina platform (Figures 2A and 2B). Overall, 33 genes were found to have mutations, and BRAF mutation results were generally concordant (18 of 19; 95% agreement) between Illumina genotyping and RGI. Among the 19 samples reported as BRAF-mutant melanomas according to local laboratories, both RGI and Illumina genotyping did not identify a BRAF mutation in three (16%). Of these three samples, Illumina genotyping identified mutations in NRAS (n=2) and KRAS (n=1). In general, tumour reduction appeared greatest in BRAF-mutant patients with no or few additional genetic aberrations.
Figure 2. Efficacy and exploratory genetics in melanoma subgroups.



Exploratory genetics and waterfall plots depicting best unconfirmed response for patients with BRAF-mutant melanoma (Panel A), BRAF wild-type melanoma (Panel B), and BRAF wild-type melanoma with NRAS status (Panel C). Patients in each panel are grouped based on results from local laboratories and/or RGI. Genes commonly implicated in tumourogenesis that were evaluated on the Illumina platform are listed beneath the waterfall plots (Panels A and B); dark squares indicate a mutation determined by Illumina genotyping. Patients who received prior BRAF-inhibitor therapy are highlighted (Panel A) and those who had non-BRAFV600 mutations identified by Illumina genotyping are marked with an asterisk (Panel C). BRAFV600WT/NRASmut patients are represented by hatched bars, and BRAFV600WT/NRASWT patients are represented by solid bars (Panel C). Illumina genotyping identified six NRAS mutations among 23 patients evaluated; two of these six mutations were known from clinical data and four were not. Among the 31 patients who had either clinical or Illumina data for NRAS, 11 (35%) were NRAS-mutant. Scan data were unavailable for three patients (NRAS wild-type, n=1; and NRAS-mutant, n=2) due to progressive disease prior to the first disease assessment.
Two non-BRAFV600 mutations (L597V, intermediate activity; G469A, low activity) were identified by Illumina genotyping in the 23 BRAF wild-type melanoma samples. Only the G469A mutation was verified by DNA sequence analysis. The patient with L597V had a confirmed PR with 60% tumour reduction and received study treatment for >2 years. In general, no correlation was suggested between genomic aberration frequency and tumour reduction in this BRAF wild-type subset (Supplementary Table I).
Exploratory and clinical NRAS data were available for 31 of the 39 BRAF wild-type patients and were combined for analysis (Figure 2C). Patients with concurrent BRAFV600WT/NRASWT (n=20) had a trend of higher RR (20%) than BRAFV600WT/NRAS-mutant patients (n=11; RR=0%; p=0·27), as well as a trend of higher percentage of patients on study at Week 24 or at 1 year (40% vs. 18% [p=0·26] and 30% vs. 0% [p=0·07], respectively).
Combined exploratory and clinical genetics for patients with uveal melanoma identified GNAQ and GNA11 mutations in six patients, in which three SD and three PD were the best responses. One patient with a GNA11 mutation identified by Illumina genotyping stayed on study treatment for >40 weeks.
Discussion
The 2 mg QD dose of trametinib, chosen for further evaluation in Phase II and III trials,12 was well tolerated and resulted in acceptable and manageable toxicities common to MEK inhibitors. AEs in melanoma patients mirrored those of the parent study patient population. Rash and diarrhoea, the most common treatment-related AEs, were controlled with conservative measures in most patients. No treatment-related retinal complications (e.g. RVO, CSR) or grade 3 left ventricular systolic dysfunction was observed in this patient population, although these were observed at low frequency in the parent study population.12
Notably absent was the occurrence of proliferative skin lesions, including squamous cell carcinoma, which have been associated with selective BRAF inhibitors,10 presumably by the paradoxical activation of the MAPK pathway resulting in increased pERK in normal cells.20,21 The absence of secondary malignancies distinguishes MEK inhibitors from BRAF inhibitors. This may provide a distinct advantage as these agents are explored in adjuvant and neoadjuvant settings in patients with earlier stage disease. Moreover, combining a MEK inhibitor and a BRAF inhibitor, presumably due to their opposing effects on pERK in normal cells,22 appears to reduce the frequency of BRAF inhibitor-induced skin lesions and MEK inhibitor-induced rash.23
To our knowledge, this is the first time a MEK inhibitor has demonstrated substantial clinical activity, achieving a 40% RR (33% confirmed), including two CRs, and a median PFS of 5·7 months (95% CI, 4·0–7·4) in BRAF-mutant, BRAF inhibitor-naïve melanoma. The subset of patients without brain metastases demonstrated a median PFS of 7·4 months (95% CI, 1·9–9·2). These median PFS results are an improvement over historical benchmarks24 and over the 1·6 months recently reported for BRAF-mutant melanoma patients treated with dacarbazine.10 The encouraging results with trametinib may be due to its unique pharmacokinetic profile12 that allows sustained target inhibition and distinguishes it from other clinically tested MEK inhibitors.
Durable responses to trametinib were observed in patients with both BRAFV600E and BRAFV600K mutations. Exploratory genetic data revealed one patient with a non-V600 BRAF mutation (L597V) who achieved a confirmed PR with 60% tumour reduction and received study treatment for >2 years, which could suggest that other less common non-V600 BRAF mutant tumours may be sensitive to MEK inhibition.
The exploratory genetic analysis may also suggest that the frequency of genetic aberrations in relevant cancer genes may impact tumour response. These aberrations could provide insight into mechanisms of drug resistance, both primary and secondary, although other factors (e.g., prior therapy, tumour microenvironment) must also be considered. A priori knowledge of a patient's mutation profile beyond BRAFV600 could influence therapeutic choices going forward. However, existence of tumour heterogeneity25 and sampling variability may confound efforts to determine reliable mutation profiles and may explain discordant results in this study.
Durable responses were also observed in BRAF wild-type melanoma patients, several of whom received study treatment for over 1 year. NRAS-mutant melanoma, constituting approximately 10–15% of all cutaneous melanoma, carries a poor prognosis. Uveal melanoma accounts for approximately 5% of all melanomas and frequently has MAPK pathway activation, most commonly via mutation in GNAQ and GNA11.7,8 The durable tumour reduction and SD observed in these two melanoma subpopulations is noteworthy given their limited treatment options and because BRAF inhibitors are ineffective in these patients. Based on the combined clinical and exploratory genetic analysis, the RR and duration on treatment appeared to be greater in patients with BRAFV600WT/NRASWT melanoma. Historical data for these populations are sparse, but confirmed RRs of 3% and 14% were reported for first-line patients with concurrent BRAFWT/NRASWT melanoma after treatment with either the MEK inhibitor selumetinib (AZD6244) or temozolomide, respectively.26
Trametinib demonstrated a trend of lower response rate and treatment duration in BRAFV600WT/NRAS-mutant melanoma or uveal melanoma, compared with other BRAF and NRAS subsets. It is unclear why NRAS-mutant melanoma appears less responsive to trametinib than BRAF-mutant melanoma, but activation of alternative signalling pathways is likely a factor. These results reinforce the need for comprehensive mutational analysis in future trials. Additional studies will be required to validate these initial findings.
The MAPK pathway plays a critical role in the proliferation of melanoma. The discovery of BRAFV600-activating mutations and the development of selective, small-molecule inhibitors have revolutionised melanoma treatment.10 The parent, first-in-human study of trametinib was conducted prior to the approval of vemurafenib for the treatment of metastatic melanoma with BRAFV600E mutation. Prior to our study, several MEK inhibitors had been clinically evaluated in human cancers, including melanoma, with limited success.17-19
The clinical activity observed with trametinib suggests that MEK is a valid therapeutic target and provides a novel mechanism by which to treat BRAF-mutant melanoma. Trametinib demonstrated considerable clinical activity in BRAF-mutant melanoma as well as activity in BRAF wild-type and uveal melanoma. This broad activity distinguishes trametinib from selective BRAF inhibitors that are active only in BRAF-mutated tumours and are potentially harmful in BRAF wild-type melanoma.27 Further, treatment with trametinib does not induce secondary cutaneous squamous cell carcinoma. Acquired mutations in MEK to both MEK and BRAF inhibitor therapy have now been described.28,29 Emerging data regarding varied mechanisms of resistance support the paradigm that melanoma treatment can be molecularly tailored. The low risk of drug–drug interaction, an acceptable safety profile, and clinical activity make trametinib an attractive agent for combinational therapy. For instance, combining trametinib with a BRAF inhibitor could increase clinical efficacy in BRAF-mutant melanoma while improving the toxicity profile.30 The optimal agent pairings (within or across signalling pathways) and timing (concomitant or sequential) will be evaluated in future clinical trials. Phase II and III monotherapy studies and novel combinatorial studies are ongoing. Additional studies to be considered include treatment of earlier-stage disease and treatment in the adjuvant setting.
Supplementary Material
Research in Context.
Systematic Review
We searched Medline with the search terms “MEK inhibitor” and “clinical trial” for reports of results from any clinical trial of MEK inhibitors in patients with cancer published between January 1, 2002, and May 19, 2012. We identified relevant studies of several MEK inhibitors which had been clinically evaluated in human cancers, including melanoma, with limited success.17-19 The Phase II trial of CI-1040 in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer demonstrated no complete or partial responses.17 The Phase I study of PD-0325901 showed that three of 48 evaluable patients with melanoma achieved confirmed partial responses.18 In the randomized Phase II trial of selumetinib versus temozolomide in patients with advanced melanoma, progression-free survival (PFS) did not differ significantly between selumetinib and temozolomide; objective response was observed in six (5.8%) patients receiving selumetinib and nine (9.4%) patients in the temozolomide group.19
Interpretation
Our study shows that trametinib has an acceptable safety profile and antitumor activity in patients with BRAF-mutant melanoma, BRAF wild-type melanoma, and uveal melanoma. To our knowledge, this is the first time a MEK inhibitor has demonstrated substantial clinical activity, achieving a 40% response rate (33% confirmed), including two complete responses, and a median PFS of 5·7 months (95% CI, 4·0–7·4) in BRAF-mutant, BRAF inhibitor-naïve melanoma. The subset of patients without brain metastases demonstrated a median PFS of 7·4 months (95% CI, 1·9–9·2). These median PFS results are an improvement over historical benchmarks24 and over the 1·6 months recently reported for BRAF-mutant melanoma patients treated with dacarbazine.10 Durable responses to trametinib were observed in patients with both BRAFV600E and BRAFV600K mutations. The exploratory genetic analysis may suggest that the frequency of genetic aberrations in relevant cancer genes may impact tumour response. Durable tumour reduction and stable disease was also observed in patients with BRAF wild-type cutaneous melanoma and uveal melanoma, which is noteworthy given the limited treatment options for these melanoma subpopulations and because BRAF inhibitors are ineffective in these patients. The clinical activity observed with trametinib suggests that MEK is a valid therapeutic target. The broad activity in melanoma subpopulations distinguishes trametinib from selective BRAF inhibitors that are active only in BRAF-mutated tumours and potentially harmful in BRAF wild-type melanoma.27 Phase II and III monotherapy studies and novel combinatorial studies are ongoing. Additional studies to be considered include treatment of earlier-stage disease and treatment in the adjuvant setting.
Acknowledgments
We thank the patients and their families, Drs. Lynn Schuchter and Ravi Amaravadi of the University of Pennsylvania Abramson Cancer Center, Jenny Jiang from the U.T. MD Anderson Cancer Center, and the entire GSK MEK111054 study team. Editorial support in the form of development of draft outline, development of manuscript first draft, collating comments, fact-checking and graphic services was provided by MediTech Media and was funded by GlaxoSmithKline.
Research support: The research in this manuscript was supported by GlaxoSmithKline
GSF, KBK and LAF have received research funding and travel reimbursement from GlaxoSmithKline. KDL, PJOD and RK have received research funding from GlaxoSmithKline. JRI served as an uncompensated advisor to GlaxoSmithKline. MSG has received research funding and other remuneration from GlaxoSmithKline. NJV served as Vice-Chair on the GU committee of SWOG, and as a compensated advisor to GlaxoSmithKline. WM served as a compensated advisor, and received research funding from GlaxoSmithKline. KF served as a consultant to GlaxoSmithKline. RG has received research support and honoraria from GlaxoSmithKline. DJD, PS, CM, SAS, LTR, VGRP and NTL are employees of GlaxoSmithKline. PFL was an employee of GlaxoSmithKline.
Footnotes
This study has been presented in part at the following conferences: 22nd AACR-NCI-EORTC Symposium on Molecular Targets and Cancer Therapeutics 2010, ESMO 2010, Perspectives in Melanoma 2010, ASCO 2010, and AACR 2011.
Conflicts of Interest: All other authors declare no conflicts of interest.
Authors' contributions: KF, JI and WM contributed to the study design. GSF and KL contributed to data collection. GSH and KL contributed to data analysis and interpretation.
All authors contributed to writing or review of the manuscript, and gave their approval of the final manuscript.
Reference List
- 1.Tsao H, Atkins MB, Sober AJ. Management of cutaneous melanoma. N Engl J Med. 2004;351:998–1012. doi: 10.1056/NEJMra041245. [DOI] [PubMed] [Google Scholar]
- 2.Hodi FS, O'Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brunet A, Pages G, Pouyssegur J. Constitutively active mutants of MAP kinase kinase (MEK1) induce growth factor-relaxation and oncogenicity when expressed in fibroblasts. Oncogene. 1994;9:3379–87. [PubMed] [Google Scholar]
- 4.Jiveskog S, Ragnarsson-Olding B, Platz A, Ringborg U. N-ras mutations are common in melanomas from sun-exposed skin of humans but rare in mucosal membranes or unexposed skin. J Invest Dermatol. 1998;111:757–61. doi: 10.1046/j.1523-1747.1998.00376.x. [DOI] [PubMed] [Google Scholar]
- 5.Houben R, Becker JC, Kappel A, et al. Constitutive activation of the Ras-Raf signaling pathway in metastatic melanoma is associated with poor prognosis. J Carcinog. 2004;3:6. doi: 10.1186/1477-3163-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 7.Van Raamsdonk CD, Griewank KG, Crosby MB, et al. Mutations in GNA11 in uveal melanoma. N Engl J Med. 2010;363:2191–9. doi: 10.1056/NEJMoa1000584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Raamsdonk CD, Bezrookove V, Green G, et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457:599–602. doi: 10.1038/nature07586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Long GV, Kefford RF, Carr PJA, et al. Phase I/II study of GSK2118436, a selective inhibitor of V600 mutant BRAF kinase: evidence of activity in melanoma brain metastases. Ann Oncol. 2010;21(Suppl.8):viii12. [Google Scholar]
- 10.Chapman PB, Hauschild A, Robert C, J, et al. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. N Engl J Med. 2011;364:2507–16. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gilmartin AG, Bleam MR, Groy A, et al. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res. 2011;17:989–1000. doi: 10.1158/1078-0432.CCR-10-2200. [DOI] [PubMed] [Google Scholar]
- 12.Falchook G, Infante JR, Fecher LA, et al. The oral MEK 1/2 inhibitor GSK1120212 demonstrates early efficacy signals. Ann Oncol. 2011;21(Suppl.8):viii162. [Google Scholar]
- 13.National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE) v3.0. http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcaev3.pdf.
- 14.Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–16. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 15.Moy C, Aziz MU, Greshock J, et al. Mutation and copy number detection in human cancers using a custom genotyping assay. Genomics. 2011;98:296–301. doi: 10.1016/j.ygeno.2011.04.008. [DOI] [PubMed] [Google Scholar]
- 16.Bamford S, Dawson E, Forbes S, et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br J Cancer. 2004;91:355–8. doi: 10.1038/sj.bjc.6601894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rinehart J, Adjei AA, Lorusso PM, et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol. 2004;22:4456–62. doi: 10.1200/JCO.2004.01.185. [DOI] [PubMed] [Google Scholar]
- 18.Lorusso PM, Krishnamurthi SS, Rinehart JJ, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral MAPK/ERK kinase inhibitor PD-0325901 in patients with advanced cancers. Clin Cancer Res. 2010;16:1924–37. doi: 10.1158/1078-0432.CCR-09-1883. [DOI] [PubMed] [Google Scholar]
- 19.Kirkwood JM, Bastholt L, Robert C, et al. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin Cancer Res. 2012;18:555–67. doi: 10.1158/1078-0432.CCR-11-1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431–5. doi: 10.1038/nature08833. [DOI] [PubMed] [Google Scholar]
- 21.Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–30. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Poulikakos PI, Solit DB. Resistance to MEK inhibitors: should we co-target upstream? Sci Signal. 2011;4:e16. doi: 10.1126/scisignal.2001948. [DOI] [PubMed] [Google Scholar]
- 23.Infante JR, Falchook GS, Lawrence DP, et al. Phase I/II study to assess safety, pharmacokinetics, and efficacy of the oral MEK 1/2 inhibitor GSK1120212 (GSK212) dosed in combination with the oral BRAF inhibitor GSK2118436 (GSK436) J Clin Oncol. 2011;29:CRA8503. [Google Scholar]
- 24.Korn EL, Liu PY, Lee SJ, et al. Meta-analysis of phase II cooperative group trials in metastatic stage IV melanoma to determine progression-free and overall survival benchmarks for future phase II trials. J Clin Oncol. 2008;26:527–34. doi: 10.1200/JCO.2007.12.7837. [DOI] [PubMed] [Google Scholar]
- 25.Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–92. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dummer M, Robert C, Chapman PB, et al. AZD6244 (ARRY-142886) vs temozolomide (TMZ) in patients (pts) with advanced melanoma: An open-label, randomized, multicentre, phase II study. J Clin Oncol. 2008;26 Abstract 9033. [Google Scholar]
- 27.Heidorn SJ, Milagre C, Whittaker S, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209–21. doi: 10.1016/j.cell.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Emery CM, Vijayendran KG, Zipser MC, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci U S A. 2009;106:20411–6. doi: 10.1073/pnas.0905833106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wagle N, Emery C, Berger MF, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol. 2011;29:3085–96. doi: 10.1200/JCO.2010.33.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Flaherty K, Infante JR, Falchook GS, et al. Phase I/II expansion cohort of BRAF inhibitor GSK2118436 + MEK inhibitor GSK1120212 in patients with BRAF mutant metastatic melanoma who progressed on a prior BRAF inhibitor. Pigment Cell Melanoma Res. 2011;24:1022. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.