

Abstract
Autism spectrum disorder (ASD) represents a heterogeneous group of disorders, which presents a substantial challenge to diagnosis and treatment. Over the past decade, considerable progress has been made in the identification of genetic risk factors for ASD that define specific mechanisms and pathways underlying the associated behavioural deficits. In this Review, we discuss how some of the latest advances in the genetics of ASD have facilitated parsing of the phenotypic heterogeneity of this disorder. We argue that only through such advances will we begin to define endophenotypes that can benefit from targeted, hypothesis-driven treatments. We review the latest technologies used to identify and characterize the genetics underlying ASD and then consider three themes—single-gene disorders, the gender bias in ASD, and the genetics of neurological comorbidities—that highlight ways in which we can use genetics to define the many phenotypes within the autism spectrum. We also present current clinical guidelines for genetic testing in ASD and their implications for prognosis and treatment.
Introduction
Autism spectrum disorder (ASD) represents a heterogeneous group of neurodevelopmental disorders that are characterized by a clinical dyad of impaired social–communication function and the presence of a restricted, repetitive pattern of behaviour or interests.1 Within the autism spectrum exists tremendous phenotypic heterogeneity in adaptive function, cognitive and language abilities, and neurological comorbidities, leading some researchers to refer to these various disorders as ‘the autisms’.2 For example, despite similar presentations at the time of diagnosis, approximately 30% of children with ASD remain nonverbal into adulthood,3 whereas 30% demonstrate a reasonably normal verbal IQ, with primary deficits in language pragmatics.4 Over the past decade, research in ASD has focused on understanding the biological basis for this clinical variability, and has made considerable breakthroughs in the identification of genetic risk factors that define specific mechanisms and pathways underlying the behavioural deficits in the disorder.
Somewhat lagging behind advances in genetics has been our ability to characterize the specific phenotypes associated with these risk genes. Thus far, the association between genes, brain and behaviour in ASD has mostly occurred in a unidirectional manner, with identification of specific risk genes facilitating characterization of common pathways and phenotypes. We argue, however, that detection of behavioural and biological endophenotypes, particularly those that precede ASD diagnosis, could eventually facilitate identification of common genetic syndromes (Figure 1). Ultimately, as discussed in this article, insights gained from genotype–phenotype correlations can greatly inform prognosis and treatment targets.
Figure 1.

From genes to brain to behaviour—a conceptual framework. The key notion is that genes contribute to behaviour and cognition in ASD via their effects on brain structure and development. Abbreviation: ASD, autism spectrum disorder.
One consequence of more complete genetic information that is tied to phenotype data could be the development of genetic classifiers for diagnosis, prognosis, and treatment stratification, as has been implemented in the management of some forms of cancer.5 Given the genetic heterogeneity of ASD, sample sizes in current studies are unlikely to permit development of widely generalizable classifiers, and caution is warranted to avoid misinterpretation of results.6 However, we expect that owing to the strong genetic component of ASD, development of genetic classifiers to identify specific groups of high-risk individuals will be possible once sufficient sample sizes are studied.
Advances in genetic methods
The heritability of ASD has been recognized from the earliest twin studies,7 but only more recently has the term ‘idiopathic autism’ potentially been rendered obsolete through technological advances in genetic methods. This is because contributory mutations in more than 20% of individuals with ASD have been identified, and several hundred major mutations are predicted.8,9 Initially, the standard test in children comprised karyotyping alone, which could only identify abnormalities larger than about 3–5 million base pairs, which are visible under a light microscope. Over the past decade, chromosomal microarray analysis (CMA) technology has facilitated investigation of chromosomal deletions and duplications with much greater resolution, defining an important role for smaller chromosomal structural variation in human disease.10
Any structural chromosomal variation that causes deviation from the control copy number, either through duplications or deletions that are larger than 1 kb, is considered a copy number variant (CNV). CNVs can be inherited or sporadic (de novo), with the latter type of mutation considered more likely to be pathogenic. The two types of CMA technologies that are most widely used are array-based comparative genomic hybridization (aCGH) and single nucleotide polymorphism (SNP) arrays, both of which permit high-resolution molecular analysis of chromosome copy number. The SNP array performs SNP genotyping in addition to detecting gene dose and has the advantage, therefore, of being able to detect specific inheritance patterns, such as uniparental disomy, which cannot be detected by aCGH.11
Use of high-resolution, high-throughput technologies such as SNP arrays or next-generation (NextGen) sequencing has led to a greater understanding of the role of both common polymorphisms, which have a small effect on ASD susceptibility, and rare genetic variation, which has a larger effect on the development of ASD.12 Currently, few common polymorphisms have been reproducibly identified. NextGen sequencing of the exome has expanded our appreciation of the contribution of rare genetic variation to ASD.8,9,13 Exome or genome sequencing moves genetic analysis of patients to the level of the single base pair, expanding testing beyond individual or small groups of related genes, such as those usually detected by CMA. Exome sequencing has proven promising for identification of genetic conditions that are not clinically evident and for identification of partial loss of gene function in ASD.14
The combination of CMA and exome sequencing has identified dozens of putative ASD risk genes and revealed a previously unappreciated role for rare, de novo mutations in ASD susceptibility. 10–20% of individuals with ASD have de novo mutations that are identifiable using current genetic testing; this high yield means that CMA and exome sequencing are appropriate clinical tests for ASD. In fact, methods are being developed for assessing copy number using exome sequencing methods, which will allow clinicians to proceed straight to exome sequencing as part of the diagnostic work-up. Notwithstanding our ability to detect genetic mutations in about 20% of cases, none of the mutations individually accounts for more than 1% of ASD cases—a pattern consistent with extreme genetic heterogeneity among cases. Parsing ASD genetic risk and implicated genes have been recently reviewed15 and are not the focus of this article.
Insights from single-gene disorders
Several Mendelian disorders— including fragile X syndrome, neurofibromatosis, Rett syndrome and tuberous sclerosis complex (TSC)—confer a high risk of social–communication deficits. Such single-gene disorders provide an important opportunity to investigate specific molecular mechanisms underlying aberrant neurodevelopment through use of mouse models and, in turn, to identify treatment targets to modify development.
TSC is an autosomal dominant disorder that serves as a model disorder for such translational investigations. It is characterized by benign tumours (hamartomas) in most organ systems, including the brain. The genes that are mutated in TSC, TSC1 and TSC2, encode hamartin and tuberin, respectively, which regulate the mTORC1 protein complex. mTOR is involved in a molecular pathway that is crucial for protein synthesis, cell growth and axon formation.16,17 Inactivation of TSC1 or TSC2 upregulates this mTORC1 pathway, resulting in an increase in protein synthesis, aberrant axon formation, and tumour growth.
Children with TSC have a wide range of neurodevelopmental disabilities, including ASD in up to 50%, and mild to profound cognitive impairment in 45%.18,19 Early neuropathological and imaging studies investigating the pathogenesis of ASD in TSC focused on the location and burden of cortical tubers. Most regions of the brain, including temporal, frontal and occipital cortex, as well as the cerebellum, were found to be involved.20–23 Increasing evidence from TSC heterozygous mouse models, however, has suggested that TSC pathology exists outside of the tubers themselves, in the form of disorganized axonal tracts, increased axonal growth, abnormal myelination, and aberrant synapse formation.17,24,25 These findings have facilitated the transition from a model based on tuber localization to one that implicates aberrant connectivity in ASD–TSC. Using diffusion tensor imaging (DTI) to quantify white matter integrity, including organization and robustness of white matter tracts, abnormalities in TSC have been reported in so-called normal-appearing white matter on traditional MRI, in regions including the corpus callosum, internal capsule and cerebellum. Several studies have also demonstrated correlations between white matter pathology and severity of ASD symptoms in patients with TSC.26–29 The fact that no one single brain region has been implicated in the development of ASD in TSC suggests that, even in a single-gene disorder, neuro-biological hetero geneity exists that can be reflected in subtle individual differences in phenotype.
Cognitive and social deficits occur in mouse models of TSC. Specifically, TSC1+/− mice show impaired hippocampal learning and atypical social behaviour, whereas TSC2+/− mice have impairments in spatial learning and contextual discrimination.30,31 Moreover, mice with heterozygous and homozygous loss of TSC1 localized to the cerebellum show abnormal social interaction, as well as repetitive behaviours and vocalizations.32 These phenotypes provide the first strong evidence that pure cerebellar dysfunction can have a profound impact on social behaviour.
The findings in mouse models also provide quantifiable outcomes for molecularly driven therapeutic trials. In the past 5 years, on the basis of the known mechanisms of mTOR pathway regulation by TSC1 and TSC2, mTOR inhibitors have been studied extensively in mouse models of TSC. These studies have revealed that mTOR inhibitors can reverse the cognitive and social impairments described above in mouse models after surprisingly short courses of treatment.30
The promising findings in mouse models have inspired the investigation of mTOR inhibitors, such as rapamycin, in patients with TSC. In 2010, everolimus received FDA approval for treatment of subependymal giant cell astrocytoma in children with TSC, and other studies have reported efficacy of mTOR inhibitors for non-neurological manifestations of the disorder, particularly renal angiomyolipomas.33,34 Now, with safety profiles established, several international studies are investigating the use of mTOR inhibitors for neurocognitive deficits in children with TSC.35 The main challenges to successful treatment with mTOR inhibitors lie in the adverse effects of immunosuppressants and potential restrictions to treatment of infants or young children with TSC. A question faced by investigators is whether treating children in late childhood will improve cognitive and social deficits that have been present since early infancy, and that have affected subsequent learning and further neurodevelopment.
Several other Mendelian disorders associated with ASD have recently been targeted in mechanism-based treatment trials.36–40 For example, in fragile X syndrome, absence of the fragile X mental retardation protein (FMRP) leads to enhanced glutamatergic signalling via the metabotropic glutamate receptor 5 (mGluR5), causing defects in synaptic plasticity. Several preclinical studies have investigated the effectiveness of mGluR5 antagonists to improve neurological and behavioural deficits of fragile X syndrome.41 Such work represents an important effort in successfully applying mechanism-based treatments targeted at specific patients’ genetic aetiology in a genetically homogeneous population of children at high risk for ASD. Additionally, these populations provide a crucial opportunity to investigate early risk markers and developmental trajectories in infancy, before diagnosis of ASD, as many Mendelian disorders are diagnosed in utero.
The model of translating known genetic and biological mechanisms to the development of informed treatments has been applied to several genes identified through genetic association studies. For example, CNTNAP2 variants have been identified as risk factors for ASD and related neurodevelopmental disorders, with a specific association with aberrant language development.42,43 Neuroimaging during an implicit language-learning task demonstrated immature neuronal network connectivity patterns as well as differential frontostriatal activity as a function of CNTNAP2 genotype,44 and neuropathological studies showed increased expression of the gene in frontostriatal circuits.45 CNTNAP2-mutant mouse models recapitulate the phenotype in humans both behaviourally and neuropathologically.46 Treatment of these mice with risperidone—a partial dopamine antagonist that is FDA-approved for reducing irritability in ASD—reverses their repetitive behaviours but not their social deficits. This response suggests not only specificity in the action of risperidone, but also CNTNAP2-dependent specificity in the response to ASD treatment.46 We suggest that this mouse model could be used pre-clinically to screen drugs that might ameliorate social deficits in ASD. The overall goal in patients would be to use genetics to inform the choice of pharmacotherapy and identify predictors of treatment response.
Insights from gender bias
Like many childhood neuropsychiatric disorders, ASD has a strong male bias, with reported male:female ratios ranging from 1.33:1 to 15.7:1.47,48 Although overall autism severity does not seem to be associated with gender,49,50 clear evidence exists of gender differences in presentation, particularly in the presence of comorbid features. Specifically, males have more externalizing symptoms such as aggression, stereotypies and hyperactivity, whereas females have more internalizing symptoms of mood disorders, such as anxiety and depression.51–55 Additionally, females with ASD tend to show greater cognitive impairment, with the male:female ratio being close to 1:1 in severely intellectually disabled populations.56
These findings beg the question of whether gender differences in the ASD phenotype are driven by differences in biological mechanisms, or by diagnostic biases that result from the patient’s disease profile at presentation.57 ASD diagnosis is not treated as a quantitative continuum, but rather a categorical trait.1 This factor might interact substantially with differences in male and female behaviour and cognitive styles, as opposed to cognitive ability. For example, given that the behaviours of girls with ASD are typically less disruptive and overt, one could speculate that only girls with more-severe impairment are brought to diagnostic attention. Awareness of these differences in presentation is crucial for clinical management, both in screening for specific comorbidities and in therapeutically targeting the more debilitating symptoms.
Some researchers argue that regardless of whether a gender bias exists in diagnosis, certain sex-specific biological mechanisms do play a part in the gender gap found in ASD.58 The most compelling theory, based on existing evidence, is the female-protective effect (FPE), which suggests that specific factors protect females from developing ASD and, as a consequence, that females have a higher threshold for reaching clinical impairment. Support for this hypothesis was provided by studies demonstrating a greater ASD-related genetic load in females with ASD than in males with ASD, and in clinically unaffected female relatives (compared with unaffected male relatives) of individuals with ASD.59,60
A compelling case for the FPE was provided by a study that characterized autistic traits and comorbidities in a large sample of dizygotic twins through use of the Childhood Autism Spectrum Test and the Autism—Tics, Attention Deficit Hyperactivity Disorder and other Comorbidities inventory. The researchers found that siblings of female probands had higher autism symptom scores than the siblings of male probands.61 Another large-scale study of siblings found similar differences specific to the domain of repetitive behaviours.51 A related contention supporting the FPE is that in the setting of comparable genetic risk, males are more likely to meet clinical criteria for ASD than are females. In individuals with a microdeletion of SHANK1, rigorous assessment using standard methodologies revealed that males more often met clinical criteria for ASD, whereas females with the same mutation showed evidence of anxiety, but not ASD.62
The mechanism underlying the FPE is probably based on two factors: genes found on the sex chromosomes, and sex hormones. Although ASD is not X-linked, it has been suggested that either the Y chromosome is a risk factor or a second X chromosome is protective, as supported by an increased rate of ASD in Turner syndrome (XO) and 47,XYY syndrome.63,64 With regard to sex hormones, tremendous interest has been shown in the role of testosterone in early brain development in children with ASD. Several studies have found correlations between fetal testosterone levels and the presence or severity of systematizing traits, social impairments and reduced empathy, and adults with ASD have increased levels of testosterone metabolites compared with unaffected individuals.65 An important area for future study lies in designing measures that are more gender-specific in order to elucidate the effect of gender-specific factors not only on diagnosis, but also on treatment approaches and response. To date, no hypothesis-driven treatment studies have specifically targeted females with ASD.
Insights from comorbidity risk factors
Neurological comorbidities—namely motor impairment, sleep disturbances and epilepsy—are common in ASD and contribute to the severity of core deficits, non-neurological comorbidities, and impairments to adaptive function.66 Although somewhat challenging to characterize, motor impairments in ASD include delayed motor milestones, apraxia, hypotonia, and malcoordination.67–70 The prevalence of insomnia in ASD ranges from 53% to 78%,71 with a clear association between sleep impairment and behavioural disturbances.72,73 Finally, epilepsy occurs in 5–46% of children with ASD, with epileptiform abnormalities found in up to 60%.74 Genetic risk factors for each of these comorbidities continue to be identified (Box 1), which has important clinical implications with regard to screening for and treating these clinical factors.
Box 1. Genetics and ASD neurological comorbidities.
Phenotype–genotype correlations in ASD are in the early stages. However, specific mutations have been associated with certain major clinical neurological phenotypes— namely, epilepsy, motor impairment, and sleep disturbance.
Epilepsy
Tuberous sclerosis complex (TSC1 and TSC2)
Rett syndrome (MECP2)
CNTNAP2
SYN1
Fragile X syndrome
1q21.1 deletion
7q11.23 duplication
15q11.1–q13.3 duplication
16p11.2 deletion
18q12.1 duplication
22q11.2 deletion
Phelan–McDermid syndrome (SHANK3, 22q.13.3 deletion)
Angelman syndrome (UBE3A)
Motor impairment
Rett syndrome: hypotonia, severe stereotypies
Phelan–McDermid syndrome (SHANK3, 22q.13.3 deletion): hypotonia
AUTS2: motor delay
Fox1 (A2BP1): motor asymmetry
NRXN1 deletion: hypotonia
2q23.1 deletion and duplication: hypotonia and motor delay
15q11.1–q13.3 duplication: hypotonia
Sleep disturbance
Rett syndrome
Smith–Magenis syndrome
Phelan–McDermid syndrome (SHANK3, 22q.13.3 deletion)
1q21.1 deletion
15q11.1–q13.3 duplication
18q12.1 deletion
Abbreviation: ASD, autism spectrum disorder.
Here, we focus on the genetics of epilepsy and ASD, as knowledge of the genetic underpinnings of this association has provided insight into common neural mechanisms that underlie the disorders, and has important implications for prognosis and screening. Several genetic syndromes—including TSC, Rett syndrome and fragile X syndrome—as well as mutations such as those in the neurexin family (CNTNAP2), are characterized by a high rate of ASD and epilepsy. A possible reason for such a large phenotypic overlap could be that both ASD and epilepsy represent disorders of synaptic plasticity, with mechanisms that result in an imbalance of excitation and inhibition.75 ASD and epilepsy could represent symptoms of a common process of aberrant neurodevelopment, but the relationship between the two disorders is probably more dynamic, with seizures further injuring a vulnerable neural system, thereby facilitating the developmental of aberrant cognitive and social development. Emerging evidence suggests that seizures in childhood alter brain development at the cellular and molecular level through excitatory and inhibitory neuro-transmitter systems (γ-aminobutyric acid and glutamate, respectively), neuronal membrane integrity, and neuromodulatory pathways such as cAMP, all of which affect synaptic plasticity, long-term potentiation, and memory formation (Figure 2).76,77
Figure 2.
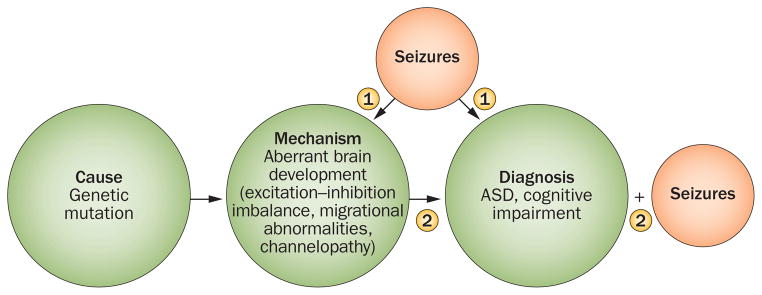
Proposed mechanism underlying the relationship between epilepsy and ASD. The comorbidity of ASD with other neurodevelopmental disorders, such as epilepsy, can be conceptualized in two main ways. In pathway 1, a genetic variant or mutation causes aberrant brain development or function, leading to seizures, which in turn impair early cognitive and social development. In pathway 2, ASD and epilepsy represent two sequelae of a common process, starting from a genetic variant or mutation that leads to aberrant brain development. These scenarios are not mutually exclusive. Abbreviation: ASD, autism spectrum disorder.
The effects of epilepsy on brain development support the need for studies to investigate the effect of epilepsy prevention on developmental outcomes in syndromes that confer a high risk of epilepsy and ASD, such as TSC. In the only published study of prophylactic antiepileptic therapy in TSC, vigabatrin was given to infants with TSC, with one group receiving the medication preventatively, before the onset of seizures, and the other therapeutically, after the onset of seizures. Limitations of this open-label study included the lack of randomization, discrepant sample sizes, and nonstandardized recruitment methods. The preliminary results nevertheless showed that not only epilepsy severity, but also the rate of intellectual disability, was reduced in the group receiving preventative treatment compared with the group receiving therapeutic vigabatrin.78
Early identification of genetic mutations that place children with ASD at higher risk of epilepsy also has tremendous implications for prognosis. A large cross-sectional study of 5,815 children with ASD found that epilepsy was associated with older age (peak prevalence at age 10 years), lower cognitive, adaptive, and language abilities, and greater autism severity.79 As clinical genetic testing (discussed below) precedes the onset of epilepsy in most children, clinicians can justify the recommendation of more-intensive services—for example, those targeting adaptive skills or language development—in children with epilepsy-associated genetic mutations. No formal guidelines have yet been developed for EEG monitoring of these high-risk children, but working groups of clinical experts are currently trying to standardize assessments and evaluations of these children. With growing understanding of the specific comorbidities associated with individual CNVs, over time CNV-specific monitoring and treatment guidelines will be established.
Future studies need to identify the exact developmental and cognitive characteristics associated with epilepsy in individual genetic syndromes and, conversely, to identify more-specific characteristics of the interictal EEG and of epilepsy subtypes associated with individual mutations. For example, a large case series of children with the interstitial duplication 15q11.2–q13 syndrome reported an unusual EEG variant characterized by excessive beta activity in the absence of epilepsy.80 The sample size was too small to enable correlations between the EEG and phenotype to be made, but in future such information could greatly inform more-tailored developmental screening and intervention, as well as the choice of antiepileptic drugs once a diagnosis has been made.
Recommendations for genetic testing
The evolution in recommendations for clinical genetic testing reflects the scientific advances made in our understanding of genetic aetiologies of ASD. In 2000, the American Academy of Neurology (AAN) and Child Neurology Society published guidelines on the screening and diagnosis of autism, with the consensus that “high-resolution chromosome studies (karyotype) and DNA analysis for fragile X should be performed in the presence of mental retardation … or if dysmorphic features are present.”81
The AAN has not updated these guidelines, but in 2008 and 2013 the American College of Medical Genetics (ACMG) published more-current recommendations.82,83 As ‘first-tier’ testing, all children with ASD should undergo investigation through CMA, involving either aCGH or SNP arrays. ‘Second-tier’ testing should include: testing for fragile X in all males; MECP2 sequencing in all females and in males if the clinical presentation suggests MECP2 involvement; and PTEN sequencing in all children with macrocephaly. Notably, metabolic or mitochondrial studies are not indicated unless a child has multiple signs of these disorders, such as anaemia, gastrointestinal dysfunction, cyclic vomiting, lactic acido sis, microcephaly, or seizures. Family history, physical examination, or dysmorphology can inform further studies on an individual basis. All children with a diagnosis of ASD, therefore, should undergo CMA testing, with further testing being contingent on gender, family history, and clinical features. We expect that clinical exome sequencing (CES), which is nearing the same cost as CMA, will supplant CMA testing within the next 2 years (Figure 3). At our institution, CES is already performed in children with a wide range of developmental disabilities (S. Nelson, personal communication).
Figure 3.

Recommendations for clinical genetic testing in children with ASD. Genetic screening in autism should be undertaken in stepwise fashion to integrate clinical history with state-of-the-art testing in the most efficient manner. In all children with ASD, chromosomal microarray analysis should be conducted, as well as detailed family history and clinical examination for signs of known or cryptic genetic syndromes. At this stage, other testing can recommended depending on other phenotypic features, such as the sex of the child or presence of macrocephaly. Genetic counselling is a key component of any clinical genetic analyses. Abbreviations: ASD, autism spectrum disorder; HC, head circumference.
After testing, genetic counselling of the family is of crucial importance. Parents often ask about the clinical utility of genetic testing and about the likelihood of occurrence of ASD in siblings. The ACMG estimates that the total diagnostic yield (percentage of children in whom a test will yield positive, clinically relevant information) of performing the above recommended genetic testing in children with ASD is 40%. This yield is markedly higher than that of any other study—including EEG, neuroimaging and metabolic profiling—that can be performed in children with ASD in the absence of a clear a priori clinical concern (for example, a history of epilepsy, or results of focal neurological examination) that would direct the clinician to a specific test. The diagnostic yield of CES remains to be determined, but is estimated at an additional 15–25%.12 Furthermore, as discussed above, the identification of risk genes has led to greater understanding of more-specific phenotypes. The clinical characteristics associated with particular CNVs—such as the high incidence of hypotonia and epilepsy in children with 15q11.2 duplications, or the high rate of intellectual disability in children with 16p11.2 deletions—greatly informs screening during development, prognostication and treatment.
A particularly important question for parents is related to the risk of ASD in siblings of affected individuals: what is the risk of my next child having autism? This risk varies considerably, depending at least partially on family composition. Multiple affected siblings and female probands also increase the risk that the next child will have ASD.84 For example, a mother with two boys with ASD has a 32% risk of the next male child being autistic, whereas in a family with only one male autistic child, the risk that the next female child will have ASD is close to 10%.84 A population-based Danish study in twins reported that the risk of a second child having ASD is approximately 6% on average, which suggests that even prospective studies of infant siblings might suffer from ascertainment bias.85 So, although these general population figures can help families understand the risk of ASD in future offspring, the wide risk ranges limit the value of such information to an individual family. We expect that knowing the precise form of the genetic factors will substantially improve this type of prognostication.
Conclusion and future directions
In this Review, we have highlighted important themes that emerge from the field of autism genetics. After a diagnosis of ASD has been made, the primary goal from a clinical standpoint is to maximize a child’s potential for cognitive and functional gains. The rapid advances in genetics have facilitated an understanding of developmental trajectories, comorbidities and biological mechanisms underlying the deficits in ASD which, in turn, will open the door to the development of more mechanism-based, phenotype-specific treatments for these children. Population-level genetic screening tied to well-curated, longitudinal phenotype data will be necessary to gain a more complete understanding of genotype–phenotype relationships and realize this promise.
Key points.
Over the past 5 years, researchers have identified many genetic factors that increase the risk of autism spectrum disorder (ASD), and that might shed light on more-homogeneous subgroups within the spectrum
The most robustly identified genetic risks for ASD are rare mutations with large effect; studies have been underpowered to detect common genetic variation
The role of rare genetic variants supports the relevance of studying monogenic disorders, such as tuberous sclerosis complex, for understanding ASD pathophysiology
The most parsimonious explanation for the male predominance in ASD involves the presence of protective factors that reduce the risk of ASD in females
Many genetic mutations associated with ASD also confer high risk of comorbidities including epilepsy, motor impairment and sleep disturbance
Genetic testing including chromosomal microarray analysis is warranted and clinically indicated for all suspected cases of ASD
Review criteria.
Literature searches were performed in PubMed using the search terms “autism,” “autism spectrum disorder”, “pervasive developmental disorders,” “genetics”, “copy number variation”, “exome sequencing”, “clinical guidelines”, “practice parameters”, “single gene disorders”, “tuberous sclerosis complex”, “CNTNAP2”, “gender bias”, “epilepsy”, “sleep impairment”, “insomnia”, “motor delay”, “motor impairment”, “neurology” and “neurological comorbidities” from January 1990 to August 2013, with a focus on papers published in the past 2–3 years. We searched the reference lists of retrieved papers to identify additional articles. Only full-text manuscripts written in English were considered.
Footnotes
Competing interests
The authors declare no competing interests.
Author contributions
S. S. Jeste researched data for the article. Both authors made substantial contributions to discussion of the content, writing of the article, and to review and/or editing of the manuscript before submission.
Contributor Information
Shafali S. Jeste, Semel Institute for Neuroscience and Human Behavior, 760 Westwood Plaza, Suite 68-237, Los Angeles, CA 90064, USA
Daniel H. Geschwind, Gonda Research Building, Room 2506, 695 Charles E. Young Drive South, University of California Los Angeles, Los Angeles, CA 90095, USA
References
- 1.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5. American Psychiatric Association; 2013. (DSM-5) [Google Scholar]
- 2.Geschwind DH, Levitt P. Autism spectrum disorders: developmental disconnection syndromes. Curr Opin Neurobiol. 2007;17:103–111. doi: 10.1016/j.conb.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 3.Hus V, Pickles A, Cook EH, Jr, Risi S, Lord C. Using the autism diagnostic interview—revised to increase phenotypic homogeneity in genetic studies of autism. Biol Psychiatry. 2007;61:438–448. doi: 10.1016/j.biopsych.2006.08.044. [DOI] [PubMed] [Google Scholar]
- 4.Anderson DK, et al. Patterns of growth in verbal abilities among children with autism spectrum disorder. J Consult Clin Psychol. 2007;75:594–604. doi: 10.1037/0022-006X.75.4.594. [DOI] [PubMed] [Google Scholar]
- 5.Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013;501:338–345. doi: 10.1038/nature12625. [DOI] [PubMed] [Google Scholar]
- 6.Belgard TG, Jankovic I, Lowe JK, Geschwind DH. Population structure confounds autism genetic classifier. Mol Psychiatry. doi: 10.1038/mp.2013.34. http://dx.doi.org/10.1038/mp.2013.34. [DOI] [PMC free article] [PubMed]
- 7.Smalley SL, Asarnow RF, Spence MA. Autism and genetics. A decade of research. Arch Gen Psychiatry. 1988;45:953–961. doi: 10.1001/archpsyc.1988.01800340081013. [DOI] [PubMed] [Google Scholar]
- 8.Neale BM, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Roak BJ, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malhotra D, Sebat J. CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell. 2012;148:1223–1241. doi: 10.1016/j.cell.2012.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heil KM, Schaaf CP. The genetics of autism spectrum disorders—a guide for clinicians. Curr Psychiatry Rep. 2013;15:334. doi: 10.1007/s11920-012-0334-3. [DOI] [PubMed] [Google Scholar]
- 12.Stein JL, Parikshak NN, Geschwind DH. Rare inherited variation in autism: beginning to see the forest and a few trees. Neuron. 2013;77:209–211. doi: 10.1016/j.neuron.2013.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sanders SJ, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu TW, et al. Using whole-exome sequencing to identify inherited causes of autism. Neuron. 2013;77:259–273. doi: 10.1016/j.neuron.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berg JM, Geschwind DH. Autism genetics: searching for specificity and convergence. Genome Biol. 2012;13:247. doi: 10.1186/gb-2012-13-7-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang J, Manning BD. The TSC1–TSC2 complex: a molecular switchboard controlling cell growth. Biochem J. 2008;412:179–190. doi: 10.1042/BJ20080281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choi YJ, et al. Tuberous sclerosis complex proteins control axon formation. Genes Dev. 2008;22:2485–2495. doi: 10.1101/gad.1685008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Curatolo P, Bombardieri R, Jozwiak S. Tuberous sclerosis. Lancet. 2008;372:657–668. doi: 10.1016/S0140-6736(08)61279-9. [DOI] [PubMed] [Google Scholar]
- 19.Jeste SS, Sahin M, Bolton P, Ploubidis GB, Humphrey A. Characterization of autism in young children with tuberous sclerosis complex. J Child Neurol. 2008;23:520–525. doi: 10.1177/0883073807309788. [DOI] [PubMed] [Google Scholar]
- 20.Bolton PF, Park RJ, Higgins JN, Griffiths PD, Pickles A. Neuro-epileptic determinants of autism spectrum disorders in tuberous sclerosis complex. Brain. 2002;125:1247–1255. doi: 10.1093/brain/awf124. [DOI] [PubMed] [Google Scholar]
- 21.Jambaque I, et al. Neuropsychological aspects of tuberous sclerosis in relation to epilepsy and MRI findings. Dev Med Child Neurol. 1991;33:698–705. doi: 10.1111/j.1469-8749.1991.tb14947.x. [DOI] [PubMed] [Google Scholar]
- 22.Eluvathingal TJ, et al. Cerebellar lesions in tuberous sclerosis complex: neurobehavioral and neuroimaging correlates. J Child Neurol. 2006;21:846–851. doi: 10.1177/08830738060210100301. [DOI] [PubMed] [Google Scholar]
- 23.Weber AM, Egelhoff JC, McKellop JM, Franz DN. Autism and the cerebellum: evidence from tuberous sclerosis. J Autism Dev Disord. 2000;30:511–517. doi: 10.1023/a:1005679108529. [DOI] [PubMed] [Google Scholar]
- 24.Nie D, et al. Tsc2–Rheb signaling regulates EphA-mediated axon guidance. Nat Neurosci. 2010;13:163–172. doi: 10.1038/nn.2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meikle L, et al. A mouse model of tuberous sclerosis: neuronal loss of Tsc1 causes dysplastic and ectopic neurons, reduced myelination, seizure activity, and limited survival. J Neurosci. 2007;27:5546–5558. doi: 10.1523/JNEUROSCI.5540-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Widjaja E, et al. Diffusion tensor imaging identifies changes in normal-appearing white matter within the epileptogenic zone in tuberous sclerosis complex. Epilepsy Res. 2010;89:246–253. doi: 10.1016/j.eplepsyres.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 27.Garaci FG, et al. Increased brain apparent diffusion coefficient in tuberous sclerosis. Radiology. 2004;232:461–465. doi: 10.1148/radiol.2322030198. [DOI] [PubMed] [Google Scholar]
- 28.Krishnan ML, et al. Diffusion features of white matter in tuberous sclerosis with tractography. Pediatr Neurol. 2010;42:101–106. doi: 10.1016/j.pediatrneurol.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peng SS, Lee WT, Wang YH, Huang KM. Cerebral diffusion tensor images in children with tuberous sclerosis: a preliminary report. Pediatr Radiol. 2004;34:387–392. doi: 10.1007/s00247-004-1162-3. [DOI] [PubMed] [Google Scholar]
- 30.Ehninger D, et al. Reversal of learning deficits in a Tsc2+/− mouse model of tuberous sclerosis. Nat Med. 2008;14:843–848. doi: 10.1038/nm1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goorden SM, van Woerden GM, van der Weerd L, Cheadle JP, Elgersma Y. Cognitive deficits in Tsc1+/− mice in the absence of cerebral lesions and seizures. Ann Neurol. 2007;62:648–655. doi: 10.1002/ana.21317. [DOI] [PubMed] [Google Scholar]
- 32.Tsai PT, et al. Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature. 2012;488:647–651. doi: 10.1038/nature11310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bissler JJ, et al. Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet. 2013;381:817–824. doi: 10.1016/S0140-6736(12)61767-X. [DOI] [PubMed] [Google Scholar]
- 34.Bissler JJ, et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med. 2008;358:140–151. doi: 10.1056/NEJMoa063564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.ClinicalTrials.gov. US National Library of Medicine. 2013 [online], http://clinicaltrials.gov/show/NCT01289912.
- 36.Sahin M. Targeted treatment trials for tuberous sclerosis and autism: no longer a dream. Curr Opin Neurobiol. 2012;22:895–901. doi: 10.1016/j.conb.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berry-Kravis E, et al. A pilot open label, single dose trial of fenobam in adults with fragile X syndrome. J Med Genet. 2009;46:266–271. doi: 10.1136/jmg.2008.063701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berry-Kravis E, et al. Open-label treatment trial of lithium to target the underlying defect in fragile X syndrome. J Dev Behav Pediatr. 2008;29:293–302. doi: 10.1097/DBP.0b013e31817dc447. [DOI] [PubMed] [Google Scholar]
- 39.Berry-Kravis EM, et al. Effects of STX209 (arbaclofen) on neurobehavioral function in children and adults with fragile X syndrome: a randomized, controlled, phase 2 trial. Sci Transl Med. 2012;4:152ra127. doi: 10.1126/scitranslmed.3004214. [DOI] [PubMed] [Google Scholar]
- 40.Paribello C, et al. Open-label add-on treatment trial of minocycline in fragile X syndrome. BMC Neurol. 2010;10:91. doi: 10.1186/1471-2377-10-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pop AS, Gomez-Mancilla B, Neri G, Willemsen R, Gasparini F. Fragile X syndrome: a preclinical review on metabotropic glutamate receptor 5 (mGluR5) antagonists and drug development. Psychopharmacology (Berl) doi: 10.1007/s00213-013-3330-3. http://dx.doi.org/1007/s00213-013-3330-3. [DOI] [PubMed]
- 42.Alarcon M, et al. Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am J Hum Genet. 2008;82:150–159. doi: 10.1016/j.ajhg.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arking DE, et al. A common genetic variant in the neurexin superfamily member CNTNAP2 increases familial risk of autism. Am J Hum Genet. 2008;82:160–164. doi: 10.1016/j.ajhg.2007.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scott-Van Zeeland AA, et al. Altered functional connectivity in frontal lobe circuits is associated with variation in the autism risk gene. CNTNAP2 Sci Transl Med. 2010;2:56ra80. doi: 10.1126/scitranslmed.3001344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abrahams BS, et al. Genome-wide analyses of human perisylvian cerebral cortical patterning. Proc Natl Acad Sci USA. 2007;104:17849–17854. doi: 10.1073/pnas.0706128104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Penagarikano O, Geschwind DH. What does CNTNAP2 reveal about autism spectrum disorder? Trends Mol Med. 2012;18:156–163. doi: 10.1016/j.molmed.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fombonne E. Epidemiology of pervasive developmental disorders. Pediatr Res. 2009;65:591–598. doi: 10.1203/PDR.0b013e31819e7203. [DOI] [PubMed] [Google Scholar]
- 48.Prevalence of autism spectrum disorders—Autism and Developmental Disabilities Monitoring Network, 14 sites, United States, 2008. MMWR Surveill Summ. 2012;61:1–19. [PubMed] [Google Scholar]
- 49.Lai MC, et al. A behavioral comparison of male and female adults with high functioning autism spectrum conditions. PLoS ONE. 2011;6:e20835. doi: 10.1371/journal.pone.0020835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zwaigenbaum L, et al. Sex differences in children with autism spectrum disorder identified within a high-risk infant cohort. J Autism Dev Disord. 2012;42:2585–2596. doi: 10.1007/s10803-012-1515-y. [DOI] [PubMed] [Google Scholar]
- 51.Szatmari P, et al. Sex differences in repetitive stereotyped behaviors in autism: implications for genetic liability. Am J Med Genet B Neuropsychiatr Genet. 2012;159B:5–12. doi: 10.1002/ajmg.b.31238. [DOI] [PubMed] [Google Scholar]
- 52.Hattier MA, Matson JL, Tureck K, Horovitz M. The effects of gender and age on repetitive and/or restricted behaviors and interests in adults with autism spectrum disorders and intellectual disability. Res Dev Disabil. 2011;32:2346–2351. doi: 10.1016/j.ridd.2011.07.028. [DOI] [PubMed] [Google Scholar]
- 53.Bolte S, Duketis E, Poustka F, Holtmann M. Sex differences in cognitive domains and their clinical correlates in higher-functioning autism spectrum disorders. Autism. 2011;15:497–511. doi: 10.1177/1362361310391116. [DOI] [PubMed] [Google Scholar]
- 54.Solomon M, Miller M, Taylor SL, Hinshaw SP, Carter CS. Autism symptoms and internalizing psychopathology in girls and boys with autism spectrum disorders. J Autism Dev Disord. 2012;42:48–59. doi: 10.1007/s10803-011-1215-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dworzynski K, Ronald A, Bolton P, Happe F. How different are girls and boys above and below the diagnostic threshold for autism spectrum disorders? J Am Acad Child Adolesc Psychiatry. 2012;51:788–797. doi: 10.1016/j.jaac.2012.05.018. [DOI] [PubMed] [Google Scholar]
- 56.Giarelli E, et al. Sex differences in the evaluation and diagnosis of autism spectrum disorders among children. Disabil Health J. 2010;3:107–116. doi: 10.1016/j.dhjo.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Constantino JN, Charman T. Gender bias, female resilience, and the sex ratio in autism. J Am Acad Child Adolesc Psychiatry. 2012;51:756–758. doi: 10.1016/j.jaac.2012.05.017. [DOI] [PubMed] [Google Scholar]
- 58.Werling DM, Geschwind DH. Understanding sex bias in autism spectrum disorder. Proc Natl Acad Sci USA. 2013;110:4868–4869. doi: 10.1073/pnas.1301602110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wiznitzer M. Autism and tuberous sclerosis. J Child Neurol. 2004;19:675–679. doi: 10.1177/08830738040190090701. [DOI] [PubMed] [Google Scholar]
- 60.Harrison JE, Bolton PF. Annotation: tuberous sclerosis. J Child Psychol Psychiatry. 1997;38:603–614. doi: 10.1111/j.1469-7610.1997.tb01687.x. [DOI] [PubMed] [Google Scholar]
- 61.Robinson EB, Lichtenstein P, Anckarsater H, Happe F, Ronald A. Examining and interpreting the female protective effect against autistic behavior. Proc Natl Acad Sci USA. 2013;110:5258–5262. doi: 10.1073/pnas.1211070110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sato D, et al. SHANK1 deletions in males with autism spectrum disorder. Am J Hum Genet. 2012;90:879–887. doi: 10.1016/j.ajhg.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Donnelly SL, et al. Female with autistic disorder and monosomy X (Turner syndrome): parent-of-origin effect of the X chromosome. Am J Med Genet. 2000;96:312–316. doi: 10.1002/1096-8628(20000612)96:3<312::aid-ajmg16>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 64.van Rijn S, Bierman M, Bruining H, Swaab H. Vulnerability for autism traits in boys and men with an extra X chromosome (47,XXY): the mediating role of cognitive flexibility. J Psychiatr Res. 2012;46:1300–1306. doi: 10.1016/j.jpsychires.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 65.Geier DA, Kern JK, King PG, Sykes LK, Geier MR. An evaluation of the role and treatment of elevated male hormones in autism spectrum disorders. Acta Neurobiol Exp (Wars) 2012;72:1–17. doi: 10.55782/ane-2012-1876. [DOI] [PubMed] [Google Scholar]
- 66.Jeste SS. The neurology of autism spectrum disorders. Curr Opin Neurol. 2011;24:132–139. doi: 10.1097/WCO.0b013e3283446450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Green D, et al. Impairment in movement skills of children with autistic spectrum disorders. Dev Med Child Neurol. 2009;51:311–316. doi: 10.1111/j.1469-8749.2008.03242.x. [DOI] [PubMed] [Google Scholar]
- 68.Fournier KA, Hass CJ, Naik SK, Lodha N, Cauraugh JH. Motor coordination in autism spectrum disorders: a synthesis and meta-analysis. J Autism Dev Disord. 2010;40:1227–1240. doi: 10.1007/s10803-010-0981-3. [DOI] [PubMed] [Google Scholar]
- 69.Dziuk MA, et al. Dyspraxia in autism: association with motor, social, and communicative deficits. Dev Med Child Neurol. 2007;49:734–739. doi: 10.1111/j.1469-8749.2007.00734.x. [DOI] [PubMed] [Google Scholar]
- 70.Hilton CL, Zhang Y, Whilte MR, Klohr CL, Constantino J. Motor impairment in sibling pairs concordant and discordant for autism spectrum disorders. Autism. 2012;16:430–441. doi: 10.1177/1362361311423018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Krakowiak P, Goodlin-Jones B, Hertz-Picciotto I, Croen LA, Hansen RL. Sleep problems in children with autism spectrum disorders, developmental delays, and typical development: a population-based study. J Sleep Res. 2008;17:197–206. doi: 10.1111/j.1365-2869.2008.00650.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wiggs L, Stores G. Severe sleep disturbance and daytime challenging behaviour in children with severe learning disabilities. J Intellect Disabil Res. 1996;40:518–528. doi: 10.1046/j.1365-2788.1996.799799.x. [DOI] [PubMed] [Google Scholar]
- 73.Malow BA, et al. A practice pathway for the identification, evaluation, and management of insomnia in children and adolescents with autism spectrum disorders. Pediatrics. 2012;130 (Suppl 2):S106–S124. doi: 10.1542/peds.2012-0900I. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Spence SJ, Schneider MT. The role of epilepsy and epileptiform EEGs in autism spectrum disorders. Pediatr Res. 2009;65:599–606. doi: 10.1203/01.pdr.0000352115.41382.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brooks-Kayal A. Epilepsy and autism spectrum disorders: are there common developmental mechanisms? Brain Dev. 2010;2:731–738. doi: 10.1016/j.braindev.2010.04.010. [DOI] [PubMed] [Google Scholar]
- 76.Ben-Yizhak N, et al. Pragmatic language and school related linguistic abilities in siblings of children with autism. J Autism Dev Disord. 2011;41:750–760. doi: 10.1007/s10803-010-1096-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Swann JW. The effects of seizures on the connectivity and circuitry of the developing brain. Ment Retard Dev Disabil Res Rev. 2004;10:96–100. doi: 10.1002/mrdd.20018. [DOI] [PubMed] [Google Scholar]
- 78.Jozwiak S, et al. Antiepileptic treatment before the onset of seizures reduces epilepsy severity and risk of mental retardation in infants with tuberous sclerosis complex. Eur J Paediatr Neurol. 2011;15:424–431. doi: 10.1016/j.ejpn.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 79.Viscidi EW, et al. Clinical characteristics of children with autism spectrum disorder and co-occurring epilepsy. PLoS ONE. 2013;8:e67797. doi: 10.1371/journal.pone.0067797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Urraca N, et al. The interstitial duplication 15q11.2–q13 syndrome includes autism, mild facial anomalies and a characteristic EEG signature. Autism Res. 2013;6:268–279. doi: 10.1002/aur.1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Filipek PA, et al. Practice parameter: screening and diagnosis of autism: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Child Neurology Society. Neurology. 2000;55:468–479. doi: 10.1212/wnl.55.4.468. [DOI] [PubMed] [Google Scholar]
- 82.Schaefer GB, Mendelsohn NJ. Genetics evaluation for the etiologic diagnosis of autism spectrum disorders. Genet Med. 2008;10:4–12. doi: 10.1097/GIM.0b013e31815efdd7. [DOI] [PubMed] [Google Scholar]
- 83.Schaefer GB, Mendelsohn NJ. Clinical genetics evaluation in identifying the etiology of autism spectrum disorders: 2013 guideline revisions. Genet Med. 2013;15:399–407. doi: 10.1038/gim.2013.32. [DOI] [PubMed] [Google Scholar]
- 84.Ozonoff S, et al. Recurrence risk for autism spectrum disorders: a Baby Siblings Research Consortium study. Pediatrics. 2011;128:e488–e495. doi: 10.1542/peds.2010-2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bolton PF, Griffiths PD. Association of tuberous sclerosis of temporal lobes with autism and atypical autism. Lancet. 1997;349:392–395. doi: 10.1016/S0140-6736(97)80012-8. [DOI] [PubMed] [Google Scholar]