

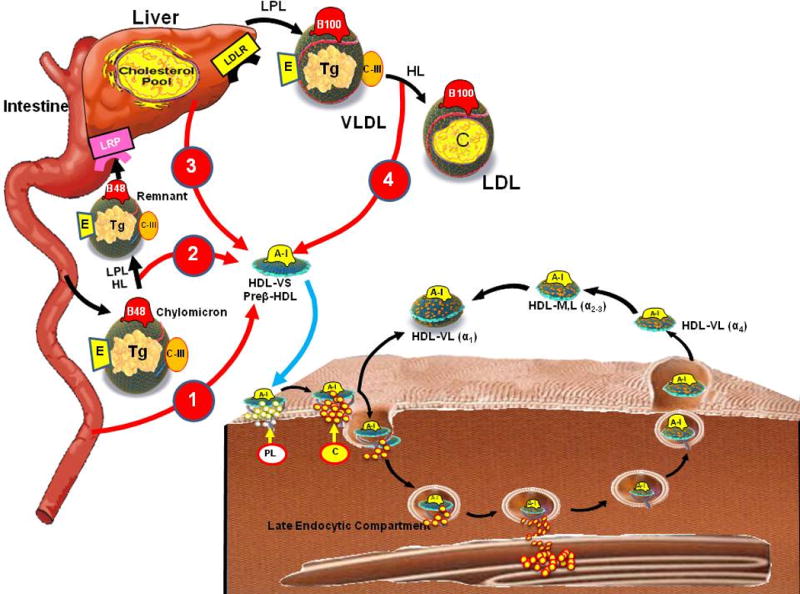
High density lipoprotein has been proposed to have several anti-atherosclerotic properties, including mediating macrophage cholesterol efflux, anti-oxidant capacity, anti-inflammatory properties, nitric oxide promoting activity and transport of proteins with their own intrinsic biological activities (1). HDL particles are critical acceptors of cholesterol from lipid-laden macrophages, and thereby participate in the maintenance of net cholesterol balance in the arterial wall and in the reduction of pro-inflammatory responses by arterial cholesterol loaded macrophages. The pathways that regulate HDL-mediated macrophage cholesterol efflux and disposition of cholesterol involve cell membrane bound transporters, plasma lipid acceptors, plasma proteins and enzymes, and hepatic cellular receptors (Figure 1). From the earliest proposed concept for HDL mediated cholesterol efflux (2,3), the concentration of the cholesterol content in HDL particles has been considered a surrogate measurement for the efficiency of the “reverse cholesterol transport” (RCT) process; however, macrophage-derived cholesterol represents a minor component of the cholesterol transported by HDL particles (4–7). One important pathway for cholesterol-mediated efflux from macrophage foam cells involves interaction between the ATP-binding cassette transporter A1 (ABCA1) with cholesterol-deficient and phospholipid-depleted apoA-I complexes (preβ migrating HDL or very-small HDL [HDL-VS]) (Figure 2) (1,8). Subsequently, the ATP-binding cassette transporter G1 (ABCGI) mediates macrophage cholesterol efflux through interactions with spherical, cholesterol-containing alpha HDL particles (small HDL, medium HDL [HDL-M], large HDL [HDL-L] and very large (HDL-VL) (Figure 3) (1). In contrast, the scavenger receptor class B type I (SR-BI) is a multifunctional receptor, which mediates bidirectional lipid transport in the macrophage, which is dependent upon the content of cholesterol in lipid-laden macrophages. A more established role for SR-BI in cholesterol trafficking involves selective uptake of cholesteryl esters from mature HDL by the liver. Recent studies suggest that polymorphisms in SR-BI contribute to the functional capacity of this cholesterol disposition pathway (9), thereby providing important insights concerning involvement of this receptor in RCT.
Figure 1.

Metabolic pathways that regulate HDL
Figure 2.
Schematic diagram of ABCA1 transporter
Figure 3.

Schematic diagram of cholesterol efflux via ABCG1, SRB1 and passive diffusion
In this review, we discuss the molecular and cellular pathways involved in macrophage cholesterol efflux and cholesterol disposition, as well as recent clinical trials aimed at better understanding HDL function. Specifically, we address new advances in HDL biology that challenge long-standing misperceptions regarding HDL and cholesterol efflux from arterial wall and non-arterial wall sites through the use of specific terminology that addresses the various tissue locations involved in cholesterol efflux and elimination (Table 1). However, non-macrophage arterial wall efflux and non-arterial wall cholesterol efflux will not be addressed in this review. Critical to understanding the processes that facilitate arterial cholesterol efflux, recent experimental studies establish that increased fecal sterol excretion is not necessarily a pre-requisite for HDL mediated macrophage cholesterol efflux and atheroprotection (10,11). Thus, this new information necessitates revision of initial models of RCT in order to foster accurate description of the critical steps required for effective HDL-mediated atheroprotection.
Table 1.
Key Concepts
|
Evolution of the Reverse Cholesterol Transport Concept
Nearly 50 years ago, Glomset proposed a concept of RCT in overall flux of cholesterol from the entire periphery to the liver and its ultimate fecal excretion (2,3). RCT is also sometimes used to refer to specific steps or specific cell types in this process, thus sometimes causing confusion. In order to enhance communication concerning cholesterol efflux pathways, we propose specific terminology that distinguishes between the various macrophage and non-macrophage sites involved in cholesterol efflux and elimination (Table 1).
Emergence of HDL cholesterol as a Biomarker of Reverse Cholesterol Transport
The emphasis on the cholesterol content of HDL in Glomset’s initial model of RCT (2,3) promulgated the use of HDL-C concentration as a biomarker for cholesterol efflux from tissues (3). Even though cholesterol efflux from the lipid-laden macrophage contributes a minor amount of the cholesterol content to the HDL particles, HDL-C has been found to be inversely related to cardiovascular disease risk (12) and to be positively correlated with the beneficial effect of lipid modifying therapies, including statins (13). Yet, the value of HDL-C as a biomarker may be limited because: 1) a small percentage of HDL-C is derived from peripheral tissues, 2) inaccuracy of direct HDL-C measurements routinely used by most clinical laboratories versus more established but technically more demanding analytical methods, and 3) potential confounding of the cardiovascular risk relationships after statistical adjustment for LDL particle concentration (1).
Efforts to refine biomarkers of RCT have encompassed measurement of specific HDL subclasses based on physical and chemical properties (1). However, the use of multiple static measures of the pool size of HDL-C is likely an inadequate approach for the characterization of a dynamic process that involves the active flux and passive transport mechanisms from lipid-laden macrophages, HDL modifying enzymes and transporters, as well as hepatic cholesterol receptors (1). For these reasons, there is a critical need to identify new and clinically measurable biomarkers that accurately relay information concerning flux through the RCT pathway.
HDL Structure and Composition
HDL Structure
Localized conformational features in apoA-I structure may be of key importance for the interaction of HDL with plasma enzymes, receptors or exposure to oxidants (Table 2). Analysis of different size spherical particles reconstituted with a neutral lipid ester core, revealed almost identical patterns, suggesting similar structures (14) that were consistent with the ‘trefoil’ model in which three apoA-I molecules arrange into a 3-D cage structure that stabilizes HDL lipids (Figure 1). Data consistent with these models was also obtained using HDL isolated directly from human plasma and separation into five distinct density subfractions, using density gradient ultracentrifugation (15). HDL particles predominantly containing apoA-I (LpA-I) isolated by immunoaffinity chromatography from plasma, showed a strikingly similarity to the cross-linking pattern observed with reconstituted HDL forms, indicating a similar apoA-I secondary and tertiary structure. Also, despite variations in particle size and apoA-I molecules per particle, there were no systematic changes in cross-linking patterns between the density subfractions. Combining this information with detailed compositional studies of the LpA-I subpopulations, human plasma HDL particle structure appears to be primarily based on the common intermolecular interactions inherent to the double belt and trefoil models. To account for changes in particle size, a twisting motion of the resident apoA-I molecules has been postulated similar to what has been described in molecular dynamics studies (16). These models hold interesting implications for apoA-I conformation and how it interacts with HDL and plasma-remodeling factors, as well as for the association of other HDL associated proteins that may also alter particle function.
Table 2.
Composition of HDL Lipoprotein Acceptors
| HDL-VL | HDL-L | HDL-M | HDL-S | HDL-VS | Free-Apo | ||
|---|---|---|---|---|---|---|---|
| HDL2b | HDL2a | HDL3a | HDL3b | HDL3c | Pre-b HDL | Apo AI | |
| Density (g/mL) Composition (%) | 1,063–1,090 | 1,090–,120 | 1,120–1,150 | 1,150–1,180 | 1,180–1,210 | x | x |
| TG | 4 | 4 | 3 | 2 | 1 | 0 | 0 |
| FC | 4 | 3 | 2 | 1 | 0.1 | 0.1 | 0 |
| CE | 29 | 27 | 25 | 23 | 17 | 0 | 0 |
| PL | 30 | 33 | 29 | 24 | 16 | 16 | 0 |
| Proteins | 33 | 34 | 41 | 50 | 66 | 84 | 100 |
| Apolipoproteins | |||||||
| ApoA-I (mol/mol HDL) | 3.6 | 4.1 | 4.4 | 3.7 | 3 | 1 | 1 |
| ApoA-II (mol/mol HDL) | 0.8 | 1.1 | 1.4 | 1 | 0.4 | 0 | 0 |
| Other Apo | ApoC, D, E | ApoC, D | x | ApoE | |||
Reviewed in reference 1.
HDL Protein Composition
Approximately 65% of HDL protein mass is apoA-I with another 12–15% apoA-II; however, recent proteomic studies (17–20) have identified up to 50 less abundant proteins in HDL, with many linked to functions such as complement regulation, protease inhibition and innate immunity (1). One view of the complexity of the HDL proteome is that apoA-I acts as an organizing scaffold that stabilizes lipids and allows the attachment of these minor proteins onto the particles. Alternatively, although apoA-I containing particles may represent a majority of HDL, other proteins could potentially generate independent particles that co-isolate with HDL particles, and are therefore similarly classified as “HDL”. The majority of these minor proteins associate with the densest (and hence smallest) HDL populations (21). About 13% of the HDL particle surface is phospholipid-covered and is available for binding by protein-lipid interaction. However, the complete particle surface is available for binding because protein-apoA-I interactions are also possible. This suggests that the term “HDL” refers to a constellation of distinct protein and lipid constructs that share a similar density (20,21), but may vary widely in function (22), as originally suggested by Alaupovic and colleagues (23). Indeed, it is now recognized that specific HDL subpopulations serve highly specialized functions (22). It follows that some of these subspecies may play particularly protective roles in cardiovascular disease and therefore represent important targets for mechanistic investigations.
HDL Lipid Composition
HDL is also heterogeneous in regard to its lipidome and exists in distinct subpopulations that differ in their lipid content (1,15,24–26 (Table 2). Among the minor bioactive lipid components, the abundance of sphingosine-1 phosphate (S1P) per HDL particle is asymmetric across the HDL spectrum, with preferential enrichment in HDL-S (small HDL) as compared to HDL-M (medium HDL), HDL-L (large HDL) and HDL-VL (very-large HDL) subfractions. Sphingomyelin (SM) decreases binding of apoE to HDL, thus the reduced content of sphingomyelin in HDL-L and HDL-VL may explain their role in apoE-mediated cholesterol efflux. Additionally, increased SM/phosphatidylcholine (PC) ratio decreased the binding and activity of LCAT in reconstituted HDL particles (27–29).
Cellular Cholesterol Efflux
The removal of excess cholesterol from macrophage derived foam cells present in the atherosclerotic plaque (2) has been incorporated into the concept of RCT (Figure 1). There are multiple pathways by which excess cholesterol in foam cells can be removed by HDL: 1) aqueous diffusion, 2) ABCA1 (Figure 2), 3) ABCG1, 4) SR-BI, and 5) by endogenous production of lipid-poor apolipoprotein E (apoE) (Table 3, Figure 3) (30). The relative contributions of these pathways to cholesterol efflux from mouse peritoneal macrophages and from cholesterol-loaded human macrophages have been determined (Table 3) (31,32). In cholesterol-normal murine macrophages, the aqueous diffusion pathway is predominant, whereas in cholesterol-loaded macrophages the contributions of the ABC transporters predominate (31). Relative contributions of the efflux pathways are ABCA1 35%, aqueous diffusion 35%, ABCG1 21% and SR-BI 9%. In cholesterol-loaded human macrophages the relative contributions of these pathways to cholesterol efflux are different; the ABCA1 pathway remains predominant but ABCG1 does not contribute to efflux and the SR-BI pathway is relatively more important (32).
Table 3.
Pathways for Macrophage-Specific Cholesterol Efflux
| Efflux Pathway | Energetics | Preferred HDL Acceptor | Characteristics |
|---|---|---|---|
| Aqueous diffusion | Passive | HDL-L~ HDL-M~ HDL-S |
|
| SR-BI | Passive | HDL-L>HDL-M>HDL-S |
|
| ABCG1 | Active | HDL-L~ HDL-M~ HDL-S E-HDL |
|
| ABCA1 | Active | HDL-VS (preβ1-HDL) Lipid poor apoE |
|
Aqueous Diffusion
The rate-limiting step is desorption of free cholesterol (FC) molecules from the cell plasma membrane into the surrounding aqueous phase. Interactions of FC molecules with neighboring phospholipid (PL) molecules influence the rate (the rate is increased by higher PL unsaturation and decreased by a higher membrane SM content (33). Collision of the desorbed FC molecules with HDL particles diffusing in the extracellular aqueous space leads to their rapid uptake into the lipoprotein acceptor. The human HDL subclasses HDL-L, HDL-M and HDL-S are equally effective acceptors in this pathway, because the efflux process is not affected significantly by alterations in HDL particle size (34).
ABCA1-dependent cholesterol efflux pathway
The concentration of the ABCA1 transporter in the plasma membrane determines the rates of PL and FC efflux and nascent HDL particle formation. The primary acceptor for cellular cholesterol efflux via the ABCA1 pathway includes cholesterol-deficient and phospholipid depleted apoA-I complexes (preβ-1-HDL mobility on two-dimensional gel electrophoresis or HDL-VS) (35,36) (Figure 2). The binding of apoA-I to ABCA1 prevents its intracellular degradation and increases the level of ABCA1 transporter in the plasma membrane. The ABCA1 transporter and apoA-I have an intracellular pathway, which facilitates the mobilization of cholesterol from the late endocytic compartment to the cell membrane (37). The ABCA1 transporter-apoA-I interaction has been proposed to lead to solubilization of an exovesiculated membrane domain, which results in the formation of a heterogeneous population of nascent HDL particles that are discoidal in shape and contain FC molecules (38,39).
Elucidation of the ABCA1 transporter and HDL-VS (preβ-HDL), as the major ligand for the ABCA1 transporter, provided a unique opportunity to quantitate the effect of expanding the HDL-VS (preβ-HDL) pool size by intravenous infusion on cholesterol efflux and coronary atherosclerosis quantitated by the IVUS technique (40–43). HDL, as well as the reconstituted lipid-poor apoA-I complex, are also effective anti-inflammatory agents (44,45). Thus, the infusion of preβ-HDL also reduces vascular inflammation providing a second potential mechanism for intravenous HDL therapy in reducing coronary atherosclerosis (44,45).
The initial clinical data to suggest that infusions of HDL-VS (preβ-HDL) would be effective in decreasing coronary atherosclerosis was provided by the apoA-I Milano infusion study (40). In the combined 36 patients who received apoA-I Milano infusions, total atheroma volume and the 10 mm most diseased segment decreased when compared to the atherosclerosis quantitated at baseline. However, the data were not significant compared to the control saline infusion, possibly because of the limited sample size.
Two additional HDL infusion clinical trials, ERASE Trial (41) and Selective Delipidation Trial (42) confirmed that infusion of preβ-HDL reduced coronary atherosclerosis. The combined results from the acute HDL infusions studies add support to the concept that increasing preβ-HDL may be an effective approach to reduce coronary atheroma burden, with stabilization of the plaque and reduction in clinical cardiovascular events.
HDL is normally cardioprotective because of its ability to promote cellular cholesterol efflux and mediate RCT. However, HDL can become dysfunctional for example by posttranslational modification of apoA-I (46). Myeloperoxidase-induced oxidative damage of apoA-I, which is seen in patients with established cardiovascular disease, impairs its ability to promote cholesterol efflux via the ABCA1 pathway (47).
ABCG1-dependent cholesterol efflux pathway
A second transporter, ABCG1 is also involved in controlling intracellular cholesterol homeostasis. Expression of ABCG1 enhances efflux of cholesterol to HDL by increasing the pool of plasma membrane FC and reorganizing it so that it can desorb more readily into the extracellular medium (Figure 1) (48,49). Larger HDL2 (HDL-VL, HDL-L) are similarly effective to smaller HDL3 (HDL-M, HDL-S, HDL-VS) particles as acceptors from ABCG1 (Table 3). Unlike ABCA1, FC efflux does not require direct binding of HDL to ABCG1 (49, 50).
The ATP binding cassette transporters, ABCA1 and ABCG1, are increased by LXR transcription factors (51–53), which play a pivotal role in modulating cholesterol efflux by both the ABCA1 and ABCG1 transporters. In vivo, LXRs are activated by specific oxysterols in cholesterol-loaded cells (54). ABCA1 and ABCG1 are key target genes of LXRs in macrophages. While ABCA1 promotes cholesterol efflux to cholesterol-deficient and phospholipid-depleted apolipoprotein A-I and E complexes, ABCG1 promotes efflux to HDL particles (35,36,50). Increased expression of the ABCA1 and ABCG1 transporters is associated with redistribution of cholesterol from the inner to the outer leaflet of the plasma membrane, facilitating cholesterol efflux from cholesterol-loaded foam cells to HDL particles (55). LXRs also increase the endogenous expression of macrophage apoE, which increase cholesterol efflux by interaction with the ABCA1 transporter (53).
New insights into the coordinated participation of ABCA1 and ABCG1 in mediating macrophage cholesterol efflux have recently evolved from animal studies. Single deficiency of ABCA1 in mice results in a moderate increase in atherosclerosis, and deficiency of ABCG1 has no effect; however, combined deficiency resulted in markedly accelerated lesion development (56). Double knockout (DKO) macrophages showed markedly defective cholesterol efflux to HDL and apoA-I (57), and also increased inflammatory responses when treated with lipopolysaccharide (57).
Cholesterol homeostasis has also recently been investigated with microRNAs (miRNA), which are small endogenous non-protein coding RNAs that are post-transcriptional regulators of genes involved in physiological processes (58–60). MiR-33, an intronic miRNA located within the gene encoding sterol-regulatory element binding factor-2 (SREBF-2), inhibits both hepatic expression of ABCA1 and ABCG1, reducing HDL-C concentrations (58,61), as well as ABCA1 expression in macrophages, thus resulting in decreased cholesterol efflux (57). Antagonism of MiR-33 by oligonucleotides raised HDL-C and reduced atherosclerosis in a mouse model, and thus may represent a novel approach to enhancing macrophage cholesterol efflux and raising HDL-C levels in the future (62).
Scavenger Receptor Class B Type 1
SR-BI is an 82-kDa integral membrane protein that controls the structure and composition of plasma HDL. SR-BI mediates bidirectional flux (e.g., efflux) of unesterified, or ‘free’ cholesterol (FC) between cells and HDL or other acceptors (63–65).
While SR-BI bone marrow deficiency leads to accelerated atherosclerosis (66–68), the physiological role of SR-BI-mediated arterial macrophage-specific cholesterol efflux in vitro is unclear (65,68,69); At least, SR-BI did not contribute to macrophage RCT in vivo (method described below) (70) and thus, a role for SR-BI in other bone marrow cell populations, such as platelets, may finally emerge to be the relevant role of SR-BI to atherosclerosis (71). By contrast, hepatic SR-BI clearly has an indirect role in atherosclerosis by modulating changes in the composition and structure of HDL particles (72).
Carriers of the first reported mutation of SR-BI in humans (P297S mutation) had increased HDL-C levels but reduced capacity for cholesterol efflux from macrophages without increased severity of atherosclerosis that may have resulted from the small numbers of carriers studied (9).
Endogenous production of apolipoprotein E
In addition to the apoA-I containing HDL, apoE-HDL (E-HDL) plays a pivotal role in RCT. Major sites of synthesis of apoE include the liver and macrophages. E-HDL facilitates both the efflux of cholesterol from the macrophage as well as the delivery of cholesterol to the liver. Lipid poor apoE secreted by the macrophage binds to the ABCA1 transporter and removes excess macrophage cholesterol. E-HDL also facilitates cholesterol efflux from the macrophage by the ABCG1 transporter and LCAT present on the E-HDL particle converts cholesterol to cholesteryl esters. E-HDL particles can deliver cholesterol to the liver through the interaction with both the SR-B1 and LDL receptors. The molecular structure of the E-HDL particles permits the expansion of the CE core and the development of large E-HDL particles. An increase in E-HDL is seen in CETP-deficient patients and in patients treated with CETP inhibitors. These large E-HDL particles containing LCAT are very effective ligands for cholesterol efflux by the ABCG1 pathway and may play an important role in reducing atherosclerosis in the patients treated with CETP inhibitors (73,74).
Enzymatic Modulation of HDL Remodeling in the Various Stages of Reverse Cholesterol Transport
Role of Lecithin:Cholesterol Acyl Transferase
LCAT, a 63 kDa hepatically-synthesized glycoprotein, mediates a two-step reaction in which a fatty acid is cleaved from the sn-2 position of phospholipids and transesterified to the 3-β-hydroxyl group on the A-ring to form cholesteryl ester. Apo A-I activates LCAT (75,76), and the LCAT reaction largely occurs on very-small HDL (HDL-VS) and small HDL (HDL-S) particles (1), transforming these particles into the larger spherical alpha-migrating forms of HDL (HDL-M, HDL-L and HDL-VL) (Figure 3).
Because of the role of LCAT in maintaining HDL-C levels, it is generally believed to be anti-atherogenic. As discussed earlier, Glomset proposed that the esterification of cholesterol would drive the net efflux or removal of cholesterol from cells, because esterification would ‘trap lipoprotein bound cholesterol” and prevent the back exchange of cholesterol from HDL to cells (2,3). LCAT enhances cholesterol efflux by ABCG1 (50) and by passive exchange (77).
However, evidence from various animal models of either over or under expression of LCAT has been controversial in regard to fecal excretion and the atheroprotective role of LCAT as extensively discussed elsewhere (78). The reason of these discrepancies in animal models have not yet been fully understood but may involve levels of overexpression of LCAT, and difference in species, including lack of CETP expression in mice (see paragraph below).
It has also been speculated that the overall effect of LCAT on atherosclerosis may be modulated by other lipid modifying genes or by different diets. Indeed, LCAT-KO mice exhibited decreased atherosclerosis when fed on a high cholesterol-cholate diet (79) but increased atherosclerosis in an apoE-KO background fed a more “Western” high fat diet (80). In addition to its effect on HDL-C, increased LCAT expression in various animal models was found to lower LDL-C (81,82), which often better correlated with atherosclerosis. For example, LCAT transgenic rabbits defective in the LDL-receptor had high HDL-C and LDL-C and did not show protection against diet-induced atherosclerosis (83).
Thus, by contrast to the initial hypothesis proposed by Glomset (2,3), it remains uncertain whether targeting LCAT activity might become a pharmaceutical approach in humans to raise HDL-C, promote macrophage RCT and prevent atherosclerosis. Indeed, similar to what was observed in animal models, LCAT deficiency in humans has not been consistently associated with accelerated atherosclerosis (78) even when HDL-C levels are low. This lack of association has been attributed to the associated low LDL-C levels in these patients. Recent carotid imaging studies in heterozygous patients with LCAT deficiency showed, however, increased atherosclerosis and higher CRP levels (84,85), although the opposite finding was reported in an earlier study (86). Development of new vascular imaging as measures of HDL functionality, as proposed in this manuscript, may potentially clarify the role of this key enzyme of the RCT in humans (87).
Role of Cholesterol Ester Transfer Protein
Human plasma CETP is a 476-residue hydrophobic glycoprotein that catalyzes the transfer of cholesteryl ester (CE), generated by lecithin:cholesterol acyltransferase (LCAT) in HDL, to other lipoproteins. A CETP gene mutation due to an intron 14 splicing defect was the first mutation described in the Japanese population (88,89). Homozygosity for this mutation results in absence of CETP in plasma, dramatic elevations in HDL-C and apoA-I levels, and moderate reductions in LDL-C and apoB levels (73). Based on this phenotype, CETP inhibition was proposed as a possible strategy to increase HDL levels in humans and reduce atherosclerosis (88,89).
Similar to LCAT animal models, variable results on atherosclerosis have been obtained in mice most likely because of the need to overexpress CETP in this animal model naturally lacking CETP (90,91). In contrast, in almost all instances CETP inhibition in the rabbit, a species naturally containing CETP has resulted in reduced lesions (90).
In humans, potent CETP inhibitors have been developed that cause marked increase in HDL-C levels and smaller decrease in LDL-C levels (92–95). The premature termination of the ILLUMINATE study with torcetrapib created concerns regarding CETP mediated increases in HDL-C as a viable therapeutic approach. Based on current data, the increase in adverse outcomes observed with torcetrapib was most likely due to off target toxicity rather than the inhibition of CETP. Post hoc analysis have shown that despite the adverse off-target effect of torcetrapib, there was a significant inverse relationship between the change in HDL-C levels and the primary measure of atherosclerosis, i.e., the percent of atheroma volume (96). The future role for CETP inhibitors without the same off-target effects, such as dalcetrapib and anacetrapib (97), is under investigation in phase III clinical trials. However, these two molecules have significant different effects on plasma lipoprotein metabolism (97).
While anacetrapib is a potent inhibitor of CETP activity, dalcetrapib is a partial inhibitor or modulator of CETP activity. The reason for this difference is not fully understood but could be related to the mode of binding of both molecules (covalent vs. noncovalent binding of dalcetrapib to CETP Cys13 vs. noncovalent biding of anacetrapib to CETP) (97–99). As a consequence, by promoting strong inhibition of CETP-mediated CE/TG transfer activity within HDL (97,98), in vitro anacetrapib treatment was reported to reduce preβ-HDL (HDL-VS) formation (98). There has been concern that the absence of increased small preβ HDL in CETP deficiency (100) may not mediate efficient cholesterol efflux to ABCA1, since preβ HDL (HDL VS) is the key receptor for the ABCA1 transporter. Indeed, the hydrolysis of triglycerides secondary to CETP-mediated CE-TG exchange results in the release of lipid-poor apoA-I. It is possible that the regeneration of these acceptors for ABCA1 is impaired in CETP deficiency (101). However, in vivo studies with enhanced formation of large spherical HDL enriched in LCAT and apoE was associated with effective ABCG1-dependent net cholesterol efflux (102), similar to what was observed in plasma samples from CETP deficiency patients (73). Although cell culture studies have limitations, these data provide evidence that anacetrapib enhanced function of HDL in humans.
In contrast, by being a modulator of CETP-mediated CE/TG transfer, dalcetrapib treatment only modestly promotes formation large spherical HDL enriched in apoE (personal communication Eric Niesor). However, dalcetrapib maintains efficient preβ HDL particle formation (98). This could explain why dalcetrapib enhances RCT in hamsters (98) and prevents atherosclerosis in other animal models with CETP expression (99).
Role of Phospholipid Transfer Protein
PLTP, a member of lipid transfer/lipopolysaccharide binding proteins (103), mediates transfer of phospholipids from apoB-containing lipoproteins to HDL. Active and inactive forms of PLTP are present in the circulation (104,105). The active form of PLTP is associated with apoAI but not apoE, whereas the inactive form is associated with apoA-I and apoE (106). The various PLTP complexes contribute to variation in plasma PLTP activity (107).
In transgenic and gene knockout mice, the association between PLTP and HDL cholesterol levels has been inconsistent (108,109). Furthermore, murine macrophage cholesterol efflux models have not clarified the involvement of PLTP in RCT (110–112). However, several investigators have shown that recombinant PLTP promotes cholesterol efflux in the presence of HDL (113). An in vivo analysis suggests that systemic elevations in PLTP concentrations and not macrophage specific PLTP expression are responsible for impaired macrophage RCT (104). The contribution of hepatic synthesis of PLTP on plasma lipids was investigated in a murine model that specifically expresses PLTP in the liver (114). On a PLTP-null background, hepatic overexpression of PLTP was responsible for increased plasma PLTP activity, and increased VLDL production and circulating concentrations of apoB-containing lipoproteins and HDL-C. In this study, there were no changes in apoA-I levels. In a recent study, human PLTP transgenic rabbits fed a cholesterol-rich diet showed a significant increase in non-HDL-C, no change in HDL-C, and increased formation of aortic fatty streaks as compared to non-transgenic littermates (115).
Role of Endothelial lipase
Endothelial lipase (EL) is a phospholipase, which belongs to the lipoprotein lipase (LPL) gene family (116). The sequence homology of the lid domain, which determines the specificity of lipases, largely differs between EL and LPL or HL. EL is unique among these lipases, because it is primarily synthesized by vascular endothelial cells, and to a lesser extent by macrophages and smooth muscle cells.
EL acts primarily as a phospholipase and hydrolyzes HDL-phospholipids at the sn-1 position. EL overexpression accelerates renal apoAI catabolism (114). In mice, inhibition of EL increases plasma HDL-C levels (117), whereas HDL-C levels decrease in mice that overexpress EL (118). In humans, plasma EL mass was inversely correlated with plasma HDL-C levels and positively correlated with atherosclerosis and features of the metabolic syndrome (119).
EL appears to have a variety of functions, which may modulate atherosclerosis. Thus, the net effect from EL inhibition on atherosclerosis may be complicated and varied among tissues or cells where EL is expressed, or the presence of inflammation. However, a low frequency EL coding variant in humans that was shown to be loss-of-function and is strongly associated with increased HDL-C levels was not associated with reduced CVD risk. (120).
Role of Hepatic Lipase
Hepatic lipase (HL), a 65-kDa sized glycoprotein, is synthesized primarily by the liver where it is bound to proteoglycans (121) and also by macrophages (122). HL has two principal effects on lipoprotein metabolism (Fig. 2). HL promotes the conversion of chylomicrons to remnants as well as VLDL to LDL. It appears, however, to primarily act on smaller size apoB-containing lipoproteins (121). Increased HL activity is associated with increased amount of the more pro-atherogenic small dense LDL (123). In addition, HL acts on large lipid-rich forms of HDL-L (α-HDL) and converts them into smaller subspecies, including preβ-HDL (121). Consistent with these findings, patients with a genetic deficiency of HL have increased concentrations of IDL-C and higher HDL-C levels (124,125).
Data from various animal models of either over or under expression of HL have been inconsistent with regard to atherosclerosis (126), although as expected HDL-C was inversely related to HL activity. An in vivo RCT model in mice involving intraperitonal macrophages radiolabeled with cholesterol showed that HL deficiency in mice increased the amount of the radiotracer in plasma and raised HDL-C but there was no increase fecal cholesterol excretion (127).
In patients, the role of HL in atherosclerosis has been controversial, because it has properties that can be considered both pro-atherogenic and anti-atherogenic, namely increasing small dense LDL and increasing the cholesterol content of HDL respectively. Because of the smaller number of patients reported, it is difficult to assess the impact of HDL in patients with a genetic deficiency of HL (121,128). Because of the role of HL in promoting hepatic uptake of lipids, it may be that higher levels of HDL-C in subjects with lower HL activity may inhibit the hepatic uptake step of the RCT pathway (129).
Liver Receptors
Hepatic expression of scavenger receptor class B type I (SR-BI) is a positive regulator of macrophage reverse cholesterol transport in vivo (130). CETP-mediated HDL cholesterol exchange to LDL particles, and subsequent clearance by hepatic LDL receptors is a second arm of the RCT pathway. SR-BI binds to HDL via apoA-I and mediates selective uptake of HDL-C, primarily from large cholesterol-enriched HDL particles, by a poorly understood process that differs markedly from classic cellular endocytic uptake of lipoproteins (e.g., LDL binding and internalization via LDL receptors) (126,131–133) (Figure 1). The lipid-depleted HDL particles then dissociate and re-enter the circulation. SR-BI also enhances efflux to HDL by creating a pool of activated cholesterol in the membrane.
Experimental Animal Models of RCT
In order to selectively assay the macrophage-specific RCT pathway, a technique in mice was developed in which macrophages are loaded with cholesterol and labeled with a 3H-cholesterol tracer ex vivo and injected intraperitoneally. Plasma specimens are obtained periodically for measurement of radioactivity levels in plasma and HDL fractions; and feces are collected continuously for measurement of fecal [3H] neutral sterols and [3H] bile acids. The 3H-cholesterol is effluxed to HDL-based acceptor particles that enter the plasma compartment. The appearance of tracer can be followed in plasma over time and the amount of macrophage-derived radiolabeled tracer excreted in feces can be quantitated as a estimate of macrophage-to-feces RCT. Quantification of bile acids and neutral sterols can be incorporated in the study to ascertain the net balance of fecal bile acids and neutral sterols. A variation of this model involves injection of [3H] cholesterol macrophages into the peritoneal cavity of Golden Syrian hamsters (98), an attractive model because unlike mice hamsters naturally express CETP. Both cholesterol loaded J774 macrophages as well as macrophages isolated from the mouse peritoneal cavity following [3H] cholesterol injection have been employed in these hamster studies. The immunologic response of the hamsters to the murine macrophages and the potential impact on the RCT process has not been assessed.
Using this macrophage to feces approach, apoA-I overexpression was shown to promote (130) and apoA-I deficiency to impair (134) macrophage RCT, consistent with the atheroprotective role of apoA-I. Multiple studies have assessed macrophage-specific RCT in the setting of genetic and pharmacologic manipulation of mice, and the effects on RCT (in contrast to the effects on HDL-C) are largely consistent with the effects of the same interventions on atherosclerosis (135). Overall, the dynamic rate of macrophage RCT correlates much better than steady-state plasma HDL-C level with atherosclerosis, suggesting that it is measuring an atheroprotective process and could be used to predict the anti-atherosclerotic effects of a novel HDL-targeted intervention. Thus, methods for assessing the integrated rate of macrophage RCT in animals have provided insights into the molecular regulation of the process. Animal studies of macrophage RCT suggest that promotion of cholesterol efflux from macrophages is a potential therapeutic approach to preventing or regressing atherosclerotic vascular disease.
Recently, however, two experimental studies demonstrate fecal cholesterol excretion is not always a pre-requisite for macrophage cholesterol efflux. For instance, infusion of rHDL stimulated cholesterol efflux from extrahepatic tissues and resulted in net flux to the liver without increasing fecal cholesterol excretion (10). Intravenous infusion of the 5A peptide, an apoA-I mimetic peptide, complexed with phospholipid increased cholesterol efflux, measured in vivo by a stable isotope kinetic technique, from ABCA1 and ABCG1-transfected macrophages and reduced atherosclerosis in apoE-KO mice, without changing net fecal excretion of neutral sterols or bile acids (11).
Human Models of Cholesterol Efflux
Robust and sensitive methods for assessing integrated RCT in humans are needed in order to advance insights gained in animals into the human realm and to assess novel therapies targeted toward HDL and RCT (136,137). A comparison of the contribution of efflux pathways of high and low efficiency HDL demonstrated that the increased cholesterol efflux observed in the high efficiency sera was largely attributed to greater efflux via the ABCA1 pathway (137). These observations indicate that HDL quality, particular in terms of HDL subfraction distribution, is a more important parameter in regulating efflux than is total HDL-C or apoA-I. Thus, the demonstration that cholesterol efflux capacity is associated with atherosclerosis further support the use of the measurement of HDL efflux in guiding the development of new HDL-target therapies for humans. While this ex vivo approach supports the concept that the capacity of HDL in promoting efflux is more important than its concentration, a more effective approach would be to develop a clinical method to determine cholesterol flux in humans and in particular the efflux from cholesterol loaded macrophages in coronary plaques. As discussed above, at present one potential approach is the quantification of the mass of fecal sterol and bile acid excretion as a surrogate for RCT. For example, an acute intravenous bolus infusion of pro-apoA-I in humans was found to result in a significant increase in fecal sterol excretion, suggesting promotion of RCT (138), however no increase in fecal sterol excretion in the steady state was reported in patients receiving CETP inhibitors (139). However, this approach is not macrophage-specific, is unlikely to be very sensitive, and may have limited utility in the chronic steady state setting due to counter-regulatory pathways involved in biliary cholesterol excretion and fecal sterol absorption.
An isotope kinetic modeling technique has been developed that involves the intravenous infusion of stable isotopically labeled cholesterol for ~24 hours, with frequent blood sampling for analysis of plasma free cholesterol, red blood cell cholesterol and plasma cholesterol-ester isotope enrichments by mass spectrometry. In addition, stool is collected over subsequent days during oral sitostanol intake, for isolation of fecal bile acid and neutral sterols for mass and isotope enrichments. Simulation modeling (SAAM) is used to calculate the rate of whole body efflux of free cholesterol from tissues into the plasma compartment, after correcting for red blood cell exchange with plasma free cholesterol. Also calculated are the esterification rates of free cholesterol to cholesterol-ester, clearance of cholesterol-ester from the blood; and flux from plasma cholesterol into fecal bile acids and neutral sterols (140). Absolute and relative RCT flux rates in humans have proven to be of interest. Esterification is ~1.3 mg/kg/hr (~2 g/d), which is of similar magnitude as total fecal excretion of sterols, suggesting that the irreversible LCAT step in plasma may be flux generating for sterol excretion from the whole body. Moreover, esterification represents a substantial fate (~40%) of the 3.2 mg/kg/hr (~5g/d) non-erythrocyte-derived free cholesterol efflux into plasma. Interestingly, exchange of red blood cell with plasma free cholesterol (~2.5mg/kg/hr) is of comparable magnitude as non-red cell efflux, potentially providing a non-lipoprotein fallback mechanism for cholesterol carriage and transport in the RCT pathway. Flux of plasma free cholesterol into fecal sterols was 3.4% and 13.4% over 7 days into fecal bile acids and neutral sterols, respectively. Coefficient of variation of the measurement was 10–15% in humans. The consequences of hypoalphalipoprotemia due to ABCAI or ApoAI genetic alterations were explored in a Dutch population (141). Among low HDL-C participants, there was ~40% reduction in efflux despite normal esterification and fecal excretion rates. However, not all hypoalphalipoproteinemia subjects had low efflux.
As with fecal sterol excretion, this method is not macrophage-specific. Accordingly, it remains to be established whether this method, which measures whole body cholesterol efflux and therefore includes non-arterial wall and non-macrophage arterial wall efflux (Table 1), will have utility in determining cholesterol efflux specifically from cholesterol loaded coronary macrophages in humans following therapeutic intervention. Furthermore, its ability to differentiate between hepatic and peripheral (non-hepatic) tissues as the source of cholesterol efflux has not been definitively proven, although prior human data with radiolabeled cholesterol (142) suggests that hepatic and plasma free cholesterol pools are likely to be fully equilibrated after 24 hours of continuous labeled cholesterol infusion, so that the liver will not contribute to measured flux rates. Other metrics of the RCT pathway generated by this stable isotope kinetic modeling approach, including plasma cholesterol-ester production and clearance rates, plasma cholesterol flux into specific neutral sterols and bile acids, and the quantitative role of the red cell membrane as a lipoprotein-independent pathway for cholesterol carriage, are also potentially useful. The combination of a macrophage-specific assay with other components of whole-body RCT has resulted in an improved understanding of cholesterol efflux pathways.
Vascular Imaging of Atherosclerosis
Although certain ex vivo HDL measures (qualitative and quantitative) are more highly correlated with atherosclerosis than HDL-C, new vascular imaging modalities provide opportunities for direct in vivo assessment of HDL-mediated cholesterol efflux. The ideal invasive tool for characterization of vascular plaque would provide a complete roadmap of atherosclerotic burden throughout the coronary tree and provide per plaque lesion specific data characterizing the architecture, composition and dynamic biology of each plaque. Specific parameters should include: (1) extent of luminal stenosis; (2) lesion length; (3) coronary blood flow reserve through any given narrowing; (4) intramural plaque architecture including atheroma burden, eccentricity, and local vascular remodeling; (5) plaque composition, specifically lipid content; (6) fibrous cap thickness; and (7) presence of inflammation (143–145).
Certain vascular imaging modalities may be particularly useful techniques for investigation into the processes that regulate cholesterol flux within the vessel wall (143). Structural changes in the vessel wall include overall measures of vessel wall thickness to specific measures of plaque size, composition and inflammatory burden (143–145). The composition and the inflammatory activity of the atheromatous plaque have important implications for clinical events (154). The studies that have evaluated the effects of HDL modifying therapies on non-invasive and invasive imaging modalities of atherosclerosis are summarized in Appendix 1.
Non-invasive imaging of atherosclerosis can be utilized to assess efficacy as well as provide pathophysiological information on the mechanism of action of novel of HDL-C–raising drugs. Much effort has been made over the past decades to develop ultrasound, MRI, and most recently FDG-PET to provide efficacy measures that can be used to assist in decision-making and whether or not to proceed with novel anti-atherogenic drugs in costly and time-consuming mortality and morbidity trials. The current armamentarium of imaging modalities is capable of providing insight in drug effects on atherosclerosis progression and composition, endothelial function, and vessel wall inflammation and will play a crucial role in evaluating emerging HDL-C–raising compounds. Recently, advanced vascular imaging was used to investigate the effects of the CETP inhibitor/modulator dalcetrapib on carotid plaque progression and inflammation (146). After 24 months, there was a significant reduction in carotid total vessel area with dalcetrapib compared to placebo. Although there were there were no treatment differences in carotid inflammation by FDG-PET, reductions in plaque inflammatory index were correlated with increases in HDL-C.
Prospective clinical studies will indicate which of these features identified by vascular imaging provides an accurate prediction of future coronary events. The High-Risk Plaque (HRP) BioImage Study, a total of 6,104 men 55 to 80 years of age and women 60 to 80 years of age without evidence of atherothrombotic disease but presumed to be at increased risk for near-term atherothrombotic events have been enrolled in a prospective trial of comprehensive risk assessment in a dedicated mobile facility equipped with advanced imaging tools (147).
Proposed Updated Concept of Reverse Cholesterol Transport
In this review, we endorse the continued use of the terminology “reverse cholesterol transport” to describe whole body cholesterol efflux from peripheral tissues to its ultimate disposition in the feces. However, this terminology is imprecise with regard to an assessment of changes in the cholesterol loaded vascular macrophage and its use has led to substantial confusion in the field of HDL biology. The adoption of a nomenclature that identifies the specific component of RCT will improve communication of experimental animal models and human translational studies that investigate various aspects of tissue-specific cholesterol efflux. The use of the term, “macrophage RCT”, more precisely describes the cholesterol removal from the cholesterol-loaded macrophage that is ultimately removed in the feces. Quantitation of fecal sterols and bile acids rather than radioactivity alone provides a more definitive analysis of sterol balance in these studies. At present, quantitation of macrophage RCT can only be ascertained in animal models; however, studies are underway to develop methods for use in humans. The ultimate choice in the evaluation of therapeutic agents for this aspect of HDL functionality is direct quantitation of cholesterol efflux from the vascular macrophage. Recent studies have established that the fraction of HDL-C coming from these cells is a very small and most likely does not change HDL-C levels. Furthermore, cholesterol efflux from macrophages because of is relatively small pool size would not be expected to significantly alter the total mass of fecal sterol or bile acid excretion on a mass basis. Thus, both HDL-C or fecal sterol and bile acid loss may be either insensitive or ineffective methods to quantitate cholesterol changes in vascular macrophage, which has been proposed to reduce cardiovascular events. At present in vitro quantitation of serum efflux capacity or plasma preβ-HDL assays have been use to assess the effectiveness of plasma HDL to reduce macrophage cholesterol content. However, the recycling and flux through the preβ-HDL to HDL-L (α1-HDL) pathway cannot be determined by the static quantitation of serum efflux capacity or preβ-HDL assays thus limiting the effectiveness of this approach. At this time, the most effective approach to determine the effect of agents on vascular cholesterol content will involve invasive and noninvasive imaging techniques. Great progress is currently being made in imaging methodology and effective approaches to the quantitation of vascular plaque may be available in the near future. Ultimately, these advanced vascular imaging techniques will require validation with clinical cardiovascular events.
Supplementary Material
Acknowledgments
Funding sources
This work emanated from a CME activity sponsored by Medical Education Resources in Littleton CO. MER received funding from Genentech, Inc. The funding agency had no input in the content of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: Dr. Rosenson has served on advisory boards and received consulting fees and honorarium from Abbott Labs (modest), Amgen (modest), Amaryn (modest), Astra Zeneca (modest), Genentech (modest), Grain Foods Advisory Council (modest), LipoScience (modest), GlaxoSmithKline (modest), Residual Risk Reduction Initiative (modest) and Sanofi Aventis (significant), and he has stock ownership in LipoScience, Inc (significant). Dr. Brewer, Jr. serves on advisory boards, received consulting fees and honoraria from Abbott (modest), Eli Lilly & Co (modest), Merck (significant), Pfizer, Roche (significant), and Sanofi-Aventis (modest); speaker’s bureau for Astra Zeneca (modest), Abbott (modest) Merck (modest) Pfizer (modest) and Roche (modest); and ownership interest in InfraRedDx Inc. (significant), Medicines Company (significant) and NDL Therapeutics (significant). Dr. Davidson serves on the Speaker’s Bureau and received honorarium from Abbott Labs and Merck (moderate). Dr. Fayad has served on advisory boards and received honorarium from Roche (significant) and research support from Bristol Myers Squibb (significant), GlaxoSmithKline (significant), Merck (significant), Roche (significant) and VBL (significant). Dr. Fuster reports no disclosures. Dr. Goldstein serves on advisory board and receives honorarium from InfraReDx, Inc. (significant) and has stock ownership in InfraReDx Inc. (significant). Dr. Hellerstein is co-founder and chief of the scientific advisory board for KineMed Inc. (significant) and has stockownership in KineMed Inc (significant). Dr. Jiang reports no disclosures. Dr. Phillips reports no disclosures. Dr. Remaley receives research support from Alpha Care-CRADA (modest) and KineMed, Inc. (significant). Dr. Rader serves as a consultant and receives honorarium from Ainlam (moderate), Astra Zeneca (moderate), Daiichi Sankyo (moderate), Eli Lilly & Co (moderate), Glaxo Smith Kline (moderate), Johnson & Johnson (significant), Merck (significant), Novartis (moderate), Omthera (moderate), Pfizer (moderate), Regeneron (moderate), Sanofi Aventis (moderate), and has has stock ownership in Aegerion Pharmaceuticals (significant) and Vascular Strategies (significant). Dr. Rothblat has stock ownership in Vascular Strategies (significant). Dr. Tall serves on advisory boards for CSL (modest), Merck (modest), Regulus (modest) and Roche (modest), speaker’s bureau for Merck (modest), and receives honorarium form Novartis (modest). Dr. Yvan-Charvet reports no disclosures.
References
- 1.Rosenson RS, Brewer HB, Chapman J, Fazio S, Hussain M, Kontush A, Krauss RM, Otvos JD, Remaley AT, Schaefer EJ. HDL measures, particle heterogeneity, proposed nomenclature, and relation to atherosclerotic cardiovascular events. Clin Chem. 2011;57:392–410. doi: 10.1373/clinchem.2010.155333. [DOI] [PubMed] [Google Scholar]
- 2.Glomset JA, Wright JL. Some properties of a cholesterol esterifying enzyme in human plasma. Biochim Biophys Acta. 1964;89:266–276. doi: 10.1016/0926-6569(64)90215-9. [DOI] [PubMed] [Google Scholar]
- 3.Glomset JA. The plasma lecithin: cholesterol acyltransferase reaction. J Lipid Res. 1968;9:155–167. [PubMed] [Google Scholar]
- 4.Haghpassand M, Bourassa PA, Francone OL, Aiello RJ. Monocyte/macrophage expression of ABCA1 has minimal contribution to plasma HDL Levels. J Clin Invest. 2001;108:1315–1320. doi: 10.1172/JCI12810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vaisman BL, Lambert G, Amar M, Joyce C, Ito T, Shamburek RD, Cain WJ, Fruchart-Najib J, Neufeld ED, Remaley AT, Brewer HB, Jr, Santamarina-Fojo S. ABCA1 overexpression leads to hyperalphalipoproteinemia. J Clin Invest. 2001;108:303–309. doi: 10.1172/JCI12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wellington CL, Brunham LR, Zhou S, Singaraja RR, Visscher H, Gelfer A, Ross C, James E, Liu G, Huber MT, Yang YZ, Parks RJ, Groen A, Fruchart-Najib J, Hayden MR. Alterations of plasma lipids in mice via adenoviral-mediated hepatic over-expression of human ABCA1. J Lipid Res. 2003;44:1470–1480. doi: 10.1194/jlr.M300110-JLR200. [DOI] [PubMed] [Google Scholar]
- 7.Timmins JM, Lee JY, Boudyguina E, Kluckman KD, Brunham LR, Mulya A, Gebre AK, Coutinho JM, Colvin PL, Smith TL, Hayden MR, Maeda N, Parks JS. Targeted inactivation of hepatic ABCA1 causes profound hypoalphalipoproteinemia and kidney hypercatabolism of ApoA-I. J Clin Invest. 2005;115:1333–1342. doi: 10.1172/JCI23915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brunham LR, Kruit JK, Iqbal J, Fievet C, Timmins JM, Pape TD, Coburn BA, Bissada N, Staels B, Groen AK, Hussain MM, Parks JS, Kuipers F, Hayden MR. Intestinal ABCA1 directly contributes to HDL biogenesis in vivo. J Clin Invest. 2006;116:1052–1062. doi: 10.1172/JCI27352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vergeer M, Korporaal SJA, Franssen R, Meurs I, Out R, Hovingh GK, Hoekstra M, Sierts JA, Dallinga-Thie GM, Motazacker MM, Holleboom AG, Van Berkel TJC, Kastelein JJP, Van Eck M, Kuivenhoven JA. Genetic variant of the scavenger receptor B1 in humans. N Engl J Med. 2011;364:136–145. doi: 10.1056/NEJMoa0907687. [DOI] [PubMed] [Google Scholar]
- 10.Alam K, Meidell RS, Spady DK. Effect of up-regulating individual steps in the reverse cholesterol transport pathway on reverse cholesterol transport in normolipidemic mice. J Biol Chem. 2001;276:15641–15649. doi: 10.1074/jbc.M010230200. [DOI] [PubMed] [Google Scholar]
- 11.Amar MJ, D’Souza W, Turner S, Demosky S, Sviridov D, Stonik J, Luchoomun J, Voogt J, Hellerstein M, Sviridov D, Remaley AT. 5A apolopoprtoein mimetic peptide promotes cholesterol efflux and reduces atherosclerosis in mice. J Pharmacol Exp Ther. 2010;334:634–641. doi: 10.1124/jpet.110.167890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Emerging Risk Factors Collaboration. Di Angelantonio E, Sarwar N, Perry P, Kaptoge S, Ray KK, Thompson A, Wood AM, Lewington S, Sattar N, Packard CJ, Collins R, Thompson SG, Danesh J. Major lipids, apolipoproteins and risk of vascular disease. JAMA. 2009;302:1993–2000. doi: 10.1001/jama.2009.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baigent C, Blackwell L, Emberson J, Holland LE, Reith C, Bhala N, Peto R, Barnes EH, Keech A, Simes J, Collins R, Cholesterol Treatment Trialists’ (CTT) Collaborators Efficacy and safety of more intensive loweirng of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials of statins. Lancet. 2010;376:1670–1681. doi: 10.1016/S0140-6736(10)61350-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Silva RA, Huang R, Morris J, Fang J, Gracheva EO, Ren G, Kontush A, Jerome WG, Rye KA, Davidson WS. Structure of apolipoprotein A-I in spherical high density lipoproteins of different sizes. Proc Natl Acad Sci USA. 2008;105:12176–12181. doi: 10.1073/pnas.0803626105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang R, Silva RAGD, Jerome WG, Kontush A, Chapman MJ, Curtiss LK, Hodges TJ, Davidson WS. Apolipoprotein A-I structural organization in high-density lipoproteins isolated from human plasma. Nat Struct Mol Biol. 2011;18:416–422. doi: 10.1038/nsmb.2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Catte A, Patterson JC, Bashtovyy D, Jones MK, Gu F, Li L, Rampioni A, Sengupta D, Vuorela T, Niemelä P, Karttunen M, Marrink SJ, Vattulainen I, Segrest JP. Structure of spheroidal HDL particles revealed by combined atomistic and coarse-grained simulations. Biophys J. 2008;94:2306–2319. doi: 10.1529/biophysj.107.115857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rezaee F, Casetta B, Levels JHM, Speijer D, Meijers JCM. Proteomic analysis of high-density lipoprotein. Proteomics. 2006;6:721–730. doi: 10.1002/pmic.200500191. [DOI] [PubMed] [Google Scholar]
- 18.Karlsson H, Leanderson P, Tagesson C, Lindahl M. Lipoproteomics II: mapping of proteins in high-density lipoprotein using two-dimensional gel electrophoresis and mass spectrometry. Proteomics. 2005;5:1431–1445. doi: 10.1002/pmic.200401010. [DOI] [PubMed] [Google Scholar]
- 19.Heller M, Stalder D, Schlappritzi E, Hayn G, Matter U, Haeberli A. Mass spectrometry-based analytical tools for the molecular protein characterization of human plasma lipoproteins. Proteomics. 2005;5:2619–2630. doi: 10.1002/pmic.200401233. [DOI] [PubMed] [Google Scholar]
- 20.Vaisar T, Pennathur S, Green PS, Gharib SA, Hoffnagle AN, Cheung MC, Byun J, Vuletic S, Kassim S, Singh P, Chea H, Knopp RH, Brunzell J, Geary R, Chait A, Zhao X, Elkon K, Marcovina S, Ridker P, Oram JF, Heinecke JW. Shotgun proteomics implicates protease inhibition and complement activation in the antiinflammatory properties of HDL. J Clin Invest. 2007;117:746–756. doi: 10.1172/JCI26206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davidson WS, Silva GD, Chantepie S, Lagor WR, Chapman MJ, Kontush A. Proteomic analysis of defined HDL subpopulations reveals particle-specific protein clusters: relevance to antioxidative function. Arterioscler Thromb Vasc Biol. 2009;29:870–876. doi: 10.1161/ATVBAHA.109.186031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vanhamme L, Paturiaux-Hanosq F, Poelvoorde P, Nolan DP, Lins L, Den Abbeele JV, Pays A, Tebabi P, Xong HV, Jacquet A, Moguilevsky N, Dieu M, Kane JP, Baetselier PD, Brasseur R, Pays E. Apolipoprotein L-I is the trypanosome lytic factor of human serum. Nature. 2003;422:83–87. doi: 10.1038/nature01461. [DOI] [PubMed] [Google Scholar]
- 23.Alaupovic P. The concept of apolipoprotein-defined lipoprotein families and its clinical significance. Curr Atheroscler Rep. 2003;5:459–67. doi: 10.1007/s11883-003-0036-8. [DOI] [PubMed] [Google Scholar]
- 24.Chapman MJ, Goldstein S, Lagrange D, Laplaud PM. A density gradient ultracentrifugal procedure for the isolation of the major lipoprotein classes from human serum. J Lipid Res. 1981;22:339–358. [PubMed] [Google Scholar]
- 25.Sviridov D, Nestel P. Dynamics of reverse cholesterol transport: protection against atherosclerosis. Atherosclerosis. 2002;161:245–254. doi: 10.1016/s0021-9150(01)00677-3. [DOI] [PubMed] [Google Scholar]
- 26.Kontush A, Chantepie S, Chapman MJ. Small, dense HDL particles exert potent protection of atherogenis LDL against oxidative stress. Arterioscler Thromb Vasc Biol. 2003;23:1881–1888. doi: 10.1161/01.ATV.0000091338.93223.E8. [DOI] [PubMed] [Google Scholar]
- 27.Morita SY, Okuhira K, Tsuchimoto N, Vertut-Doi A, Saito H, Nakano M, Handa T. Effects of sphingomyelin on apolipoprotein E- and lipoprotiein lipase-mediated cell uptake of lipid particles. Biochim Biophys Acta. 2003;163:169–176. doi: 10.1016/s1388-1981(02)00365-7. [DOI] [PubMed] [Google Scholar]
- 28.Bolin DJ, Jonas A. Sphinomyelin inhibits the lecithin-cholesterol acyltransferase reaction with reconstituted high density lipoproteins by decreasing enzyme binding. J Biol Chem. 1996;271:19152–191528. doi: 10.1074/jbc.271.32.19152. [DOI] [PubMed] [Google Scholar]
- 29.Subbaiah PV, Liu M. Role of sphingomyelin in the regulation of cholesterol esterification in the plasma lipoproteins. Inhibiton of lecithin-cholesterol acyltransferase reaction. J Biol Chem. 1993;268:20156–20163. [PubMed] [Google Scholar]
- 30.Rothblat GH, Phillips MC. High-density lipoprotein heterogeneity and function in reverse cholesterol transport. Curr Opin Lipidol. 2010;21:229–238. doi: 10.1097/mol.0b013e328338472d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adorni MP, Zimetti F, Billheimer JT, Wang N, Rader DJ, Phillips MC, Rothblat GH. The roles of different pathways in the release of cholesterol from macrophages. J Lipid Res. 2007;48:2453–2462. doi: 10.1194/jlr.M700274-JLR200. [DOI] [PubMed] [Google Scholar]
- 32.Larrede S, Quinn CM, Jessup W, Frisdal E, Olivier M, Hsieh V, Kim M-J, Van Eck M, Couvert P, Carrie A, Giral P, Chapman MJ, Guerin M, Le Goff W. Stimulation of cholesterol efflux by LXR agonists in cholesterol-loaded human macrophages is ABCA1-dependent but ABCG1-independent. Arterioscler Thromb Vasc Biol. 2009;29:1930–1936. doi: 10.1161/ATVBAHA.109.194548. [DOI] [PubMed] [Google Scholar]
- 33.Phillips MC, Johnson WJ, Rothblat GH. Mechanisms and consequences of cellular cholesterol exchange and transfer. Biochim Biophys Acta. 1987;906:223–276. doi: 10.1016/0304-4157(87)90013-x. [DOI] [PubMed] [Google Scholar]
- 34.Davidson WS, Rodrigueza WV, Lund-Katz S, Johnson WJ, Rothblat GH, Phillips MC. Effects of acceptor particle size on the efflux of cellular free cholesterol. J Biol Chem. 1995;270:17106–17113. doi: 10.1074/jbc.270.29.17106. [DOI] [PubMed] [Google Scholar]
- 35.Duong PT, Collins HL, Nickel M, Lund-Katz S, Rothblat GH, Phillips MC. Characterization of nascent HDL particles and microparticles formed by ABCA1-mediated efflux of cellular lipids to apoA-I. J Lipid Res. 2006;47:832–843. doi: 10.1194/jlr.M500531-JLR200. [DOI] [PubMed] [Google Scholar]
- 36.Mulya A, Lee J-Y, Gebre AK, Thomas MJ, Colvin PL, Parks JS. Minimal lipidation of pre-β HDL by ABCA1 results in reduced ability to interact with ABCA1. Arterioscler Thromb Vasc Biol. 2007;27:1828–1836. doi: 10.1161/ATVBAHA.107.142455. [DOI] [PubMed] [Google Scholar]
- 37.Neufeld EB, Remaley AT, Demosky SJ, Stonik JA, Cooney A, Comly M, Dwyer NK, Zhang M, Blanchette-Mackie EJ, Santamarina-Fojo S, Brewer HB., Jr Cellular localization and trafficking of the human ABCA1 transporter. J Biol Chem. 2001;276:27584–27590. doi: 10.1074/jbc.M103264200. [DOI] [PubMed] [Google Scholar]
- 38.Vedhachalam C, Duong PT, Nickel M, Nguyen D, Dhanasekaran P, Saito H, Rothblat GH, Lund-Katz S, Phillips MC. Mechanism of ATP-binding cassette transporter A-mediated cellular lipid efflux to apolipoprotein A-I and formation of high density lipoprotein particles. J Biol Chem. 2007;282:25123–23130. doi: 10.1074/jbc.M704590200. [DOI] [PubMed] [Google Scholar]
- 39.Nicholls SJ, Cutri B, Worthley SG, Kee P, Rye K-A, Bao S, Barter PJ. Impact of short-term administration of high-density lipoproteins and atorvastatin on atherosclerosis in rabbits. Arterioscler Thromb Vasc Biol. 2005;25:2416–2421. doi: 10.1161/01.ATV.0000184760.95957.d6. [DOI] [PubMed] [Google Scholar]
- 40.Nissen SE, Tsunoda T, Tuzcu EM, Schoenhagen P, Cooper CJ, Yasin M, Eaton GM, Lauer MA, Sheldon WS, Grines CL, Halpern S, Crowe T, Blankenship JC, Kerensky R. Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: a randomized controlled trial. JAMA. 2003;290:2292–2300. doi: 10.1001/jama.290.17.2292. [DOI] [PubMed] [Google Scholar]
- 41.Tardif JC, Grégoire J, L’Allier PL, Ibrahim R, Lespérance J, Heinonen TM, Kouz S, Berry C, Basser R, Lavoie MA, Guertin MC, Rodés-Cabau J. Effect of rHDL on Atherosclerosis-Safety and Efficacy (ERASE) Investigators. Effects of reconstituted high-density lipoprotein infusions on coronary atherosclerosis: a randomized controlled trial. JAMA. 2007;297:1675–1682. doi: 10.1001/jama.297.15.jpc70004. [DOI] [PubMed] [Google Scholar]
- 42.Sacks FM, Rudel LL, Conner A, Akeefe H, Kostner G, Baki T, Rothblat G, de la Llera-Moya M, Asztalos B, Perlman T, Zheng C, Alaupovic P, Maltais JA, Brewer HB., Jr Selective delipidation of plasma HDL enhances reverse cholesterol transport in vivo. J Lipid Res. 2009;50:894–907. doi: 10.1194/jlr.M800622-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Waksman R, Torguson R, Kent KM, Pichard AD, Suddath WO, Satler LF, Martin BD, Perlman TJ, Maltais J, Weissman NJ, Fitzgerald PJ, Brewer HB., Jr A first-in-man, randomized, placebo-controlled study to evaluate the safety and feasibility of autologous delipidated HDL plasma infusions in patients with acute coronary syndrome. J Am Coll Cardiol. 2010;55:2727–2735. doi: 10.1016/j.jacc.2009.12.067. [DOI] [PubMed] [Google Scholar]
- 44.Van Lenten BJ, Wagner AC, Navab M, Anantharamaiah GM, Hui EK, Nayak DP, Fogelman AM. D-4F, an apolipoprotein A-I mimetic peptide, inhibits the inflammatory response induced by influenza A infection of human type II pneumocytes. Circulation. 2004;110:3252–3258. doi: 10.1161/01.CIR.0000147232.75456.B3. [DOI] [PubMed] [Google Scholar]
- 45.Van Lenten BJ, Wagner AC, Jung CL, Ruchala P, Waring AJ, Lehrer RI, Watson AD, Hama S, Navab M, Anantharamaiah GM, Fogelman AM. Anti-inflammatory ApoA-I-mimetic peptides bind oxidized lipids with much higher affinity than human ApoA-I. J Lipid Res. 2008;49:2302–2311. doi: 10.1194/jlr.M800075-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kontush A, Chapman MJ. Functionally defective high-density lipoprotein: a new therapeutic target at the crossroads of dyslipidemia, inflammation, and atherosclerosis. Pharmacol Rev. 2006;58:342–374. doi: 10.1124/pr.58.3.1. [DOI] [PubMed] [Google Scholar]
- 47.Shao B, Oda MN, Oram JF, Heinecke Myeloperoxidase: an oxidative pathway for generating dysfunctional high-density lipoprotein. Chem Res Toxicol. 2010;23:447–454. doi: 10.1021/tx9003775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vaughan J, Oram JF. ABCG1 redistributes cell cholesterol to domains removable by high density lipoprotein but not by lipid-depleted apolipoproteins. J Biol Chem. 2005;280:30150–30157. doi: 10.1074/jbc.M505368200. [DOI] [PubMed] [Google Scholar]
- 49.Sankaranarayanan S, Oram JF, Asztalos BF, Vaughan AM, Lund-Katz S, Adorni MP, Phillips MC, Rothblat GH. Effects of acceptor composition and mechanism of ABCG1-mediated cellular free cholesterol efflux. J Lipid Res. 2009;50:275–284. doi: 10.1194/jlr.M800362-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang N, Lan D, Chen W, Matsuura F, Tall AR. ATP-binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high-density lipoproteins. Proc Natl Acad Sci USA. 2004;101:9774–9779. doi: 10.1073/pnas.0403506101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Costet P, Luo Y, Wang N, Tall AR. Sterol-dependent transactivation of the ABC1 promoter by the liver X receptor/retinoid X receptor. J Biol Chem. 2000;275:28240–8245. doi: 10.1074/jbc.M003337200. [DOI] [PubMed] [Google Scholar]
- 52.Kennedy MA, Venkateswaran A, Tarr PT, Xenarios I, Kudoh J, Shimizu N, Edwards PA. Characterization of the human ABCG1 gene: liver X receptor activates an internal promoter that produces a novel transcript encoding an alternative form of the protein. J Biol Chem. 2001;276:39438–39447. doi: 10.1074/jbc.M105863200. [DOI] [PubMed] [Google Scholar]
- 53.Laffitte BA, Repa JJ, Joseph SB, Wilipitz DC, Kast HR, Mangelsdorf DJ, Totonoz P. LXRs control lipid-inducible expression of the apolipoprotein E gene in macrophages and adipocytes. Proc Natl Acad Sci USA. 2001;98:507–512. doi: 10.1073/pnas.021488798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jankowski BA, Willy PJ, Devi TR, Falck JR, Mangelsdorf DJ. An oxyserol signaling pathway mediated by the nuclear receptor LXR alpha. Nature. 1996;383:728–731. doi: 10.1038/383728a0. [DOI] [PubMed] [Google Scholar]
- 55.Pagler TA, Wang M, Mondal M, Murphy AJ, Westerterp M, Moore KJ, Maxfield FR, Tall AR. Deletion of ABCA1 and ABCG1 impairs macrophage migration because of increased Rac1 signaling. Circ Res. 2011;108:194–200. doi: 10.1161/CIRCRESAHA.110.228619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yvan-Charvet L, Ranalletta M, Wang N, Han S, Terasaka N, Li R, Welch C, Tall AR. Combined deficiency of ABCA1 and ABCG1 promotes foam cell accumulation and accelerates atherosclerosis in mice. J Clin Invest. 2007;117:3900–3908. doi: 10.1172/JCI33372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yvan Charvet L, Welch C, Pagler TA, Ranaletta M, Lamkanfi M, Han S, Ishibashi M, Li R, Wang N, Tall AR. Increased inflammatory gene expression in ABC transporter-deficient macrophages: free cholesterol accumulation, increased signaling via toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation. 2008;118:1837–1847. doi: 10.1161/CIRCULATIONAHA.108.793869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rayner KJ, Suarez Y, Davalos A, Parathath S, Fitzgerald ML, Tamehiro N, Fisher EA, Moore KJ, Fernandez-Hernando C. MiR contributes to the regulation of cholesterol homeostasis. Science. 2010;328:1570–1573. doi: 10.1126/science.1189862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Najafi-Shoushtari SH, Kristo F, Li Y, Shioda T, Cohen DE, Gerszten RE, Näär AM. MicroRNA-33 and the SREBP host genes cooperate to control cholesterol homeostasis. Science. 2010;328:1566–1569. doi: 10.1126/science.1189123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Marquart TJ, Allen RM, Ory DS, Baldan A. miR-33 links SREBP-2 induction to repression of sterol transporters. Proc Natl Acad Sci USA. 2010;107:12228–12232. doi: 10.1073/pnas.1005191107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Horie T, Ono K, Horiguchi M, Nishi H, Nakamura T, Nagao K, Kinosita M, Kuwabara Y, Marusawa H, Iwanaga Y, Hasegawa K, Yokode M, Kimura T, Kita T. MicroRNA-33 encoded by an intron of sterol regulatory element-binding protein 2 (SREBP2) regulates HDL in vivo. Proc Natl Acad Sci USA. 2010;107:17321–17326. doi: 10.1073/pnas.1008499107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rayner KJ, Sheedy FJ, Esau CC, Hussain FN, TGemel RE, Paratath S, van Gils JM, Rayner AJ, Chang AN, Suarez Y, Fernandez-Hernancdo C, Fisher EA, Moore KJ. Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J Clin Invest. 2011;121:2921–2931. doi: 10.1172/JCI57275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ji Y, Jian B, Wang N, Sun Y, de la Llera Moya M, Phillips MC, Rothblat GH, Swaney JB, Tall AR. Scavenger receptor BI promotes high density lipoprotein-mediated cellular cholesterol efflux. J Biol Chem. 1997;272:20982–20985. doi: 10.1074/jbc.272.34.20982. [DOI] [PubMed] [Google Scholar]
- 64.Jian B, de la Llera-Moya M, Ji Y, Wang N, Phillips MC, Swaney JB, Tall AR, Rothblat GH. Scavenger receptor class B type I as a mediator of cellular cholesterol efflux to lipoproteins and phospholipid acceptors. J Biol Chem. 1998;273:5599–5606. doi: 10.1074/jbc.273.10.5599. [DOI] [PubMed] [Google Scholar]
- 65.Zhang Y, Zanotti I, Reilly MP, Glick JM, Rothblat GH, Rader DJ. Overexpression of apolipoprotein A-I promotes reverse transport of cholesterol from macrophages to feces in vivo. Circulation. 2003;108:661–663. doi: 10.1161/01.CIR.0000086981.09834.E0. [DOI] [PubMed] [Google Scholar]
- 66.Covey SD, Krieger M, Wang W, Penman M, Trigatti BL. Scavenger receptor class B type I-mediated protection against atherosclerosis in LDL receptor-negative mice involves its expression in bone marrow-derived cells. Arterioscler Thromb Vasc Biol. 2003;23:1589–1594. doi: 10.1161/01.ATV.0000083343.19940.A0. [DOI] [PubMed] [Google Scholar]
- 67.Van Eck M, Bos IS, Hildebrand RB, Van Rij BT, Van Berkel TJ. Dual role for scavenger receptor class B, type I on bone marrow-derived cells in atherosclerotic lesion development. Am J Pathol. 2004;165:785–794. doi: 10.1016/S0002-9440(10)63341-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brundert M, Heeren J, Bahar-Bayansar M, Ewert A, Moore KJ, Rinninger F. Selective uptake of HDL cholesteryl esters and cholesterol efflux from mouse peritoneal macrophages independent of SR-BI. J Lipid Res. 2006;47:2408–2421. doi: 10.1194/jlr.M600136-JLR200. [DOI] [PubMed] [Google Scholar]
- 69.Yvan-Charvet L, Pagler TA, Wang N, Senokuchi T, Brundert M, Li H, Rinninger F, Tall AR. SR-BI inhibits ABCG1-stimulated net cholesterol efflux from cells to plasma HDL. J Lipid Res. 2008;49:107–114. doi: 10.1194/jlr.M700200-JLR200. [DOI] [PubMed] [Google Scholar]
- 70.Wang X, Collins HL, Ranalletta M, Fuki IV, Rothblat GH, Tall AR, Rader DJ. Macrophage ABCA1 and ABCG1, but not SR-BI, promote macrophage reverse cholesterol transport in vivio. J Clin Invest. 2007;117:2216–2224. doi: 10.1172/JCI32057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ma Y, Ashraf MZ, Podrez EA. Scavenger receptor BI modulates platelet reactivity and thrombosis in dyslipidemia. Blood. 2010;116:1932–1941. doi: 10.1182/blood-2010-02-268508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.El Bouhassani M, Gilibert S, Moreau M, Saint-Charles F, Trequier M, Poti F, Chapman MJ, LeGoff W, Lesnik P, Huby T. Cholesteryl ester transfer protein expression partially attenuates the adverse effects of SR-BI receptor deficiency on cholesterol metabolism and atherosclerosis. J Biol Chem. 2011;286:17227–17238. doi: 10.1074/jbc.M111.220483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Matsuura F, Wang N, Chen W, Jiang X, Tall AR. HDL from CETP-deficient subjects shows enhanced ability to promote cholesterol efflux from macrophages in an apoE- and ABCG1-dependent pathway. J Clin Invest. 2006;116:1435–1442. doi: 10.1172/JCI27602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mahley RW, Huang Y, Weisgraber KH. Putting cholesterol in its place:apoE and reverse cholesterol transport. J Clin Invest. 2006;116:1226–1229. doi: 10.1172/JCI28632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fielding CJ, Shore VG, Fielding PE. A protein cofactor of lecithin:cholesterol acyltransferase. Biochem Biophys Res Commun. 1972;46:1493–1498. doi: 10.1016/0006-291x(72)90776-0. [DOI] [PubMed] [Google Scholar]
- 76.Jonas A. Lecithin-cholesterol acyltransferase in the metabolism of high-density lipoproteins. Biochim Biophys Acta. 1991;1084:205–220. doi: 10.1016/0005-2760(91)90062-m. [DOI] [PubMed] [Google Scholar]
- 77.Czarnecka H, Yokoyama S. Regulation of cellular cholesterol efflux by lecithin:cholesterol acyltransferase reaction through nonspecific lipid exchange. J Biol Chem. 1996;271:2023–8. doi: 10.1074/jbc.271.4.2023. [DOI] [PubMed] [Google Scholar]
- 78.Rousset X, Rhamburek R, Vaisman B, Amar M, Remaley AT. Lecithin Cholesterol Acyltransferase: An anti- or pro-atherogenic Factor? Curr Athero Rep. 2011;13:249–256. doi: 10.1007/s11883-011-0171-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hoeg JM, Santamarina-Fojo S, Berard AM, Cornhill JF, Herderick EE, Feldman SH, Haudenschild CC, Vaisman BL, Hoyt RF, Demosky SJ, Kauffman RD, Hazel CM, Marcovina SM, Brewer HB. Overexpression of lecithin:cholesterol acyltransferase in transgenic rabbits prevents diet-induced atherosclerosis. Proc Natl Acad Sci USA. 1996;93:11448–11453. doi: 10.1073/pnas.93.21.11448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lambert G, Sakai N, Vaisman BL, Neufeld EB, Marteyn B, Chan CC, Paigen B, Lupia E, Thomas A, Striker LJ, Blanchette-Mackie J, Csako G, Brady JN, Costello R, Striker GE, Remaley AT, Brewer HB, Jr, Santamarina-Fojo S. Analysis of glomerulosclerosis and atherosclerosis in lecithin cholesterol acyltransferase-deficient mice. J Biol Chem. 2001;276:15090–15098. doi: 10.1074/jbc.M008466200. [DOI] [PubMed] [Google Scholar]
- 81.Furbee JW, Jr, Sawyer JK, Parks JS. Lecithin:cholesterol acyltransferase deficiency increases atherosclerosis in the low density lipoprotein receptor and apolipoprotein E knockout mice. J Biol Chem. 2002;277:3511–3519. doi: 10.1074/jbc.M109883200. [DOI] [PubMed] [Google Scholar]
- 82.Brousseau ME, Santamarina-Fojo S, Vaisman BL, pplebaum-Bowden D, Berard AM, Talley GD, Brewer HB, Jr, Hoeg JM. Overexpression of human lecithin:cholesterol acyltransferase in cholesterol-fed rabbits: LDL metabolism and HDL metabolism are affected in a gene dose-dependent manner. J Lipid Res. 1997;38:2537–2547. [PubMed] [Google Scholar]
- 83.Amar MJ, Shamburek RD, Vaisman B, Knapper CL, Foger B, Hoyt RF, Santamarina-Fojo S, Brewer HB, Remaley AT. Adenoviral expression of human lecithin-cholesterol acyltransferase in nonhuman primates leads to an antiatherogenic lipoprotein phenotype by increasing high-density lipoprotein and lowering low-density lipoprotein. Metabolism. 2009;58:568–575. doi: 10.1016/j.metabol.2008.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Brousseau ME, Kauffman RD, Herderick EE, Demosky SJ, Jr, Evans W, Marcovina S, Santamarina-Fojo S, Brewer HB, Jr, Hoeg JM. LCAT modulates atherogenic plasma lipoproteins and the extent of atherosclerosis only in the presence of normal LDL receptors in transgenic rabbits. Arterioscler Thromb Vasc Biol. 2000;20:450–458. doi: 10.1161/01.atv.20.2.450. [DOI] [PubMed] [Google Scholar]
- 85.Hovingh GK, Hutten BA, Holleboom AG, Peterson W, Rol P, Stalenhoef A, Zwinderman AH, de Groot E, Kastelein JJ, Kuivenhoven JA. Compromised LCAT function is associated with increased atherosclerosis. Circulation. 2005;112:879–884. doi: 10.1161/CIRCULATIONAHA.105.540427. [DOI] [PubMed] [Google Scholar]
- 86.Duivenvoorden R, Holleboom AG, Bogaard B, Groot E, Nederveen AJ, Lameris JS, Kastelein JJP, Kuivenhoven JA, Stroes ESG. Carriers of LCAT Gene Mutations Have Increased Atherosclerosis: A 3.0 Tesla MRI Study. Atherosclerosis Suppl. 2010;11:E1634–E1636. [Google Scholar]
- 87.Calabresi L, Baldassarre D, Castelnuovo S, Conca P, Bocchi L, Candini C, Frigerio B, Amato M, Sirtori CR, Alessandrini P, Arca M, Boscutti G, Cattin L, Gesualdo L, Sampietro T, Vaudo G, Veglia F, Calandra S, Francescini Functional lecithin: cholesterol acyltransferase is not required for efficient atheroprotection in humans. Circulation. 2009;120:628–635. doi: 10.1161/CIRCULATIONAHA.108.818143. [DOI] [PubMed] [Google Scholar]
- 88.Inazu A, Brown ML, Hesler CB, Agellon LB, Koizumi J, Takata K, Maruhama Y, Mabuchi H, Tall AR. Increased high-density lipoprotein levels caused by a common cholesteryl-ester transfer protein gene mutation. N Engl J Med. 1990;323:1234–1238. doi: 10.1056/NEJM199011013231803. [DOI] [PubMed] [Google Scholar]
- 89.Inazu A, Jiang XC, Haraki T, Yagi K, Kamon N, Koizumi J, Mabuchi H, Takeda R, Takata K, Moriyama Y. Genetic cholesteryl ester transfer protein deficiency caused by two prevalent mutations as a major determinant of increased levels of high density lipoprotein cholesterol. J Clin Invest. 1994;94:1872–1882. doi: 10.1172/JCI117537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Brown ML, Inazu A, Hesler CB, Agellon LB, Mann C, Whitlock ME, Marcel YL, Milne RW, Koizumi J, Mabuchi H, Takeda R, Tall AR. Molecular basis of lipid transfer protein deficiency in a family with increased high-density lipoproteins. Nature. 1989;342:448–451. doi: 10.1038/342448a0. [DOI] [PubMed] [Google Scholar]
- 91.Barter PJ, Brewer HB, Jr, Chapman MJ, Hennekens CH, Rader DJ, Tall AR. Cholesteryl ester transfer protein: a novel target for raising HDL and inhibiting atherosclerosis. Arterioscler Thromb Vasc Biol. 2003;23:160–167. doi: 10.1161/01.atv.0000054658.91146.64. [DOI] [PubMed] [Google Scholar]
- 92.Tall AR, Yvan-Charvet L, Terasaka N, Pagler T, Wang N. HDL, ABC transporters, and cholesterol efflux: implications for the treatment of atherosclerosis. Cell Metab. 2008;7:365–375. doi: 10.1016/j.cmet.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 93.Clark RW, Sutfin TA, Ruggeri RB, Willauer AT, Sugarman ED, Magnus-Aryitey G, Cosgrove PG, Sand TM, Wester RT, Williams JA, Perlman ME, Bamberger MJ. Raising high-density lipoprotein in humans through inhibition of cholesteryl ester transfer protein. Arterioscler Thromb Vasc Biol. 2004;24:490–497. doi: 10.1161/01.ATV.0000118278.21719.17. [DOI] [PubMed] [Google Scholar]
- 94.De Grooth GJ, Kuivenhoven JA, Stalenhoef AFH, de Graaf J, Zwinderman AH, Posma JL, van Tol A, Kastelein JJP. Efficacy and safety of a novel cholesteryl ester transfer protein inhibitor, JTT-705, in humans. Circulation. 2002;105:2159–2165. doi: 10.1161/01.cir.0000015857.31889.7b. [DOI] [PubMed] [Google Scholar]
- 95.Krishna R, Anderson MS, Bergman AJ, Jin B, Fallon M, Cote J, Rosko K, Chavez-End C, Lutz R, Bloomfield DM, Gutierrez M, Doherty J, Bieberdorf F, Chodakewitz J, Gottesdiener KM, Wagner JA. Effect of the cholesteryl ester transfer protein inhibitor, anacetrapib, on lipoproteins in patients with dyslipidaemia and on 24-h ambulatory blood pressure in healthy individuals: two double-blind, randomised placebo-controlled phase I studies. Lancet. 2007;370:1907–1914. doi: 10.1016/S0140-6736(07)61813-3. [DOI] [PubMed] [Google Scholar]
- 96.Nicholls SJ, Tuzcu EM, Brennan DM, Tardif J-C, Nissen SE. Cholesteryl ester transfer protein inhibition, high-density lipoprotein raising, and progression of coronary atherosclerosis. Circulation. 2008;118:2506–2514. doi: 10.1161/CIRCULATIONAHA.108.790733. [DOI] [PubMed] [Google Scholar]
- 97.Ranalletta M, Bierilo KK, Chen Y, Milot D, Chen Q, Tung E, Houde C, Elowe NH, Garcia-Calvo M, Porter G, Eveland S, Frantz-Wattley B, Kavana M, Addona G, Sinclair P, Sparrow C, O’Neill EA, Koblan KS, Sitlani A, Hubbard B, Fisher TS. Biochemical characterization of cholesteryl ester transfer protein inhibitors. J Lipid Res. 2010;51:2739–2752. doi: 10.1194/jlr.M007468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Niesor EJ, Magg C, Ogawa N, Okamoto H, von der Mark E, Matile H, Schmid G, Clerg RG, Chaput E, Blum-Kaelin D, Huber W, Thoma R, Pflieger P, Kakutani M, Takahashi D, Dernick G, Maugeais C. Modulating cholesteryl ester transfer protein activity maintains efficient pre-beta HDL formation and increases reverse cholesterol transport. J Lipid Res. 2010;51:3443–3454. doi: 10.1194/jlr.M008706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Okamoto H, Yonemori F, Wakitani K, Minowa T, Maeda K, Shinkai H. A cholesteryl ester transfer protein inhibitor attenuates atherosclerosis in rabbits. Nature. 2000;406:203–207. doi: 10.1038/35018119. [DOI] [PubMed] [Google Scholar]
- 100.Rye K-A, Barter PJ. Formulation and metabolism of prebeta-migrating, lipid-poor apolipoprotein A-I. Arterioscler Thromb Vasc Biol. 2004;24:421–428. doi: 10.1161/01.ATV.0000104029.74961.f5. [DOI] [PubMed] [Google Scholar]
- 101.Tall AR, Yvan-Charvet L, Wang N. The failure of torcetrapib: was it the molecule or the mechanism? Areterioscler Thromb Vasc Biol. 2007;27:257–260. doi: 10.1161/01.ATV.0000256728.60226.77. [DOI] [PubMed] [Google Scholar]
- 102.Yvan-Charvet L, King J, Pagler T, Li H, Hubbard B, Fisher T, Sparrow CP, Taggart AK, Tall AR. Cholesterol efflux potential and antiinflammatory properties of high-density lipoprotein after treatment with niacin or anacetrapib. Arterioscler Thromb Vasc Biol. 2010;30:1430–1438. doi: 10.1161/ATVBAHA.110.207142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bruce C, Beamer LJ, Tall AR. The implications of the structure of the bactericidal/permeability-increasing protein on the lipid-transfer function of the cholesteryl ester transfer protein. Curr Opin Struct Biol. 1998;8:426–434. doi: 10.1016/s0959-440x(98)80118-8. [DOI] [PubMed] [Google Scholar]
- 104.Oka T, Kujiraoka T, Ito M, Egashira T, Takahashi S, Nanjee MN, Miller NE, Metso J, Olkkonen VM, Ehnholm C, Jauhiainen M, Hattori H. Distribution of phospholipid transfer protein in human plasma: presence of two forms of phospholipid transfer protein, one catalytically active and the other inactive. J Lipid Res. 2000;41:1651–1657. [PubMed] [Google Scholar]
- 105.Siggins S, Kärkkäinen M, Tenhunen J, Metso J, Tahvanainen E, Olkkonen VM, Jauhiainen M, Ehnholm C. Quantitation of the active and low-active forms of human plasma phospholipid transfer protein by ELISA. J Lipid Res. 2004;45:387–395. doi: 10.1194/jlr.D300023-JLR200. [DOI] [PubMed] [Google Scholar]
- 106.Cheung MC, Albers JJ. Active plasma phospholipid transfer protein is associated with apoA-I- but not apoE-containing lipoproteins. J Lipid Res. 2006;47:1315–1321. doi: 10.1194/jlr.M600042-JLR200. [DOI] [PubMed] [Google Scholar]
- 107.Cheung MC, Wolfbauer G, Albers JJ. Different phospholipid transfer protein complexes contribute to the variation in plasma PLTP specific activity. Biochim Biophys Acta. 2011;1811:343–347. doi: 10.1016/j.bbalip.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Van Haperen R, van Tol A, Vermeulen P, Jauhiainen M, van Gent T, van den Berg P, Ehnholm S, Grosveld F, van der Kamp A, de Crom R. Human plasma phospholipid transfer protein increases the antiatherogenic potential of high density lipoproteins in transgenic mice. Arterioscler Thromb Vasc Biol. 2000;20:1082–1088. doi: 10.1161/01.atv.20.4.1082. [DOI] [PubMed] [Google Scholar]
- 109.Jiang X-C, Bruce C, Mar J, Lin M, Ji Y, Francone OL, Tall AR. Targeted mutation of plasma phospholipid transfer protein gene markedly reduces high-density lipoprotein levels. J Clin Invest. 1999;103:907–914. doi: 10.1172/JCI5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Vikstedt R, Metso J, Hakala J, Olkkonen VM, Ehnholm C, Jauhiainen M. Cholesterol efflux from macrophage foam cells is enhanced by active phospholipid transfer protein through generation of two types of acceptor particles. Biochemistry. 2007;46:11979–11986. doi: 10.1021/bi700833h. [DOI] [PubMed] [Google Scholar]
- 111.Cao G, Beyer TP, Yang XP, Schmidt RJ, Zhang Y, Bensch WR, Kauffman RF, Gao H, Ryan TP, Liang Y, Eacho PI, Jiang XC. Phospholipid transfer protein is regulated by liver X receptors in vivo. J Biol Chem. 2002;277:39561–39565. doi: 10.1074/jbc.M207187200. [DOI] [PubMed] [Google Scholar]
- 112.Samyn H, Moerland M, van Gent T, van Haperen R, Grosveld F, van Tol A, de Crom R. Elevation of systemic PLTP, but not macrophage-PLTP, impairs macrophage reverse cholesterol transport in transgenic mice. Atherosclerosis. 2009;204:429–434. doi: 10.1016/j.atherosclerosis.2008.10.020. [DOI] [PubMed] [Google Scholar]
- 113.Oram JF, Wolfbauer G, Vaughan AM, Tang C, Albers JJ. Phospholipid transfer protein interacts with and stabilizes ATP-binding cassette transporter A1 and enhances cholesterol efflux from cells. J Biol Chem. 2003;278:52379–52385. doi: 10.1074/jbc.M310695200. [DOI] [PubMed] [Google Scholar]
- 114.Jiang X-C, Li Z, Liu R, Yang XP, Pan M, Lagrost L, Fisher EA, Williams KJ. Phospholipid transfer protein deficiency impairs apolipoprotein-B secretion from hepatocytes by stimulating a proteolytic pathway through a relative deficiency of vitamin E and an increase in intracellular oxidants. J Biol Chem. 2005;280:18336–18340. doi: 10.1074/jbc.M500007200. [DOI] [PubMed] [Google Scholar]
- 115.Masson D, Deckert V, Gautier T, Klein A, Desrumaux C, Viglietta C, Pais de Barros JP, Le Guern N, Grober J, Labbe J, Menetrier F, Ripoll PJ, Leroux-Coyau M, Jolivet G, Houdebine LM, Lagrost L. Worsening of diet-induced atherosclerosis in a new model of transgenic rabbit expressing the human plasma phospholipid transfer protein. Arterioscler Thromb Vasc Biol. 2011;31:766–774. doi: 10.1161/ATVBAHA.110.215756. [DOI] [PubMed] [Google Scholar]
- 116.Jaye M, Lynch KJ, Krawiec J, Marchadier D, Maugeais C, Doan K, South V, Amin D, Perrone M, Rader DJ. A novel endothelial-derived lipase that modulates HDL metabolism. Nat Genet. 1999;21:424–428. doi: 10.1038/7766. [DOI] [PubMed] [Google Scholar]
- 117.Jin W, Millar JS, Broedl U, Glick JM, Rader DJ. Inhibition of endothelial lipase causes increased HDL cholesterol levels in vivo. J Clin Invest. 2003;111:357–362. doi: 10.1172/JCI16146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Maugeais C, Tietge UJ, Broedl UC, Marchadier D, Cain W, McCoy MG, Lund-Katz S, Glick JM, Rader DJ. Dose-dependent acceleration of high-density lipoprotein catabolism by endothelial lipase. Circulation. 2003;108:2121–2126. doi: 10.1161/01.CIR.0000092889.24713.DC. [DOI] [PubMed] [Google Scholar]
- 119.Badellino KO, Wolfe ML, Reilly MP, Rader DJ. Endothelial lipase concentrations are increased in metabolic syndrome and associated with coronary atherosclerosis. PLoS Med. 2006;3:e22. doi: 10.1371/journal.pmed.0030022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, Pirruccello JP, Ripatti S, Chasman DI, Willer CJ, Johansen CT, Fouchier SW, Isaacs A, Peloso GM, Barbalic M, Ricketts SL, Bis JC, Aulchenko YS, Thorleifsson G, Feitosa MF, Chambers J, Orho-Melander M, Melander O, Johnson T, Li X, Guo X, Li M, Shin Cho Y, Jin Go M, Jin Kim Y, Lee JY, Park T, Kim K, Sim X, Twee-Hee Ong R, Croteau-Chonka DC, Lange LA, Smith JD, Song K, Hua Zhao J, Yuan X, Luan J, Lamina C, Ziegler A, Zhang W, Zee RY, Wright AF, Witteman JC, Wilson JF, Willemsen G, Wichmann HE, Whitfield JB, Waterworth DM, Wareham NJ, Waeber G, Vollenweider P, Voight BF, Vitart V, Uitterlinden AG, Uda M, Tuomilehto J, Thompson JR, Tanaka T, Surakka I, Stringham HM, Spector TD, Soranzo N, Smit JH, Sinisalo J, Silander K, Sijbrands EJ, Scuteri A, Scott J, Schlessinger D, Sanna S, Salomaa V, Saharinen J, Sabatti C, Ruokonen A, Rudan I, Rose LM, Roberts R, Rieder M, Psaty BM, Pramstaller PP, Pichler I, Perola M, Penninx BW, Pedersen NL, Pattaro C, Parker AN, Pare G, Oostra BA, O’Donnell CJ, Nieminen MS, Nickerson DA, Montgomery GW, Meitinger T, McPherson R, McCarthy MI, McArdle W, Masson D, Martin NG, Marroni F, Mangino M, Magnusson PK, Lucas G, Luben R, Loos RJ, Lokki ML, Lettre G, Langenberg C, Launer LJ, Lakatta EG, Laaksonen R, Kyvik KO, Kronenberg F, König IR, Khaw KT, Kaprio J, Kaplan LM, Johansson A, Jarvelin MR, Janssens AC, Ingelsson E, Igl W, Kees Hovingh G, Hottenga JJ, Hofman A, Hicks AA, Hengstenberg C, Heid IM, Hayward C, Havulinna AS, Hastie ND, Harris TB, Haritunians T, Hall AS, Gyllensten U, Guiducci C, Groop LC, Gonzalez E, Gieger C, Freimer NB, Ferrucci L, Erdmann J, Elliott P, Ejebe KG, Döring A, Dominiczak AF, Demissie S, Deloukas P, de Geus EJ, de Faire U, Crawford G, Collins FS, Chen YD, Caulfield MJ, Campbell H, Burtt NP, Bonnycastle LL, Boomsma DI, Boekholdt SM, Bergman RN, Barroso I, Bandinelli S, Ballantyne CM, Assimes TL, Quertermous T, Altshuler D, Seielstad M, Wong TY, Tai ES, Feranil AB, Kuzawa CW, Adair LS, Taylor HA, Jr, Borecki IB, Gabriel SB, Wilson JG, Holm H, Thorsteinsdottir U, Gudnason V, Krauss RM, Mohlke KL, Ordovas JM, Munroe PB, Kooner JS, Tall AR, Hegele RA, Kastelein JJ, Schadt EE, Rotter JI, Boerwinkle E, Strachan DP, Mooser V, Stefansson K, Reilly MP, Samani NJ, Schunkert H, Cupples LA, Sandhu MS, Ridker PM, Rader DJ, van Duijn CM, Peltonen L, Abecasis GR, Boehnke M, Kathiresan S. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–13. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Connelly PW. The role of hepatic lipase in lipoprotein metabolism. Clin Chim Acta. 1999;286:243–255. doi: 10.1016/s0009-8981(99)00105-9. [DOI] [PubMed] [Google Scholar]
- 122.Nong Z, Gonzalez-Navarro H, Amar M, Freeman L, Knapper C, Neufeld EB, Paigen BJ, Hoyt RF, Fruchart-Najib J, Santamarina-Fojo S. Hepatic lipase expression in macrophages contributes to atherosclerosis in apoE-deficient and LCAT-transgenic mice. J Clin Invest. 2003;112:367–378. doi: 10.1172/JCI16484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Jansen H. Hepatic Lipase: Friend or foe and under what circumstances. Curr Athero Rep. 2004;6:343–47. doi: 10.1007/s11883-004-0044-3. [DOI] [PubMed] [Google Scholar]
- 124.Demant T, Carlson LA, Holmquist L, Karpe F, Nilsson-Ehle P, Packard CJ, Shepherd J. Lipoprotein metabolism in hepatic lipase deficiency: studies on the turnover of apolipoprotein B and on the effect of hepatic lipase on high density lipoprotein. J Lipid Res. 1988;29:1603–1611. [PubMed] [Google Scholar]
- 125.Ruel IL, Couture P, Cohn JS, Bensadoun A, Marcil M, Lamarche B. Evidence that hepatic lipase deficiency in humans is not associated with proatherogenic changes in HDL composition and metabolism. J Lipid Res. 2004;45:528–537. doi: 10.1194/jlr.M400090-JLR200. [DOI] [PubMed] [Google Scholar]
- 126.Annema W, Tietge UJF. Role of hepatic lipase and endothelial lipase in high-density lipoprotein-mediated reverse cholesterol transport. Curr Athero Rep. 2011;13:257–265. doi: 10.1007/s11883-011-0175-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Brown RJ, Lagor WR, Sankaranaravanan S, Yasuda T, Quertermous T, Rothblat GH, Rader DJ. Impact of combined deficiency of hepatic lipase and endothelial lipase on the metabolism of both high-density lipoproteins and apoB-containing lipoprotiens. Circ Res. 2010;107:357–364. doi: 10.1161/CIRCRESAHA.110.219188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hegele RA, Little JA, Vezina C, Maguire GF, Tu L, Wolever TS, Jenkins DJ, Connelly PW. Hepatic lipase deficiency. Clinical, biochemical and molecular genetic characteristics. Arterioscler Thromb. 1993;13:720–728. doi: 10.1161/01.atv.13.5.720. [DOI] [PubMed] [Google Scholar]
- 129.Jin W, Marchadler D, Rader DJ. Lipases and HDL metabolism. Trends Endocrinol Metab. 2002;13:174–178. doi: 10.1016/s1043-2760(02)00589-1. [DOI] [PubMed] [Google Scholar]
- 130.Zhang Y, DaSilva JR, Reilly M, Billheimer JT, Rothblat GH, Rader DJ. Hepatic expression of scavenger receptor class B type I (SR-BI) is a positive regulator of macrophage reverse cholesterol transport in vivo. J Clin Invest. 2005;115:2870–2874. doi: 10.1172/JCI25327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Scavenger receptor B1 (SR-B1) substrates inhibit the selective uptake of high-density-lipoprotein cholesteryl esters by rat parenchymal liver cells. Science. 1996;271:518–520. [Google Scholar]
- 132.Krieger M. Charting the fat of the “good cholesterol”: identification and characterization of the high-density lipoprotein receptor SR-BI. Ann Rev Biochem. Ann Rev Biochem. 1999;68:523–558. doi: 10.1146/annurev.biochem.68.1.523. [DOI] [PubMed] [Google Scholar]
- 133.Glass C, Pittman RC, Weinstein DB, Steinberg D. Dissociation of tissue uptake of cholesterol ester from that of apoprotein A-1 of rat plasma high density lipoprotein: selective delivery of cholesterol ester to liver, adrenal, and gonad. Proc Natl Acad Sci USA. 1983;80:5435–5439. doi: 10.1073/pnas.80.17.5435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Moore RE, Navab M, Millar JS, Zimetti F, Hama S, Rothblat GH, Rader DJ. Increased atherosclerosis in mice lacking apolipoprotein A-I attributable to both impaired reverse cholesterol transport and increased inflammation. Circ Res. 2005;97:763–771. doi: 10.1161/01.RES.0000185320.82962.F7. [DOI] [PubMed] [Google Scholar]
- 135.Rader DJ, Alexander ET, Weibel GL, Billheimer J, Rothblat GH. Role of reverse cholesterol transport in animals and humans and relationship to atherosclerosis. J Lipid Res. 2008;50(Suppl):S189–194. doi: 10.1194/jlr.R800088-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Khera AV, Cuchel M, de la Llera-Moya M, Rodrigues A, M Burke MF, Jafri K, French BC, Phillips JA, Mucksavage ML, Wilensky RL, Mohler ER, Rothblat GH, Rader DJ. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med. 2011;364:127–135. doi: 10.1056/NEJMoa1001689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.de la Llera-Moya M, Drazul-Schrader D, Asztalos BF, Cuchel M, Rader DJ, Rothblat GH. The ability to promote efflux via ABCA1 determines the capacity of serum specimens with similar HDL-C to remove cholesterol from macrophages. Arterioscler Thromb Vasc Biol. 2010;30:796–801. doi: 10.1161/ATVBAHA.109.199158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Eriksson M, Carlson LA, Miettinen TA, Angelin B. Stimulation of fecal steroid excretion after infusion of recombinant proapolipoprotein A-I: Potential reverse cholesterol transport in humans. Circulation. 1999;100:594–598. doi: 10.1161/01.cir.100.6.594. [DOI] [PubMed] [Google Scholar]
- 139.Brousseau ME, Diffenderfer MR, Millar JS, Nartsupha C, Asztalos BF, Welty FK, Wolfe ML, Rudling M, Bjorkhem I, Angelin B, Mancuso JP, Digenio AG, Rader DJ, Schaefer EJ. Effects of cholesteryl ester transfer protein inhibition on high-density lipoprotein subspecies, apolipoprotein A-I metabolism, and fecal sterol excretion. Arterioscler Thromb Vasc Biol. 2005;25:1057–1064. doi: 10.1161/01.ATV.0000161928.16334.dd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zhou M, Sawyer J, Kelley K, Fordstrom P, Chan J, Tonn G, Carlson T, Retter M, Meininger D, Cheng J, Gates A, Woodward A, DeJaney J, Zhang R, Tang J, Liu Q, Cao P, Luchoomum J, Voog J, Turner S, Shan B, Boone T, Rudel L, Schwartz M. Lecithin cholesterol acyltransferase promotes reverse cholesterol transport and attenuates atherosclerosis progression in New Zealand white rabbits. Circulation. 2009;120:S1175. (Abst.) [Google Scholar]
- 141.Holleboom AG, Franssen R, Jakulj L, Decaris J, Vergeer M, Koetsveld J, Hellerstein MK, Kastelein JJ, Groen AK, Turner SM, Stroes ES. In vivo tissue cholesterol efflux is impaired in carriers of mutations in Apo A1 and ABCA1. Circulation. 2011;122:A17881. (Abst.) [Google Scholar]
- 142.Schwartz CC, VandenBroek JM, Cooper PS. Lipoprotein cholesteryl ester production, transfer, and output in vivo in humans. J Lipid Res. 2004;45:1594–1607. doi: 10.1194/jlr.M300511-JLR200. [DOI] [PubMed] [Google Scholar]
- 143.Owen DR, Lindsay AC, Choudhury RP, Fayad ZA. Imaging of atherosclerosis. Annu Rev Med. 2011;62:25–40. doi: 10.1146/annurev-med-041709-133809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Vancraeynest D, Pasquet A, Roelants V, Gerber BL, Vanoverschelde J-LJ. Imaging of the vulnerable plaque. J Am Coll Cardiol. 2011;57:1961–1979. doi: 10.1016/j.jacc.2011.02.018. [DOI] [PubMed] [Google Scholar]
- 145.Muller JE, Weissman NJ, Tuzcu EM. The year in intracoronary imaging. J Am Coll Cardiol Img. 2010;3:881–891. doi: 10.1016/j.jcmg.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 146.Fayad ZA, Mani M, Woodward M, Kallend D, Abt M, Burgess T, Fuster V, Ballantyne CM, Stein EA, Tardif JC, Rudd JHF, Farkouh ME, Tawakol A. Safety and efficacy of dalcetrapib on atherosclerotic disease using novel non-invasive multimodality imaging (dal-PLAQUE): a randomized cliical trial. Lancet. 2011;378:1547–1559. doi: 10.1016/S0140-6736(11)61383-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Muntendam P, McCall C, Sanz J, Falk E, Fuster V, High-Risk Plaque Initiative The Biolmage Study: novel approaches to risk assessment in the primary prevention of atherosclerotic cardiovascular disease-study design and objectives. Am Heart J. 2010;160:49–57. doi: 10.1016/j.ahj.2010.02.021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.