

Summary
Introduction
Platelet activation via the Fcγ receptor IIa (FcγRIIa) is implicated in the pathogenesis of immune complex (IC)-mediated thrombocytopenia and thrombosis (ITT). We previously showed that ICs composed of antigen and antibodies targeting CD40 ligand (CD40L) or β2 Glycoprotein I (β2GPI) induce ITT in mice transgenic for human FcγRIIa (hFcR) but not wild-type controls (which lack FcγRIIa). Here we evaluated the contribution of the guanine nucleotide exchange factor, CalDAG-GEFI, and P2Y12, key regulators of Rap1 signaling in platelets, to ITT induced by these clinically relevant ICs.
Methods
Pre-formed anti-CD40L or anti-β2GPI ICs were injected into hFcR/Caldaggef1+/+ or hFcR/Caldaggef1-/- mice, with or without clopidogrel pre-treatment. Animals were observed for symptoms of shock for 30 minutes, during which time core body temperature was monitored. Platelet counts were obtained before and 30 minutes after IC injection. Lungs were assessed for thrombosis by histology or near-infrared imaging.
Results
Both CD40L and β2GPI ICs rapidly induced severe thrombocytopenia, shock and a reduction in body temperature in hFcR/Caldaggef1+/+ mice. hFcR/Caldaggef1-/- mice were protected from CD40L and β2GPI IC-induced thrombocytopenia and shock, whereas P2Y12 inhibition had only a modest effect on IC-induced ITT. Consistent with these findings, IC-induced integrin activation in vitro and the accumulation of activated platelets in the lungs of IC-challenged mice was strongly dependent on CalDAG-GEFI.
Conclusions
Our studies demonstrate that CalDAG-GEFI plays a critical role in platelet activation, thrombocytopenia and thrombosis induced by clinically relevant ICs in mice. Thus, CalDAG-GEFI may be a promising target for the intervention of IC-associated, FcγRIIa-mediated thrombotic conditions.
Keywords: Platelet activation, Thrombosis, Thrombocytopenia, Antigen-antibody complex, Receptors, IgG
Introduction
Platelets are essential components of the hemostatic response to vascular injury. However, platelets also play a role in pathologic atherothrombotic thrombosis and in immune-mediated thrombocytopenia and thrombosis (ITT) syndromes, including heparin-induced thrombocytopenia and thrombosis (HIT) [1,2], thrombocytopenia and disseminated intravascular coagulation associated with bacterial sepsis [3], and antiphospholipid syndrome [4,5]. Thrombotic complications have also been observed with the use of therapeutic IgG antibodies, such as bevacizumab and anti-CD40 ligand (anti-CD40L) monoclonal antibodies [6,7]. Critical to antibody-induced activation is FcγRIIa, a receptor for IgG immune complexes [8].
Clustering of platelet FcγRIIa triggers a signaling response that is typical for immunoreceptor tyrosine-based activation motif (ITAM) receptors. Binding of the non-receptor tyrosine kinase Syk initiates the formation of a signalosome, consisting of various adapter and effector proteins, which is critical for the activation of phospholipase Cγ2 (PLCγ2), and thus the generation of the second messengers calcium (Ca2+) and diacylglycerol (DAG) [9]. The small GTPase Rap1 orchestrates platelet activation downstream of PLCγ2. Two independent yet synergistic signaling pathways control the activation state of Rap1. Rapid but reversible activation of Rap1 depends on the guanine nucleotide exchange factor, CalDAG-GEFI (RasGRP2), which responds to increases in cytosolic Ca2+. Sustained Rap1 activation, however, requires signaling by protein kinase C and the Gi-coupled receptor for ADP, P2Y12, the target of the anti-platelet drug clopidogrel bisulfate (Plavix). In our recent work, we showed that platelet activation by ITAM receptors is strongly dependent on CalDAG-GEFI [10,11]. However, the contribution of Rap1 signaling to ITT induced by clinically relevant ICs has not been determined.
We recently established novel mouse models for ITT induced by CD40L/anti-CD40L and β2-GPI/anti β2-GPI ICs [12,13]. Both anti-CD40L (monoclonal) and anti-β2-GPI (polyclonal) antibodies form multivalent ICs when combined with their respective antigens. CD40L antibody was chosen for its capacity to bind a therapeutically relevant antigen, whereas β2-GPI antibody was selected for the importance of this antigen in autoimmunity. In these models of acute ITT, ICs injected intravenously into mice transgenic for FcγRIIA [14] cause rapid onset shock, a fall in platelet count, and deposition of fibrin rich thrombi in the lungs [15,12,13]. In an effort to identify an effective therapeutic strategy against ITT in this model, we tested anticoagulant and anti-platelet drugs by means of pretreatment prior to IC challenge. Bivalirudin, heparin, and aspirin, however, were largely ineffective in this regard (unpublished data). In search of a more effective target, we here assessed the role of CalDAG-GEFI and P2Y12, critical regulators of Rap1 signaling in platelets, in ITT.
Methods
Reagents and antibodies
Hybridoma cells were obtained from American Type Culture Collection (Manassas, VA). M90 (monoclonal anti-human CD40L IgG1 mAb, ATCC HB-12055) was purified into azide-free PBS from conditioned media by protein-G chromatography. Polyclonal goat anti-human β2GPI antibody (which does not bind mouse β2GPI) was from Affinity Biologicals (Ancaster, ON, Canada). Anti-human-FcγRIIa mAb, IV.3, was from StemCell Technologies (Vancouver, BC, Canada). The phycoerythrin (PE)-labeled anti-mouse αIIbβ3 mAb, JON/A-PE, was from Emfret Analytics (Eibelstadt, Germany). Antibodies against mouse P-selectin were obtained from BD Biosciences and labeled with Alexa-800 (Invitrogen, Grand Island, NY). Purified human β2GPI was from Haemotologic Technologies (Essex Junction, VT). Recombinant soluble human CD40L was from PeproTech (Rocky Hill, NJ). Clopidogrel (Plavix) was from Sanofi-Aventis (Bridgewater NJ). 2-methylthio-adenosine 5′-monophosphate triethylammonium salt hydrate (2-MeSAMP, P2Y12 inhibitor) was from Biolog (Farmingdale, NY). Preformed ICs were prepared in a microfuge tube by mixing the antibody (anti-CD40L or anti- β2GPI) with antigen (CD40L or β2GPI) in PBS at balanced stoichiometry and incubated 5 minutes at room temperature prior to use.
Mice
Mice transgenic for human FcγRIIa (FCGR2A, or “hFcR”) [14] and Caldaggef1-deficient mice [16], as well as littermate wild type (WT) controls, were bred on a C57BL6/J strain background. hFcR and Caldaggef1-deficient mice were crossed to create hFcR/Caldaggef1-/- (hFcR/CDGI /-) mice. FcγRIIa and Caldaggef1 genotypes were verified by PCR analysis. All experimental procedures were approved by the Animal Care and Use Committee of the University of North Carolina. Where indicated, mice were treated (by oral gavage) with clopidogrel (75 mg/kg) 24 and 3 hours before the experiment.
Flow cytometry
Platelet surface FcγRIIa expression was measured in blood (50 μl) drawn from the retro-orbital plexus of anesthetized mice into heparin-coated capillary tubes (VWR, Arlington Heights IL). Samples were stained with a PE-labeled antibody against GPIbα and an Alexa488-labeled antibody against FcγRIIa (IV.3). FcγRIIa surface expression was determined as the mean Alexa 488 fluorescence intensity for all GPIbα posivite events. For counting platelets, blood samples were stained with anti-GPIbα-PE, and platelets were counted by flow cytometry gating for PE-positive events. Platelet counts at t = 0 were defined as 100%. For measurement of ADP or IC-induced platelet activation ex vivo, diluted blood samples were activated with ADP (10 μM) or IC (500nM) in the presence of JON/A-PE for 10 minutes and analyzed immediately.
Rap1 activation assay
To assess IC-induced Rap1 activation in platelets from hFcR/CDGI+/+ and hFcR/CDGI-/- mice, washed platelets (2.5 × 108/ml; 250 μl vol) were stimulated with anti-CD40L or anti-β2GPI IC in standard aggregometry for 3 minutes in the presence or absense of the P2Y12 inhibitor 2-MeSAMP (100 μM). Reactions were stopped and platelets were lysed with ice-cold 2× lysis buffer (100mM Tris/HCl, pH 7.4, 400mM NaCl, 5mM MgCl2, 2% Nonidet P-40, 20% glycerol, and complete protease inhibitor cocktail lacking ethylenediaminetetraacetic acid; 15 minutes). Rap1-GTP was precipitated from platelet lysates using RalGDS-RBD beads as described previously [16]. Pellets were solubilized in sample buffer (75mM Tris/HCl pH 6.8, 10% SDS, 10% glycerol, 5% 2-mercaptoethanol, 0.004% bromophenol blue), proteins separated by SDS-PAGE, and Rap1 detected by Western blotting. Small aliquots were saved to control that each sample contained equal amounts of proteins.
Assessment of immune complex-induced thrombotic thrombocytopenia
Fifteen minutes prior to IC injection, mice were anesthetized with isoflurane and Alexa800-coupled antibodies to P-selectin (1 μg/g body weight) were administered by retro-orbital injection for in vivo labeling of activated platelets. Preformed ICs were prepared by mixing 120 μg Ab (anti-CD40L or anti-β2-GPI in PBS) with corresponding Ag (hCD40L [8 μg] or hβ2-GPI [20 μg] in PBS) respectively. Solutions (200 μl) were incubated for 5 minutes at RT prior to injection. Central body temperature of each animal was recorded immediately prior to IC injection. Animals received tail vein IC injections (200 μl) and were observed continuously for 30 minutes. Apparent symptoms of thrombotic shock for each animal were assessed based on observations of balance, mobility and respiration, and recorded as severe (complete immobility, loss of consciousness), moderate (impaired mobility, irregular respiration), mild (lethargy, shallow respiration), or none.
In addition, post IC body temperatures were measured every 10 minutes. At 30 minutes blood was drawn retro-orbitally and platelets were counted as described above. The lungs were carefully flushed with 1 ml PBS by left ventricular cardiac puncture, removed en bloc, and incubated in 4% paraformaldehyde for 24 hours before analysis on a LI-COR Odyssey scanner (LI-COR Biosciences, Lincoln NE). The lungs were also processed by H&E staining of 5 μm paraffin sections for the visualization of intravascular thrombi.
Statistical analysis
Results are reported as mean ± standard deviation. Differences between groups were analyzed by two-way Analysis of Variance (ANOVA). P values of < 0.05 were considered statistically significant.
Results and discussion
In order to evaluate the role of CalDAG-GEFI in IC-induced FcγRIIa-mediated thrombocytopenia and thrombosis, mice transgenic for human FcγRIIa either having or lacking Caldaggef1 (hFcR/CDGI+/+ or hFcR/CDGI-/-, respectively) were challenged with anti-CD40L+hCD40L or anti-β2GPI+hβ2GPI immune complexes. To evaluate the contribution of P2Y12 activation pathway, select groups of hFcR/CDGI+/+ or hFcR/CDGI-/- mice were given clopidogrel before IC injection. The expression level of hFcγRIIa was similar between hFcR/CDGI+/+ and hFcR/CDGI-/- mice (not shown). P2Y12 function as assessed by measuring ADP-induced αIIbβ3 activation ex vivo, was abolished in clopidogrel-treated mice (not shown). Consistent with previous work [16], ADP-induced integrin activation was also abolished in platelets lacking CalDAG-GEFI (hFcR/CDGI-/-).
Intravenous injection of anti-CD40L or anti-β2GPI immune complexes in hFcR/CDGI+/+ mice rapidly induced moderate to severe symptoms of thrombotic shock as evidenced by disorientation, shallow irregular breathing, and impaired mobility (Fig 1A,B). Shock symptoms correlated with a significant drop in central body temperature (Fig 1C,D). We previously showed that injection of either antibody or antigen alone does not cause any symptoms in hFcR/CDGI+/+ mice. Also, we have determined that platelets are the primary mediators of IC-induced thrombotic shock in this model since both ICs failed to cause thrombotic shock and hypothermia in thrombocytopenic hFcR mice [13].
Figure 1.


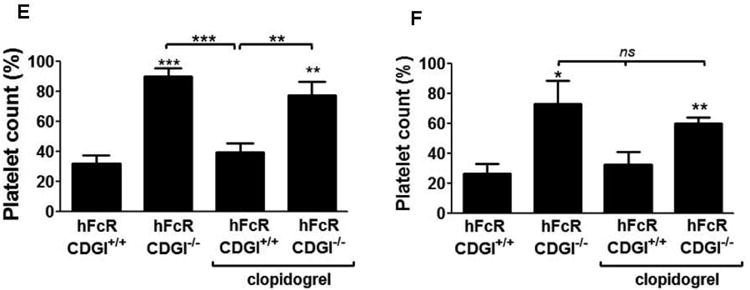
CalDAG-GEFI deficiency protects mice from IC-induced thrombocytopenia and thrombotic shock. Symptoms of thrombotic shock following the intravenous injection of anti-CD40L (A) or anti-β2GPI (B) immune complexes in hFcR/CDGI+/+ but not hFcR/CDGI-/- mice. Central body temperature of mice was measured before and at 10, 20, and 30 minutes after the injection of anti-CD40L (C) or antiβ2GPI (D) in hFcR/CDGI+/+ (open circles; [n=8] for anti-CD40L IC; [n=4] for anti-β2GPI IC), hFcR/CDGI-/- (open triangles; n=8 for anti-CD40L IC; n=5 for anti-β2GPI IC), hFcR/CDGI+/+ mice with clopidogrel (closed circles; [n=8] for anti-CD40L IC; [n=4] for anti-β2GPI IC), and hFcR/CDGI-/- mice with clopidogrel (closed triangles; [n=7] for anti-CD40L IC; [n=4] for anti-β2GPI IC). (*) indicates statistically significant difference. Platelet counts were determined by flow cytometry before and thirty minutes after the injection of anti-CD40L (E) or antiβ2GPI (F) ICs. Post injection values are presented as percent of pre-injection values.
hFcR/CDGI-/- mice were largely protected from shock and hypothermia induced by anti-CD40L ICs (Fig 1A,C), whereas the protection was partial when challenged with anti-β2GPI ICs (Fig 1B,D). In contrast, clopidogrel treatment provided little to no protection from ITT-induced shock and hypothermia, both in hFcR/CDGI+/+ and hFcR/CDGI-/- mice.
Immune complex injection also induced profound thrombocytopenia in hFcR/CDGI+/+ mice (Figure 1E,F). Consistent with the hypothesis that FcγRIIa-mediated platelet activation is critical to ITT, peripheral platelet counts were significantly higher in hFcR/CDGI-/- mice challenged with anti-CD40L and anti-β2GPI ICs (Figure 1E,F). In contrast, IC-induced thrombocytopenia was only minimally affected by pre-treatment with clopidogrel.
To confirm that hFcR/CDGI-/- mice are protected from ITT due to impaired platelet activation and thrombosis, we evaluated IC-induced platelet activation in vitro and in vivo. We first added anti-CD40L or anti-β2GPI ICs to platelets obtained from hFcR/CDGI+/+ and hFcR/CDGI-/- mice in the presence or absence of the P2Y12 inhibitor 2-MeSAMP. Both ICs strongly activated hFcR/CDGI+/+ platelets, as evidenced by Jon/A-PE binding (Fig 2A,B). IC-induced platelet activation was significantly inhibited by CalDAG-GEFI deficiency and, to a much lesser extent, by P2Y12 inhibition. The combination of CalDAG-GEFI deficiency and P2Y12 inhibition resulted in a further reduction in IC-induced platelet activation. Consistently, IC-induced aggregation (Fig. 2C,D) and Rap1 activation (Fig. 2E) were markedly reduced in hFcR/CDGI-/- platelets, and to a lesser degree in 2-MeSAMP-treated hFcR/CDGI+/+ platelets. Taken together, these results demonstrate a correlation between reduced Rap1 activation and IC-induced platelet activation.
Figure 2.

CalDAG-GEFI deficiency inhibits IC-induced platelet activation and aggregation as well as IC-induced Rap1 activation (A-B). Flow cytometric analysis of platelet αIIbβ3 activation (Jon/A-PE; [22]) induced by anti-CD40L (A) or anti-β2GPI (B) ICs (C-D). Aggregation of washed platelets in reponse to anti-CD40L (C), or anti-β2GPI (D) ICs. Arrows point to the time point when the indicated concentration of IC was added to the reaction mixture. (E) Determination of Rap1-GTP levels in lysates from platelets aggregated with anti-CD40L or anti-β2GPI ICs. Total Rap1 levels in platelet lysates are shown as loading controls.
To quantify and visualize platelet activation in vivo, we used near-infrared imaging to monitor the accumulation of P-selectin-positive, activated platelets in the lungs of animals (Fig. 3A,B). Injection of anti-CD40L or anti-β2GPI ICs caused a marked accumulation of activated platelets in the lungs of hFcR/CDGI+/+ mice, consistent with the pervasive intravascular thrombi observed in H&E tissue sections (Fig 3C,D). In mice challenged with anti-CD40L IC, CalDAG-GEFI deficiency virtually abolished pulmonary thrombosis, whereas clopidogrel treatment was only partially effective (Fig 3E,F). CalDAG-GEFI deficiency but not clopidogrel treatment conferred partial protection from β2GPI IC-induced thrombosis. It is unclear why CalDAG-GEFI deficiency had a greater impact in vivo with CD40L ICs than with β2GPI ICs. It is possible that the differences in activity between the two ICs could be a consequence of the more heterogeneous nature of polyclonal β2GPI IC structures (CD40L antibody is monoclonal). For example, we have observed with HPLC SEC that β2GPI antibodies appear to create a significantly wider range of IC sizes (0.5 to <2 mega Daltons) than CD40L mAb (<1 mega Dalton; not shown).
Figure 3.
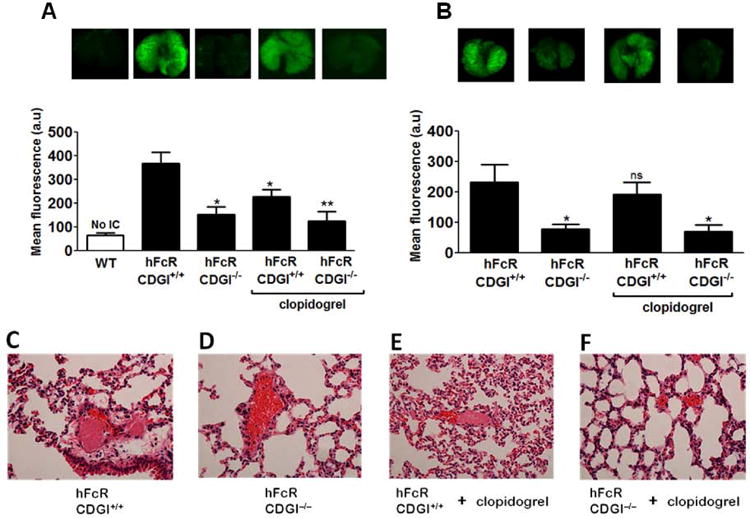
Quantitative analysis of activated platelet accumulation in the lungs of mice following the injection of anti-CD40L (A) or anti-β2GPI (B) ICs; (*) indicates statistically significant difference compared to the hFcR/CD+/+ group. Representative near-infrared images of lungs harvested from respective mice are shown. Intravascular microthrombi were analyzed by H&E staining histology. Representative sections of lungs harvested from hFcR/CDGI+/+ and hFcR/CDGI-/- mice injected with anti-CD40L ICs (C-F) are shown.
Our results showing impaired FcγRIIa-dependent activation of platelets isolated from mice treated with clopidogrel are in agreement with previous in vitro work suggesting that commonly used P2Y12 inhibitors may prevent ITT/HIT [17]. This defect in activation, however, only led to a mild protection from IC-induced ITT in vivo, a finding that is in line with clinical observations [18].
In contrast, deficiency in CalDAG-GEFI, a critical mediator of P2Y12-independent activation of Rap1 and integrin αIIbβ3 in platelets, provided significant protection with both ICs tested. These findings are consistent with our previous studies, where we identified a critical role for CalDAG-GEFI in ITAM-dependent platelet activation, both in vitro and in vivo [10,19,11]. ITAM-coupled receptors rely on the ability of the CalDAG-GEFI/Rap1 signaling module to respond to small increases in the cytosolic Ca2+ concentration, facilitating granule release and engagement of the P2Y12 signaling pathway [20]. Importantly, however, CalDAG-GEFI is less critical for thrombin-dependent platelet activation, and the hemostatic response in Caldaggef1-/- mice was significantly better when compared to WT mice treated clopidogrel [21]. Thus, targeting CalDAG-GEFI may be a viable strategy to safely prevent thrombotic complications in ITT.
Acknowledgments
We thank Agnieszka Cholka for providing excellent animal husbandry services; Caterina Casari for help with intravenous injections, and Hina Desai and the Florida Hospital Pathology Department for preparing slides for histological analysis. This work was supported by the American Heart Association (12POST12040088 to Y.B. and 10GRNT4460011 to A.A.), and the National Heart, Lung, and Blood Institute, NIH, grant HL106009 (W.B., S.E.McK).
Addendum
A. Amirkhosravi, Y. Boulaftali and W. Bergmeier designed the study and wrote the manuscript; A. Amirkhosravi, Y. Boulaftali, and L. Robles-Carrillo performed experiments and analyzed data; T. Meyer contributed to experimental design; S. E. McKenzie provided transgenic mice and reviewed the manuscript; J. L. Francis participated in data interpretation and manuscript review.
Footnotes
Disclosure of Conflict of Interests: The authors state that they have no conflicts of interest.
References
- 1.Amiral J, Bridey F, Dreyfus M, Vissoc AM, Fressinaud E, Wolf M, Meyer D. Platelet factor 4 complexed to heparin is the target for antibodies generated in heparin-induced thrombocytopenia. Thromb Haemost. 1992;68:95–6. [PubMed] [Google Scholar]
- 2.Warkentin TE, Sheppard JI. Generation of platelet-derived microparticles and procoagulant activity by heparin-induced thrombocytopenia IgG/serum and other IgG platelet agonists: a comparison with standard platelet agonists. Platelets. 1999;10:319–26. doi: 10.1080/09537109975960. [DOI] [PubMed] [Google Scholar]
- 3.Yin H, Stojanovic-Terpo A, Xu W, Corken A, Zakharov A, Qian F, Pavlovic S, Krbanjevic A, Lyubimov AV, Wang ZJ, Ware J, Du X. Role for platelet glycoprotein Ib-IX and effects of its inhibition in endotoxemia-induced thrombosis, thrombocytopenia, and mortality. Arterioscler Thromb Vasc Biol. 2013;33:2529–37. doi: 10.1161/ATVBAHA.113.302339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chong BH, Brighton TC, Chesterman CN. Antiphospholipid antibodies and platelets. Semin Thromb Hemost. 1995;21:76–84. doi: 10.1055/s-2007-1000381. [DOI] [PubMed] [Google Scholar]
- 5.Espinosa G, Cervera R, Font J, Shoenfeld Y. Antiphospholipid syndrome: pathogenic mechanisms. Autoimmun Rev. 2003;2:86–93. doi: 10.1016/s1568-9972(02)00144-1. [DOI] [PubMed] [Google Scholar]
- 6.Nalluri SR, Chu D, Keresztes R, Zhu X, Wu S. Risk of venous thromboembolism with the angiogenesis inhibitor bevacizumab in cancer patients: a meta-analysis. JAMA. 2008;300:2277–85. doi: 10.1001/jama.2008.656. [DOI] [PubMed] [Google Scholar]
- 7.Sidiropoulos PI, Boumpas DT. Lessons learned from anti-CD40L treatment in systemic lupus erythematosus patients. Lupus. 2004;13:391–7. doi: 10.1191/0961203304lu1032oa. [DOI] [PubMed] [Google Scholar]
- 8.Rosenfeld SI, Looney RJ, Leddy JP, Phipps DC, Abraham GN, Anderson CL. Humanplatelet Fc receptor for immunoglobulin G. Identification as a 40,000-molecular-weight membrane protein shared by monocytes. J Clin Invest. 1985;76:2317–22. doi: 10.1172/JCI112242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mócsai A, Ruland J, Tybulewicz VL. The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat Rev Immunol. 2010;10:387–402. doi: 10.1038/nri2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stefanini L, Roden RC, Bergmeier W. CalDAG-GEFI is at the nexus of calcium-dependent platelet activation. Blood. 2009;114:2506–14. doi: 10.1182/blood-2009-04-218768. [DOI] [PubMed] [Google Scholar]
- 11.Stolla M, Stefanini L, André P, Ouellette TD, Reilly MP, McKenzie SE, Bergmeier W. CalDAG-GEFI deficiency protects mice in a novel model of Fcγ RIIA-mediated thrombosis and thrombocytopenia. Blood. 2011;118:1113–20. doi: 10.1182/blood-2011-03-342352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robles-Carrillo L, Meyer T, Hatfield M, Desai H, Dávila M, Langer F, Amaya M, Garber E, Francis JL, Hsu YM, Amirkhosravi A. Anti-CD40L immune complexes potently activate platelets in vitro and cause thrombosis in FCGR2A transgenic mice. J Immunol. 2010;185:1577–83. doi: 10.4049/jimmunol.0903888. [DOI] [PubMed] [Google Scholar]
- 13.Amirkhosravi A, Meyer T, Robles-Carrillo L, Hatfield M, Desai H, Amaya M, Langer F, McKenzie SE, Francis JL. β2-Glycoprotein 1 antibodies directly activate the platelet IgG receptor, FcγRIIa, and cause thrombosis in FCGR2A transgenic but not in wild type mice. Blood. 2012;120 Abstract 106. [Google Scholar]
- 14.McKenzie SE, Taylor SM, Malladi P, Yuhan H, Cassel DL, Chien P, Schwartz E, Schreiber AD, Surrey S, Reilly MP. The role of the human Fc receptor Fc gamma RIIA in the immune clearance of platelets: a transgenic mouse model. J Immunol. 1999;162:4311–8. [PubMed] [Google Scholar]
- 15.Meyer T, Robles-Carrillo L, Robson T, Langer F, Desai H, Davila M, Amaya M, Francis JL, Amirkhosravi A. Bevacizumab immune complexes activate platelets and induce thrombosis in FCGR2A transgenic mice. J Thromb Haemost. 2009;7:171–81. doi: 10.1111/j.1538-7836.2008.03212.x. [DOI] [PubMed] [Google Scholar]
- 16.Crittenden JR, Bergmeier W, Zhang Y, Piffath CL, Liang Y, Wagner DD, Housman DE, Graybiel AM. CalDAG-GEFI integrates signaling for platelet aggregation and thrombus formation. Nat Med. 2004;10:982–6. doi: 10.1038/nm1098. [DOI] [PubMed] [Google Scholar]
- 17.Polgár J, Eichler P, Greinacher A, Clemetson KJ. Adenosine diphosphate (ADP) and ADP receptor play a major role in platelet activation/aggregation induced by sera from heparin-induced thrombocytopenia patients. Blood. 1998;91:549–54. [PubMed] [Google Scholar]
- 18.Denomme GA. The platelet Fc Receptor in Heparin-induced thrombocytopenia. In: Warkentin TE, Greinacher A, editors. Heparin-Induced Thrombocytopenia: Third Edition, Revised and Expanded. Marcel Dekker, Inc; 2004. pp. 223–250. [Google Scholar]
- 19.Stefanini L, Boulaftali Y, Ouellette TD, Holinstat M, Désiré L, Leblond B, Andre P, Conley PB, Bergmeier W. Rap1-Rac1 circuits potentiate platelet activation. Arterioscler Thromb Vasc Biol. 2012;32:434–41. doi: 10.1161/ATVBAHA.111.239194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bergmeier W, Stefanini L. Platelet ITAM signaling. Curr Opin Hematol. 2013;20:445–50. doi: 10.1097/MOH.0b013e3283642267. [DOI] [PubMed] [Google Scholar]
- 21.Stolla M, Stefanini L, Roden RC, Chavez M, Hirsch J, Greene T, Ouellette TD, Maloney SF, Diamond SL, Poncz M, Woulfe DS, Bergmeier W. The kinetics of αIIbβ3 activation determines the size and stability of thrombi in mice: implications for antiplatelet therapy. Blood. 2011;117:1005–13. doi: 10.1182/blood-2010-07-297713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bergmeier W, Schulte V, Brockhoff G, Bier U, Zirngibl H, Nieswandt B. Flow cytometric detection of activated mouse integrin αIIbβ3 with a novel monoclonal antibody. Cytometry. 2002;48:80–6. doi: 10.1002/cyto.10114. [DOI] [PubMed] [Google Scholar]